 Anh T. N. Nguyen
Anh T. N. Nguyen Quan L. Tran1
Quan L. Tran1 Samantha M. McNeill
Samantha M. McNeill Lauren T. May
Lauren T. May- 1Drug Discovery Biology, Monash Institute of Pharmaceutical Sciences, Monash University, Parkville, VIC, Australia
- 2Department of Information Technology, Faculty of Engineering and Technology, Vietnam National University, Hanoi, Vietnam
G protein-coupled receptors (GPCRs) represent the target for approximately a third of FDA-approved small molecule drugs. The adenosine A1 receptor (A1R), one of four adenosine GPCR subtypes, has important (patho)physiological roles in humans. A1R has well-established roles in the regulation of the cardiovascular and nervous systems, where it has been identified as a potential therapeutic target for a number of conditions, including cardiac ischemia-reperfusion injury, cognition, epilepsy, and neuropathic pain. A1R small molecule drugs, typically orthosteric ligands, have undergone clinical trials. To date, none have progressed into the clinic, predominantly due to dose-limiting unwanted effects. The development of A1R allosteric modulators that target a topographically distinct binding site represent a promising approach to overcome current limitations. Pharmacological parameters of allosteric ligands, including affinity, efficacy and cooperativity, can be optimized to regulate A1R activity with high subtype, spatial and temporal selectivity. This review aims to offer insights into the A1R as a potential therapeutic target and highlight recent advances in the structural understanding of A1R allosteric modulation.
Introduction
The adenosine A1 receptor (A1R) belongs to the adenosine family of G protein-coupled receptors (GPCRs). The A1R is highly expressed in the central nervous system (CNS), particularly in the cerebral cortex, cerebellum, basal ganglia, thalamus, hypothalamus, midbrain, pons, medulla oblongata, hippocampal formation, spinal cord, white matter, and amygdala (1–4). In the periphery, A1R has moderate expression in the heart (5), parathyroid gland, salivary gland, pancreas, kidney, testis, placenta, and spleen (2, 6–9).
A1R regulate numerous physiological events. A1R activation reduces heart rate and contractility and confers potent cardioprotection following ischemia-reperfusion injury (10, 11). A1R inhibit presynaptic neurotransmitter release and induce neuronal hyperpolarisation at postsynaptic terminals (12). Additional physiological functions influenced by A1R activation include sleep regulation, inhibition of insulin release, and reducing renal blood flow (13–16). This review aims to offer insights into the importance of the A1R as a therapeutic target and will address recent advances in A1R allosteric ligands as a promising mechanism to selectively modulate A1R activity with spatial and temporal selectivity.
Endogenous adenosine
Endogenous adenosine is a ubiquitous signaling molecule, acting via both autocrine and paracrine mechanisms to modulate physiology. Adenosine concentrations are largely maintained by dynamic but tightly regulated enzymatic processes that drive its formation, degradation, and transport. In the extracellular space, hydrolysis of adenine nucleotides can increase local adenosine concentrations. The enzyme CD39 (also known as ectonucleoside triphosphate diphosphohydrolase) converts extracellular ATP and ADP to form AMP, which can then be hydrolyzed by the enzyme CD73 (ecto-5′-nucleotidase) to form adenosine (17–19). Extracellular adenosine degradation involves the deamination of adenosine to inosine by adenosine deaminase (20). Intracellular adenosine can be produced through the conversion of AMP to adenosine by intracellular nucleotidases, primarily 5’-nucelotidases (21), or through the hydrolysis of S-adenosylhomocysteine (SAH) to adenosine and homocysteine by SAH-hydrolase (22). Intracellular adenosine degradation involves the conversion of adenosine to AMP by adenosine kinase (23). Throughout the body, the presence of bidirectional equilibrative nucleoside transporters 1 and 2 (ENT 1 & 2) preserve the concentration of adenosine both intra- and extracellularly via facilitated diffusion (24, 25). High adenosine concentrations can be maintained against a concentration gradient by unidirectional concentrative nucleoside transporters 1 and 2 (26).
Overall, these biochemical processes maintain the extracellular concentration of adenosine in a reported range of approximately 20-300 nM (27–29) However, in conditions associated with limited oxygen availability, including hypoxia, ischemia, cellular stress and damage, the local concentration of extracellular adenosine can dramatically rise to micromolar concentrations (30–33). As such, while concentrations of extracellular adenosine are tightly regulated under physiological conditions, coordinated mechanisms facilitate sensitive and dynamic responses under disease conditions.
A1 receptor: a vital GPCR in health and disease
A1R typically modulates physiological effects through coupling to Gi/o proteins (34). Cardiac effects of A1R activation include a reduction in heart rate, through direct Gβγ-coupling to inwardly rectifying potassium channels or Gαi/o inhibition of β-adrenoceptor-mediated cAMP production, antiarrhythmic effects, and atrial contractility (35, 36). Adenosine is used in the clinic to treat supraventricular tachycardia (37). Furthermore, A1R activation during ischemia-reperfusion injury promotes significant cardioprotection through stimulation of cardioprotective pathways, such as the reperfusion injury salvage kinase pathway (38–42). Cardiac overexpression of A1R in transgenic mice has been shown to confer significant protection against ischemia-reperfusion injury, an effect that was suggested to be mediated by mitochondrial channel activation (31, 43–45). The challenge for the development of cardioprotective agents that enhance A1R activity remains the need to separate the cardioprotective signaling from unwanted hemodynamic effects. Adenosine and the non-selective agonist AMP579 entered clinical trials to treat myocardial infarction (46–48). Unfortunately, the agonist doses administered in these trials were limited to avoid on-target adverse effects. Neladenoson, an A1R partial agonist, was evaluated in heart failure patients with reduced ejection fraction (HFrEF) or preserved ejection fraction (HFpEF) (49, 50). Neither clinical trial identified a significant drug effect on the primary endpoint, likely due to the partial agonism of neladenoson. However, recent advances in understanding GPCR biology, such as biased agonism and allostery, provide the opportunity to fine-tune receptor activity to overcome these limitations (11, 51).
Neuronal A1R activation can induce hyperpolarization and inhibition of neurotransmitter release, dampening neuronal excitability and promoting neuroprotection (3, 52). The A1R also plays an important role in cognition and can regulate sleep (16). Systemic administration of adenosine in animals reduces pain sensations through adenosine receptor stimulation, particularly A1R (53, 54). Enhanced A1R activation in the spinal cord of rats with neuropathic pain conferred significant analgesic effects (54, 55), likely through modulation of potassium channels (56) that reduced neuronal activity in sensory nerve terminals (57). An A1R positive allosteric modulator, T-62, was used in clinical trial to treat pain associated with postherpetic neuralgia (ClinicalTrials.gov Identifier: NCT00809679). Unfortunately, this trial was terminated due to a subset of patients having asymptomatic, transient elevations in liver transaminases. This trial highlighted the requirement for more efficacious and tailored modulation of A1R activity, alongside a detailed mechanistic understanding of the drug-receptor-effector interactions, to facilitate clinical translation.
Additional peripheral effects of the A1R include decreased lipolysis and increased glucose uptake in adipocytes, reduced renal blood flow and tubuloglomerular feedback, inhibition of renin release, increased sodium and water reabsorption, and inhibition of insulin and glucagon release (58–66). Given the role of the A1R in the renal system, it has been suggested that the A1R may represent a therapeutic target, as its stimulation can protect against acute renal ischemia-reperfusion injury (67), whilst A1R antagonism may be useful in treating acute renal disorders in patients with congestive heart failure (68–70). Clinical trials assessed whether A1R inhibition can improve renal function in heart failure patients and airway hyperreactivity and late allergic response in asthmatics (71, 72). The A1R antagonist rolofylline was evaluated in a large multicenter, double-blind, placebo-controlled Phase III clinical trial in acute heart failure patients to prevent the commonly associated deterioration of renal function. The trial was conducted following positive results from smaller trials that indicated A1R antagonism increased the glomerular filtration rate and urine output (73). However, the Phase III trial observed no benefit of rolofylline with respect to the primary clinical composite endpoint or renal function (74). Moreover, rolofylline was associated with an increased incidence of seizure, an effect attributed to the A1R antagonism.
A1R activation has been suggested to promote inflammation, bronchoconstriction, and mucous secretion (75). Thus, inhibition of the A1R may be useful in patients with asthma (76). Promising results have been presented for Phase IIa trials in mild-to-moderate atopic asthmatics, where the ability of the orally available A1R antagonist PBF-680 to inhibit the late allergic response or adenosine monophosphate airway hyperresponsiveness was assessed (71, 72). Recruitment is ongoing for another Phase IIa trial of PBF-680 in patients with chronic obstructive pulmonary disease (ClinicalTrials.gov Identifier: NCT05262218). As such, the A1R remains a promising novel therapeutic target for several respiratory diseases.
Clearly, A1R represents a promising therapeutic target for the treatment of major global health burdens. However, the transition of A1R small molecule agonists and antagonists into the clinic has thus far failed. Common challenges of A1R drug discovery include the design of highly subtype-selective molecules due to conservation in the adenosine-binding pocket across the adenosine receptor family and modulation of A1R signaling beyond the scope of the desired effect (10, 11, 54, 77, 78). Unwanted outcomes of A1R activation can include bradycardia, atrioventricular block, a reduction in atrial contractility and sedation, whereas over-inhibition of A1R can increase seizure liability (79, 80). Such on-target unwanted effects are typically associated with targeting the A1R orthosteric binding site. In contrast, drugs targeting the allosteric binding site can be tailored for an optimal therapeutic profile, providing exquisite selectivity of effect, acting to “fine-tune” endogenous agonist tone in a tissue- and disease-specific manner.
Allosteric modulation to “tailor” A1R activity with tissue- & disease-specificity
A1R possess allosteric sites that are spatially distinct from the adenosine-binding pocket, known as the orthosteric site (81, 82). Allosteric ligands can influence the binding and/or function of orthosteric ligands. Positive allosteric modulators (PAMs) enhance, whereas negative allosteric modulators (NAMs) inhibit, the affinity and/or efficacy of an endogenous ligand for its cognate receptor. Neutral allosteric ligands (NAL) have neutral cooperativity with the orthosteric ligand. Allosteric ligands have the capacity to stabilize the active or inactive receptor conformation thereby having intrinsic efficacy in the absence of orthosteric ligand, adding an additional level of texture to their profile (83).
The A1R was the first GPCR for which a PAM was identified. This seminal discovery identified a series of 2-amino-3-benzoylthiophenes, including the well characterized A1R PAM, PD 81,723 (1; 2-amino-4,5-dimethyl-3-thienyl-[3-(trifluoromethyl)phenyl]methanone) (Figure 1) (84, 84). Pharmacological studies demonstrated that several 2-amino-3-benzoylthiophenes had an allosteric mechanism of action at the A1R, decreasing the rate of agonist dissociation and enhancing agonist binding and function. Subsequent studies have demonstrated that A1R PAMs enhance the actions of endogenous adenosine in the heart, including cardioprotection (85–87) and atrioventricular nodal function (88, 89). A1R PAMs have also been shown to promote anti-nociception, selectively reducing hypersensitivity (54, 55, 90, 91). Recent breakthroughs in the structural understanding of A1R PAMs provide the opportunity to optimize PAM design to enhance allosteric enhancer potency whilst removing the adverse effects observed for T-62 (54). As such, the rational design of allosteric modulators will facilitate the future therapeutic translation of this class of small molecules.
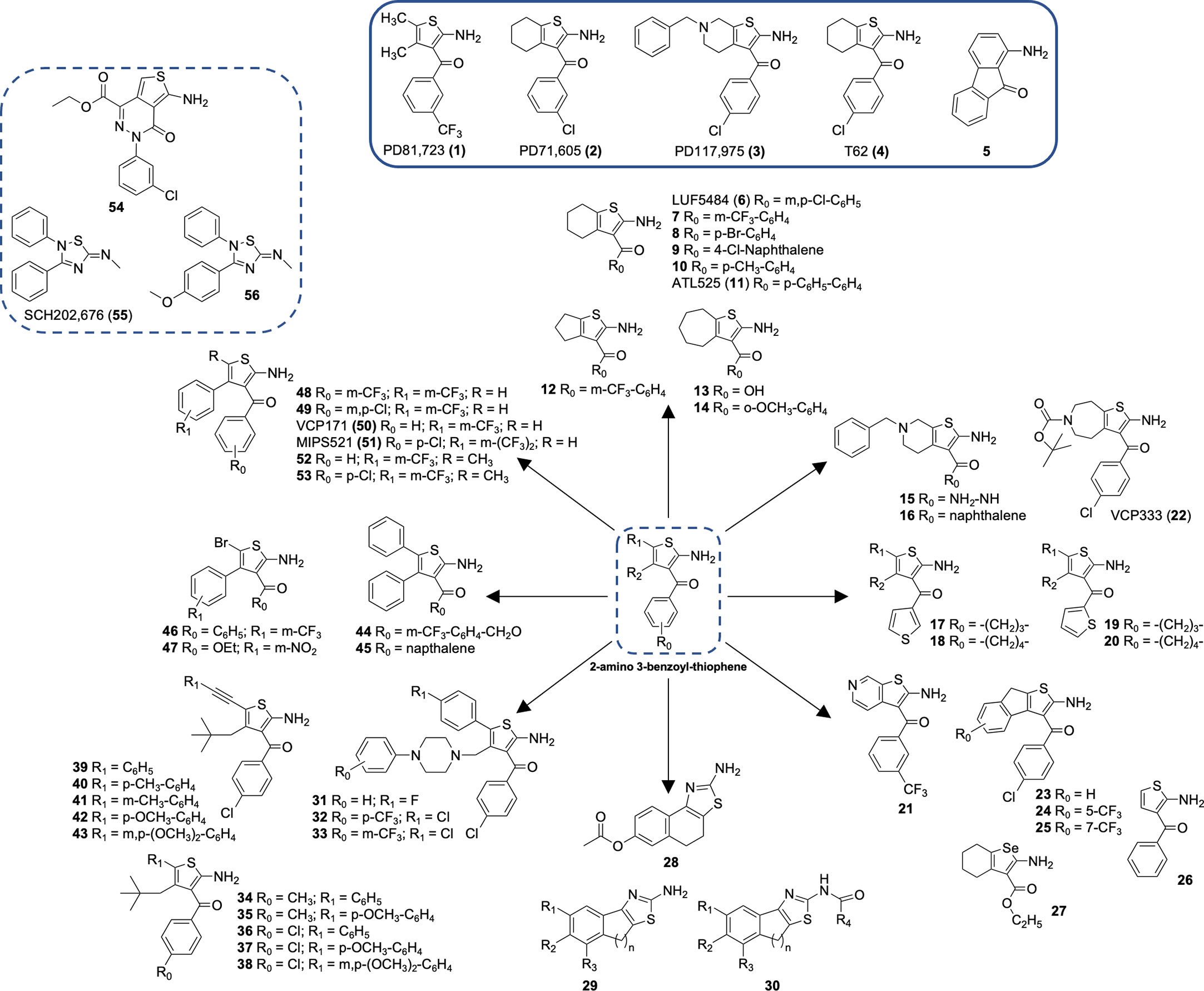
Figure 1 A select subset of adenosine A1 receptor allosteric modulators.
Advantages of allosteric modulation
A1R allosteric binding sites provide a mechanism to target the A1R with subtype, spatial, and temporal selectivity. As such, A1R allostery is a promising approach to overcome current therapeutic limitations of orthosteric ligands. Allosteric ligands can display high subtype selectivity as a direct consequence of their allosteric nature. Allosteric sites are spatially distinct from the orthosteric site, which typically exhibit significant sequence conservation across receptor subtypes due to evolutionary pressure (81, 92, 93). Allosteric sites typically show greater sequence divergence compared to orthosteric sites, facilitating the design of small molecules with high subtype-selectivity and, as such, minimizing the potential for off-target side effects.
The reciprocal nature of the binding cooperativity enables allosteric modulators to “sense” the (patho)physiological concentration of the endogenous agonist within a specific tissue (81, 94). A1R allosteric modulators will predominantly exert their effects at the time and location of significant cytoprotective adenosine release and have little effect in the presence of a low adenosine concentration (95). This feature is particularly promising for the development A1R PAMs that modulate receptor activity with tissue and disease-specificity, as the local adenosine concentration has been shown to increase significantly for several disease conditions associated with cellular stress or hypoxia (96, 97). Under these conditions, PAMs will primarily act at the site of injury, avoiding the classical on-target systemic side effects associated with prototypical A1R agonists.
The influence of an allosteric modulator on orthosteric ligand binding is saturable, a feature that contrasts competitive interactions. The influence of an allosteric modulator on orthosteric ligand affinity is defined by the cooperativity between the allosteric and orthosteric sites when both ligands are co-bound to the receptor. This property enables allosteric modulators to ‘fine-tune’ orthosteric ligand activity by scaling up or down the binding cooperativity as appropriate whilst avoiding the over-stimulation or complete inhibition often observed with orthosteric agonists and antagonists, respectively (81).
Advantages of allosteric ligands that deserve greater exploration at the A1R include the ability of allosteric ligands to promote biased signaling and regulate receptor trafficking from the allosteric site. Allosteric ligands may stabilize unique receptor conformations, influencing the spectrum of pathways stimulated or inhibited by an orthosteric ligand. Indeed, A1R PAMs have been shown to stimulate biased agonism in the absence of an orthosteric ligand or mediate biased allosteric modulation of an orthosteric agonist (98). Although relatively unexplored, A1R PAMs have been suggested to cause less receptor desensitization compared to A1R orthosteric agonists, an effect that may be therapeutically beneficial (99). Such features of A1R allosteric ligands require further in-depth exploration due to potential therapeutic implications.
Structure-activity relationship of A1R Allosteric Modulators
2-amino 3-benzoylthiophenes as A1R PAMs
A1R is the first GPCR for which the allosteric modulators were identified in the 1990s (84) The first representatives were three compounds PD81723 (1), PD71605 (2), and PD117975 (3). Since then, benzoylthiophene derivatives have been intensively investigated (Table 1, Figure 1) for their allosteric mechanism of action, typically slowing the rate of orthosteric agonist dissociation and enhancing agonist activity in functional assays (84). A1R PAM 2-amino-3-benzoylthiophene (2A3BT) scaffolds, represented by PD81723 (1), typically display significant A1R selectivity and the best ratio of enhancement to inhibition (100, 120). The extension of this family, followed by additional analogs represented by T62 (4), had substituents in 4- and 5-positions in the thiophene moiety bridged by a methylene chain (100, 120, 121). However, these compounds were inhibitors at higher concentrations and displayed significant intrinsic activity by causing a functional response in the absence of an agonist.
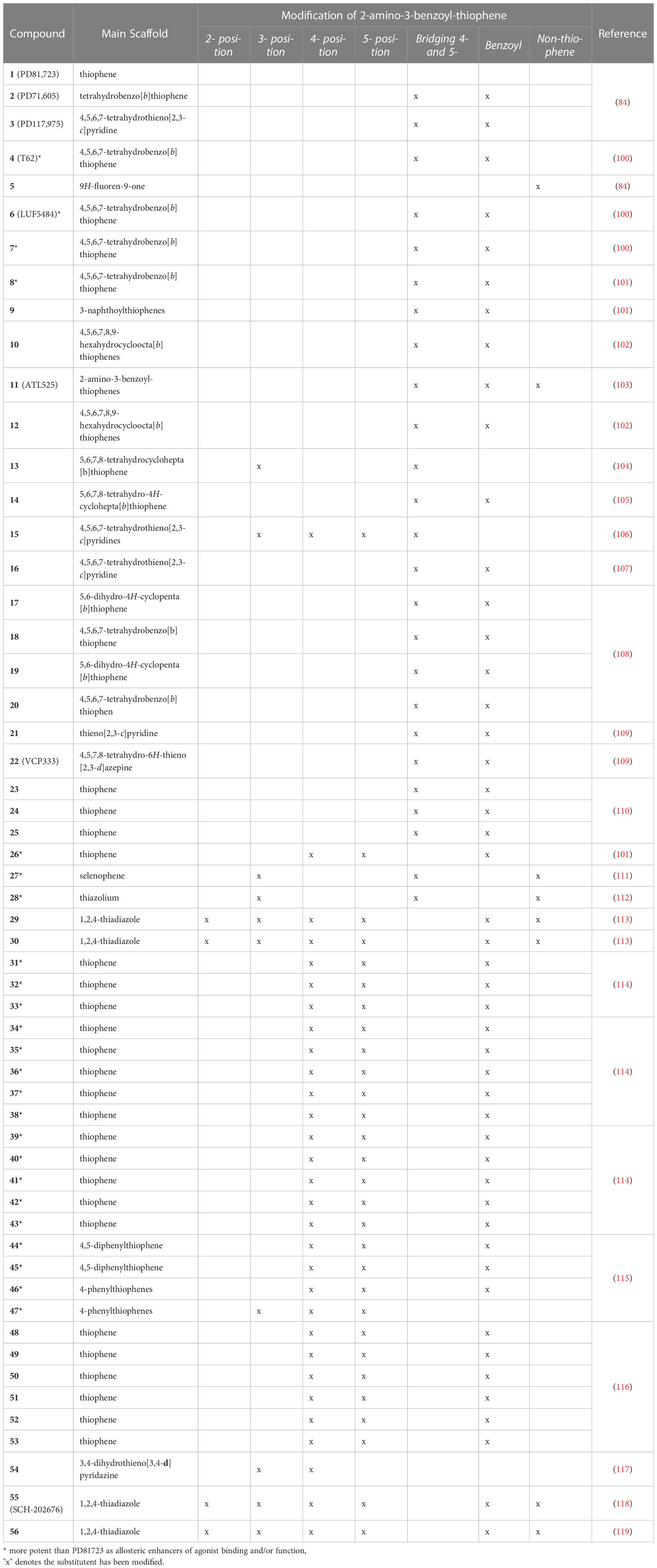
Table 1 An overview of structural modifications of A1R allosteric modulators.
SAR studies on the 2A3BT scaffold have identified general structural requirements for the PAM effects (122). The omission of the 2-amino and 3-keto groups of the 2-aminothiophene core resulted in a notable decrease of allosteric enhancer (AE) activity, highlighting the importance of these groups for PAM activity. An intramolecular hydrogen bond between these two groups is proposed to create an additional coplanar ring with the thiophene ring in the active conformation of the PAMs. Supporting this hypothesis, 1-aminofluoren-9-one (5), which conformationally locked the amino and keto groups with a hydrogen bond, was reported to have good AE activity (122). 4- and 5- substituents in 2A3BT, such as large hydrophobic alkyl to aryl groups, have been shown to increase the AE activity of 2A3BT derivatives (116), suggesting the inclusion of such substituents in the design of more potent A1R PAMs.
SAR of the thiophene ring of A1R PAMs
The thiophene ring in the PAM scaffold with substituents in the 4- and 5-position plays an important role in conferring allosteric activity. Increased A1R AE activity was observed for compounds with a long carbon chain bridging the 4- and 5-positions, such as LUF 5484 (6), 7-20, or 21-25 (98, 100, 102, 104, 105, 107–110). The optimal size of the 4,5-cycloalkyl ring varied from five to seven-membered rings to maintain PAM activity measured by in vitro dissociation kinetic binding assays (105). Compound 26, without 4- and 5-substituents in the thiophene ring, displayed lower allosteric agonism than PD81723 (1) and increased antagonism (101).
Selenophenes and thiazoles have been characterized as A1R PAMs. Preliminary data for 2-aminoselenophene-3-carboxylate (27) suggested superior A1R AE activity compared to PD81723 (1), although the compound was unstable under mildly acidic conditions (111). Introduction of a nitrogen atom into the thiophene ring was also shown to retain A1R PAM activity, as demonstrated by the synthesis of a series of 2-aminothiazolium salts, which identified 28 as a relatively potent and efficacious A1R PAM (112). However, synthesis of another series of 2-aminothiazoles (29, 30) did not display A1R PAM activity (113). In fact, several 2-aminothiazoles inhibited A1R activity.
The presence of hydrophobic groups at the 4-position of thiophene ring, such as arylpiperazine moieties (31-33) or neopentyl (34-43), with 39-43 having an addition (hetero)aryl moiety in the 5-position of the thiophene ring, are suggested to be important for hydrophobic interactions within the A1R allosteric binding site (123–125). Such compounds had superior AE potency compared to the well-studied PD81723 (1). Furthermore, the two-carbon linker between hetero aryl or akyl moiety at the 5-position of thiophene ring (39-43) allowed for exploration of the hydrophobic domains within the A1R allosteric pocket (125).
SAR of 3-benzoyl moiety of A1R PAMs
Numerous modifications in the 3-benzoyl ring of the original PD81723 (1) scaffold have been investigated, some of which are well tolerated. For example, substitutions of the 3-benzoyl moiety as trifluoromethyl in PD81723 (1); chloro in LUF5484 (6), or phenyl in ATL525 (11) have been suggested to improve the magnitude of positive allosteric modulation. 3-benzoyl moiety modification combined with the introduction of large hydrophobic domains in the 4-position of the thiophene ring enhanced compound AE activity, suggesting the A1R allosteric pocket can accommodate a range of substituents.
Substitution of the 3-benzoyl group by a 3-carboxylate (13) or 3-hydrazide (15) can maintain A1R PAM activity comparable to PD81723 (1), although these compounds were reported as a novel class of A1R antagonists that recognized the allosteric site at higher concentrations (105, 107). Replacement of the phenyl ring at the 3-position with a naphthoyl (9, 16, 30) (107, 115) or unsubstituted thienyl ring (17-20) (108) was well tolerated with a number of derivatives favoring A1R PAM activity. 2-aminothiophene with a carboxylic acid (13) or ester (27, 47) substituents in the 3-position or incorporation of phenyl groups in the 4- and 5-position retained A1R AE activity in functional assays (104, 111, 115). Only continuous replacement of 3-benzoyl moiety with a benzyl-ester (44) or 1-naphthoyl group (45); or the addition of a halo substituent (Br) in the 5-position of the thiophene (46, 47) significantly improved the A1R enhancer effect (115).
In addition, the nature of the 3-benzoyl moiety has been shown to influence A1R PAM bias signaling (116). Among the evaluated series of potent A1R PAMs (48-53), MIPS521 (51) displayed high AE activity alongside considerable allosteric agonism (116). Compared to VCP171 (50), which induced weak analgesic effects in rats (55), MIPS521 (51) stimulated significant analgesia efficacy in an in-vivo rat model, in the absence of notable side effects (54). Structural derivatives of 51, specifically 52 and 53, displayed biased profiles (116). The structure of 52 and 53 differed only by the presence of one electron-withdrawing group (p-chloro) on the 3-benzoyl ring. However, they displayed distinct A1R signaling profiles in functional assays, with 52 behaving as a biased allosteric agonist and 53 conferring biased allosteric modulation (116)
Interestingly, a PD81723 (1) derivative with bridged 3- and 4- positions displayed A1R antagonism at low concentrations, but PAM activity at higher concentrations (54) (117). Similarly, the scaffold 2-amino-4,5,6,7-tetrahydrothieno[2,3-c]pyridines scaffold with a carbohydrazide substitution at the 3- position (15) was reported as a novel class of A1R antagonists that recognized the allosteric site at relatively high concentrations (106).
Thiadiazole compounds
A thiadiazole compound, SCH-202676 (55) (N-(2,3-diphenyl-[1,2,4]-thiadiazol-5-(2H)-ylidene)-methanamine) was reported to act as a non-selective allosteric inhibitor of several Family A GPCRs, including A1R (118). Synthesis of 2,3,5-substituted [1,2,4]-thiadiazole analogues of SCH-202676 identified (56), which appeared to act as an allosteric inhibitor of agonist binding (119). However, subsequent studies suggested that these compounds acted as non-selective protein modifiers, with their GPCR effects resulting from sulfhydryl modification. Some amiloride analogs have also been reported to act as A1R negative allosteric modulators (NAMs), increasing the dissociation rate of the antagonist [3H]DPCPX without affecting the dissociation rate of [3H]R-PIA (126, 127).
A1R allosteric binding site
Mutagenesis and computational modeling studies
Prior to the determination of a high-resolution structure of A1R bound to an allosteric modulator, efforts have been made to map the location of the A1R allosteric site using indirect approaches such as mutagenesis and computational modeling. Several structure-function studies have identified important residues for A1R allosteric modulation, with the second extracellular loop (ECL2) playing a crucial role (128–130). Alanine substitution of residue E172ECL2 caused a significant decrease in the binding affinity for the unoccupied A1R of two allosteric modulators, PD81723 and VCP171 (130). This study also predicted that residues involved in allosteric ligand intrinsic efficacy were relatively conserved between the two modulators and the hydrogen-bonding networks within A1R extracellular vestibule may facilitate the transmission of cooperativity between orthosteric and allosteric sites.
A1R X-ray crystallography and cryo-electron microscopy structures
Inactive and active state A1R structures have been solved, providing important insights into A1R ligand binding and activation. A1R structures in complex with the orthosteric antagonists DU172 and PSB36 (PDB: 5UEN and 5N2S, respectively) (77, 131) or the endogenous agonist, adenosine (PDB: 6D9H) (78) revealed common interaction with conserved residues F171ECL2 (via a π- π stacking interaction) and N2546.55 (via a double hydrogen bond) superscript denoting Ballesteros-Weinstein residue numbering (132). In both inactive and active structures, A1R ECL2 was found to adopt a unique conformation compared to its relative A2AR with a longer helix and almost perpendicular to the plane of the membrane. The inactive A1R is characterized by a wider extracellular vestibule (compared to the A2AR structures) that may hold an orthosteric and allosteric site. Residue T2707.35 within this pocket was found to be responsible for the selectivity of the antagonist DU272 (77). Due to the movement of the transmembrane domain 1 and 2, the orthosteric binding site of the active A1R was found to have a contraction on the extracellular surface compared to the inactive A1R (78). An all-atom Gaussian accelerated molecular dynamics (GaMD) simulation was performed using A1R inactive structure (PDB: 5UEN) to predict the binding modes of two A1R PAMs, PD71723 1 and VCP171 27 (133). The simulation further supported the role of residue E172ECL2 as a key binding determinant of the two PAMs and showed that the presence of PAMs stabilized the bound agonist within the transmembrane bundle.
Most recently, the cryoEM structure of A1R-Gi2 complex co-bound with adenosine and the PAM (MIPS521; 51) was solved (PDB: 7LD3) (54). MIPS521 bound to a unique, extrahelical lipid-facing pocket, harbored by hydrogen bonds between its 2-amino substituent of the thiophene ring and residues in TM6 and TM7 (S2466.47 and L2767.41). The binding site was well supported by site-directed mutagenesis studies where mutation of these two residues to alanine significantly decreased MIPS521 binding affinity, owing to the loss of a hydrogen bond with the 2-amino group (54). All-atom GaMD simulations in combination with deep learning and free energy profiling workflow (GLOW) of the active A1R structure co-bound to adenosine in the presence or absence of MIPS521 were employed to determine conformational changes mediated by A1R activation and allosteric modulation (134). Interestingly, the study confirmed that ECL2 has a significant impact on A1R allosteric modulation.
Future applications of computational advances in the development of A1R allosteric ligands as novel therapeutics
Artificial intelligence to facilitate the development of novel selective and drug like A1R PAMs
Artificial intelligence (AI), including machine learning and deep learning methods, are widely used in GPCR drug discovery. In 2021, 34% of publications related to GPCR drug discovery mentioned AI, and the number increased to more than 40% in 2022 based on Google Scholar search results. AI applications vary from simply detecting properties and outcomes (e.g., active/inactive states) to recognizing the characteristics and patterns of activity (e.g., finding functional sites) and generation of de novo drug candidates. Predicting properties is often done via classification models using traditional machine learning methods or deep learning techniques like convolutional neural network (CNN) and recurrent neural network (RNN) (134, 135). Generative models, either trained via deep learning or reinforcement learning, are the most advanced in the field (136–141). They can be used not only to generate de novo structures (i.e., design new drug candidates) but also to predict the properties and inspect the functional sites of a structure. In these generative methods, the chemical space of small molecules is constructed and explored iteratively under constrained objectives, namely desired 3D structures or desired functions.
Multistage virtual screening
Compared with the traditional experimental high-throughput screening, virtual screening (structure-based or ligand-based approaches) has emerged as a more direct and rational drug discovery approach to screen large libraries of chemical structures. Approximately 60 GPCR structures with small-molecule allosteric ligands have been determined using X-ray crystallography or cryo-EM (142, 143). GPCR allosteric sites have been identified in a range of locations, including within the seven transmembrane domains (corticotropin-releasing factor receptor 1, metabotropic glutamate receptors 1,2, and 5, smoothened receptor, luteinizing hormone-choriogonadotropin receptor, calcium-sensing receptor), within the extracellular vestibule (M2 and M4 muscarinic acetylcholine receptor, protease-activated receptor-2), outside the seven-transmembrane domain (C5a anaphylatoxin chemotactic receptor 1, GPR40 receptor, P2Y1 receptor, glucagon receptor, protease-activated receptor-2, glucagon-like peptide-1 receptor, adenosine A1 receptor, cannabinoid CB1 receptor), overlapping with cholesterol-binding sites (bile acid receptor, D1 dopamine receptor, glucagon-like peptide 1 receptor), or on intracellular surface (β2 adrenergic receptor, CC chemokine receptor 2 and 9) (142, 143). Such structural determination has provided key insights into allosteric binding across various GPCRs, which in turns aids the discovery of new allosteric modulators using the structure-based drug discovery (SBDD) approach. Several SBDD studies have successfully employed to find new allosteric modulators for GPCRs, including M2 muscarinic acetylcholine receptors (144, 145), glucagon-like peptide 1 receptor (146–148), metabotropic glutamate receptor 5 (149), and proton-sensing receptors GPR68 and GPR65 (150). These studies demonstrate the power of SBDD to identify new allosteric modulators for GPCRs, paving the way for the development of novel therapeutics with improved selectivity and reduced side effects.
The recent breakthroughs in solving A1R structures have enriched our understanding of A1R ligand binding and signal transduction and offered a great opportunity to leverage SBDD approaches. The A1R structure co-bound with a PAM and agonist has provided significant structural detail, which will facilitate the design of novel A1R allosteric modulators as new therapeutic agents. Molecular dynamics (MD) simulations and site-directed mutagenesis can enhance our understanding of the activation mechanism and generate multiple conformations of dynamic receptors co-bound with orthosteric and allosteric ligands. The rapid development of pharmacophore and molecular docking methods also enabled the screening of very large databases from millions to billions of molecules via ligand-based drug discovery (LBDD) approaches. Combining SBDD and LBDD, the multistage virtual screening approaches are now possible for drug discovery (135).
Multistage methods can be designed as a workflow of several filters against large databases. Workflows have been developed to use deep learning to classify A1/A2AR antagonists among ChemDiv database, then pharmacophore modeling and docking for additional filtering, and finally MD to observe drug-receptor interactions to support compound activity (135). Alternative workflows can be designed using MD to gain insights into drug candidate interactions at A1R, then applying pharmacophore modeling to predict key ligand features and screening of small molecule databases using molecular docking. A different workflow could first integrate a generative deep learning method to obtain ligands or drug candidates by exploring chemical space, then apply a permutation of virtual screening methods and property prediction methods to further filter through the candidate sets. The profound understanding of A1R structures, SAR of A1R PAMs, alongside the growing accessibility of data sources, public AI tools, and source codes, have paved the way for the imminent discovery of novel A1R PAMs.
Author contributions
AN and LM designed the structure of the manuscript. AN, QT prepared Figure 1 and Table 1. All authors contributed to writing and reviewing the article and approved the submitted version.
Funding
Research on adenosine receptor signaling in Dr May’s lab was supported by a National Heart Foundation Future Leader Fellowship (101857) and National Health and Medical Research Council (Australia) project grant: APP1147291. Ongoing research into adenosine receptor signaling is supported by NHMRC Ideas grant APP2013629 and a Department of Health and Aged Care (MRFF) Stem Cell Therapies Mission grant (MRF2015957).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Chen J-F, Eltzschig HK, Fredholm BB. Adenosine receptors as drug targets–what are the challenges? Nat Rev Drug Discov (2013) 12:265–86. doi: 10.1038/nrd3955
2. Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Tissue-based map of the human proteome. Science (2015) 347:1260419. doi: 10.1126/science.1260419
3. Ballesteros-Yáñez I, Castillo CA, Merighi S, Gessi S. The role of adenosine receptors in psychostimulant addiction. Front Pharmacol (2018) 8:985. doi: 10.3389/fphar.2017.00985
4. Peleli M, Fredholm BB, Sobrevia L, Carlström M. Pharmacological targeting of adenosine receptor signaling. Mol Aspects Med (2017) 55:4–8. doi: 10.1016/j.mam.2016.12.002
5. Headrick JP, Ashton KJ, Rose’Meyer RB, Peart JN. Cardiovascular adenosine receptors: expression, actions and interactions. Pharmacol Ther (2013) 140:92–111. doi: 10.1016/j.pharmthera.2013.06.002
6. Sun C-X, Young HW, Molina JG, Volmer JB, Schnermann J, Blackburn MR. A protective role for the A1 adenosine receptor in adenosine-dependent pulmonary injury. J Clin Invest (2005) 115:35–43. doi: 10.1172/jci22656
7. Sachdeva S, Gupta M. Adenosine and its receptors as therapeutic targets: an overview. Saudi Pharm J (2013) 21:245–53. doi: 10.1016/j.jsps.2012.05.011
8. Boros D, Thompson J, Larson DF. Adenosine regulation of the immune response initiated by ischemia reperfusion injury. Perfusion (2016) 31:103–10. doi: 10.1177/0267659115586579
9. Soni H, Peixoto-Neves D, Buddington RK, Adebiyi A. Adenosine A1 receptor-operated calcium entry in renal afferent arterioles is dependent on postnatal maturation of TRPC3 channels. Am J Physiol Renal Physiol (2017) 313:F1216–22. doi: 10.1152/ajprenal.00335.2017
10. Valant C, May LT, Aurelio L, Chuo CH, White PJ, Baltos J-A, et al. Separation of on-target efficacy from adverse effects through rational design of a bitopic adenosine receptor agonist. Proc Natl Acad Sci (2014) 111:4614–9. doi: 10.1073/pnas.1320962111
11. McNeill SM, Baltos J-A, White PJ, May LT. Biased agonism at adenosine receptors. Cell Signal (2021) 82:109954. doi: 10.1016/j.cellsig.2021.109954
12. Borea PA, Gessi S, Merighi S, Varani K. Adenosine as a multi-signalling guardian angel in human diseases: when, where and how does it exert its protective effects? Trends Pharmacol Sci (2016) 37:419–34. doi: 10.1016/j.tips.2016.02.006
13. Hillaire-Buys D, Bertrand G, Gross R, Loubatières-Mariani M-M. Evidence for an inhibitory A1 subtype adenosine receptor on pancreatic insulin-secreting cells. Eur J Pharmacol (1987) 136:109–12. doi: 10.1016/0014-2999(87)90786-2
14. Bertrand G, Petit P, Bozem M, Henquin JC. Membrane and intracellular effects of adenosine in mouse pancreatic beta-cells. Am J Physiol (1989) 257:E473–8. doi: 10.1152/ajpendo.1989.257.4.e473
15. Just A, Arendshorst WJ. A novel mechanism of renal blood flow autoregulation and the autoregulatory role of A1 adenosine receptors in mice. Am J Physiol-renal (2007) 293:F1489–500. doi: 10.1152/ajprenal.00256.2007
16. Krueger J. The role of cytokines in sleep regulation. Curr Pharm Design (2008) 14:3408–16. doi: 10.2174/138161208786549281
17. Zimmermann H. Extracellular purine metabolism. Drug Dev Res (1996) 39:337–52. doi: 10.1002/(sici)1098-2299(199611/12)39:3/4<337::aid-ddr15>3.0.co;2-z
18. Resta R, Yamashita Y, Thompson LF. Ecto-enzyme and signaling functions of lymphocyte CD73. Immunol Rev (1998) 161:95–109. doi: 10.1111/j.1600-065x.1998.tb01574.x
19. Zimmermann H, Braun N, Kegel B, Heine P. New insights into molecular structure and function of ecto-nucleotidases in the nervous system. Neurochem Int (1998) 32:421–5. doi: 10.1016/s0197-0186(97)00126-5
20. Cristalli G, Costanzi S, Lambertucci C, Lupidi G, Vittori S, Volpini R, et al. Adenosine deaminase: functional implications and different classes of inhibitors. Med Res Rev (2001) 21:105–28. doi: 10.1002/1098-1128(200103)21:2<105::aid-med1002>3.0.co;2-u
21. Sträter N. Ecto-5’-nucleotidase: structure function relationships. Purinerg Signal (2006) 2:343. doi: 10.1007/s11302-006-9000-8
22. Turner MA, Yang X, Yin D, Kuczera K, Borchardt RT, Howell PL. Structure and function of s-adenosylhomocysteine hydrolase. Cell Biochem Biophys (2000) 33:101–25. doi: 10.1385/cbb:33:2:101
23. Park J, Gupta RS. Adenosine kinase and ribokinase – the RK family of proteins. Cell Mol Life Sci (2008) 65:2875–96. doi: 10.1007/s00018-008-8123-1
24. Baldwin SA, Beal PR, Yao SYM, King AE, Cass CE, Young JD. The equilibrative nucleoside transporter family, SLC29. Pflügers Archiv (2004) 447:735–43. doi: 10.1007/s00424-003-1103-2
25. Young JD, Yao SYM, Sun L, Cass CE, Baldwin SA. Human equilibrative nucleoside transporter (ENT) family of nucleoside and nucleobase transporter proteins. Xenobiotica (2008) 38:995–1021. doi: 10.1080/00498250801927427
26. Young JD, Yao SYM, Baldwin JM, Cass CE, Baldwin SA. The human concentrative and equilibrative nucleoside transporter families, SLC28 and SLC29. Mol Aspects Med (2013) 34:529–47. doi: 10.1016/j.mam.2012.05.007
27. Zetterström T, Vernet L, Ungerstedt U, Tossman U, Jonzon B, Fredholm BB. Purine levels in the intact rat brain. studies with an implanted perfused hollow fibre. Neurosci Lett (1982) 29:111–5. doi: 10.1016/0304-3940(82)90338-x
28. Ballarín M, Fredholm BB, Ambrosio S, Mahy N. Extracellular levels of adenosine and its metabolites in the striatum of awake rats: inhibition of uptake and metabolism. Acta Physiol Scand (1991) 142:97–103. doi: 10.1111/j.1748-1716.1991.tb09133.x
29. Pedata F, Corsi C, Melani A, Bordoni F, Latini S. Adenosine extracellular brain concentrations and role of A2A receptors in ischemia. Ann Ny Acad Sci (2001) 939:74–84. doi: 10.1111/j.1749-6632.2001.tb03614.x
30. Blay J, White TD, Hoskin DW. The extracellular fluid of solid carcinomas contains immunosuppressive concentrations of adenosine. Cancer Res (1997) 57:2602–5.
31. Harrison GJ, Willis RJ, Headrick JP. Extracellular adenosine levels and cellular energy metabolism in ischemically preconditioned rat heart. Cardiovasc Res (1998) 40:74–87. doi: 10.1016/s0008-6363(98)00123-0
32. Latini S, Pedata F. Adenosine in the central nervous system: release mechanisms and extracellular concentrations. J Neurochem (2001) 79:463–84. doi: 10.1046/j.1471-4159.2001.00607.x
33. Dworak M, Diel P, Voss S, Hollmann W, Strüder HK. Intense exercise increases adenosine concentrations in rat brain: implications for a homeostatic sleep drive. Neuroscience (2007) 150:789–95. doi: 10.1016/j.neuroscience.2007.09.062
34. Linden J. Adenosine in tissue protection and tissue regeneration. Mol Pharmacol (2005) 67:1385–7. doi: 10.1124/mol.105.011783
35. Fraser H, Gao Z, Ozeck MJ, Belardinelli L. N-[3-(R)-tetrahydrofuranyl]-6-aminopurine riboside, an A1 adenosine receptor agonist, antagonizes catecholamine-induced lipolysis without cardiovascular effects in awake rats. J Pharmacol Exp Ther (2003) 305:225–31. doi: 10.1124/jpet.102.046821
36. Mustafa SJ, Morrison RR, Teng B, Pelleg A. Adenosine receptors and the heart: role in regulation of coronary blood flow and cardiac electrophysiology. Handb Exp Pharmacol (2009) (193):161–88. doi: 10.1007/978-3-540-89615-9_6
37. Jacobson KA, Tosh DK, Jain S, Gao Z-G. Historical and current adenosine receptor agonists in preclinical and clinical development. Front Cell Neurosci (2019) 13:124. doi: 10.3389/fncel.2019.00124
38. Baxter GF, Marber MS, Patel VC, Yellon DM. Adenosine receptor involvement in a delayed phase of myocardial protection 24 hours after ischemic preconditioning. Circulation (1994) 90:2993–3000. doi: 10.1161/01.cir.90.6.2993
39. Dana A, Baxter GF, Walker JM, Yellon DM. Prolonging the delayed phase of myocardial protection: repetitive adenosine A1 receptor activation maintains rabbit myocardium in a preconditioned state. J Am Coll Cardiol (1998) 31:1142–9. doi: 10.1016/S0735-1097(98)00054-0
40. Mozzicato S, Joshi BV, Jacobson KA, Liang BT. Role of direct RhoA-phospholipase D1 interaction in mediating adenosine-induced protection from cardiac ischemia. FASEB J (2004) 18:406–8. doi: 10.1096/fj.03-0592fje
41. Fredholm BB, Chen J-F, Masino SA, Vaugeois J-M. Actions of adenosine at its receptors in the CNS: insights from knockouts and drugs. Annu Rev Pharmacol Toxicol (2005) 45:385–412. doi: 10.1146/annurev.pharmtox.45.120403.095731
42. Hausenloy DJ, Yellon DM. Reperfusion injury salvage kinase signalling: taking a RISK for cardioprotection. Heart Fail Rev (2007) 12:217–34. doi: 10.1007/s10741-007-9026-1
43. Matherne GP, Linden J, Byford AM, Gauthier NS, Headrick JP. Transgenic A1 adenosine receptor overexpression increases myocardial resistance to ischemia. Proc Natl Acad Sci USA (1997) 94:6541–6. doi: 10.1073/pnas.94.12.6541
44. Gauthier NS, Headrick JP, Matherne GP. Myocardial function in the working mouse heart overexpressing cardiac A1 adenosine receptors. J Mol Cell Cardiol (1998) 30:187–93. doi: 10.1006/jmcc.1997.0585
45. Yang Z, Cerniway RJ, Byford AM, Berr SS, French BA, Matherne GP. Cardiac overexpression of A1-adenosine receptor protects intact mice against myocardial infarction. Am J Physiol Heart Circ Physiol (2002) 282:H949–55. doi: 10.1152/ajpheart.00741.2001
46. Mahaffey KW, Puma JA, Barbagelata NA, DiCarli MF, Leesar MA, Browne KF, et al. Adenosine as an adjunct to thrombolytic therapy for acute myocardial infarction results of a multicenter, randomized, placebo-controlled trial: the acute myocardial infarction STudy of ADenosine (AMISTAD) trial. J Am Coll Cardiol (1999) 34:1711–20. doi: 10.1016/s0735-1097(99)00418-0
47. Kopecky SL, Aviles RJ, Bell MR, Lobl JK, Tipping D, Frommell G, et al. A randomized, double-blinded, placebo-controlled, dose-ranging study measuring the effect of an adenosine agonist on infarct size reduction in patients undergoing primary percutaneous transluminal coronary angioplasty (AmP579 delivery for myocardial infarction REduction) study. Am Heart J (2003) 146:146–52. doi: 10.1016/s0002-8703(03)00172-8
48. Ross AM, Gibbons RJ, Stone GW, Kloner RA, Alexander RW, Investigators A-I. A randomized, double-blinded, placebo-controlled multicenter trial of adenosine as an adjunct to reperfusion in the treatment of acute myocardial infarction (AMISTAD-II). J Am Coll Cardiol (2005) 45:1775–80. doi: 10.1016/j.jacc.2005.02.061
49. Shah SJ, Voors AA, McMurray JJV, Kitzman DW, Viethen T, Wirtz AB, et al. Effect of neladenoson bialanate on exercise capacity among patients with heart failure with preserved ejection fraction. Jama (2019) 321:2101–12. doi: 10.1001/jama.2019.6717
50. Voors AA, Bax JJ, Hernandez AF, Wirtz AB, Pap AF, Ferreira AC, et al. Safety and efficacy of the partial adenosine A1 receptor agonist neladenoson bialanate in patients with chronic heart failure with reduced ejection fraction: a phase IIb, randomized, double-blind, placebo-controlled trial. Eur J Heart Fail (2019) 21:1426–33. doi: 10.1002/ejhf.1591
51. Vecchio EA, Baltos J, Nguyen ATN, Christopoulos A, White PJ, May LT. New paradigms in adenosine receptor pharmacology: allostery, oligomerization and biased agonism. Brit J Pharmacol (2018) 175:4036–46. doi: 10.1111/bph.14337
52. Dunwiddie TV, Masino SA. The role and regulation of adenosine in the central nervous system. Annu Rev Neurosci (2001) 24:31–55. doi: 10.1146/annurev.neuro.24.1.31
53. Sawynok J, Liu XJ. Adenosine in the spinal cord and periphery: release and regulation of pain. Prog Neurobiol (2003) 69:313–40. doi: 10.1016/s0301-0082(03)00050-9
54. Draper-Joyce CJ, Bhola R, Wang J, Bhattarai A, Nguyen ATN, Cowie-Kent I, et al. Positive allosteric mechanisms of adenosine A1 receptor-mediated analgesia. Nature (2021) 597:571–6. doi: 10.1038/s41586-021-03897-2
55. Imlach WL, Bhola RF, May LT, Christopoulos A, Christie MJ. A positive allosteric modulator of the adenosine A1 receptor selectively inhibits primary afferent synaptic transmission in a neuropathic pain model. Mol Pharmacol (2015) 88:460–8. doi: 10.1124/mol.115.099499
56. Ocaña M, Cendán CM, Cobos EJ, Entrena JM, Baeyens JM. Potassium channels and pain: present realities and future opportunities. Eur J Pharmacol (2004) 500:203–19. doi: 10.1016/j.ejphar.2004.07.026
57. Zambrowicz BP, Turner CA, Sands AT. Predicting drug efficacy: knockouts model pipeline drugs of the pharmaceutical industry. Curr Opin Pharmacol (2003) 3:563–70. doi: 10.1016/j.coph.2003.04.002
58. Palmer TM, Stiles GL. Adenosine receptors. Neuropharmacology (1995) 34:683–94. doi: 10.1016/0028-3908(95)00044-7
59. Porkka-Heiskanen T, Strecker RE, Thakkar M, Bjorkum AA, Greene RW, McCarley RW. Adenosine: a mediator of the sleep-inducing effects of prolonged wakefulness. Science (1997) 276:1265–8. doi: 10.1126/science.276.5316.1265
60. Haas HL, Selbach O. Functions of neuronal adenosine receptors. Naunyn Schmiedebergs Arch Pharmacol (2000) 362:375–81. doi: 10.1007/s002100000314
61. Brown R, Ollerstam A, Johansson B, Skøtt O, Gebre-Medhin S, Fredholm B, et al. Abolished tubuloglomerular feedback and increased plasma renin in adenosine A1 receptor-deficient mice. Am J Physiol Regul Integr Comp Physiol (2001) 281:R1362–7. doi: 10.1152/ajpregu.2001.281.5.r1362
62. Johansson B, Halldner L, Dunwiddie TV, Masino SA, Poelchen W, Gimenez-Llort L, et al. Hyperalgesia, anxiety, and decreased hypoxic neuroprotection in mice lacking the adenosine A1 receptor. PNAS (2001) 98:9407–12. doi: 10.1073/pnas.161292398
63. Sun D, Samuelson LC, Yang T, Huang Y, Paliege A, Saunders T, et al. Mediation of tubuloglomerular feedback by adenosine: evidence from mice lacking adenosine 1 receptors. PNAS (2001) 98:9983–8. doi: 10.1073/pnas.171317998
64. Yang JN, Tiselius C, Daré E, Johansson B, Valen G, Fredholm BB. Sex differences in mouse heart rate and body temperature and in their regulation by adenosine A1 receptors. Acta Physiol (Oxf) (2007) 190:63–75. doi: 10.1111/j.1365-201x.2007.01690.x
65. Johansson SM, Salehi A, Sandström ME, Westerblad H, Lundquist I, Carlsson P-O, et al. A1 receptor deficiency causes increased insulin and glucagon secretion in mice. Biochem Pharmacol (2007) 74:1628–35. doi: 10.1016/j.bcp.2007.08.006
66. Johansson SM, Yang JN, Lindgren E, Fredholm BB. Eliminating the antilipolytic adenosine A1 receptor does not lead to compensatory changes in the antilipolytic actions of PGE2 and nicotinic acid. Acta Physiol (Oxf) (2007) 190:87–96. doi: 10.1111/j.1365-201x.2007.01692.x
67. Lee HT, Emala CW. Protective effects of renal ischemic preconditioning and adenosine pretreatment: role of A(1) and A(3) receptors. Am J Physiol Renal Physiol (2000) 278:F380–7. doi: 10.1152/ajprenal.2000.278.3.f380
68. Suzuki F, Shimada J, Mizumoto H, Karasawa A, Kubo K, Nonaka H, et al. Adenosine A1 antagonists. 2. structure-activity relationships on diuretic activities and protective effects against acute renal failure. J Med Chem (1992) 35:3066–75. doi: 10.1021/jm00094a022
69. Wilcox CS, Welch WJ, Schreiner GF, Belardinelli L. Natriuretic and diuretic actions of a highly selective adenosine A1 receptor antagonist. J Am Soc Nephrol JASN (1999) 10:714–20. doi: 10.1681/asn.v104714
70. Gottlieb SS, Brater DC, Thomas I, Havranek E, Bourge R, Goldman S, et al. BG9719 (CVT-124), an A1 adenosine receptor antagonist, protects against the decline in renal function observed with diuretic therapy. Circulation (2002) 105:1348–53. doi: 10.1161/hc1102.105264
71. Ramos-Barbon D, Brienza NS, Rodríguez TB, Medina É.FM, Saladich IG, Rodríguez MP, et al. PBF-680, an oral A1 adenosine receptor antagonist, inhibits the late allergic response (LAR) in mild-to-moderate atopic asthmatics: a phase-IIa trial. Eur Res J (2020) 56 (suppl 64):4784. doi: 10.1183/13993003.congress-2020.4784
72. Ramos-Barbon D, Brienza NS, Rodríguez TB, Saladich IG, Rodríguez MP, Arbos RMA, et al. PBF-680, an oral A1 adenosine receptor antagonist, inhibits adenosine monophosphate (AMP) airway hyperresponsiveness (AHR) in mild-to-moderate asthma: a phase-IIa proof-of-concept trial. Eur Res J (2020) 56 (suppl 64):2279. doi: 10.1183/13993003.congress-2020.2279
73. Voors AA, Dittrich HC, Massie BM, DeLucca P, Mansoor GA, Metra M, et al. Effects of the adenosine A1 receptor antagonist rolofylline on renal function in patients with acute heart failure and renal dysfunction: results from PROTECT (Placebo-controlled randomized study of the selective adenosine A1 receptor antagonist rolofylline for patients hospitalized with acute decompensated heart failure and volume overload to assess treatment effect on congestion and renal function). J Am Coll Cardiol (2011) 57:1899–907. doi: 10.1016/j.jacc.2010.11.057
74. Massie BM, O’Connor CM, Metra M, Ponikowski P, Teerlink JR, Cotter G, et al. Rolofylline, an adenosine A1–receptor antagonist, in acute heart failure. New Engl J Med (2010) 363:1419–28. doi: 10.1056/nejmoa0912613
75. Polosa R, Blackburn MR. Adenosine receptors as targets for therapeutic intervention in asthma and chronic obstructive pulmonary disease. Trends Pharmacol Sci (2009) 30:528–35. doi: 10.1016/j.tips.2009.07.005
76. Gao Z-G, Jacobson KA. Purinergic signaling in mast cell degranulation and asthma. Front Pharmacol (2017) 8:947. doi: 10.3389/fphar.2017.00947
77. Glukhova A, Thal DM, Nguyen AT, Vecchio EA, Jörg M, Scammells PJ, et al. Structure of the adenosine A1 receptor reveals the basis for subtype selectivity. Cell (2017) 168:867–877.e13. doi: 10.1016/j.cell.2017.01.042
78. Draper-Joyce CJ, Khoshouei M, Thal DM, Liang Y-L, Nguyen ATN, Furness SGB, et al. Structure of the adenosine-bound human adenosine A1 receptor–gi complex. Nature (2018) 558:559–63. doi: 10.1038/s41586-018-0236-6
79. Jacobson KA, Gao Z-G. Adenosine receptors as therapeutic targets. Nat Rev Drug Discovery 2018 17:4 (2006) 5:247–64. doi: 10.1038/nrd1983
80. Greene SJ, Sabbah HN, Butler J, Voors AA, Albrecht-Küpper BE, Düngen H-D, et al. Partial adenosine A1 receptor agonism: a potential new therapeutic strategy for heart failure. Heart Fail Rev (2016) 21:95–102. doi: 10.1007/s10741-015-9522-7
81. May LT, Leach K, Sexton PM, Christopoulos A. Allosteric modulation of G protein-coupled receptors. Annu Rev Pharmacol Toxicol (2007) 47:1–51. doi: 10.1146/annurev.pharmtox.47.120505.105159
82. Changeux J-P, Christopoulos A. Allosteric modulation as a unifying mechanism for receptor function and regulation. Diabetes Obes Metab (2017) 19 Suppl 1:4–21. doi: 10.1111/dom.12959
83. Leach K, Sexton PM, Christopoulos A. Allosteric GPCR modulators: taking advantage of permissive receptor pharmacology. Trends Pharmacol Sci (2007) 28:382–9. doi: 10.1016/j.tips.2007.06.004
84. Bruns RF, Fergus JH. Allosteric enhancement of adenosine A1 receptor binding and function by 2-amino-3-benzoylthiophenes. Mol Pharmacol (1990) 38:939–49.
85. Mudumbi RV, Montamat SC, Bruns RF, Vestal RE. Cardiac functional responses to adenosine by PD 81,723, an allosteric enhancer of the adenosine A1 receptor. Am J Physiol (1993) 264:H1017–22. doi: 10.1152/ajpheart.1993.264.3.h1017
86. Mizumura T, Auchampach JA, Linden J, Bruns RF, Gross GJ. PD 81,723, an allosteric enhancer of the A1 adenosine receptor, lowers the threshold for ischemic preconditioning in dogs. Circ Res (1996) 79:415–23. doi: 10.1161/01.res.79.3.415
87. Butcher A, Scammells PJ, White PJ, Devine SM, Rose’Meyer RB. An allosteric modulator of the adenosine A1 receptor improves cardiac function following ischaemia in murine isolated hearts. Pharm (2013) 6:546–56. doi: 10.3390/ph6040546
88. Dennis DM, Raatikainen MJ, Martens JR, Belardinelli L. Modulation of atrioventricular nodal function by metabolic and allosteric regulators of endogenous adenosine in guinea pig heart. Circulation (1996) 94:2551–9. doi: 10.1161/01.cir.94.10.2551
89. Kollias-Baker C, Ruble J, Dennis D, Bruns RF, Linden J, Belardinelli L. Allosteric enhancer PD 81,723 acts by novel mechanism to potentiate cardiac actions of adenosine. Circ Res (2018) 75:961–71. doi: 10.1161/01.res.75.6.961
90. Pan HL, Xu Z, Leung E, Eisenach JC. Allosteric adenosine modulation to reduce allodynia. Anesthesiology (2001) 95:416–20. doi: 10.1097/00000542-200108000-00025
91. Li X, Conklin D, Ma W, Zhu X, Eisenach JC. Spinal noradrenergic activation mediates allodynia reduction from an allosteric adenosine modulator in a rat model of neuropathic pain. Pain (2002) 97:117–25. doi: 10.1016/s0304-3959(02)00011-8
92. Wootten D, Christopoulos A, Sexton PM. Emerging paradigms in GPCR allostery: implications for drug discovery. Nat Rev Drug Discovery (2013) 12:630–44. doi: 10.1038/nrd4052
93. Gentry PR, Sexton PM, Christopoulos A. Novel allosteric modulators of G protein-coupled receptors*. J Biol Chem (2015) 290:19478–88. doi: 10.1074/jbc.r115.662759
94. Bradley SJ, Bourgognon J-M, Sanger HE, Verity N, Mogg AJ, White DJ, et al. M1 muscarinic allosteric modulators slow prion neurodegeneration and restore memory loss. J Clin Invest (2017) 127:487–99. doi: 10.1172/jci87526
95. Gao Z-G, Kim S-K, IJzerman A, Jacobson K. Allosteric modulation of the adenosine family of receptors. Mini-rev Med Chem (2005) 5:545–53. doi: 10.2174/1389557054023242
96. Mubagwa K, Flameng W. Adenosine, adenosine receptors and myocardial protection: an updated overview. Cardiovasc Res (2001) 52:25–39. doi: 10.1016/S0008-6363(01)00358-3
97. Bright R, Mochly-Rosen D. The role of protein kinase c in cerebral ischemic and reperfusion injury. Stroke (2005) 36:2781–90. doi: 10.1161/01.str.0000189996.71237.f7
98. Valant C, Aurelio L, Urmaliya VB, White P, Scammells PJ, Sexton PM, et al. Delineating the mode of action of adenosine A1 receptor allosteric modulators. Mol Pharmacol (2010) 78:444–55. doi: 10.1124/mol.110.064568
99. Bhattacharya S, Linden J. Effects of long-term treatment with the allosteric enhancer, PD81,723, on Chinese hamster ovary cells expressing recombinant human A1 adenosine receptors. Mol Pharmacol (1996) 50:104–11.
100. Klein PAM, Kourounakis AP, IJzerman AP. Allosteric modulation of the adenosine A1 receptor. synthesis and biological evaluation of novel 2-Amino-3-benzoylthiophenes as allosteric enhancers of agonist binding. J Med Chem (1999) 42:3629–35. doi: 10.1021/jm991051d
101. Baraldi PG, Zaid AN, Lampronti I, Fruttarolo F, Pavani MG, Tabrizi MA, et al. Synthesis and biological effects of a new series of 2-amino-3-benzoylthiophenes as allosteric enhancers of A1-adenosine receptor. Bioorg Med Chem Lett (2000) 10:1953–7. doi: 10.1016/s0960-894x(00)00379-6
102. Aurelio L, Christopoulos A, Flynn BL, Scammells PJ, Sexton PM, Valant C. The synthesis and biological evaluation of 2-amino-4,5,6,7,8,9-hexahydrocycloocta[b]thiophenes as allosteric modulators of the A1 adenosine receptor. Bioorganic Medicinal Chem Lett (2011) 21:3704–7. doi: 10.1016/j.bmcl.2011.04.080
103. Figler H, Olsson RA, Linden J. Allosteric enhancers of A1 adenosine receptors increase receptor-G protein coupling and counteract guanine nucleotide effects on agonist binding. Mol Pharmacol (2003) 64:1557–64. doi: 10.1124/mol.64.6.1557
104. Nikolakopoulos G, Figler H, Linden J, Scammells PJ. 2-Aminothiophene-3-carboxylates and carboxamides as adenosine A1 receptor allosteric enhancers. Bioorgan Med Chem (2006) 14:2358–65. doi: 10.1016/j.bmc.2005.11.018
105. Tranberg CE, Zickgraf A, Giunta BN, Luetjens H, Figler H, Murphree LJ, et al. 2-Amino-3-aroyl-4,5-alkylthiophenes: agonist allosteric enhancers at human A1 adenosine receptors. J Med Chem (2002) 45:382–9. doi: 10.1021/jm010081p
106. Aurelio L, Valant C, Figler H, Flynn BL, Linden J, Sexton PM, et al. 3- and 6-substituted 2-amino-4,5,6,7-tetrahydrothieno[2,3-c]pyridines as A1 adenosine receptor allosteric modulators and antagonists. Bioorg Med Chem (2009) 17:7353–61. doi: 10.1016/j.bmc.2009.08.024
107. Baraldi PG, Romagnoli R, Pavani MG, Nuñez M, del C, Tabrizi MA, et al. Synthesis and biological effects of novel 2-Amino-3-naphthoylthiophenes as allosteric enhancers of the A1 adenosine receptor. J Med Chem (2003) 46:794–809. doi: 10.1021/jm0210212
108. Baraldi PG, Pavani MG, Shryock JC, Moorman AR, Iannotta V, Borea PA, et al. Synthesis of 2-amino-3-heteroaroylthiophenes and evaluation of their activity as potential allosteric enhancers at the human A1 receptor. Eur J Med Chem (2004) 39:855–65. doi: 10.1016/j.ejmech.2004.06.009
109. Romagnoli R, Baraldi PG, Moorman AR, Iaconinoto MA, Carrion MD, Cara CL, et al. Microwave-assisted synthesis of thieno[2,3-c]pyridine derivatives as a new series of allosteric enhancers at the adenosine A1 receptor. Bioorg Med Chem Lett (2006) 16:5530–3. doi: 10.1016/j.bmcl.2006.08.041
110. Aurelio L, Valant C, Flynn BL, Sexton PM, White JM, Christopoulos A, et al. Effects of conformational restriction of 2-amino-3-benzoylthiophenes on A(1) adenosine receptor modulation. J Med Chem (2010) 53:6550–9. doi: 10.1021/jm1008538
111. Aumann KM, Scammells PJ, White JM, Schiesser CH. On the stability of 2-aminoselenophene-3-carboxylates: potential dual-acting selenium-containing allosteric enhancers of A1 adenosine receptor binding. Org Biomol Chem (2007) 5:1276–81. doi: 10.1039/b700812k
112. Chordia MD, Murphree LJ, Macdonald TL, Linden J, Olsson RA. 2-aminothiazoles: a new class of agonist allosteric enhancers of A1 adenosine receptors. Bioorg Med Chem Lett (2002) 12:1563–6. doi: 10.1016/s0960-894x(02)00236-6
113. Göblyös A, Santiago SN, Pietra D, Mulder-Krieger T, Künzel J, von FD, et al. Synthesis and biological evaluation of 2-aminothiazoles and their amide derivatives on human adenosine receptors. lack of effect of 2-aminothiazoles as allosteric enhancers. Bioorgan Med Chem (2005) 13(6):2079–87. doi: 10.1016/j.bmc.2005.01.006
114. Romagnoli R, Baraldi PG, Carrion MD, Cara CL, Cruz-Lopez O, Iaconinoto MA, et al. Synthesis and biological evaluation of 2-Amino-3-(4-Chlorobenzoyl)-4-[N-(Substituted) piperazin-1-yl]Thiophenes as potent allosteric enhancers of the a 1 adenosine receptor. J Med Chem (2008) 51:5875–9. doi: 10.1021/jm800586p
115. Lütjens H, Zickgraf A, Figler H, Linden J, Olsson RA, Scammells PJ. 2-Amino-3-benzoylthiophene allosteric enhancers of A1 adenosine agonist binding: new 3, 4-, and 5-modifications. J Med Chem (2003) 46:1870–7. doi: 10.1021/jm020295m
116. Valant C, Aurelio L, Devine SM, Ashton TD, White JM, Sexton PM, et al. Synthesis and characterization of novel 2-amino-3-benzoylthiophene derivatives as biased allosteric agonists and modulators of the adenosine A(1) receptor. J Med Chem (2012) 55:2367–75. doi: 10.1021/jm201600e
117. Ferguson GN, Valant C, Horne J, Figler H, Flynn BL, Linden J, et al. 2-aminothienopyridazines as novel adenosine A1 receptor allosteric modulators and antagonists. J Med Chem (2008) 51:6165–72. doi: 10.1021/jm800557d
118. Fawzi AB, Macdonald D, Benbow LL, Smith-Torhan A, Zhang H, Weig BC, et al. SCH-202676: an allosteric modulator of both agonist and antagonist binding to G protein-coupled receptors. Mol Pharmacol (2001) 59:30–7. doi: 10.1124/mol.59.1.30
119. Nieuwendijk AMCH, Pietra D, Heitman L, Göblyös A, IJzerman AP. Synthesis and biological evaluation of 2,3,5-substituted [1,2,4]thiadiazoles as allosteric modulators of adenosine receptors. J Med Chem (2004) 47:663–72. doi: 10.1021/jm030863d
120. Baraldi PG, Iaconinoto MA, Moorman AR, Carrion MD, Cara CL, Preti D, et al. Allosteric enhancers for A1 adenosine receptor. Mini-rev Med Chem (2007) 7:559–69. doi: 10.2174/138955707780859459
121. Romagnoli R, Baraldi PG, Moorman AR, Borea PA, Varani K. Current status of A1 adenosine receptor allosteric enhancers. Future Med Chem (2015) 7:1247–59. doi: 10.4155/fmc.15.65
122. Bruns RF, Fergus JH, Coughenour LL, Courtland GG, Pugsley TA, Dodd JH, et al. Structure-activity relationships for enhancement of adenosine A1 receptor binding by 2-amino-3-benzoylthiophenes. Mol Pharmacol (1990) 38:950–8.
123. Romagnoli R, Baraldi PG, Carrion MD, Cara CL, Cruz-Lopez O, Salvador MK, et al. Synthesis and biological evaluation of 2-Amino-3-(4-chlorobenzoyl)-4-[(4-arylpiperazin-1-yl)methyl]-5-substituted-thiophenes. effect of the 5-modification on allosteric enhancer activity at the A1 adenosine receptor. J Med Chem (2012) 55:7719–35. doi: 10.1021/jm3007504
124. Romagnoli R, Baraldi PG, Carrion MD, Cruz-Lopez O, Cara CL, Saponaro G, et al. Synthesis and biological evaluation of novel 2-amino-3-aroyl-4-neopentyl-5-substituted thiophene derivatives as allosteric enhancers of the A1 adenosine receptor. Bioorgan Med Chem (2014) 22:148–66. doi: 10.1016/j.bmc.2013.11.043
125. Romagnoli R, Baraldi PG, IJzerman AP, Massink A, Cruz-Lopez O, Lopez-Cara LC, et al. Synthesis and biological evaluation of novel allosteric enhancers of the A1 adenosine receptor based on 2-Amino-3-(4′-Chlorobenzoyl)-4-Substituted-5-Arylethynyl thiophene. J Med Chem (2014) 57:7673–86. doi: 10.1021/jm5008853
126. Garritsen A, IJzerman AP, Beukers MW, Cragoe EJ, Soudijn W. Interaction of amiloride and its analogues with adenosine A1 receptors in calf brain. Biochem Pharmacol (1990) 40:827–34. doi: 10.1016/0006-2952(90)90323-d
127. Gao Z-G, Melman N, Erdmann A, Kim SG, Müller CE, IJzerman AP, et al. Differential allosteric modulation by amiloride analogues of agonist and antagonist binding at A1 and A3 adenosine receptors. Biochem Pharmacol (2003) 65:525–34. doi: 10.1016/s0006-2952(02)01556-3
128. Peeters MC, Wisse LE, Dinaj A, Vroling B, Vriend G, IJzerman AP. The role of the second and third extracellular loops of the adenosine A1 receptor in activation and allosteric modulation. Biochem Pharmacol (2012) 84:76–87. doi: 10.1016/j.bcp.2012.03.008
129. Kennedy DP, McRobb FM, Leonhardt SA, Purdy M, Figler H, Marshall MA, et al. The second extracellular loop of the adenosine A1 receptor mediates activity of allosteric enhancers. Mol Pharmacol (2014) 85:301–9. doi: 10.1124/mol.113.088682
130. Nguyen ATN, Vecchio EA, Thomas T, Nguyen TD, Aurelio L, Scammells PJ, et al. Role of the second extracellular loop of the adenosine A1 receptor on allosteric modulator binding, signaling, and cooperativity. Mol Pharmacol (2016) 90:715–25. doi: 10.1124/mol.116.105015
131. Cheng RKY, Segala E, Robertson N, Deflorian F, Doré AS, Errey JC, et al. Structures of human A1 and A2A adenosine receptors with xanthines reveal determinants of selectivity. Structure (2017) 25:1275–1285.e4. doi: 10.1016/j.str.2017.06.012
132. Ballesteros JA, Weinstein H. Integrated methods for the construction of three dimensional models and computational probing of structure function relations in G-protein-coupled receptors. Methods Neurosci (1995) 25:366–428. doi: 10.1016/S1043-9471(05)80049-7
133. Miao Y, Bhattarai A, Nguyen ATN, Christopoulos A, May LT. Structural basis for binding of allosteric drug leads in the adenosine A1 receptor. Sci Rep (2018) 8:829. doi: 10.1038/s41598-018-35266-x
134. Do HN, Wang J, Bhattarai A, Miao Y. GLOW: a workflow integrating Gaussian-accelerated molecular dynamics and deep learning for free energy profiling. J Chem Theory Comput (2022) 18(3):1423–36. doi: 10.1021/acs.jctc.1c01055
135. Wang M, Hou S, Wei Y, Li D, Lin J. Discovery of novel dual adenosine A1/A2A receptor antagonists using deep learning, pharmacophore modeling and molecular docking. PloS Comput Biol (2021) 17:e1008821. doi: 10.1371/journal.pcbi.1008821
136. Segler MHS, Kogej T, Tyrchan C, Waller MP. Generating focused molecule libraries for drug discovery with recurrent neural networks. ACS Cent Sci (2018) 4:120–31. doi: 10.1021/acscentsci.7b00512
137. Jensen JH. A graph-based genetic algorithm and generative model/Monte Carlo tree search for the exploration of chemical space. Chem Sci (2019) 10:3567–72. doi: 10.1039/c8sc05372c
138. Anishchenko I, Pellock SJ, Chidyausiku TM, Ramelot TA, Ovchinnikov S, Hao J, et al. De novo protein design by deep network hallucination. Nature (2021) 600:547–52. doi: 10.1038/s41586-021-04184-w
139. Wang J, Lisanza S, Juergens D, Tischer D, Watson JL, Castro KM, et al. Scaffolding protein functional sites using deep learning. Science (2022) 377:387–94. doi: 10.1126/science.abn2100
140. Liu X, Ye K, Vlijmen HWT, IJzerman AP, van Westen GJP. DrugEx v3: scaffold-constrained drug design with graph transformer-based reinforcement learning. J Cheminformatics (2023) 15:24. doi: 10.1186/s13321-023-00694-z
141. Yeh AH-W, Norn C, Kipnis Y, Tischer D, Pellock SJ, Evans D, et al. De novo design of luciferases using deep learning. Nature (2023) 614:774–80. doi: 10.1038/s41586-023-05696-3
142. Thal DM, Glukhova A, Sexton PM, Christopoulos A. Structural insights into G-protein-coupled receptor allostery. Nature (2018) 559:45–53. doi: 10.1038/s41586-018-0259-z
143. Zhang L, Mobbs JI, May LT, Glukhova A, Thal DM. The impact of cryo-EM on determining allosteric modulator-bound structures of G protein-coupled receptors. Curr Opin Struc Biol (2023) 79:102560. doi: 10.1016/j.sbi.2023.102560
144. Miao Y, Goldfeld DA, Moo EV, Sexton PM, Christopoulos A, McCammon JA, et al. Accelerated structure-based design of chemically diverse allosteric modulators of a muscarinic G protein-coupled receptor. Proc Natl Acad Sci USA (2016) 113:E5675–84. doi: 10.1073/pnas.1612353113
145. Korczynska M, Clark MJ, Valant C, Xu J, Moo EV, Albold S, et al. Structure-based discovery of selective positive allosteric modulators of antagonists for the M2 muscarinic acetylcholine receptor. Proc Natl Acad Sci (2018) 115:E2419–28. doi: 10.1073/pnas.1718037115
146. Redij T, Chaudhari R, Li Z, Hua X, Li Z. Structural modeling and in silico screening of potential small-molecule allosteric agonists of a glucagon-like peptide 1 receptor. ACS Omega (2019) 4:961–70. doi: 10.1021/acsomega.8b03052
147. Redij T, Ma J, Li Z, Hua X, Li Z. Discovery of a potential positive allosteric modulator of glucagon-like peptide 1 receptor through virtual screening and experimental study. J Comput Aid Mol Des (2019) 33:973–81. doi: 10.1007/s10822-019-00254-4
148. Zhou Q, Guo W, Dai A, Cai X, Vass M, de Graaf C, et al. Discovery of novel allosteric modulators targeting an extra-helical binding site of GLP-1R using structure- and ligand-based virtual screening. Biomol (2021) 11:929. doi: 10.3390/biom11070929
149. Kampen S, Rodriíguez D, Jørgensen M, Kruszyk-Kujawa M, Huang X, Collins M, et al. Structure-based discovery of negative allosteric modulators of the metabotropic glutamate receptor 5. ACS Chem Biol (2022) 17:2744–52. doi: 10.1021/acschembio.2c00234
Keywords: adenosine, A1 receptor, allosteric modulation, G protein-coupled receptor, structure-activity relationship, structure-function relationship
Citation: Nguyen ATN, Tran QL, Baltos J-A, McNeill SM, Nguyen DTN and May LT (2023) Small molecule allosteric modulation of the adenosine A1 receptor. Front. Endocrinol. 14:1184360. doi: 10.3389/fendo.2023.1184360
Received: 11 March 2023; Accepted: 23 May 2023;
Published: 26 June 2023.
Edited by:
Paula Morales, Spanish National Research Council (CSIC), SpainReviewed by:
Zhenhua Shao, Sichuan University, ChinaJohn Auchampach, Medical College of Wisconsin, United States
Copyright © 2023 Nguyen, Tran, Baltos, McNeill, Nguyen and May. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anh T. N. Nguyen, YW5oLnRuLm5ndXllbkBtb25hc2guZWR1; Lauren T. May, bGF1cmVuLm1heUBtb25hc2guZWR1