 Xiaoman Zhang1†
Xiaoman Zhang1† Dequan Liu
Dequan Liu Guangzhen Wu
Guangzhen Wu- 1Department of Urology, The First Affiliated Hospital of Dalian Medical University, Dalian, China
- 2Department of Laboratory Medicine, The Faculty of Medicine and Pharmaceutical Sciences, Hainan Vocational University of Science and Technology, Haikou, China
- 3Department of Nursing, The Second Affiliated Hospital of Dalian Medical University, Dalian, China
- 4Department of Oncology, Cancer Hospital of Dalian University of Technology, Cancer Hospital of China Medical University, Liaoning Cancer Hospital and Institute, Shenyang, China
Epigenetic changes, such as DNA methylation, chromatin remodeling, and histone modifications, regulate gene expression without altering the DNA sequence. This review systematically analyzed over 500 studies including human cell line experiments (n>200), animal models (n>50), clinical cohort studies (n>100), and bioinformatics analyses retrieved from PubMed, Web of Science, and TCGA (The Cancer Genome Atlas). Studies increasingly show that genes involved in glucose and lipid metabolism, energy production, and modulation of metabolic hormones are regulated through epigenetic mechanisms. On the other hand, various metabolites participate in epigenetic modifications as coenzymes or substrates. Therefore, a greater understanding of the crosstalk between metabolism and epigenetics in cancer-related pathways could lead to the identification of key signaling molecules for targeted therapies, and raise the possibility of using dietary interventions to modulate epigenetic markers for individualized treatment. In this review, we have summarized the metabolic and epigenetic regulatory networks in cancer development, including glycolipid metabolic reprograming, the role of metabolites produced by the glut flora and tumor microenvironment, and key epigenetic drivers such as non-coding RNAs (ncRNAs). Data were curated from peer-reviewed articles, grounded in mechanistic studies using cell lines (SW480, MCF7 (Michigan cancer foundation-7)) and animal models (APC-mutant mice), with a focus on mechanistic studies, omics analyses, and translational research. Furthermore, we have discussed the potential of therapeutically targeting these pathways, along with the current challenges and future research directions, and a new strategy for reversing therapeutic drug resistance based on metabolism and epigenetic interaction was systematically explored.
1 Introduction
The occurrence and development of tumors are complex processes driven by multiple levels and dynamic molecular networks (1).With the in-depth study of epigenetic mechanisms, its central role in maintaining tumor cell identity, plasticity, and shaping intratumor heterogeneity has been revealed (2, 3). At the same time, the unique metabolic reprogramming characteristics of tumor cells, such as enhanced glycolysis, accelerated lipid synthesis and abnormal amino acid metabolism, have also had a profound impact on tumor biology on the premise of meeting the energy and biosynthetic requirements required for rapid tumor proliferation (4). These altered metabolic pathways produce a variety of specific metabolic intermediates and key substrates or cofactors that participate in and regulate epigenetic processes, such as histone modification and DNA methylation (5–7). The remodeling effect of metabolites on the epigenetic landscape plays a key connecting role in the cellular metabolic state and gene expression program (8, 9).
More critically, the interaction between metabolism and epigenetics is closely linked to the tumor microenvironment (TME) and immune regulation (10). On one hand, epigenetic modifiers regulate the expression of immune-related genes and influence the metabolic phenotype of tumor cells and the activity of immune cells (11). On the other hand, metabolites resulting from metabolic reprogramming in tumor cells, such as lactate and fatty acids, directly suppress the function of anti-tumor immune cells through epigenetic mechanisms, promote the activation of immune-suppressive cells, and facilitate tumor immune escape (12, 13). The gut microbiota, as an external driver, can promote tumor progression by regulating metabolites and immune responses when dysregulated. The interplay between metabolism, epigenetics, and immune regulation plays a key role in driving tumor progression (14). Our analysis integrated basic research (cell lines and animal models), clinical data (prospective cohorts), and omics datasets (TCGA, GEO (Gene Expression Omnibus)), including 387 cell line studies, 112 animal model investigations, and 45 clinical trials.
Metabolism and epigenetics and immune regulation play a key role in tumorigenesis, progression and treatment resistance (15, 16), the bidirectional regulatory mechanism between metabolism and epigenetics was verified in cell lines (SETD2 knockdown renal cancer cells) and mouse models (Kras mutated pancreatic cancer mice), which enabled us to have a more comprehensive understanding of their complex interactions and regulatory networks. In this review, we clarify the complex bidirectional regulatory relationship between metabolic reprogramming and epigenetic modification in tumors (17), how metabolites play the key role of regulatory factors in epigenetics, and how this interaction jointly promotes the progression of malignant tumors by influencing the immune microenvironment and non-metabolic pathways (18). We also explored targeted metabolic enzymes and epigenetic modification factors as key factors for developing new therapeutic directions, and proposed new insights for reversing treatment resistance and developing more precise anti-cancer therapies (19, 20) (Figure 1).
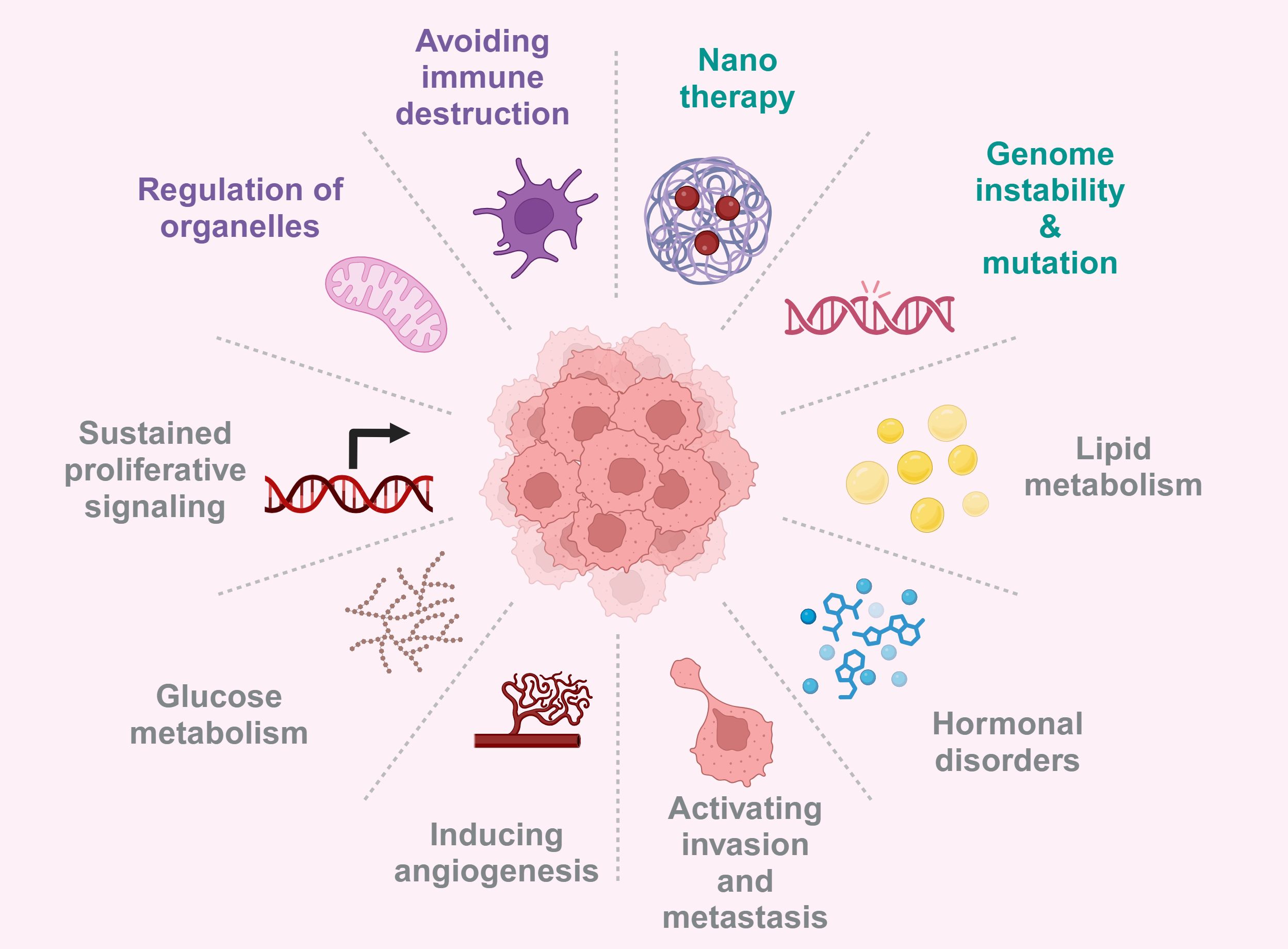
Figure 1. Tumorigenesis is driven by gene mutations, microenvironment abnormalities, metabolic reprograming, and hormonal dysregulation, thus provided multiple avenues for targeted therapies. Created with BioRender.com.
2 The modern era of epigenetic research
The study of whole-genome chromatin maps ushered in the era of modern epigenetic research (21). Epigenetics refers to inheritable changes in gene expression and cell phenotypes without altering the DNA sequence (Table 1).

Table 1. Epigenetic basis of molecular regulatory mechanisms and cellular processes.
3 Metabolic reprogramming features
Metabolic reprogramming is a phenomenon wherein cancer cells adjust their metabolic pathways to adapt to the TME and higher energy needs (Table 2).

Table 2. Molecular basis of metabolic reprogramming in cancer cells.
4 Regulation of gene expression by epigenetic modifying enzymes
4.1 The central role of SETD2-mediated H3K36me3 modification in tumor suppression and immune regulation
Trimethylation of histone H3 lysine at position 36 (H3K36me3) is an epigenetic modification that regulates gene transcription and messenger RNA (mRNA) splicing. Histone methyltransferase SET domain containing protein 2 (SETD2), the key enzyme catalyzing this modification, has been identified as a tumor suppressor and immune modulator. For example, in terms of immune regulation, SETD2’s regulation of regulatory T cells (Tregs) affects tumor control and antiviral response (30, 31). In addition, SETD2 can enhance the expression/function of GATA binding protein 3 (GATA3) in intestinal derived thymic regulatory T cells (tTreg cells), thereby promoting the expression of ST2 (interleukin-1 receptor like 1, IL1RL1) (32). The absence of SETD2 in prostate cancer (PCa) cells promotes excessive activation of enhancer-binding protein 2 (EZH2), leading to an increase in whole genome H3K27me3 and chromatin repression, which in turn inhibits expression of tumor suppressor genes and promotes metastasis (33). Furthermore, the SETD2-knockout polycystic kidney disease-clear cell renal cell carcinoma (PKD-ccRCC) mouse model exhibits increased tumorigenesis and poor survival (34). This phenomenon may be partly attributed to the impaired heme synthesis and accumulated ferroptosis-related factors, which collectively create a pro-TME conducive to malignant progression (35). In addition to renal cancer, in pancreatic cancer cells, the deletion of SETD2 will promote the reorganization of acinar to ductal metaplasia (ADM) of pancreatic acinar cells driven by the oncogene KRAS, which is mainly mediated by F-box and WD repeat domain protein 7 (Fbxw7) (36). In the SGC-7901 human gastric cancer cells, restoration of forkhead box O transcription factors (FOXO) signaling pathway agonist or S IRT1 expression reverses the increase in proliferation and migration caused by SETD2 deletion (37). Smad7 is a negative feedback regulator of the TGF-β/Smad signaling pathway, and SETD2 deficiency leads to Smad7 down-regulation, which promotes TGF-β/Smad hyperactivation and the trans differentiation of myofibroblasts (38). In addition, SETD2 deletion can promote renal fibrosis by activating the TGF-β/Smad signaling pathway, even in the absence of Von Hippel-Lindau (VHL) protein (39). Thus, histone methyltransferases have an important role in tumorigenesis and cancer progression.
4.2 The synergistic regulation of m6A modification and histone demethylation drives tumorigenesis and drug resistance
N6-methyladenosine (m6A) is the most abundant RNA modification and plays a key role in transcriptional regulation (40). Methyltransferase-like 3 (METTL3) is central to the formation of m6A, which subsequently binds to specific RNA-binding proteins that influence metabolic processes (41). The combination of a DNA hypomethylating agent (HMA) with a PARPi showed potent anti-tumor effects against SETD2-deficient RCC cells (42). In breast cancer cells (BRCA), METTL3 can stabilize and upregulate the PD-L1 mRNA upon binding to m6A-modified IGF2BP3 (43). METTL3-mediated m6A modification of uncapped mRNA2235 (DCP2) triggers its degradation, and promotes mitosis and chemoresistance in small cell lung cancer (SCLC) cells through the PINK1/Parkin pathway (44). Knockdown of METTL3 inhibited Pin1-induced clonal expansion of the breast cancer MCF7 cells, but promoted the growth of 4T1 tumors in vivo (45). While METTL3 regulates gene expression at the post-transcriptional level, histone lysine demethylases (KDMs) determine the accessibility of gene transcription through chromatin remodeling (46). For example, KDM6A regulates chromatin structure and DNA accessibility by removing methyl groups from H3K27me3, and removal of this histone repressor mark activates gene transcription (47). METTL3-mediated m6A modification of the HOXA9 oncogene promoter, and oncogene silencing by H3K27me3 in the absence of KDM6A have been shown to synergistically drive leukemogenesis (48). YTH domain family protein 2 (YTHDF2), a reading protein that recognizes m6A and promotes RNA degradation, promotes tumor growth by facilitating degradation of target mRNAs (49). The METTL3-YTHDF2 axis can accelerate colorectal carcinogenesis through epigenetic suppression of YPEL5 (50). Furthermore, the chromatin remains in the transcriptionally repressed state in the absence of KDM6A, which can complement the regulatory activity of METTL3/YTHDF2 (51). Overall, histone methyltransferases (HMTs) and KDMs mediate chromatin accessibility and gene transcriptional activity by dynamically regulating histone post-translational modifications (PTMS), such as H3K27me3, and their dysfunction can drive tumorigenesis. Therefore, these epigenetic pathways are promising targets for overcoming cancer heterogeneity and drug resistance (52, 53) (Figure 2).
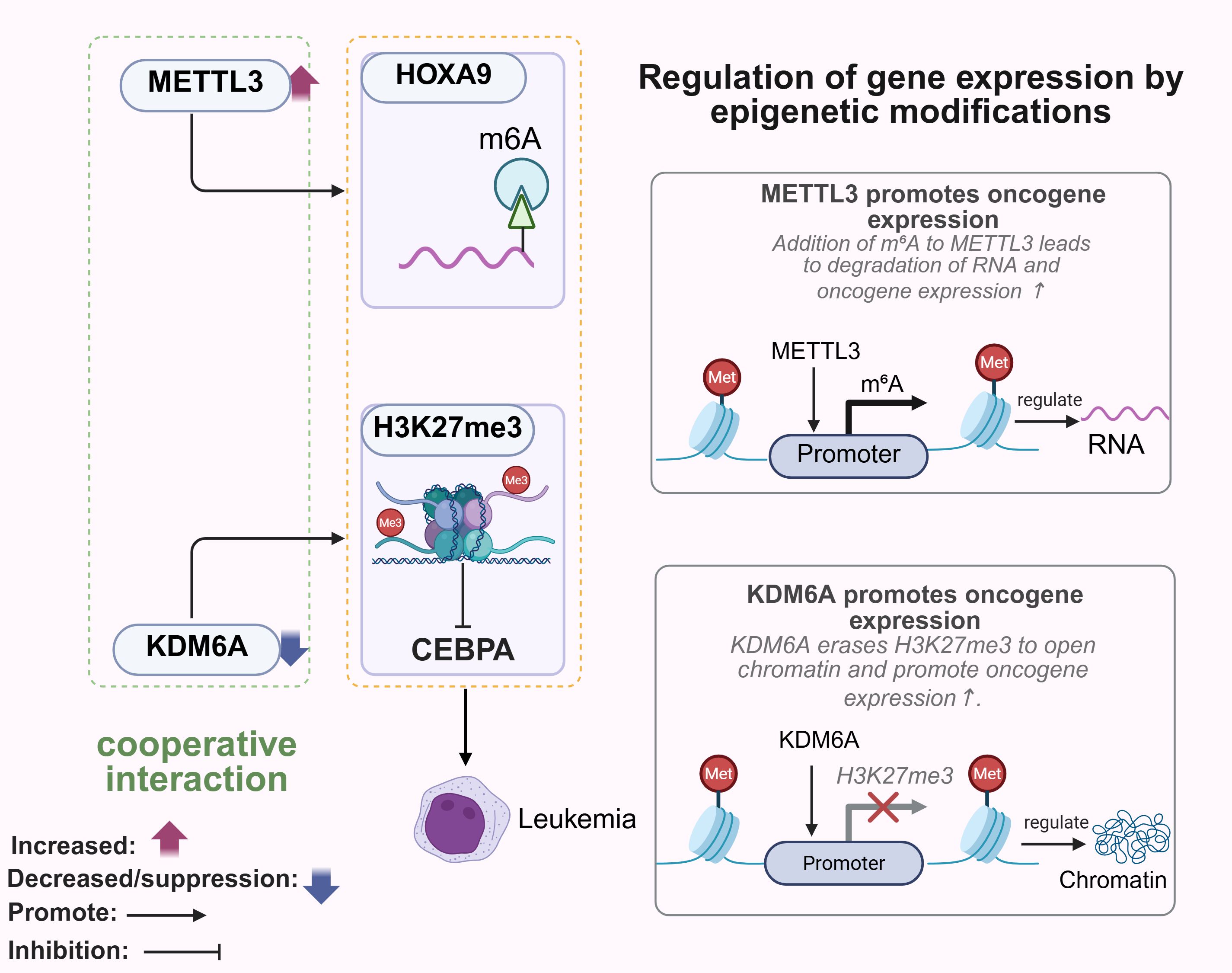
Figure 2. Role of methyltransferases and demethylases in tumorigenesis. METTL3 promotes m6A modification of HOXA9, whereas KDM6A deficiency leads to oncogene silencing by H3K27me3, which synergistically drives leukemogenesis. METTL3 affects RNA fate at the post-transcriptional level, and KDM6A determines the accessibility of gene transcripts through chromatin remodeling. METTL3, Methyltransferase-like 3, HOXA9, Homeobox A9. Created with BioRender.com.
5 Metabolites regulate epigenetic pathways as substrates and cofactors
Metabolites are important substrates and cofactors for epigenetic modifications, linking cellular metabolism with gene regulation. The metabolic phenotype of tumor cells exhibits heterogeneity, involving multiple metabolic regions. Its high metabolic demand requires H + to accumulate in cells, thereby forming an acidic and oxidative TME (54, 55). The metabolic state of a cell can influence gene expression patterns through epigenetic mechanisms (56, 57). The metabolites produced during metabolic reprogramming of tumor cells, such as acetyl-CoA, NAD+, SAM, and αKG, can specifically affect epigenetic pathways (58). Acetyl CoA is produced during the metabolism of sugars, lipids, and proteins, and is involved in energy production, biosynthesis, and epigenetic regulation (59). The chemical modification and structural remodeling of chromatin depend on metabolic cofactors, and metabolite availability is therefore a direct indicator of changes in the epigenome (60, 61). CoA provides acetyl groups, promotes H3K27ac acetylation, determines histone acetyl transferase activity, and influences the expression of metabolism-related genes (62). The fluctuation of acetyl CoA levels plays a crucial role in regulating lipid synthesis. This regulation mainly occurs through epigenetic mechanisms: acetyl CoA dependent histone acetylation alters chromatin accessibility and structure, changes the chromatin status of lipid metabolism related genes, and affects the activity of related enzymes. Therefore, these changes affect the lipid synthesis pathway, leading to cellular metabolic disorders and cancer occurrence (63, 64). Targeting metabolic epigenetic interactions offers therapeutic potential. For example, in the phosphatidylinositol signaling pathway, breviscapine prevents the progression of metabolic stress-induced nonalcoholic steatohepatitis (NASH) by directly inhibiting TAK1 signaling (65).
Metabolic regulation plays a crucial role in various cellular processes (66), and is often impaired during carcinogenesis (67). Changes in metabolite levels may also affect epigenetic modifications by regulating the activity of specific enzymes (68). The metabolites and enzymes produced during metabolic reprogramming of tumor cells activate or inhibit certain epigenetic changes (69–71), such as DNA methylation and histone modifications, thereby coordinating cellular activity with altered nutrient availability (69). For instance, the catalytic activity of N-alpha acetyltransferase 40 (NAA40), a histone acetyltransferase (HAT), depends on acetyl CoA. The latter is a direct source of acetyl groups, which are transferred to specific sites on histone H4 by NAA40 (72). Thus, the increased abundance of acetyl CoA in tumor cells with high glucose metabolism may drive aberrant NAA40-mediated histone acetylation, resulting in the activation of pro-oncogenes (63). In addition, environmental carcinogens can hijack these metabolic and epigenetic pathways to promote cancer. Benzo (a) pyrene (BAP) is a polycyclic aromatic hydrocarbon (PAH) and potent organic toxicant, which forms reactive epoxide metabolites through metabolic activation. These metabolites can react with DNA to form adducts, leading to the mutation of key tumor suppressor genes, such as p53, which is the key mechanism of BaP induced lung carcinogenesis (73). Importantly, BAP exposure can also cause epigenetic dysregulation. It is associated with genome-wide DNA hypomethylation and may be caused by DNA methylation or inhibition of HDAC activity. This hypomethylation, coupled with the finding that this inhibition reduces the activities of biotin dependent enzymes, such as biotinidase (BTD) and holocarboxylase synthetase (HCS), which are themselves regulated by other epigenetic mechanisms, represents another important way for BAP to promote cancer development (74). Taken together, the metabolic heterogeneity of tumor cells affects histone diversity and epigenetic regulation, and plays a significant role in tumor progression (Table 3).

Table 3. Epigenetic modification of specific metabolites is a key factor in the development of cancer.
5.1 Regulation of epigenetics by lipid metabolism
Increased de novo lipid synthesis is a key feature of many cancers (80). Cholesterol, an important component of the cell membrane, plays an indispensable role in cell growth and signaling (81, 82). Cholesterol, an important component of the cell membrane, plays an indispensable role in cell growth and signaling. Epigenetic mechanisms such as DNA methylation and histone modifications can regulate intracellular lipid concentration and signaling pathways (83). Recent studies have shown that the transcription factor Ikaros influences tumor development by modulating cholesterol metabolism pathways in the TME (84). Similarly, KLF10 exerts a protective effect in metabolic liver disease by regulating HNF4α-mediated metabolic pathways (85). Studies show that low-density lipoprotein (LDL) downregulates Krüppel-like factor 2 (KLF2) in endothelial cells through DNA and histone methylation, resulting in endothelial dysfunction and a hypercoagulable state (86). Furthermore, hyperlipidemia-induced coronary heart disease and peripheral artery disease have also been linked to epigenetic changes induced by circular RNAs (such as MICRA), and other RNA-level epitranscriptomics (87). Metabolite mediated epigenetic reprogramming further demonstrates this interaction. These findings collectively suggest that lipid metabolites are key epigenetic regulators. Among them, cholesterol biosynthetic intermediates, lipoproteins and acetyl CoA directly remodel chromatin structure through methylation, acetylation and RNA mediated mechanisms, thus linking metabolic disorders with tumorigenesis, vascular pathology and metabolic organ dysfunction.
5.2 Remodeling of gene expression by glucose metabolism
Glycolysis provides energy for lactate dependent epigenetic reprogramming. Elevated glycolysis in tumors generates lactate, driving lactylation – a novel post-translational modification that directly activates gene transcription through chromatin remodeling (6, 88, 89). Increased production of lactic acid in the TME is known to induce immunosuppressive conditions, which can mitigate the response to immunotherapies. In tumor cells, elevated glycolysis drives cancer cell metastasis by activating oncogenes ccl2/7 through h3k18 lactylation (h3k18la). Furthermore, lactate regulates epigenetic modifications by altering the chromatin (90). Specifically, lactate enhances the recruitment of the key homologous recombination (HR) protein MRE11 to DNA damage sites, thereby promoting DNA end resection and HR repair (91). Considering tumor metabolism and tumor immunity, exploring targeted lactoacylation has great potential for the development of cancer treatment strategies (89, 92). For example, two lactose modification sites were found in the zinc finger domain sample mettl3. Emulsification driven mettl3 mediated RNA m6A modification plays an important role in promoting tumor infiltrating myeloid cells (TIM) (93). In conclusion, glucose metabolism derived lactate regulates gene expression by remodeling the expression network of Pro metastatic genes, immunosuppressive genes, and genes related to genome stability through h3k18la, mettl3-m6a modification, and Mre11 mediated DNA repair triple epigenetic mechanism (Figure 3).
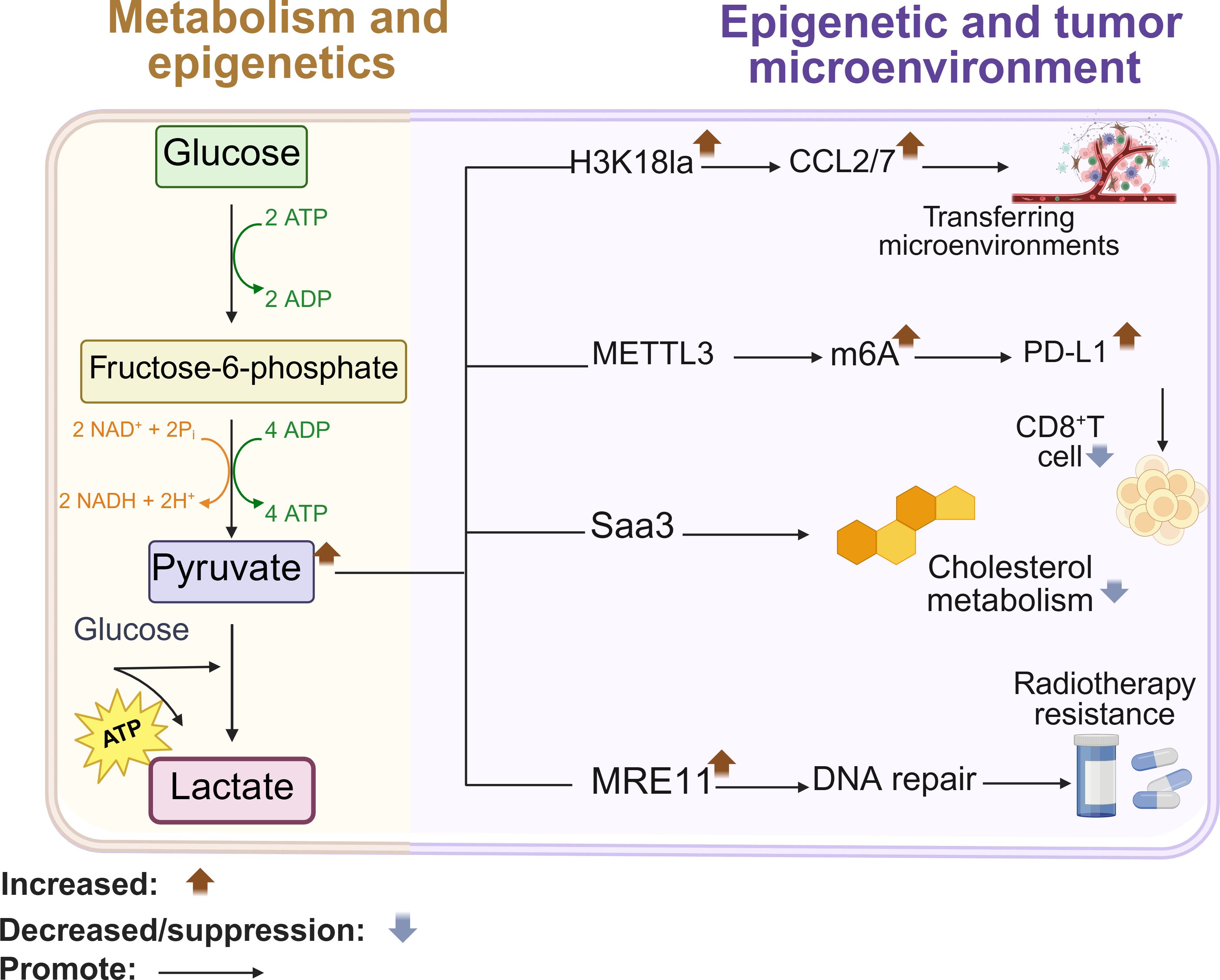
Figure 3. Glucose metabolism remodels gene expression. High glycolysis enhances transcription of the oncogene CCL2/7 through H3K18la modification, and promotes metastasis. Lactylation of METTL3 increases m6A modification in the PD-L1 mRNA promoter, resulting in increased transcript stability that eventually activates the immune checkpoint against T cells. Lactylation of MRE11 also promotes DNA repair and chemoresistance. Created with BioRender.com.
5.3 Conduct a systematic assessment of the hypothesis that “cancer is essentially a metabolic disease”
The assumption that cancer is fundamentally a metabolic disease: the assumption that metabolic reprogramming precedes and drives malignant transformation, but this assumption is still controversial at present. Although metabolic alterations are an indisputable feature of cancer, they are primary compared to gene mutations and require rigorous assessment. Studies have shown that mitochondrial dysfunction is observed in more than 80% of tumors. In models such as Kras mutant pancreatic cancer, damage to oxidative phosphorylation (OXPHOS) precedes genomic instability (94). Tumor metabolites, such as 2-HG from IDH mutations, directly disrupt epigenetic mechanisms, inducing hypermethylation and silencing tumor suppressors before significant mutations accumulate (95). Metabolic disorders, through endoplasmic reticulum stress, increase the apoptosis of cancer cells, thereby promoting the progression of endometrial cancer and inducing tumor occurrence and progression (96). However, most of the studies have limitations. For example, there are doubts about causal timing: whether metabolic changes precede driver mutations (TP53, APC) still has inferential significance (97). In addition, the Warburg effect in the hypoxic microenvironment may be a survival adaptation rather than a carcinogenic source, which is worthy of in-depth exploration (98). Although the origin remains unclear, the interaction between targeted metabolism and epigenetics shows clinical prospects: metformin takes advantage of the metabolic vulnerability of breast cancer, and its efficacy may depend on the STK11 status (99). hypothesis that cancer is essentially a metabolic disease is supported by certain evidence and shows potential in clinical applications, the causal relationship between metabolic reprogramming and genetic mutations, as well as the exact role of metabolic alterations in tumorigenesis, still require more comprehensive and in-depth research to clarify its scientific validity and practical significance.
6 Non-metabolic epigenetic mechanism based on enzyme translocation
Various intermediate metabolites can alter chromatin structure and function through chemical PTMS (100). Studies show that tumor metabolites are heavily influenced by the microenvironment (101), and enzyme translocation from the nucleus and mitochondria may alter the metabolism of cancer cells and their interactions with stromal cells in the TME (102). Apart from mediating metabolic reactions, enzyme translocation also regulates epigenetic pathways through non-metabolic effects (103). Therefore, the role of tumor metabolites in different organelles has important implications for the interplay between metabolism and epigenetics.
6.1 Nonmetabolic functions of the endoplasmic reticulum: calcium signaling and cholesterol homeostasis regulate tumor progression
The endoplasmic reticulum is the site of protein translation, folding and processing, as well as lipid secretion (104). Aberrant lipid metabolism or dysregulated ion transport in the endoplasmic reticulum can trigger organelle stress and tumorigenesis (105). Endoplasmic reticulum transmembrane protein 147(TMEM147) promotes the proliferation and metastasis of tumor cells, endows them with resistance to iron-mediated cell death, and induces polarization of M2-type macrophages by disrupting cholesterol homeostasis and increasing 27HC secretion (106). In addition, calcium ion in endoplasmic reticulum regulates the pathway related to tumor cell growth and drug resistance (107). Sigma-1 receptor (Sig-1R) is a molecular chaperone protein located in the endoplasmic reticulum, which plays a key role in regulating the endoplasmic reticulum calcium channel, which controls the growth of tumor cells and drug resistance (108). Sig-1R is located in the endoplasmic reticulum mitochondria associated membrane (MAM) domain. By sensing the change of calcium ion (CA ² + concentration) in the endoplasmic reticulum cavity, Sig-1R regulates Ca ² + signal transmission between mitochondria and cells, thereby affecting cell survival (109).
6.2 Targeted mitochondrial-related epigenetic reprogramming: a hub for metabolic adaptation and therapeutic resistance
Increased glycolysis may be related to mitochondrial dysfunction and enzymatic changes in tumor cells (110, 111). The key enzymes and intermediates of the glycolytic pathway can be used by tumor cells to synthesize proteins and nucleic acids, or protect mitochondrial function (112). On the other hand, mitochondrial intermediates initiate epigenetic pathways in the nucleus, and the resulting epigenetic marks regulate the expression of mitochondrial proteins (113, 114), a key bidirectional regulatory circuit is formed between the nucleus and mitochondria. For example, in pancreatic cancer, epigenetic defects of tumor cells (such as abnormal h3k27ac modification caused by setd2 deletion) were found to be associated with specific metabolic phenotypes (115). More importantly, epigenetic disorders (including abnormal histone modification and DNA methylation changes) can significantly change the functional state of mitochondria (116–118). Therefore, the regulation of nuclear gene expression by epigenetic mechanism directly affects the level and activity of these mitochondrial proteins, and then regulates the level of mitochondrial metabolites needed to maintain cell function (119). For instance, METTL17 regulates mitochondrial function in colorectal cancer (CRC) cells through epigenetic regulation (120). Therefore, exploring the interaction between epigenetic mechanism and mitochondrial function, especially the regulation of epigenetic mechanism on mitochondria, provides an important way for developing new targeted therapy strategies.
7 Metabolic reprogramming, sugar, lipid metabolism, crosstalk between epigenetics three relations
Metabolic reprogramming can modulate the function of intra-tumoral immune cells by altering the concentration of intracellular metabolites (121–123), making it a key feature of tumorigenesis and progression. Tumor-derived exosomes (TDE) stimulate elevated nitric oxide synthase 2 (NOS2), thereby inhibiting mitochondrial oxidative phosphorylation, and promoting conversion of pyruvate to lactate (124). Furthermore, enhanced glycolysis in cancer cells increases acetyl Coa levels, which promotes up-regulation of oncogenes (MYC) through histone acetylation (125). Furthermore, epigenetic modifications can regulate the genes involved in glycolipid metabolism and promote tumorigenesis and progression (126). For example, increased methylation of the insulin gene (INS) promoter in pancreatic beta cells promotes gene silencing in diabetes (127). On the other hand, β-hydroxybutyric acid (ketone bodies) can activate antioxidant genes like FOXO3A by inhibiting HDACs and increasing histone acetylation (128). Therefore, the crosstalk between epigenetic mechanisms, glucose metabolism, and lipid metabolism regulates gene expression in metabolic diseases and cancer (Figure 4).
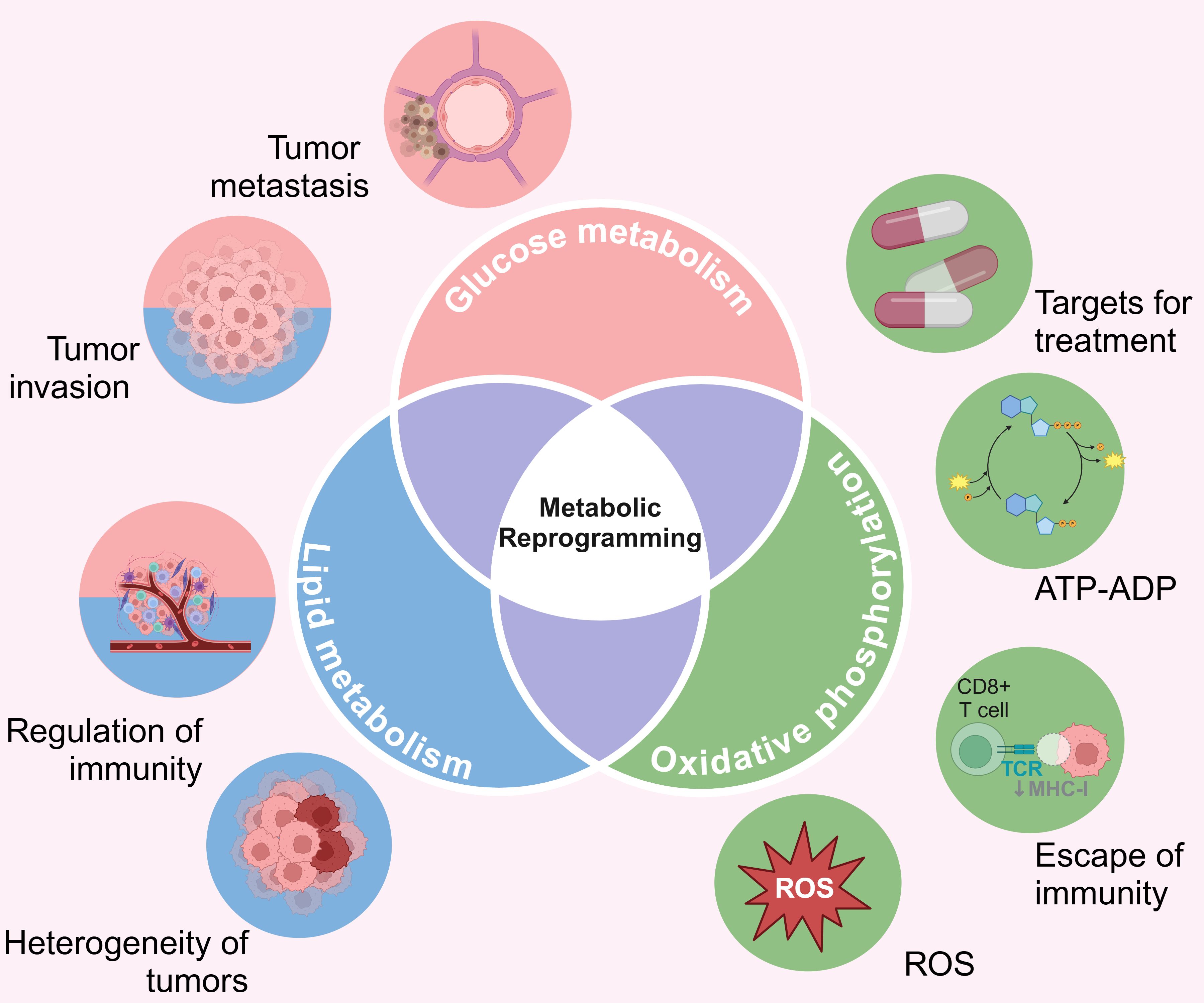
Figure 4. The cross-talk between metabolic reprogramming, glucose metabolism, lipid metabolism, and epigenetics. ATP, Adenosine triphosphate; ADP, Adenosine diphosphate; ROS, Reactive oxygen species. Created with BioRender.com.
8 Internal and external factors jointly drive tumor progression through metabolism, immunity and epigenetics
8.1 External factors (microbiome) drive cancer by affecting internal metabolism and immunity
Clinical research on the effect of microbes on cancer began in 1868 and plays an important role in maintaining the ecological balance (129). The current scientific community believes that dysbiosis of gut microbiota is a hallmark of cancer (130, 131). The process of carcinogenesis is a highly complex and involves a variety of physiological and pathological events. Multiple sequencing methods have shown that the microbiota in the lung and intestine can cross-talk and are important components of TME (132). In CRC, which is the most common cancer, gut microbiota causes CRC by altering immune function (133, 134). Found microbes have the function of the dialectic, namely in the protection of human genes will damage to human multiple genes to form at the same time, its mechanism is mainly its ecological imbalance can by changing the host susceptibility to cancer events (including pathogenic microorganisms increase load) to significantly promote this process (135, 136). Gastrointestinal cancer is one of the main causes of cancer death at present. Intestinal microorganisms can damage cells or change the tumor immune microenvironment through direct or indirect effects to promote the development of gastrointestinal tumors (137, 138). Gut microbiota may be detecting high-risk PCa as a new useful marker, intestinal bacteria and their metabolites, short chain fatty acids (SCFAs), can promote PCa mouse model of cancer growth (139). Intestinal bacteria microbes in the estrogen group, namely, to metabolism of the aggregate of the intestinal bacterial genes of estrogen, increase the risk of BRCA in women (140). Symbiotic microbes are one of the factors influencing the antitumor immunity and treatment outcomes (141). The understanding of host-microbiome interactions and the assessment of microbial composition and function in patients provide a theoretical basis for subsequent targeted regulation and targeted interventions to reduce cancer risk (142, 143).
8.2 TME internal core process (metabolic reprogramming) drives cancer by regulating epigenetics and immunity
Metabolic rewiring of the tumor cells and immune cells regulates tumor progression by shaping the epigenome in TME (100). Although cancer is initiated by genomic alterations, it is more of a metabolic disease (144). Epigenetic pathways regulate gene expression levels by integrating environmental stimuli. Normal cells and cancer cells differ considerably in their metabolic levels (145, 146). Recent evidence suggests that a significant enhancement of the glycolytic pathway is a major feature of tumor cells (147), and energy production through glycolysis and lipid synthesis is fundamental to their ability to proliferate uncontrollably (148). Epigenetic regulation is highly sensitive to metabolic cues, and the metabolites in the TME can alter key epigenetic factors or enzyme activity to allow cancer cells to quickly adapt to the dynamic environment (149, 150). For instance, lactic acid and fatty acids can epigenetically regulate the function of immune cells and affect tumor resistance (151–153). Studies show that V-ATPase V0 subunit d2 (ATP6V0d2) expressed by macrophages inhibits tumor growth in vivo, while tumor-derived lactate inhibits ATP6V0d2 in macrophages, thereby promoting HIF-2α-mediated tumor progression (154). Lactate-derived histone lysine lactylation in the TME is a novel epigenetic modification that can directly stimulate chromatin structure and gene expression (6). TME reverses the effects of immune cells through a coordinated “regulatory triad”. Cancer cells compete with normal cells for key nutrients, which damage the immune cell function. The accumulation of tumor metabolites in the TME support the immunosuppressive cells and impair T cell function. At the same time, the spatial distribution, composition, and activation state of immune cells in tumor cells can influence the outcomes of immunotherapy (155, 156).
9 New insights into reversing treatment resistance and developing precise anti-cancer therapies
9.1 New insights into reversing resistance to therapy
The dysregulation of the interaction between metabolism and epigenetics can lead to drug resistance in tumor treatment. Recent studies have revealed innovative strategies targeting this intersection: First, overcome drug resistance through the combination of epigenetics and metabolic enzymes. For example, in renal cancer cells with SETD2 deficiency, the combination of DNA hypomethylation agents (HMA) and PARP inhibitors can overcome drug resistance by inducing synthetic lethal effects (42). This strategy is supported by preclinical data from 17 cell line studies and 3 phase II trials (NCT02850058, NCT03252097), as well as single-cell RNA-seq analysis of tumor-immune cell interactions. Second, intervention through the microbiome, metabolism and epigenetic axes. For example, animal models have confirmed that intragastric administration of butyrate can enhance the ability of chemotherapy drugs to penetrate the blood-brain barrier and reverse drug resistance in glioma. Personalized probiotic intervention based on microbiome characteristics is being verified for its sensitizing effect in clinical trials (157).
9.2 Developing precision cancer therapies using ncRNA delivery systems
Analysis of TCGA has revealed significant difference in the expression of ncRNAs between tumor tissues and normal tissues (158). NcRNAs are a group of heterogeneous transcripts that are not translated into proteins, but regulate gene expression at the post-transcriptional and post-translational levels (159–161). The m6A modification in the ncRNAs has been associated with gene expression levels and the biological behavior of tumor cells, thus providing potential new targets for cancer therapy (162, 163). For instance, several ncRNAs secreted by the Tumor-Associated Macrophage (TAMs) promote tumor proliferation, metastasis, angiogenesis, chemotherapy resistance, and immunosuppression (164, 165), and may also drive M1 or M2 polarization of the macrophages (166). By influencing molecular targets, ncRNAs influence alternative splicing (AS) processes and generate AS isomers, thereby promoting or inhibiting cancer signaling pathways (167). NcRNAs are also transported via extracellular vesicle (EVs) to regulate tumor development (168). For example, studies have confirmed that targeted delivery of miR-122 to liver cancer cells by cationic lipid nanoparticles (NPs) can inhibit angiogenesis and tumor growth (169). Similarly, the antitumor effects of siRNA and miRNA inhibitors have been verified in glioblastoma (170). In addition, the migration and clonal proliferation of A549 non-small cell lung cancer (NSCLC) cells were significantly inhibited by blocking the transcription factor -1(MALAT-1) mediated by RNA interference (171). In the liver metastasis model of colorectal cancer, the ncRNA delivery system has been precisely regulated. Specific delivery of miR-122 to hepatocytes through nanoparticles can down-regulate metastasis-related genes (MMPs), significantly inhibit tumor growth and prolong survival time, which provides a new idea for precise treatment (172). These innovative strategies based on the interaction between metabolism and epigenetics can reverse drug resistance, open up new ideas for precise anti-cancer treatment, and finally realize truly personalized cancer treatment.
10 Frontier progress and future directions of metabolism and epigenetic interaction in tumor research
At present, there is a lack of systematic analysis of intra-tumor heterogeneity. Future research should integrate single-cell multi-omics data (scRNA-seq, spatial metabonomics), use the database of Human Tumor Atlas Network and Tabula Sapiens, and combine the longitudinal clinical data of prospective cohort. The role of intestinal microflora needs to be verified by large-scale metabonomic research (American Intestinal Project and Human Microbial Project), and the relationship between microbial metabolites and epigenetic characteristics should be established (94). Key directions include: verifying the causal relationship between microbial metabolites (short-chain fatty acids) and epigenetic reprogramming in clinical cohort (137). At present, the treatment strategy for a single pathway faces challenges in terms of efficacy and specificity. Future efforts should focus on developing metabolically sensitive epigenetic regulatory factors as drug targets, for example, inhibiting tumor progression dependent on lipid synthesis by targeting acetyl-CoA-NAA40 axis (72). Additionally, exploring how non-coding RNAs regulate macrophage polarization by targeting metabolic-epigenetic crosstalk could further expand the repertoire of drug targets for remodeling the pro-TME (166). This aligns with the notion that innate immune cells, particularly macrophages, dynamically integrate metabolic cues and epigenetic reprogramming to shape the TME, as highlighted in the interplay between innate immunity and cancer pathophysiology (156). Specifically, tumor-derived lactic acid, as a key metabolic cue, modulates the activation and metabolic reprogramming of fibroblastic reticular cells in draining lymph nodes, thereby cooperating with innate immune cells to foster a pre-metastatic niche conducive to tumor progression (153). The integration of metabolism and epigenetic research represents the forefront of precision oncology. Future research must combine mechanism understanding with technological innovation to solve the problem of causality and transformation, so as to release the full potential of personalized cancer treatment.
11 Conclusion
In this review, we have explored the crosstalk between epigenetics and metabolism in tumor progression. While epigenetic mechanisms can affect metabolic reprogramming and immune infiltration in the TME, the local metabolites regulate tumor progression by targeting epigenetic factors. However, our knowledge of tumor metabolomics is incomplete, and the epigenetic mechanisms controlling glycolipid metabolism pathways, and their impact on tumor growth need to be explored. Advances in genomics and proteomics can provide new insights into metabolic mechanisms and key regulatory pathways, and help in the development of more effective therapies. A deeper understanding of the relationship between epigenetics and glycolipid metabolism in cancer will be of clinical significance, and pave the way for targeted and personalized therapies.
Author contributions
XZ: Conceptualization, Data curation, Writing – original draft. DL: Formal Analysis, Writing – original draft. SY: Investigation, Writing – original draft. YG: Methodology, Writing – original draft. XL: Resources, Writing – original draft. GW: Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by funding from the Liaoning Provincial Department of Education (numbers JYTMS20230577) and funded by the Dalian Life and Health field Guidance Plan (2024ZDJH01PT068).
Acknowledgments
Figures 1-4 were created with BioRender.com. We sincerely thank DeepL for providing language services. We invited native English speakers at NGS assistant to polish the article, to assure compliance with journal academic standards in terms of style, punctuation, grammar, and spelling.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
APC, Adenomatous polyposis coli; AS, Alternative splicing; ATP6V0d2, V-ATPase V0 subunit d2; BCAT1, Branched-Chain Amino Acid Transaminase 1; BRCA, Breast cancer; CRC, Colorectal cancer; FOXO, Forkhead box O transcription factors; HDAC, Histone deacetylase; KDM, Histone lysine demethylase; m6A, N6-methyladenosine; MCF7, Michigan cancer foundation – 7; METTL3, Methyltransferase-like 3; mRNA, Messenger RNA; NAA40, N-alpha acetyltransferase 40; ncRNA, Non-coding RNA; PARPi, PARP inhibitor; PCa, Prostate cancer; PD-L1, Programmed death-ligand 1; PTMS, Post-translational modifications; SETD2, SET domain-containing protein 2; Sig-1R, Sigma-1 receptor; TCGA, The cancer Genome Atlas; TME, Tumor microenvironment; YTHDF2, YTH domain family protein 2.
References
1. Hanahan D and Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
2. Baylin SB and Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer. (2011) 11:726–34. doi: 10.1038/nrc3130
3. Flavahan WA, Gaskell E, and Bernstein BE. Epigenetic plasticity and the hallmarks of cancer. Science. (2017) 357:eaal2380. doi: 10.1126/science.aal2380
4. Vander Heiden MG and DeBerardinis RJ. Understanding the intersections between metabolism and cancer biology. Cell. (2017) 168:657–69. doi: 10.1016/j.cell.2016.12.039
5. Kinnaird A, Zhao S, Wellen KE, and Michelakis ED. Metabolic control of epigenetics in cancer. Nat Rev Cancer. (2016) 16:694–707. doi: 10.1038/nrc.2016.82
6. Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. Metabolic regulation of gene expression by histone lactylation. Nature. (2019) 574:575–80. doi: 10.1038/s41586-019-1678-1
7. Shi L and Tu BP. Acetyl-CoA and the regulation of metabolism: mechanisms and consequences. Curr Opin Cell Biol. (2015) 33:125–31. doi: 10.1016/j.ceb.2015.02.003
8. Reid MA, Dai Z, and Locasale JW. The impact of cellular metabolism on chromatin dynamics and epigenetics. Nat Cell Biol. (2017) 19:1298–306. doi: 10.1038/ncb3629
9. Katada S, Imhof A, and Sassone-Corsi P. Connecting threads: epigenetics and metabolism. Cell. (2012) 148:24–8. doi: 10.1016/j.cell.2012.01.001
10. DeBerardinis RJ and Chandel NS. We need to talk about the Warburg effect. Nat Metab. (2020) 2:127–9. doi: 10.1038/s42255-020-0172-2
11. Pfister SX, Ahrabi S, Zalmas LP, Sarkar S, Aymard F, Bachrati CZ, et al. SETD2-dependent histone H3K36 trimethylation is required for homologous recombination repair and genome stability. Cell Rep. (2014) 7:2006–18. doi: 10.1016/j.celrep.2014.05.026
12. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. (2014) 513:559–63. doi: 10.1038/nature13490
13. Watson MJ, Vignali PDA, Mullett SJ, Overacre-Delgoffe AE, Peralta RM, Grebinoski S, et al. Metabolic support of tumour-infiltrating regulatory T cells by lactic acid. Nature. (2021) 591:645–51. doi: 10.1038/s41586-020-03045-2
14. O’Sullivan D, Sanin DE, Pearce EJ, and Pearce EL. Metabolic interventions in the immune response to cancer. Nat Rev Immunol. (2019) 19:324–35. doi: 10.1038/s41577-019-0140-9
15. Leone RD and Powell JD. Metabolism of immune cells in cancer. Nat Rev Cancer. (2020) 20:516–31. doi: 10.1038/s41568-020-0273-y
16. Faubert B, Solmonson A, and DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science. (2020) 368:eaaw5473. doi: 10.1126/science.aaw5473
17. Martínez-Reyes I and Chandel NS. Cancer metabolism: looking forward. Nat Rev Cancer. (2021) 21:669–80. doi: 10.1038/s41568-021-00378-6
18. Carrer A and Wellen KE. Metabolism and epigenetics: a link cancer cells exploit. Curr Opin Biotechnol. (2015) 34:23–9. doi: 10.1016/j.copbio.2014.11.012
19. Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nat Rev Drug Discov. (2011) 10:671–84. doi: 10.1038/nrd3504
20. Dawson MA and Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. (2012) 150:12–27. doi: 10.1016/j.cell.2012.06.013
21. Allis CD and Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet. (2016) 17:487–500. doi: 10.1038/nrg.2016.59
22. Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. (2012) 13:484–92. doi: 10.1038/nrg3230
23. Zhu X, Hong S, Bu J, Liu Y, Liu C, Li R, et al. Antiviral memory B cells exhibit enhanced innate immune response facilitated by epigenetic memory. Sci Adv. (2024) 10:eadk0858. doi: 10.1126/sciadv.adk0858
24. Grover P, Asa JS, and Campos EI. H3-H4 histone chaperone pathways. Annu Rev Genet. (2018) 52:109–30. doi: 10.1146/annurev-genet-120417-031547
25. Oh ES and Petronis A. Origins of human disease: the chrono-epigenetic perspective. Nat Rev Genet. (2021) 22:533–46. doi: 10.1038/s41576-021-00348-6
26. Zaib S, Rana N, and Khan I. Histone modifications and their role in epigenetics of cancer. Curr Med Chem. (2022) 29:2399–411. doi: 10.2174/0929867328666211108105214
27. Zhou YJ, Li G, Wang J, Liu M, Wang Z, Song Y, et al. PD-L1: expression regulation. Blood Sci. (2023) 5:77–91. doi: 10.1097/BS9.0000000000000149
28. Quinn O, Kumar M, and Turner S. The role of lipid-modified proteins in cell wall synthesis and signaling. Plant Physiol. (2023) 194:51–66. doi: 10.1093/plphys/kiad491
29. Qian L, Li N, Lu XC, Xu M, Liu Y, Li K, et al. Enhanced BCAT1 activity and BCAA metabolism promotes RhoC activity in cancer progression. Nat Metab. (2023) 5:1159–73. doi: 10.1038/s42255-023-00818-7
30. Xie Y, Sahin M, Sinha S, Wang Y, Nargund AM, Lyu Y, et al. SETD2 loss perturbs the kidney cancer epigenetic landscape to promote metastasis and engenders actionable dependencies on histone chaperone complexes. Nat Cancer. (2022) 3:188–202. doi: 10.1038/s43018-021-00316-3
31. Leung W, Teater M, Durmaz C, Meydan C, Chivu AG, Chadburn A, et al. SETD2 haploinsufficiency enhances germinal center-associated AICDA somatic hypermutation to drive B-cell lymphomagenesis. Cancer Discov. (2022) 12:1782–803. doi: 10.1158/2159-8290.CD-21-1514
32. Ding Z, Cai T, Tang J, Sun H, Qi X, Zhang Y, et al. Setd2 supports GATA3(+)ST2(+) thymic-derived Treg cells and suppresses intestinal inflammation. Nat Commun. (2022) 13:7468. doi: 10.1038/s41467-022-35250-0
33. Yuan H, Han Y, Wang X, Li N, Liu Q, Yin Y, et al. SETD2 restricts prostate cancer metastasis by integrating EZH2 and AMPK signaling pathways. Cancer Cell. (2020) 38:350–65.e7. doi: 10.1016/j.ccell.2020.05.022
34. Rao H, Liu C, Wang A, Ma C, Xu Y, Ye T, et al. SETD2 deficiency accelerates sphingomyelin accumulation and promotes the development of renal cancer. Nat Commun. (2023) 14:7572. doi: 10.1038/s41467-023-43378-w
35. Xue W, Jian W, Meng Y, Wang T, Cai L, Yu Y, et al. Knockdown of SETD2 promotes erastin-induced ferroptosis in ccRCC. Cell Death Dis. (2023) 14:539. doi: 10.1038/s41419-023-06057-8
36. Niu N, Lu P, Yang Y, He R, Zhang L, Shi J, et al. Loss of Setd2 promotes Kras-induced acinar-to-ductal metaplasia and epithelia-mesenchymal transition during pancreatic carcinogenesis. Gut. (2020) 69:715–26. doi: 10.1136/gutjnl-2019-318362
37. Feng W, Ma C, Rao H, Zhang W, Liu C, Xu Y, et al. Setd2 deficiency promotes gastric tumorigenesis through inhibiting the SIRT1/FOXO pathway. Cancer Lett. (2023) 579:216470. doi: 10.1016/j.canlet.2023.216470
38. Liang S, Zheng R, Zuo B, Li J, Wang Y, Han Y, et al. SMAD7 expression in CAR-T cells improves persistence and safety for solid tumors. Cell Mol Immunol. (2024) 21:213–26. doi: 10.1038/s41423-023-01120-y
39. Liu C, Ni L, Li X, Rao H, Feng W, Zhu Y, et al. SETD2 deficiency promotes renal fibrosis through the TGF-β/Smad signalling pathway in the absence of VHL. Clin Trans Med. (2023) 13:e1468. doi: 10.1002/ctm2.1468
40. Wang Q, Geng W, Guo H, Wang Z, Xu K, Chen C, et al. Emerging role of RNA methyltransferase METTL3 in gastrointestinal cancer. J Hematol Oncol. (2020) 13:57. doi: 10.1186/s13045-020-00895-1
41. Yan C, Xiong J, Zhou Z, Li Q, Gao C, Zhang M, et al. A cleaved METTL3 potentiates the METTL3-WTAP interaction and breast cancer progression. eLife. (2023) 12:RP87283. doi: 10.7554/eLife.87283
42. Zhou X, Sekino Y, Li HT, Fu G, Yang Z, Zhao S, et al. SETD2 deficiency confers sensitivity to dual inhibition of DNA methylation and PARP in kidney cancer. Cancer Res. (2023) 83:3813–26. doi: 10.1158/0008-5472.CAN-23-0401
43. Wan W, Ao X, Chen Q, Yu Y, Ao L, Xing W, et al. METTL3/IGF2BP3 axis inhibits tumor immune surveillance by upregulating N(6)-methyladenosine modification of PD-L1 mRNA in breast cancer. Mol Cancer. (2022) 21:60. doi: 10.1186/s12943-021-01447-y
44. Sun Y, Shen W, Hu S, Lyu Q, Wang Q, Wei T, et al. METTL3 promotes chemoresistance in small cell lung cancer by inducing mitophagy. J Exp Clin Cancer Res: CR. (2023) 42:65. doi: 10.1186/s13046-023-02638-9
45. Bhattarai PY, Kim G, Lim SC, Mariappan R, Ohn T, and Choi HS. METTL3 stabilization by PIN1 promotes breast tumorigenesis via enhanced m(6)A-dependent translation. Oncogene. (2023) 42:1010–23. doi: 10.1038/s41388-023-02617-6
46. Chakraborty AA, Laukka T, Myllykoski M, Ringel AE, Booker MA, Tolstorukov MY, et al. Histone demethylase KDM6A directly senses oxygen to control chromatin and cell fate. Science. (2019) 363:1217–22. doi: 10.1126/science.aaw1026
47. Shait Mohammed MR, Zamzami M, Choudhry H, Ahmed F, Ateeq B, and Khan MI. The histone H3K27me3 demethylases KDM6A/B resist anoikis and transcriptionally regulate stemness-related genes. Front Cell Dev Biol. (2022) 10:780176. doi: 10.3389/fcell.2022.780176
48. Aryal S, Zhang Y, Wren S, Li C, and Lu R. Molecular regulators of HOXA9 in acute myeloid leukemia. FEBS J. (2023) 290:321–39. doi: 10.1111/febs.16268
49. Dixit D, Prager BC, Gimple RC, Poh HX, Wang Y, Wu Q, et al. The RNA m6A reader YTHDF2 maintains oncogene expression and is a targetable dependency in glioblastoma stem cells. Cancer Discov. (2021) 11:480–99. doi: 10.1158/2159-8290.CD-20-0331
50. Zhou D, Tang W, Xu Y, Xu Y, Xu B, Fu S, et al. METTL3/YTHDF2 m6A axis accelerates colorectal carcinogenesis through epigenetically suppressing YPEL5. Mol Oncol. (2021) 15:2172–84. doi: 10.1002/1878-0261.12898
51. Yang X, Mei C, Raza SHA, Ma X, Wang J, Du J, et al. Interactive regulation of DNA demethylase gene TET1 and m(6)A methyltransferase gene METTL3 in myoblast differentiation. Int J Biol Macromol. (2022) 223:916–30. doi: 10.1016/j.ijbiomac.2022.11.081
52. Khan FH, Bhat BA, Sheikh BA, Tariq L, Padmanabhan R, Verma JP, et al. Microbiome dysbiosis and epigenetic modulations in lung cancer: From pathogenesis to therapy. Semin Cancer Biol. (2022) 86:732–42. doi: 10.1016/j.semcancer.2021.07.005
53. Yang C, Zhang J, Ma Y, Wu C, Cui W, and Wang L. Histone methyltransferase and drug resistance in cancers. J Exp Clin Cancer Res: CR. (2020) 39:173. doi: 10.1186/s13046-020-01682-z
54. Corbet C and Feron O. Tumour acidosis: from the passenger to the driver’s seat. Nat Rev Cancer. (2017) 17:577–93. doi: 10.1038/nrc.2017.77
55. Martinez-Outschoorn UE, Peiris-Pagés M, Pestell RG, Sotgia F, and Lisanti MP. Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol. (2017) 14:11–31. doi: 10.1038/nrclinonc.2016.60
56. Ye C and Tu BP. Sink into the epigenome: histones as repositories that influence cellular metabolism. Trends Endocrinol Metab: TEM. (2018) 29:626–37. doi: 10.1016/j.tem.2018.06.002
57. Butler JS and Dent SY. The role of chromatin modifiers in normal and Malignant hematopoiesis. Blood. (2013) 121:3076–84. doi: 10.1182/blood-2012-10-451237
58. Fu ZW, Li JH, Feng YR, Yuan X, and Lu YT. The metabolite methylglyoxal-mediated gene expression is associated with histone methylglyoxalation. Nucleic Acids Res. (2021) 49:1886–99. doi: 10.1093/nar/gkab014
59. Guertin DA and Wellen KE. Acetyl-CoA metabolism in cancer. Nat Rev Cancer. (2023) 23:156–72. doi: 10.1038/s41568-022-00543-5
60. Haws SA, Leech CM, and Denu JM. Metabolism and the epigenome: A dynamic relationship. Trends Biochem Sci. (2020) 45:731–47. doi: 10.1016/j.tibs.2020.04.002
61. Rodriguez H, Rafehi H, Bhave M, and El-Osta A. Metabolism and chromatin dynamics in health and disease. Mol Aspects Med. (2017) 54:1–15. doi: 10.1016/j.mam.2016.09.004
62. Li L, Chen K, Wang T, Wu Y, Xing G, Chen M, et al. Glis1 facilitates induction of pluripotency via an epigenome-metabolome-epigenome signalling cascade. Nat Metab. (2020) 2:882–92. doi: 10.1038/s42255-020-0267-9
63. Charidemou E, Tsiarli MA, Theophanous A, Yilmaz V, Pitsouli C, Strati K, et al. Histone acetyltransferase NAA40 modulates acetyl-CoA levels and lipid synthesis. BMC Biol. (2022) 20:22. doi: 10.1186/s12915-021-01225-8
64. Demetriadou C, Pavlou D, Mpekris F, Achilleos C, Stylianopoulos T, Zaravinos A, et al. NAA40 contributes to colorectal cancer growth by controlling PRMT5 expression. Cell Death Dis. (2019) 10:236. doi: 10.1038/s41419-019-1487-3
65. Lan T, Jiang S, Zhang J, Weng Q, Yu Y, Li H, et al. Breviscapine alleviates NASH by inhibiting TGF-β-activated kinase 1-dependent signaling. Hepatology. (2021) 76:155–71. doi: 10.1002/hep.32221
66. Zhu J and Thompson CB. Metabolic regulation of cell growth and proliferation. Nat Rev Mol Cell Biol. (2019) 20:436–50. doi: 10.1038/s41580-019-0123-5
67. Diehl KL and Muir TW. Chromatin as a key consumer in the metabolite economy. Nat Chem Biol. (2020) 16:620–9. doi: 10.1038/s41589-020-0517-x
68. Pareja-Galeano H, Sanchis-Gomar F, and García-Giménez JL. Physical exercise and epigenetic modulation: elucidating intricate mechanisms. Sports Med (Auckland NZ). (2014) 44:429–36. doi: 10.1007/s40279-013-0138-6
69. Yu X and Li S. Specific regulation of epigenome landscape by metabolic enzymes and metabolites. Biol Rev Cambridge Philos Society. (2024) 99:878–900. doi: 10.1111/brv.13049
70. Baksh SC and Finley LWS. Metabolic coordination of cell fate by α-ketoglutarate-dependent dioxygenases. Trends Cell Biol. (2021) 31:24–36. doi: 10.1016/j.tcb.2020.09.010
71. Nirello VD, Rodrigues de Paula D, Araújo NVP, and Varga-Weisz PD. Does chromatin function as a metabolite reservoir? Trends Biochem Sci. (2022) 47:732–5. doi: 10.1016/j.tibs.2022.03.016
72. Demetriadou C, Raoukka A, Charidemou E, Mylonas C, Michael C, Parekh S, et al. Histone N-terminal acetyltransferase NAA40 links one-carbon metabolism to chemoresistance. Oncogene. (2022) 41:571–85. doi: 10.1038/s41388-021-02113-9
73. Stading R, Gastelum G, Chu C, Jiang W, and Moorthy B. Molecular mechanisms of pulmonary carcinogenesis by polycyclic aromatic hydrocarbons (PAHs): Implications for human lung cancer. Semin Cancer Biol. (2021) 76:3–16. doi: 10.1016/j.semcancer.2021.07.001
74. Bukowska B and Sicińska P. Influence of benzo(a)pyrene on different epigenetic processes. Int J Mol Sci. (2021) 22:13453. doi: 10.3390/ijms222413453
75. Zapata-Pérez R, Wanders RJA, van Karnebeek CDM, and Houtkooper RH. NAD(+) homeostasis in human health and disease. EMBO Mol Med. (2021) 13:e13943. doi: 10.15252/emmm.202113943
76. Yaku K and Nakagawa T. NAD(+) precursors in human health and disease: current status and future prospects. Antioxid Redox Signaling. (2023) 39:1133–49. doi: 10.1089/ars.2023.0354
77. Lv H, Lv G, Chen C, Zong Q, Jiang G, Ye D, et al. NAD(+) metabolism maintains inducible PD-L1 expression to drive tumor immune evasion. Cell Metab. (2021) 33:110–27.e5. doi: 10.1016/j.cmet.2020.10.021
78. Vincenzi M, Mercurio FA, and Leone M. Sam domains in multiple diseases. Curr Medicinal Chem. (2020) 27:450–76. doi: 10.2174/0929867325666181009114445
79. Wang X, Liu R, Qu X, Yu H, Chu H, Zhang Y, et al. α-ketoglutarate-activated NF-κB signaling promotes compensatory glucose uptake and brain tumor development. Mol Cell. (2019) 76:148–62.e7. doi: 10.1016/j.molcel.2019.07.007
80. Menendez JA and Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. (2007) 7:763–77. doi: 10.1038/nrc2222
81. Hyötyläinen T and Orešič M. Systems biology strategies to study lipidomes in health and disease. Prog Lipid Res. (2014) 55:43–60. doi: 10.1016/j.plipres.2014.06.001
82. Jones SF and Infante JR. Molecular pathways: fatty acid synthase. Clin Cancer Res: an Off J Am Assoc Cancer Res. (2015) 21:5434–8. doi: 10.1158/1078-0432.CCR-15-0126
83. Braun KV, Voortman T, Dhana K, Troup J, Bramer WM, Troup J, et al. The role of DNA methylation in dyslipidaemia: A systematic review. Prog Lipid Res. (2016) 64:178–91. doi: 10.1016/j.plipres.2016.10.002
84. Loeper S, Asa SL, and Ezzat S. Ikaros modulates cholesterol uptake: a link between tumor suppression and differentiation. Cancer Res. (2008) 68:3715–23. doi: 10.1158/0008-5472.CAN-08-0103
85. Pan X, Hu S, Xu Y, Gopoju R, Zhu Y, Cassim Bawa FN, et al. Krüppel-like factor 10 protects against metabolic dysfunction-associated steatohepatitis by regulating HNF4α-mediated metabolic pathways. Metabolism. (2024) 155:155909. doi: 10.1016/j.metabol.2024.155909
86. Kumar A, Kumar S, Vikram A, Hoffman TA, Naqvi A, Lewarchik CM, et al. Histone and DNA methylation-mediated epigenetic downregulation of endothelial Kruppel-like factor 2 by low-density lipoprotein cholesterol. Arterioscler Thromb Vasc Biol. (2013) 33:1936–42. doi: 10.1161/ATVBAHA.113.301765
87. Schiano C, Benincasa G, Franzese M, Della Mura N, Pane K, Salvatore M, et al. Epigenetic-sensitive pathways in personalized therapy of major cardiovascular diseases. Pharmacol Ther. (2020) 210:107514. doi: 10.1016/j.pharmthera.2020.107514
88. Ippolito L, Morandi A, Giannoni E, and Chiarugi P. Lactate: A metabolic driver in the tumour landscape. Trends Biochem Sci. (2019) 44:153–66. doi: 10.1016/j.tibs.2018.10.011
89. Wang T, Ye Z, Li Z, Jing DS, Fan GX, Liu MQ, et al. Lactate-induced protein lactylation: A bridge between epigenetics and metabolic reprogramming in cancer. Cell Prolif. (2023) 56:e13478. doi: 10.1111/cpr.13478
90. Yu K, Li Q, Sun X, Peng X, Tang Q, Chu H, et al. Bacterial indole-3-lactic acid affects epithelium-macrophage crosstalk to regulate intestinal homeostasis. Proc Natl Acad Sci U States A. (2023) 120:e2309032120. doi: 10.1073/pnas.2309032120
91. Chen Y, Wu J, Zhai L, Zhang T, Yin H, Gao H, et al. Metabolic regulation of homologous recombination repair by MRE11 lactylation. Cell. (2024) 187:294–311.e21. doi: 10.1016/j.cell.2023.11.022
92. Chen S, Xu Y, Zhuo W, and Zhang L. The emerging role of lactate in tumor microenvironment and its clinical relevance. Cancer Lett. (2024) 590:216837. doi: 10.1016/j.canlet.2024.216837
93. Xiong J, He J, Zhu J, Pan J, Liao W, Ye H, et al. Lactylation-driven METTL3-mediated RNA m(6)A modification promotes immunosuppression of tumor-infiltrating myeloid cells. Mol Cell. (2022) 82:1660–77.e10. doi: 10.1016/j.molcel.2022.02.033
94. Kang H, Valerio M, Feng J, Gu L, Hoang DH, Blackmon A, et al. AOH1996 targets mitochondrial dynamics and metabolism in leukemic stem cells via mitochondrial PCNA inhibition. Exp Hematol Oncol. (2024) 13:123. doi: 10.1186/s40164-024-00586-4
95. Pollyea DA, Kohrt HE, Zhang B, Zehnder J, Schenkein D, Fantin V, et al. 2-Hydroxyglutarate in IDH mutant acute myeloid leukemia: predicting patient responses, minimal residual disease and correlations with methylcytosine and hydroxymethylcytosine levels. Leuk Lymphoma. (2013) 54:408–10. doi: 10.3109/10428194.2012.701009
96. Zhou J, Lin Y, Yang X, Shen B, Hao J, Wang J, et al. Metabolic disorders sensitise endometrial carcinoma through endoplasmic reticulum stress. Cell Mol Biol Lett. (2022) 27:110. doi: 10.1186/s11658-022-00412-x
97. Yoshie T, Nishiumi S, Izumi Y, Sakai A, Inoue J, Azuma T, et al. Regulation of the metabolite profile by an APC gene mutation in colorectal cancer. Cancer Sci. (2012) 103:1010–21. doi: 10.1111/j.1349-7006.2012.02262.x
98. Burns JS and Manda G. Metabolic pathways of the Warburg effect in health and disease: perspectives of choice, chain or chance. Int J Mol Sci. (2017) 18:2755. doi: 10.3390/ijms18122755
99. Memmott RM and Dennis PA. LKB1 and mammalian target of rapamycin as predictive factors for the anticancer efficacy of metformin. J Clin Oncol. (2009) 27:e226; author reply e7. doi: 10.1200/JCO.2009.25.3963
100. Sun L, Zhang H, and Gao P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell. (2022) 13:877–919. doi: 10.1007/s13238-021-00846-7
101. Sullivan MR, Danai LV, Lewis CA, Chan SH, Gui DY, Kunchok T, et al. Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. eLife. (2019) 8:e44235. doi: 10.7554/eLife.44235
102. Carmona-Fontaine C, Deforet M, Akkari L, Thompson CB, Joyce JA, and Xavier JB. Metabolic origins of spatial organization in the tumor microenvironment. Proc Natl Acad Sci U States A. (2017) 114:2934–9. doi: 10.1073/pnas.1700600114
103. Drexler HG, Quentmeier H, and MacLeod RA. Malignant hematopoietic cell lines: in vitro models for the study of MLL gene alterations. Leukemia. (2004) 18:227–32. doi: 10.1038/sj.leu.2403236
104. Faitova J, Krekac D, Hrstka R, and Vojtesek B. Endoplasmic reticulum stress and apoptosis. Cell Mol Biol Lett. (2006) 11:488–505. doi: 10.2478/s11658-006-0040-4
105. Ozcan L and Tabas I. Role of endoplasmic reticulum stress in metabolic disease and other disorders. Annu Rev Med. (2012) 63:317–28. doi: 10.1146/annurev-med-043010-144749
106. Huang J, Pan H, Sun J, Wu J, Xuan Q, Wang J, et al. TMEM147 aggravates the progression of HCC by modulating cholesterol homeostasis, suppressing ferroptosis, and promoting the M2 polarization of tumor-associated macrophages. J Exp Clin Cancer Res: CR. (2023) 42:286. doi: 10.1186/s13046-023-02865-0
107. Luo G, Li X, Lin J, Ge G, Fang J, Song W, et al. Multifunctional calcium-manganese nanomodulator provides antitumor treatment and improved immunotherapy via reprogramming of the tumor microenvironment. ACS Nano. (2023) 17:15449–65. doi: 10.1021/acsnano.3c01215
108. Wei X, Zheng Z, Feng Z, Zheng L, Tao S, Zheng B, et al. Sigma-1 receptor attenuates osteoclastogenesis by promoting ER-associated degradation of SERCA2. EMBO Mol Med. (2022) 14:e15373. doi: 10.15252/emmm.202115373
109. Hayashi T and Su TP. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell. (2007) 131:596–610. doi: 10.1016/j.cell.2007.08.036
110. Paul S, Ghosh S, and Kumar S. Tumor glycolysis, an essential sweet tooth of tumor cells. Semin Cancer Biol. (2022) 86:1216–30. doi: 10.1016/j.semcancer.2022.09.007
111. Brandon M, Baldi P, and Wallace DC. Mitochondrial mutations in cancer. Oncogene. (2006) 25:4647–62. doi: 10.1038/sj.onc.1209607
112. Qi W, Keenan HA, Li Q, Ishikado A, Kannt A, Sadowski T, et al. Pyruvate kinase M2 activation may protect against the progression of diabetic glomerular pathology and mitochondrial dysfunction. Nat Med. (2017) 23:753–62. doi: 10.1038/nm.4328
113. Zhao Q, Liu J, Deng H, Ma R, Liao JY, Liang H, et al. Targeting Mitochondria-Located circRNA SCAR Alleviates NASH via Reducing mROS Output. Cell. (2020) 183:76–93.e22. doi: 10.1016/j.cell.2020.08.009
114. Shaughnessy DT, McAllister K, Worth L, Haugen AC, Meyer JN, Domann FE, et al. Mitochondria, energetics, epigenetics, and cellular responses to stress. Environ Health Perspect. (2014) 122:1271–8. doi: 10.1289/ehp.1408418
115. Niu N, Shen X, Wang Z, Chen Y, Weng Y, Yu F, et al. Tumor cell-intrinsic epigenetic dysregulation shapes cancer-associated fibroblasts heterogeneity to metabolically support pancreatic cancer. Cancer Cell. (2024) 42:869–84.e9. doi: 10.1016/j.ccell.2024.03.005
116. Yang J, Xu J, Wang W, Zhang B, Yu X, and Shi S. Epigenetic regulation in the tumor microenvironment: molecular mechanisms and therapeutic targets. Signal Transduct Target Ther. (2023) 8:210. doi: 10.1038/s41392-023-01480-x
117. Liang Y, Wang L, Ma P, Ju D, Zhao M, and Shi Y. Enhancing anti-tumor immune responses through combination therapies: epigenetic drugs and immune checkpoint inhibitors. Front Immunol. (2023) 14:1308264. doi: 10.3389/fimmu.2023.1308264
118. Thuwajit C, Ferraresi A, Titone R, Thuwajit P, and Isidoro C. The metabolic cross-talk between epithelial cancer cells and stromal fibroblasts in ovarian cancer progression: Autophagy plays a role. Medicinal Res Rev. (2018) 38:1235–54. doi: 10.1002/med.21473
119. Matilainen O, Quirós PM, and Auwerx J. Mitochondria and epigenetics - crosstalk in homeostasis and stress. Trends Cell Biol. (2017) 27:453–63. doi: 10.1016/j.tcb.2017.02.004
120. Li H, Yu K, Hu H, Zhang X, Zeng S, Li J, et al. METTL17 coordinates ferroptosis and tumorigenesis by regulating mitochondrial translation in colorectal cancer. Redox Biol. (2024) 71:103087. doi: 10.1016/j.redox.2024.103087
121. Tharp KM, Kersten K, Maller O, Timblin GA, Stashko C, Canale FP, et al. Tumor-associated macrophages restrict CD8(+) T cell function through collagen deposition and metabolic reprogramming of the breast cancer microenvironment. Nat Cancer. (2024) 5:1045–62. doi: 10.1038/s43018-024-00775-4
122. Van den Bossche J, O’Neill LA, and Menon D. Macrophage immunometabolism: where are we (Going)? Trends Immunol. (2017) 38:395–406. doi: 10.1016/j.it.2017.03.001
123. Xia L, Oyang L, Lin J, Tan S, Han Y, Wu N, et al. The cancer metabolic reprogramming and immune response. Mol Cancer. (2021) 20:28. doi: 10.1186/s12943-021-01316-8
124. Morrissey SM, Zhang F, Ding C, Montoya-Durango DE, Hu X, Yang C, et al. Tumor-derived exosomes drive immunosuppressive macrophages in a pre-metastatic niche through glycolytic dominant metabolic reprogramming. Cell Metab. (2021) 33:2040–58.e10. doi: 10.1016/j.cmet.2021.09.002
125. Knörlein A, Xiao Y, and David Y. Leveraging histone glycation for cancer diagnostics and therapeutics. Trends Cancer. (2023) 9:410–20. doi: 10.1016/j.trecan.2023.01.005
126. He Y, Fang Y, Zhang M, Zhao Y, Tu B, Shi M, et al. Remodeling “cold” tumor immune microenvironment via epigenetic-based therapy using targeted liposomes with in situ formed albumin corona. Acta Pharm Sin B. (2022) 12:2057–73. doi: 10.1016/j.apsb.2021.09.022
127. Lewandowski Ł, Urbanowicz I, Kepinska M, and Milnerowicz H. Concentration/activity of superoxide dismutase isozymes and the pro-/antioxidative status, in context of type 2 diabetes and selected single nucleotide polymorphisms (genes: INS, SOD1, SOD2, SOD3) - Preliminary findings. BioMed Pharmacother. (2021) 137:111396. doi: 10.1016/j.biopha.2021.111396
128. Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, et al. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science. (2013) 339:211–4. doi: 10.1126/science.1227166
129. Meng C, Bai C, Brown TD, Hood LE, and Tian Q. Human gut microbiota and gastrointestinal cancer. Genom Proteomics Bioinf. (2018) 16:33–49. doi: 10.1016/j.gpb.2017.06.002
130. Sepich-Poore GD, Zitvogel L, Straussman R, Hasty J, Wargo JA, and Knight R. The microbiome and human cancer. Sci (New York NY). (2021) 371:eabc4552. doi: 10.1126/science.abc4552
131. El Tekle G and Garrett WS. Bacteria in cancer initiation, promotion and progression. Nat Rev Cancer. (2023) 23:600–18. doi: 10.1038/s41568-023-00594-2
132. Wong CC and Yu J. Gut microbiota in colorectal cancer development and therapy. Nat Rev Clin Oncol. (2023) 20:429–52. doi: 10.1038/s41571-023-00766-x
133. Si H, Yang Q, Hu H, Ding C, Wang H, and Lin X. Colorectal cancer occurrence and treatment based on changes in intestinal flora. Semin Cancer Biol. (2021) 70:3–10. doi: 10.1016/j.semcancer.2020.05.004
134. Zou Y, Wang S, Zhang H, Gu Y, Chen H, Huang Z, et al. The triangular relationship between traditional Chinese medicines, intestinal flora, and colorectal cancer. Medicinal Res Rev. (2024) 44:539–67. doi: 10.1002/med.21989
135. Galeano Niño JL, Wu H, LaCourse KD, Kempchinsky AG, Baryiames A, Barber B, et al. Effect of the intratumoral microbiota on spatial and cellular heterogeneity in cancer. Nature. (2022) 611:810–7. doi: 10.1038/s41586-022-05435-0
136. Sun J, Chen F, and Wu G. Potential effects of gut microbiota on host cancers: focus on immunity, DNA damage, cellular pathways, and anticancer therapy. ISME J. (2023) 17:1535–51. doi: 10.1038/s41396-023-01483-0
137. Tong Y, Gao H, Qi Q, Liu X, Li J, Gao J, et al. High fat diet, gut microbiome and gastrointestinal cancer. Theranostics. (2021) 11:5889–910. doi: 10.7150/thno.56157
138. Miao Y, Tang H, Zhai Q, Liu L, Xia L, Wu W, et al. Gut microbiota dysbiosis in the development and progression of gastric cancer. J Oncol. (2022) 2022:1–15. doi: 10.1155/2022/9971619
139. Matsushita M, Fujita K, Motooka D, Hatano K, Fukae S, Kawamura N, et al. The gut microbiota associated with high-Gleason prostate cancer. Cancer Sci. (2021) 112:3125–35. doi: 10.1111/cas.14998
140. Kwa M, Plottel CS, Blaser MJ, and Adams S. The intestinal microbiome and estrogen receptor-positive female breast cancer. J Natl Cancer Institute. (2016) 108:djw029. doi: 10.1093/jnci/djw029
141. Matson V, Chervin CS, and Gajewski TF. Cancer and the microbiome-influence of the commensal microbiota on cancer, immune responses, and immunotherapy. Gastroenterology. (2021) 160:600–13. doi: 10.1053/j.gastro.2020.11.041
142. Park EM, Chelvanambi M, Bhutiani N, Kroemer G, Zitvogel L, and Wargo JA. Targeting the gut and tumor microbiota in cancer. Nat Med. (2022) 28:690–703. doi: 10.1038/s41591-022-01779-2
143. Helmink BA, Khan MAW, Hermann A, Gopalakrishnan V, and Wargo JA. The microbiome, cancer, and cancer therapy. Nat Med. (2019) 25:377–88. doi: 10.1038/s41591-019-0377-7
144. Finley LWS. What is cancer metabolism? Cell. (2023) 186:1670–88. doi: 10.1016/j.cell.2023.01.038
145. Kalyanaraman B. Teaching the basics of cancer metabolism: Developing antitumor strategies by exploiting the differences between normal and cancer cell metabolism. Redox Biol. (2017) 12:833–42. doi: 10.1016/j.redox.2017.04.018
146. Li Z, Ding Y, Liu J, Wang J, Mo F, Wang Y, et al. Depletion of tumor associated macrophages enhances local and systemic platelet-mediated anti-PD-1 delivery for post-surgery tumor recurrence treatment. Nat Commun. (2022) 13:1845. doi: 10.1038/s41467-022-29388-0
147. Zhu X, Xuan Z, Chen J, Li Z, Zheng S, and Song P. How DNA methylation affects the Warburg effect. Int J Biol Sci. (2020) 16:2029–41. doi: 10.7150/ijbs.45420
148. Duval K, Grover H, Han LH, Mou Y, Pegoraro AF, Fredberg J, et al. Modeling physiological events in 2D vs. 3D cell culture. Physiol (Bethesda Md). (2017) 32:266–77. doi: 10.1152/physiol.00036.2016
149. Sharma U and Rando OJ. Metabolic inputs into the epigenome. Cell Metab. (2017) 25:544–58. doi: 10.1016/j.cmet.2017.02.003
150. García-Martínez JM, Chocarro-Calvo A, Martínez-Useros J, Fernández-Aceñero MJ, Fiuza MC, Cáceres-Rentero J, et al. Vitamin D induces SIRT1 activation through K610 deacetylation in colon cancer. eLife. (2023) 12:RP86913. doi: 10.7554/eLife.86913
151. Apostolova P and Pearce EL. Lactic acid and lactate: revisiting the physiological roles in the tumor microenvironment. Trends Immunol. (2022) 43:969–77. doi: 10.1016/j.it.2022.10.005
152. Li X, Yang Y, Zhang B, Lin X, Fu X, An Y, et al. Lactate metabolism in human health and disease. Signal Transduct Target Ther. (2022) 7:305. doi: 10.1038/s41392-022-01151-3
153. Riedel A, Helal M, Pedro L, Swietlik JJ, Shorthouse D, Schmitz W, et al. Tumor-derived lactic acid modulates activation and metabolic status of draining lymph node stroma. Cancer Immunol Res. (2022) 10:482–97. doi: 10.1158/2326-6066.CIR-21-0778
154. Liu N, Luo J, Kuang D, Xu S, Duan Y, Xia Y, et al. Lactate inhibits ATP6V0d2 expression in tumor-associated macrophages to promote HIF-2α-mediated tumor progression. J Clin Invest. (2019) 129:631–46. doi: 10.1172/JCI123027
155. Wang S, Li F, Ye T, Wang J, Lyu C, Qing S, et al. Macrophage-tumor chimeric exosomes accumulate in lymph node and tumor to activate the immune response and the tumor microenvironment. Sci Trans Med. (2021) 13:eabb6981. doi: 10.1126/scitranslmed.abb6981
156. Maiorino L, Daßler-Plenker J, Sun L, and Egeblad M. Innate immunity and cancer pathophysiology. Annu Rev Pathol. (2022) 17:425–57. doi: 10.1146/annurev-pathmechdis-032221-115501
157. Davar D, Dzutsev AK, McCulloch JA, Rodrigues RR, Chauvin JM, Morrison RM, et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science. (2021) 371:595–602. doi: 10.1126/science.abf3363
158. Liu PF, Farooqi AA, Peng SY, Yu TJ, Dahms HU, Lee CH, et al. Regulatory effects of noncoding RNAs on the interplay of oxidative stress and autophagy in cancer Malignancy and therapy. Semin Cancer Biol. (2022) 83:269–82. doi: 10.1016/j.semcancer.2020.10.009
159. Toden S, Zumwalt TJ, and Goel A. Non-coding RNAs and potential therapeutic targeting in cancer. Biochim Biophys Acta Rev Cancer. (2021) 1875:188491. doi: 10.1016/j.bbcan.2020.188491
160. Slack FJ and Chinnaiyan AM. The role of non-coding RNAs in oncology. Cell. (2019) 179:1033–55. doi: 10.1016/j.cell.2019.10.017
161. Lin C and Li W. Noncoding RNAs as regulators of cancer. RNA Biol. (2020) 17:1521–2. doi: 10.1080/15476286.2020.1813920
162. Ma S, Chen C, Ji X, Liu J, Zhou Q, Wang G, et al. The interplay between m6A RNA methylation and noncoding RNA in cancer. J Hematol Oncol. (2019) 12:121. doi: 10.1186/s13045-019-0805-7
163. Chen Y, Lin Y, Shu Y, He J, and Gao W. Interaction between N(6)-methyladenosine (m(6)A) modification and noncoding RNAs in cancer. Mol Cancer. (2020) 19:94. doi: 10.1186/s12943-020-01207-4
164. Xu Z, Chen Y, Ma L, Chen Y, Liu J, Guo Y, et al. Role of exosomal non-coding RNAs from tumor cells and tumor-associated macrophages in the tumor microenvironment. Mol Therapy: J Am Soc Gene Ther. (2022) 30:3133–54. doi: 10.1016/j.ymthe.2022.01.046
165. Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. (2011) 12:861–74. doi: 10.1038/nrg3074
166. Mohapatra S, Pioppini C, Ozpolat B, and Calin GA. Non-coding RNAs regulation of macrophage polarization in cancer. Mol Cancer. (2021) 20:24. doi: 10.1186/s12943-021-01313-x
167. Liu Y, Liu X, Lin C, Jia X, Zhu H, Song J, et al. Noncoding RNAs regulate alternative splicing in Cancer. J Exp Clin Cancer Res: CR. (2021) 40:11. doi: 10.1186/s13046-020-01798-2
168. Pan Y, Liu Y, Wei W, Yang X, Wang Z, and Xin W. Extracellular vesicles as delivery shippers for noncoding RNA-based modulation of angiogenesis: insights from ischemic stroke and cancer. Small (Weinheim an der Bergstrasse Germany). (2023) 19:e2205739. doi: 10.1002/smll.202205739
169. Ning Q, Liu YF, Ye PJ, Gao P, Li ZP, Tang SY, et al. Delivery of Liver-Specific miRNA-122 Using a Targeted Macromolecular Prodrug toward Synergistic Therapy for Hepatocellular Carcinoma. ACS Appl Mater Interfaces. (2019) 11(11):10578–88.
170. Yoo JY, Yeh M, Kaur B, and Lee TJ. Targeted delivery of small noncoding RNA for glioblastoma. Cancer Lett. (2021) 500:274–80. doi: 10.1016/j.canlet.2020.11.004
171. Schmidt LH, Spieker T, Koschmieder S, Schäffers S, Humberg J, Jungen D, et al. The long noncoding MALAT-1 RNA indicates a poor prognosis in non-small cell lung cancer and induces migration and tumor growth. J Thorac Oncol: Off Publ Int Assoc Study Lung Cancer. (2011) 6:1984–92. doi: 10.1097/JTO.0b013e3182307eac
Keywords: cancer, glucose metabolism, lipid metabolism, epigenetics, metabolic reprogramming, tumor microenvironment
Citation: Zhang X, Liu D, Yin S, Gao Y, Li X and Wu G (2025) Metabolism and epigenetics in cancer: toward personalized treatment. Front. Endocrinol. 16:1530578. doi: 10.3389/fendo.2025.1530578
Received: 19 November 2024; Accepted: 02 July 2025;
Published: 25 July 2025.
Edited by:
Rongzhang Dou, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
Sergei Tevosian, University of Florida, United StatesPradeep Mk Nair, Mirakle Integrated Health Centre, India
Qin Xie, Wuhan University, China
Copyright © 2025 Zhang, Liu, Yin, Gao, Li and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yaru Gao, MTM4NDI2NTAxMDZAMTYzLmNvbQ==; Xiaorui Li, eGlhb3J1aV8wNTEzQDE2My5jb20=; Guangzhen Wu, d3VndWFuZ3poZW5AZmlyc3Rob3NwLWRtdS5jb20=
†These authors have contributed equally to this work