 Gaia Vincenzi1*†
Gaia Vincenzi1*† Ilenia Teresa Petralia2†Marco Abbate2Giulia Tarantola2Silvia Laura Carla Meroni1Riccardo Maggiore3Gilberto Mari3Maria Grazia Patricelli4Marco Schiavo Lena5Graziano Barera1Maria Cristina Vigone1
Ilenia Teresa Petralia2†Marco Abbate2Giulia Tarantola2Silvia Laura Carla Meroni1Riccardo Maggiore3Gilberto Mari3Maria Grazia Patricelli4Marco Schiavo Lena5Graziano Barera1Maria Cristina Vigone1- 1Department of Pediatrics, Endocrine Unit, IRCCS San Raffaele Scientific Institute, Milan, Italy
- 2Department of Pediatrics, Endocrine Unit, Vita-Salute San Raffaele University, IRCCS San Raffaele Scientific Institute, Milan, Italy
- 3Department of Surgery, Endocrine Surgery Unit, IRCCS San Raffaele Scientific Institute, Milan, Italy
- 4Medical Genetics, IRCCS San Raffaele Scientific Institute, Milan, Italy
- 5Pathology Unit, IRCCS San Raffaele Scientific Institute, Milan, Italy
We report the case of a paediatric female patient affected by Bannayan-Riley-Ruvalcaba syndrome (BRRS) and congenital hypothyroidism (CH) with homozygous mutation of the TPO gene. She underwent total thyroidectomy at the age of seven years because of the development of a multinodular goiter. BRRS patients present an increased risk of benign and malignant thyroid disease since childhood because of inactivating mutation of PTEN, an onco-suppressor gene. Instead, homozygous mutations in the TPO gene can be associated with severe forms of hypothyroidism with goiter; previous studies have described cases of follicular and papillary thyroid cancer in CH patients with TPO mutation despite a perfectly controlled thyroid function with Levothyroxine therapy. To our knowledge, this is the first case that describes the possible synergic role of coexisting mutation of both TPO and PTEN in the development of multinodular goiter underlining the importance of a tailored surveillance program in these patients, especially during childhood.
1 Introduction
Bannayan-Riley-Ruvalcaba syndrome (BRRS) is caused by germline inactivating mutations of phosphatase and tensin homolog (PTEN) gene inherited with an autosomal dominant mechanism. The PTEN gene, located on chromosome 10q23, is a tumor suppressor gene with a fundamental role in cell growth, migration, and apoptosis via the PI3K/AKT/mTOR pathway (1). An excessive activation of this pathway leads to overgrowth and an increased risk for tumor development as described in PTEN hamartoma tumor syndrome (PHTS), which encompass not only BRRS but a spectrum of syndromes including Cowden syndrome, Lhermitte-Duclos syndrome, Proteus-like syndrome and autism spectrum diseases with macrocephaly (2). BRRS is considered the PHTS form of childhood (3). Macrocephaly (head circumference > 97° ple) is the hallmark of this syndrome (4), variably associated with developmental delay, lipomas, hemangiomas, intestinal hamartomatous polyps, penile freckling and other highly variable phenotypic features as downward slanting palpebral fissures, frontal bossing, macrosomia, café au lait spots, hypotonia, joint hyperextensibility, hypoglycemia and seizures (3, 5). All patients are at high risk of tumor development especially affecting endometrium, kidneys, skin and thyroid, therefore cancer surveillance programs are recommended and needed also for paediatric population (6, 7). Thyroid diseases are common in these children: two thirds of all BRRS patients present with autoimmune thyroiditis, nodular goiter and benign or malignant thyroid tumors, either follicular or papillary thyroid carcinoma (1). In this paper, we describe the case of a girl affected by BRRS presenting with multiple thyroid nodules, previously diagnosed with severe congenital hypothyroidism (CH) due to TPO gene mutation: as far as we know this is the first description of BRRS associated with congenital hypothyroidism.
2 Case presentation
Our patient was born at 31 + 4 gestational weeks because of premature rupture of membranes. At birth her auxological parameters were adequate for gestational age (weight 0,4 SDS, length 1,5 SDS and head circumference 0,6 SDS). In the first days of life she suffered from respiratory distress, neonatal jaundice and patent ductus arteriosus pharmacologically closed. Due to prematurity, cerebral ultrasound was performed, resulting within limits. At neonatal screening, she was detected with a high blood TSH value (152 microU/mL). A severe form of congenital hypothyroidism was subsequently diagnosed (TSH 1016 μIU/mL, fT4 <0,4 ng/dL) and at ten days of life Levothyroxine (LT4) was started at a dosage of 10 mcg/kg/day. The thyroid ultrasound revealed the presence of a hyperplastic gland with inhomogeneous echotexture in the absence of thyroid antibodies. Genetic analysis was performed, revealing the presence of a homozygous mutation in the TPO gene (ins.GGCC395, exon 8) inherited from both parents. The father presented with normal thyroid function; the mother suffered from non-autoimmune hypothyroidism with right lobe hypoplasia diagnosed at the age of sixteen. At six months of age, an increasing head circumference (>97° percentile) and facial abnormalities such as triangular face, frontal bossing and hyperthelorism were observed. Karyotype was normal (46 XX) while the array-CGH showed the presence of a non-pathological variation in the copy number without chromosomal imbalance (arr(1-22,X)x2). At seven months, a brain MRI was performed showing a minimal amplification of the subarachnoid space and a slight para physiological reduction of the myelinisation signal. During the follow-up, a mild delay in neuromotor development (first steps at 18 months, first words at 24 months) was observed. Since the second year of life, multiple lipomas were diagnosed and subsequently surgically removed. At the age of six, she appeared in good clinical conditions, with regular growth both in height (25° ple) and weight (25° ple) but with a persisting important macrocrania. She also presented with an important thyromegaly, despite well controlled thyroid function values since the very first months of age. Therefore, a neck ultrasound was performed showing the presence of an enlarged thyroid (antero-posterior and transversal diameters: 16,5*15,6 mm right, 14,5*14,4 mm left, Figure 1A) with non-homogeneous echotexture and multiple nodules with intrinsic and peripheral vascularisation: five in the right lobe (diameter: 7-16 mm, Figure 1B) and four in the left lobe (diameter: 6-15mm). Six months later the nodules were increasing both in number and size: six-seven in the right lobe (diameter: 4-19 mm) and six in the left lobe (diameter: 4-17 mm). Moreover, brain MRI was repeated revealing the presence of a bone alteration in the orbital roof (1,8-2 cm) suspicious for haemangioma. Because of all the clinical peculiarities presented (macrocrania, neuromotor delay, facial abnormalities, lipomas, thyroid nodules, doubt of hemangioma), after genetic counselling, the sequencing analysis of PTEN gene was performed showing the presence of a heterozygous mutation (c.635-1G>C) coding for a truncated protein causing Bannayan-Riley-Ruvalcaba syndrome (BRRS). In consideration of the increased risk of developing thyroid cancer, the patient underwent total thyroidectomy at the age of seven; the histological exam showed 21 adenomatous nodules (0.1 to 1.7 centimetres) with microfollicular and trabecular architecture and poor in colloid. In 8/8 nodules analysed, the immunohistochemical analysis revealed a loss of nuclear expression of PTEN. Later in the follow-up, she developed an arteriovenous malformation at her right ankle that was surgically removed and genetically analysed confirming the presence of the PTEN gene alteration. Finally, at the latest follow-up (12 years), a breast nodule was observed and confirmed at the ultrasound, as well as duodenal and colic polyps at the endoscopic exam performed because of the presence of faecal occult blood.
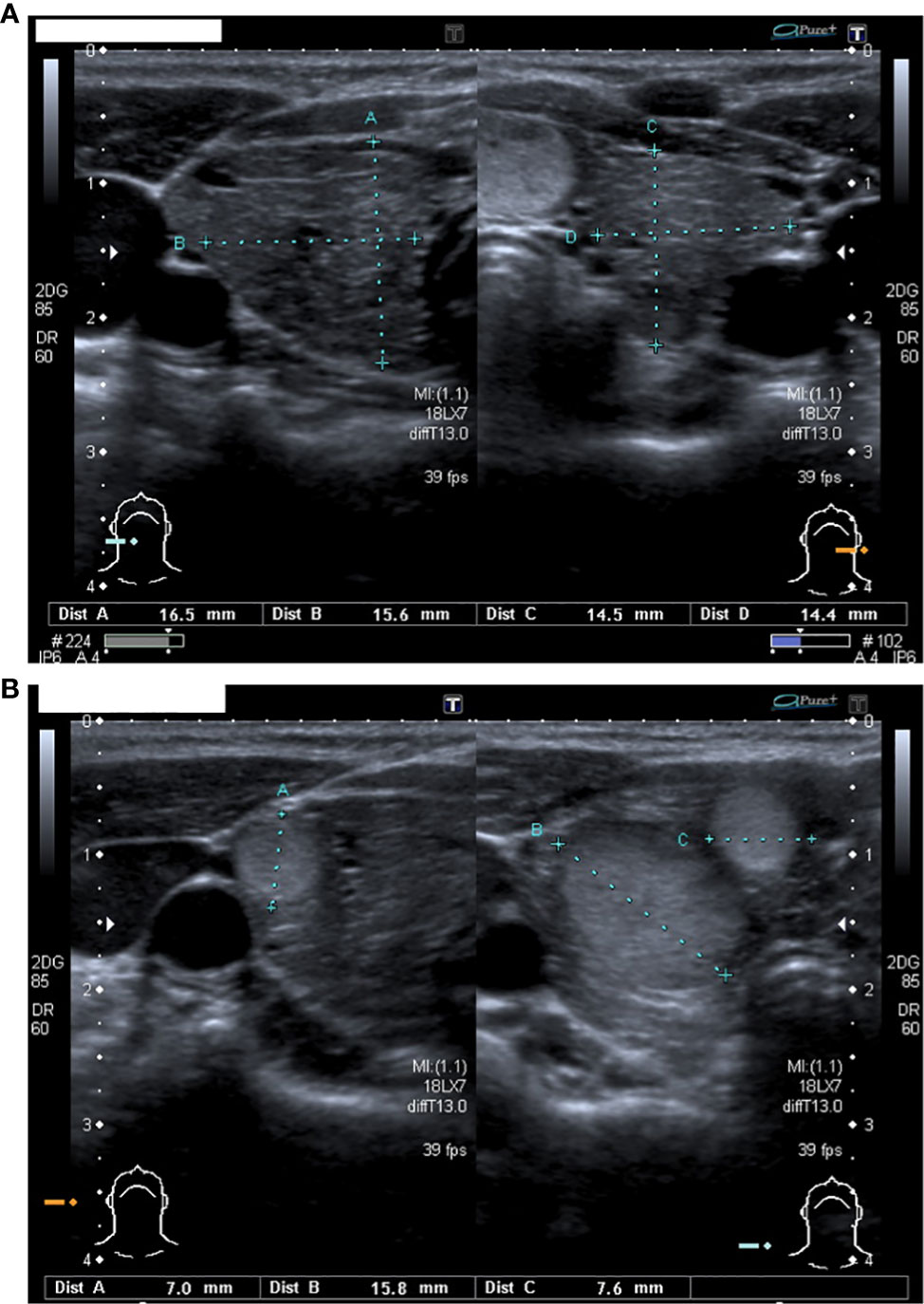
Figure 1 (A) The thyroid ultrasound performed at 6 years of age: an enlarged and non-homogeneous echotexture gland with increased antero-posterior and transversal diameters (16,5*15,6 mm for right lobe and 14,5*14,4 mm for left lobe). (B) Nodules in the right lobe.
3 Discussion
PTEN hamartoma tumor syndrome (PHTS) patients present an increased risk of developing benign and malignant thyroid disease. Nodules, goiter and autoimmune thyroiditis have been described in up to 75% of all patients affected by PHTS, including children with BRRS (1, 2). Previous studies have demonstrated that patients with PTEN gene mutations present a cumulative lifetime risk of developing thyroid carcinoma ranging from 21 to 38%, in particular the risk of paediatric differentiated thyroid carcinoma (DTC) is estimated between 4 and 12% with an incidence of 5% from the age of ten (8–11). Therefore, considering the early involvement of the thyroid gland and the possible development of thyroid cancer, clinical and ultrasound surveillance should be performed early in childhood. However, a clear consensus regarding both the time of initiation and the timepoints of thyroid cancer surveillance in paediatric patients has not been established yet. Although the youngest patient described in literature with PHTS and thyroid carcinoma is a four years old boy (10), most authors have suggested that thyroid disease surveillance should begin at age of seven or ten (10, 11). As described by Smith et al. in their retrospective study on 64 children with PHTS, 44% of patients undergoing thyroid ultrasound present with a clinically significant thyroid nodule at the mean age of 13.3 years with a later presentation in males than females according to gender pubertal period. In this cohort, nodules were rare before the age of seven years (11). On the contrary, in a recent study conducted on a small group of 12 children with PTHS, 41.7% of subjects with nodular thyroid disease were less than seven years old (3). Nonetheless, confirming the prevalence of benign nodular disease in children with PHTS, no patient developed DCT during an average follow-up period of five years, thus not suggesting the need for anticipating the starting age of thyroid cancer surveillance (3). According to the recent comprehensive review of the German paediatric guidelines by Plamper et al., patients with PHTS should undergo a comprehensive physical examination and thyroid ultrasound right after the diagnosis; the following thyroid ultrasound should then be repeated annually or more frequently in case of any suspicious result. Furthermore, children under the age of seven without proof of thyroid nodules are allowed to repeat an ultrasound after 2-3 years (1, 12). Another recent prospective study investigated the development and progression of thyroid nodules and DTC in patients with PHTS both in the presence and absence of thyroid disease at initial ultrasound, with the aim of providing stronger evidence to refine current surveillance recommendations and stratify surveillance intervals based on the initial ultrasound result (13). According to these authors, in contrast with current recommendations, patients without thyroid nodules at the first ultrasound should repeat the exam after 3-5 years; patients with nodules should instead repeat the ultrasound depending on the presence of any suspicious pattern (13). Despite the optimal thyroid function values with LT4 therapy, our patient presented with an important thyromegaly and from the age of six multiple thyroid nodules were described. Homozygous mutations in TPO gene, inherited with an autosomal recessive pattern, are associated with severe forms of hypothyroidism with goiter (14). The combination of CH and DTC is a rare condition with no established causal relationship. As recently suggested by Penna et al., the long exposure to elevated serum TSH levels, a congenital goiter as well as the presence of stimulating factors or the absence of tumor suppressor genes, the type of mutation and the iodine intake could all be factors involved in the development of DTC in patients with CH due to dyshormonogenesis (15, 16). Previous studies have described cases of follicular and papillary thyroid carcinoma in CH patients with TPO mutation and a perfectly controlled thyroid function (17, 18) suggesting that genetic and environmental factors other than TSH level might be involved in the development of thyroid cancer in dyshormonogenic multinodular goiter (MNG) and perhaps play a synergic role in the development of DTC. Tobias et al. analysing the long-term outcome of 33 CH patients with TPO mutations showed that 61% of them developed MNG over time at an average age of 8,6 years (19) without an association with higher TSH levels. In this case series, thyroidectomy was performed in eight patients (24%) leading to the diagnosis of a minimally invasive follicular carcinoma and seven cases of follicular hyperplasia or adenoma (19). Thus, the high rate of MNG development and the risk for thyroid carcinoma indicates the need for regular ultrasound and a long-term follow up in these patients (19). Although most patients affected by PTHS develop benign nodular goiter (3, 20, 21), our patient underwent total thyroidectomy at the age of seven without previously performing a cytological investigation by fine-needle aspiration. According to recent studies, the direct surgical approach could be justified because fine needle biopsy may not be sufficient to exclude malignancy in patients with more than one suspicious lesion (1). Nevertheless, paediatric patients, even more PTHS children with neuromotor delay, are often uncooperative (22) and most cases need general anaesthesia for FNA (1). Furthermore, the cytological analysis does not allow to distinguish a benign follicular adenoma from follicular carcinoma (23). Therefore, prophylactic thyroidectomy should be considered in selected patients with multiple nodules (24). However in the decision-making process, the consequences of total thyroidectomy should be always carefully analysed, such as the need of a lifelong daily replacement therapy and the risk of a surgical procedure in patients with other comorbidities. When indicated, thyroidectomy should be performed by an expert surgeon to minimize the risk of complications.
In conclusion, this is an original case where TPO and PTEN mutation could have played a synergic role in the development of multinodular goiter. Both TPO and PTEN patients need a strict ultrasound follow-up because of the risk of benign and malignant thyroid carcinoma. Further studies are needed to better understand the natural history of thyroid involvement in patients with TPO and/or PTEN mutations in order to provide stronger evidence to refine paediatric surveillance recommendations and develop a tailored diagnostic and therapeutic approach for these patients.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Acknowledgments
Thanks are given to all the clinicians providing care and management to the patients.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Plamper M, Schreiner F, Gohlke B, Kionke J, Korsch E, Kirkpatrick J, et al. Thyroid disease in children and adolescents with PTEN hamartoma tumor syndrome (PHTS). Eur J Pediatr (2018) 177:429–35. doi: 10.1007/s00431-017-3067-9
2. Yehia L, Keel E, Eng C. The clinical spectrum of PTEN mutations. Annu Rev Med (2020) 71:103–16. doi: 10.1146/annurev-med-052218-125823
3. Tuli G, Munarin J, Mussa A, Carli D, Gastaldi R, Borgia P, et al. Thyroid nodular disease and PTEN mutation in a multicentre series of children with PTEN hamartoma tumor syndrome (PHTS). Endocrine (2021) 74:632–6. doi: 10.1007/s12020.021-02805-y
4. Ciaccio C, Saletti V, D'Arrigo S, Esposito S, Alfei E, Moroni I, et al. Clinical spectrum of PTEN mutation in pediatric patients. a bicenter experience. Eur J Med Genet (2019) 62(12):103596. doi: 10.1016/j.ejmg.2018.12.001
5. Macken WL, Tischkowitz M, Lachlan KL. PTEN hamartoma tumor syndrome in childhood: a review of the clinical literature. Am J Med Genet C Semin Med Genet (2019) 181(4):591–610. doi: 10.1002/ajmg.c.31743
6. Tischkowitz M, Colas C, Pouwels S, Hoogerbrugge N, PHTS Guideline Development Group, European Reference Network GENTURIS. Cancer surveillance guideline for individuals with PTEN hamartoma tumour syndrome. Eur J Hum Genet (2020) 28(10):1387–93. doi: 10.1038/s41431-020-0651-7
7. Hendricks LAJ, Hoogerbrugge N, Schuurs-Hoeijmakers JHM, Vos JR. A review on age-related cancer risks in PTEN hamartoma tumor syndrome. Clin Genet (2021) 99:219–25. doi: 10.1111/cge.13875
8. Bubien V, Bonnet F, Brouste V, Hoppe S, Barouk-Simonet E, David A, et al. High cumulative risks of cancer in patients with PTEN hamartoma tumour syndrome. J Med Genet (2013) 50(4):255–63. doi: 10.1136/jmedgenet-2012-101339
9. Tan MH, Mester JL, Ngeow J, Rybicki LA, Orloff MS, Eng C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res (2012) 18(2):400–7. doi: 10.1158/1078-0432.CCR-11-2283
10. Jonker LA, Lebbink CA, Jongmans MCJ, Nievelstein RAJ, Merks JHM, Nieveen van Dijkum EJM, et al. Recommendation on surveillance for differentiated thyroid carcinoma in children with PTEN hamartoma tumor syndrome. Eur Thyroid J (2020) 9(5):234–42. doi: 10.1150/000508872
11. Smith JR, Liu E, Church AJ, Asch E, Cherella CE, Srivastava S, et al. Natural history of thyroid disease in children with PTEN hamartoma tumor syndrome. JCEM (2021) 106(3):e1121–30. doi: 10.1210/clinem/dgaa944
12. Plamper M, Gohlke B, Woelfle J. PTEN hamartoma tumor syndrome in childhood and adolescence-a comprehensive review and presentation of the German pediatric guideline. Mol Cell Pediatr (2022) 9(1):3. doi: 10.1186/s40348-022-00135-1
13. Plitt G, Brewer T, Yehia L, Jin J, Shin J, Eng C. Development and progression of thyroid disease in PTEN hamartoma tumor syndrome: refined surveillance recommendations. Thyroid (2022) 32(9):1094–100. doi: 10.1089/thy.2022.0181
14. Stoupa A, Kariyawasam D, Polak M, Carré A. Genetics of congenital hypothyroidism: modern concepts. Pediatr Investig (2022) 6(2):123–34. doi: 10.1002/ped4.12324
15. Penna G, Rubio IGS, Brust ES, Cazarin J, Hecht F, Alkmim NR, et al. Congenital hypothyroidism and thyroid cancer. Endocr Relat Cancer (2021) 28(9):R217–30. doi: 10.1530/ERC-21-0159
16. Cipollini M, Pastor S, Gemignani F, Castell J, Garritano S, Bonotti A, et al. TPO genetic variants and risk of differentiated thyroid carcinoma in two European populations. Int J Cancer (2013) 133:2843–51. doi: 10.1002/ijc.28317
17. Zhu H, Peng YG, Ma SG, Liu H. TPO gene mutations associated with thyroid carcinoma: case report and literature review. Cancer biomark (2015) 15(6):909–13. doi: 10.3233/CBM-150522
18. Chertok Shacham E, Ishay A, Irit E, Pohlenz J, Tenenbaum-Rakover Y. Minimally invasive follicular thyroid carcinoma developed in dyshormonogenetic multinodular goiter due to thyroid peroxidase gene mutation. Thyroid (2012) 22(5):542–6. doi: 10.1089/thy.2011.0478
19. Tobias L, Elias-Assad G, Khayat M, Admoni O, Almashanu S, Tenenbaum-Rakover Y. Long-term outcome of patients with TPO mutations. J Clin Med (2021) 10(17):3898. doi: 10.3390/jcm10173898
20. Milani D, Dolci A, Muller I, Pavesi MA, Runza L, Kuhn E, et al. Thyroid findings in pediatric and adult patients with PTEN hamartoma tumor syndrome: a retrospective analysis, and literature review. Endocrine (2023). doi: 10.1007/s12020-023-03313-x
21. Szabo Yamashita T, Baky F J, McKenzie T J, Thompson G B, Farley D R, Lyden M L, et al. Occurrence and natural history of thyroid cancer in patients with cowden syndrome. Eur Thyroid J (2020) 9:243–6. doi: 10.1159/000506422
22. Feldkamp J, Führer D, Luster M, Musholt TJ, Spitzweg C, Schott M. Fine needle aspiration in the investigation on thyroid nod- ules–indications, procedures and interpretation. Dtsch Arztebl Int (2016) 113(20):353–9. doi: 10.3238/arztebl.2016.0353
23. Ngeow J, Mester J, Rybicki LA, Ni Y, Milas M, Eng C. Incidence and clinical characteristics of thyroid cancer in prospec- tive series of individuals with cowden and cowden-like syndrome characterized by germline PTEN, SDH, or KLLN alterations. J Clin Endocrinol Metab (2011) 96(12):E2063–71. doi: 10.1210/jc.2011-1616
Keywords: case report, PTEN hamartoma tumor syndrome, congenital hypothyroidism, TPO, goiter
Citation: Vincenzi G, Petralia IT, Abbate M, Tarantola G, Meroni SLC, Maggiore R, Mari G, Patricelli MG, Schiavo Lena M, Barera G and Vigone MC (2023) Case Report - Multinodular goiter in a patient with Congenital Hypothyroidism and Bannayan-Riley-Ruvalcaba syndrome: the possible synergic role of TPO and PTEN mutation. Front. Endocrinol. 14:1205785. doi: 10.3389/fendo.2023.1205785
Received: 14 April 2023; Accepted: 15 May 2023;
Published: 08 June 2023.
Edited by:
Luisa De Sanctis, University of Turin, ItalyReviewed by:
Loredana Pagano, University of Turin, ItalyGerdi Tuli, Regina Margherita Hospital, Italy
Copyright © 2023 Vincenzi, Petralia, Abbate, Tarantola, Meroni, Maggiore, Mari, Patricelli, Schiavo Lena, Barera and Vigone. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gaia Vincenzi, dmluY2VuemkuZ2FpYUBoc3IuaXQ=
†These authors have contributed equally to this work and share first authorship