 Zhi-Hui Wang1
Zhi-Hui Wang1 Wei Tu
Wei Tu- 1First Clinical Medical College, Fujian University of Traditional Chinese Medicine, Fujian, Fuzhou, China
- 2Department of Dermatology, The Second Affiliated Hospital of Chongqing Medical University, Chongqing, China
- 3Department of Tumor and Endocrinology, The Affiliated Chinese Medicine Hospital of Chongqing Three Gorges Medical College, Chongqing, China
- 4Department of Nephrology, People’s Hospital Affiliated to Fujian University of Traditional Chinese Medicine, Fuzhou, China
Diabetic kidney disease (DKD) is one of the most serious complications of diabetes mellitus (DM) and the main cause of end-stage renal disease (ESRD). The number of affected patients is increasing annually worldwide. Therefore, it is necessary to establish new strategies to treat DKD and improve prognosis. The Notch signaling pathway is involved in multiple mechanisms in DKD, including glomerular endothelial dysfunction, filtration barrier damage, podocyte EMT and dedifferentiation, tubulointerstitial fibrosis, proximal tubule cell dedifferentiation, macrophage polarization, etc. In addition, Notch signaling interacts with other pathways involved in DKD progression, such as TGF-β, Wnt/β-catenin, mTOR, AMPK, autophagy, etc. Therefore, new ideas for the future treatment of DKD may be provided through clarification of the role of the Notch signaling pathway and development of novel drugs.
1 Introduction
In recent years, the incidence of diabetes mellitus has increased dramatically. According to estimates, the prevalence of diabetes among people aged 20 to 79 worldwide was 10.5% in 2021 and will increase to 12.2% in 2045, which represents an increase of over 50% compared to 2017 levels (1, 2). Diabetic kidney disease is one of the most serious complications of diabetes and the main cause of end-stage renal disease worldwide (3). The typical clinical course of DKD begins with microalbuminuria, followed by severe proteinuria, which in turn induces tubular damage, progressing from low-grade renal inflammation to renal fibrosis, renal sclerosis, and finally to end-stage renal disease (4). When DKD develops to the ESRD stage, dialysis or kidney transplantation is essential. Diabetic kidney disease is prevalent worldwide, causing serious health problems and huge economic burdens on global human society (5). The pathogenesis of DKD involves multiple aspects and multiple signals. Notch signaling is involved in the pathogenesis of DKD, including vascular endothelial disorders (6), renal inflammation (7), renal fibrosis and necrosis (8), and podocyte and tubular epithelial cell damage (9). This article reviews the role of the Notch signaling pathway in DKD and discusses the molecular mechanism of Notch signaling pathway regulation. It is envisioned that analysis of the functional significance of Notch signaling will be critical to the development of novel therapeutic approaches for DKD.
2 Notch signaling pathway
Notch signaling has a role in many facets of metazoan life, such as cell fate determination, embryonic development, tissue repair and function, and non-cancerous and malignant disorders (10). Multiple intermediates exist between the nuclear effectors and membrane receptors in classical Notch signaling pathways, which are mediated by G protein-coupled receptors (GPCRs) and enzyme-linked receptors (11, 12). The Notch signal is transmitted between adjacent cells via the Notch receptor, which undergoes three cleavages and translocates to the nucleus to regulate the transcription of target genes (13). Mammals, including humans, are known to possess four distinct Notch receptors: Notch 1, 2, 3, and 4 (14). Delta-like ligand 1 (DLL1), delta-like ligand 3 (DLL3), delta-like ligand 4 (DLL4), Jagged-1 (JAG1), and Jagged-2 (JAG2) are the five recognized Notch ligands in humans (15, 16). Each of these ligands performs both redundant and distinct roles. DLL1 is in charge of cell differentiation and cell-to-cell communication (16), DLL3 induces apoptosis to stop cell growth (17), DLL4 activates NF-κB signaling to improve tumor metastasis and VEGF secretion (18), JAG1 stimulates angiogenesis, and JAG2 encourages cell survival and proliferation (16). Notch signaling are transmitted via the binding of Notch receptors and ligands, which are likewise single-transmembrane proteins produced on the cell surface.
In cells receiving signals, Notch receptors are first produced in the endoplasmic reticulum (ER) and then transported to the Golgi apparatus. The EGF-like repeat domain of Notch receptors is glycosylated during transport. The Notch receptors are then split into heterodimers (S1 cleavage) in the Golgi apparatus and moved to the cell membrane. Certain Notch receptors on the cell membrane are incorporated into endosomes with the aid of ubiquitin ligases (19). Metalloproteases (ADAMs) and γ-secretase are found in an acidic environment inside endosomes. Endosome notch receptors can be broken down in lysosomes, returned to the cell membrane, or cleaved into NICD. The portion of the Notch receptor that remains after S2 cleavage is known as Notch extracellular truncation (NeXT) (20–22). NeXT can be endocytosed into endosomes or further broken down by γ-secretase on the cell membrane. NICD is released onto the cell membrane in the former way. In the latter mode, NeXT can be taken to lysosomes, where it will be degraded, or it can be broken down into NICD. In general, there are three pathways for the production of NICD, namely ligand-independent activation, ligand-dependent endocytosis-independent activation, and ligand-dependent endocytosis-activated (10). Interactions between NICD and several signaling pathways, including NF-κB, mTORC2, AKT, and WNT, are mediated by its translocation into the nucleus or retention in the cytoplasm. According to the traditional theory, CBF-1/suppressor of hairless/Lag1 CSL binds to the corepressor and prevents target gene transcription in the absence of NICD (23–25). After binding to CSL and enlisting Mastermind-like proteins (MAML), NICD can reach the nucleus and promote the transcription of Notch target genes by releasing co-repressors and enlisting co-activators (Figure 1).
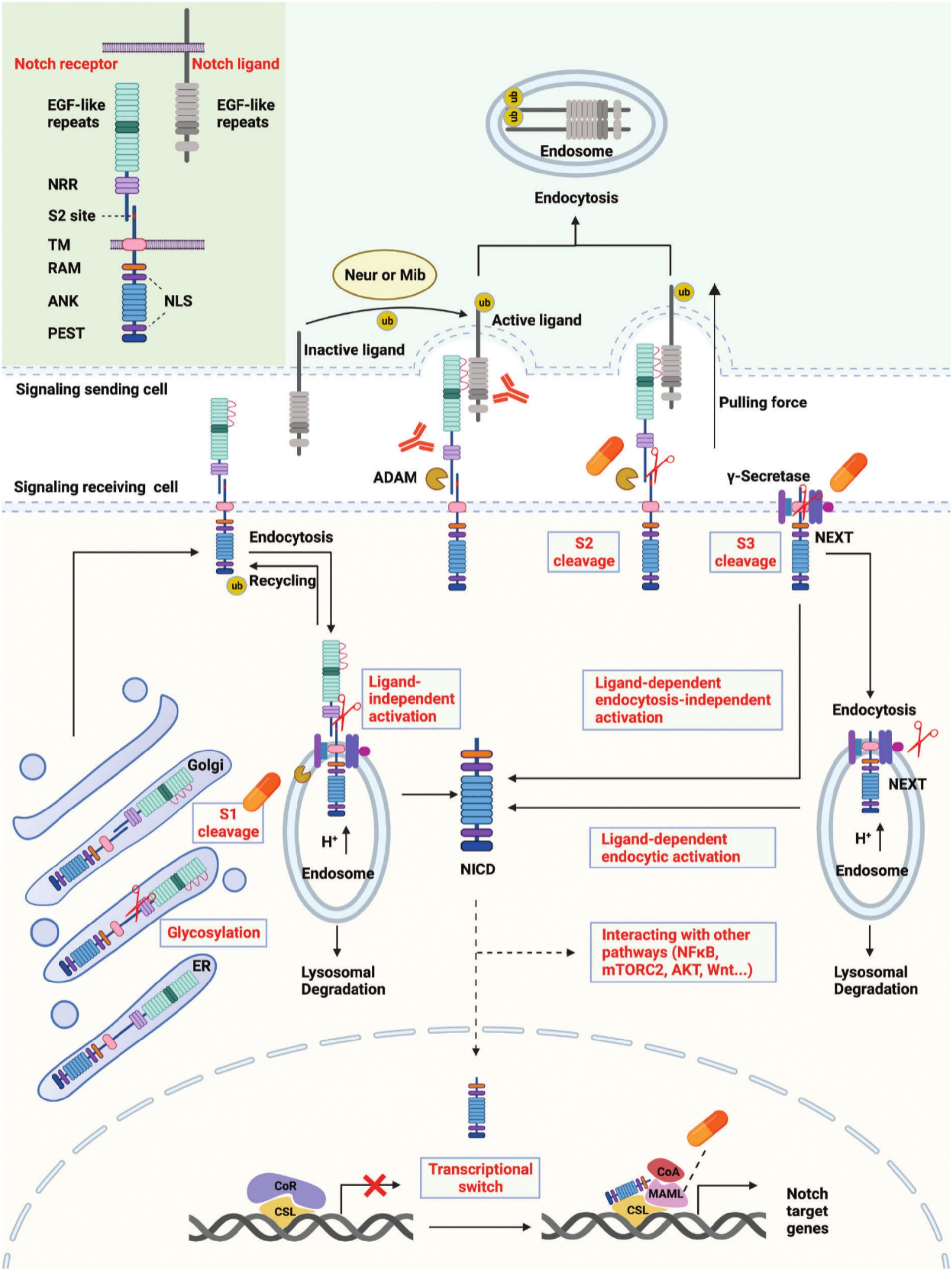
Figure 1. Overview of the Notch signaling pathway and pivotal targets.
3 Glomerulus dysfunction in typical DKD and Notch
Diabetic kidney disease is a primary microvascular complication of diabetes (26, 27). However, the exact pathways mediating endothelial dysfunction in DKD are poorly understood. Glomerular endothelial cells (GEnCs) are specialized vascular cells found in the kidney that serve as the walls of the glomerular tufts and are crucial for maintaining renal homeostasis. The glycocalyx, a network of endothelial polysaccharide layers, covers GEnCs. In animal studies, the loss of the glycocalyx is correlated with the severity of proteinuria (28). Endothelial cell dysfunction increases endothelial permeability and apoptosis and can lead to proteinuria. The Notch signaling pathway is involved in the regulation of the glomerular filtration barrier. Endothelial Notch1 signaling activation downregulates VE-cadherin levels through transcription factors SNAI1 and ERG, reducing glomerular endothelial glycocalyx, thereby leading to glomerular filtration barrier dysfunction and proteinuria (29). The Notch pathway plays a crucial role in kidney development, including guiding the differentiation of various progenitor cells, and abnormal Notch signaling leads to severe changes in cell fate (30). Endothelial ADAM10, a key regulator of Notch signaling, promotes the development and maturation of glomerular vasculature (31).
One of the main characteristics of diabetes complications is abnormal angiogenesis. Podocyte-released vascular endothelial growth factor A (VEGFA) attaches to its receptors, VEGFR1 and VEGFR2, which are expressed on GEnCs. VEGFA stimulates sprouting angiogenesis and regulates GEnC activity. Notch signaling interacts with other key factors, such as VEGF-A, to affect the health status and pathological processes of glomeruli (32, 33). The increase of Notch1 signaling in renal podocytes treated with high glucose causes VEGF production, which in turn causes nephrin inhibition and apoptosis (34). In diabetic kidneys, Notch1 knockdown results in decreased proteinuria, decreased nephrin expression, and decreased VEGF expression. The pathophysiology of endothelial dysfunction in diabetic nephropathy and retinopathy may involve Notch signaling. By modulating the susceptibility of hemangioblasts to VEGF, Notch signaling does prevent diabetic extrarenal angiogenesis (35, 36). Activation of Notch signaling in endothelial cells can promote neovascularization and increase microvascular permeability, destroy adhesion junctions between endothelial cells, mediate endothelial cell dysfunction, and eventually lead to diabetic endothelial cell dissociation (37). Activation of the Notch1 signaling pathway can cause endothelial cell leakage, damage the glomerular filtration barrier, and lead to increased urinary protein (38). Low-intensity pulsed ultrasound-induced calcium influx promotes the beneficial effects of angiogenesis, improved renal function, and Akt-eNOS phosphorylation in rats with acute kidney injury through the Notch1-Akt-eNOS signaling pathway, making Notch1 activation a therapeutic strategy for acute kidney injury targeting angiogenesis (39). Endothelial-mesenchymal transition (EndMT) is a hallmark of diabetes-related vascular complications. Intermittent high glucose exposure upregulates H3K4me3 levels in glomerular endothelial cells of Ob/Ob mice, activates Notch signaling, and induces some mesenchymal-like features in endothelial cells (40). Through Notch activation, matrix metalloproteinase-9 causes the endothelium-mesenchymal transition in human glomerular endothelial cells (41).
Podocytes are terminally differentiated cells that are unable to proliferate. Foot process effacement and podocyte hypertrophy are observed in early and middle stages of DKD, while late stages of the disease are characterized by podocyte death and dedifferentiation. An irreversible stage in the pathophysiology of DKD is the loss of more than 20% of podocytes, which results in glomerular scarring and the onset of end-stage renal disease (42, 43). In DKD, podocyte injury is a key event leading to proteinuria, nephropathy, glomerulosclerosis, and loss of renal function (44). The Notch signaling pathway plays an important role in glomerular cells, especially podocytes (32). Studies have shown that activation of Notch signaling can promote glomerular lesions and albuminuria by regulating TGF-β expression and activity (45). High-level activation of the Notch1 signaling pathway weakens the improvement effect of islet transplantation on kidney damage and the recovery of podocytes. The use of N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester (DAPT) can inhibit the excessive activation of the Notch1 pathway and improve podocyte damage under high glucose conditions (46). A study done in a lab setting found that microencapsulated islet transplantation reduced the levels of Jag-1, Notch1, and Hes-1 proteins in rat glomeruli. It also stopped Notch signaling and improved damage to podocytes in a high glucose environment (47). Abnormal activation of the Notch1 signaling pathway may affect the therapeutic effect of islet transplantation in diabetic nephropathy by changing the balance of podocyte apoptosis and autophagy (46). Under high glucose conditions, the reduction of Sirt6 leads to overactivation of Notch signaling, resulting in damage to podocytes, including inflammation, apoptosis, and reduced autophagy levels. Sirt6 deacetylates H3K9, inhibiting the transcription of Notch1 and Notch4 genes, thereby protecting podocytes from damage (48). Under hyperglycemia, MAD2B expression is upregulated in diabetic glomeruli and cultured podocytes. Upregulated MAD2B expression can lead to Numb loss and activation of the Notch1 signaling pathway during the progression of DKD, ultimately leading to podocyte injury. Podocyte-specific deletion of MAD2B can alleviate podocyte injury and renal function deterioration in diabetic nephropathy mice (49). High glucose-induced CDKN2B-AS1 promotes apoptosis and fibrosis in human podocytes and human tubular cells through the TGF-β1 signaling pathway mediated by the miR-98-5p/NOTCH2 axis (50).
Research revealed that removing the histone methylating enzyme EZH2 from podocytes reduced the levels of H3K27me3 and made animals more susceptible to glomerular illness. H3K27me3 was enriched at the promoter region of the Notch ligand Jag1 in podocytes, and derepression of Jag1 by EZH2 inhibition facilitated the activation of Notch signaling and podocyte dedifferentiation (51). METTL3 affects the stability of TIMP2 mRNA by regulating the m6A modification level, thereby enhancing the expression of TIMP2. The increase in TIMP2 further activates the Notch signaling pathway, promoting the expression of inflammatory factors and podocyte apoptosis. TIMP2 participates in the progression of diabetic nephropathy by regulating the Notch signaling pathway (52). A CARM1-AMPKα-Notch1-CB1R signaling axis mediates the high-glucose-induced podocyte apoptosis. In DKD, podocyte loss may be avoided by employing techniques to maintain CARM1 expression or lower the enzymatic activity of a ubiquitin ligase specific for CARM1 (53). In DKD, advanced glycation end products (AGEs) can damage podocytes through the Notch1 signaling pathway. AGEs directly damage podocytes through the RAGE-Notch1 signaling pathway and cytoskeletal remodeling, promoting epithelial-mesenchymal transition (EMT) and functional loss of podocytes. Notch1 signals activated by AGEs also promote podocyte mesenchymalization, leading to collagen deposition, disappearance of foot processes, and renal tubular lesions, which in turn induce proteinuria (54). Notch signaling plays a pivotal role in podocyte EMT and is intricately connected with other pathways like Wnt and TGF-β. Under hyperglycemia, Notch activation crosses with TGF-β, Wnt, and other pathways to promote the EMT process of podocytes, causing podocytes to lose their unique epithelial properties, leading to proteinuria and the progression of glomerular diseases (55). Notch signaling promotes the initiation of the cell cycle by transmitting short-range signals between cells. GH and TGF-β1 induce podocytes to re-enter the cell cycle from the quiescent phase (G0 phase) by activating the Notch signaling pathway, but these cells are abnormal when completing mitosis, showing that only karyokinesis but not cytokinesis is completed, causing the cells to become binuclear and eventually undergo mitotic catastrophe, ultimately causing podocyte death (56). Inhibition of JAK2, TGFBR1, or Notch signaling pathways can prevent re-entry of the cell cycle and protect cells from cell death associated with mitotic catastrophe (Figure 2).
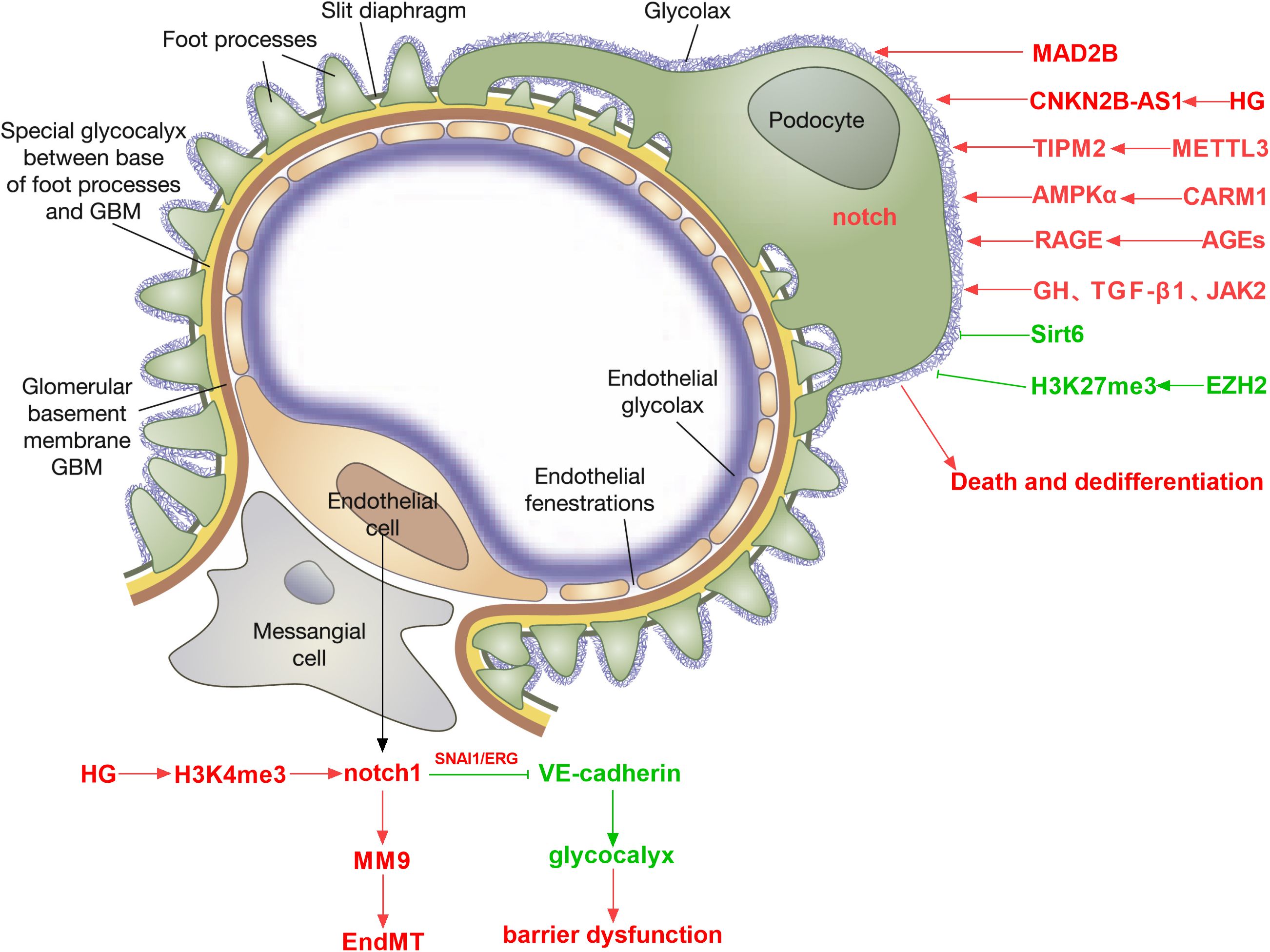
Figure 2. Role of Notch signaling in regulating DKD-associated endothelial cell dysfunction, podocyte dedifferentiation, and death.
4 Renal tubular injury in typical DKD and Notch
Common features of fibrosis-related renal function decline in DKD include tubular epithelial cell dedifferentiation, immune cell influx, and myofibroblast activation (57). The early manifestations of DKD are hyperfiltration and renal hypertrophy, and proximal tubule cell (PT) loss is the main cause of renal hypertrophy and hyperfiltration (58). The late stage of DKD is characterized by PT cell dedifferentiation, and the loss of renal PT cells is associated with a decrease in eGFR. Under high glucose stimulation, the expression of Polo-like kinase 2 (PLK2) increased in the kidney tissue of DKD mice. Silencing PLK2 significantly inhibited the activation of the Notch1 signaling pathway and reduced the expression of renal fibrosis-related markers, while overexpression of HES1 rescued the downregulation of markers induced by si-PLK2. PLK2 can mediate tubulointerstitial fibrosis in DKD by activating the Notch1 signaling pathway (59). Renal tubular fibrosis is considered to be a complex and irreversible metabolic change and one of the key signs of disease progression (60, 61). In the study of diabetic renal tubular fibrosis, multiple signaling pathways interact, including TGF-β, Wnt/β-catenin, MAPK, Notch, etc., forming a complex signal transduction network (61). The TGF-β pathway is considered to be the key to renal fibrosis, inducing extracellular matrix (ECM) accumulation by activating Smad proteins and non-Smad pathways. In renal fibrosis, the interaction between the TGF-β and Notch signaling pathways is essential (55). Notch signaling is activated through ADAM10-mediated proteolytic reactions, and the ADAM10-Notch signaling axis is associated with renal fibrosis (62). Ligand-receptor binding triggers Notch signaling, which results in the release of NICD. NICD translocation to the nucleus controls the expression of target genes, which includes involvement in the creation of the extracellular matrix (ECM) and EMT. In renal cells, the TGF-β pathway can upregulate Notch ligands, including Jagged1, which increases Notch activation. Notch target gene Hes-1 is also a direct downstream gene of the TGF-β pathway. The degree of tubulointerstitial fibrosis is strongly associated with NICD, which has the ability to directly affect downstream Smad3 (63). The Jumonji domain containing-3 (JMJD3) reduces the level of H3K27me3 and inhibits the activation of TGF-β and NOTCH signaling, thereby exerting an anti-renal fibrosis effect (64). Strategies that target either process (e.g., γ-secretase inhibitors or particular pathway inhibitors) have demonstrated potential for ameliorating renal fibrosis in experimental animals (65, 66). In rats with tissue fibrosis, TGF-β inhibitors decrease the expression of Notch and its target genes, Notch1, Hes1, and Hes5. By suppressing TGFβRII and Smad3, epigallocatechin gallate (EGCG) blocks the Notch pathway in renal cells (67, 68). By blocking the Notch pathway’s activation, TGF-β production, and Smad2 and Smad3 phosphorylation, Notch inhibitors can markedly lessen the severity of renal fibrosis.
Numb is downregulated in diabetic nephropathy tissues and high glucose-stimulated endothelial cells, while Notch1 and Hes1 are upregulated. Numb affects endothelial-mesenchymal transition (EndoMT) by negatively regulating the Notch signaling pathway, which leads to a decrease in the expression of endothelial cell markers (such as E-cadherin and CD31) and an increase in the expression of mesenchymal cell markers (such as α-SMA and vimentin), thereby participating in the pathological process of DKD and mediating renal fibrosis and disease progression (69). In addition, studies have found that the Wnt/β-catenin signaling pathway is an upstream mediator of the Notch signaling pathway. Inhibition of Wnt reduces JAG1 expression, while Wnt10b can promote the activation of Wnt and Notch signals (70–73). Downregulation of A cluster-Homeobox genes encoding a5 protein (HOXA5) by DNA methylation induces NOTCH activation and promotes renal fibrosis. HOXA5 inhibits the transcription of Jag1 by directly binding to its gene promoter, inhibiting Notch signaling and alleviating renal tubular fibrosis (74). Therefore, understanding the interplay between Notch and multiple signaling pathways could help inform potential therapeutic interventions for the management of fibrotic kidney diseases (Figure 3).
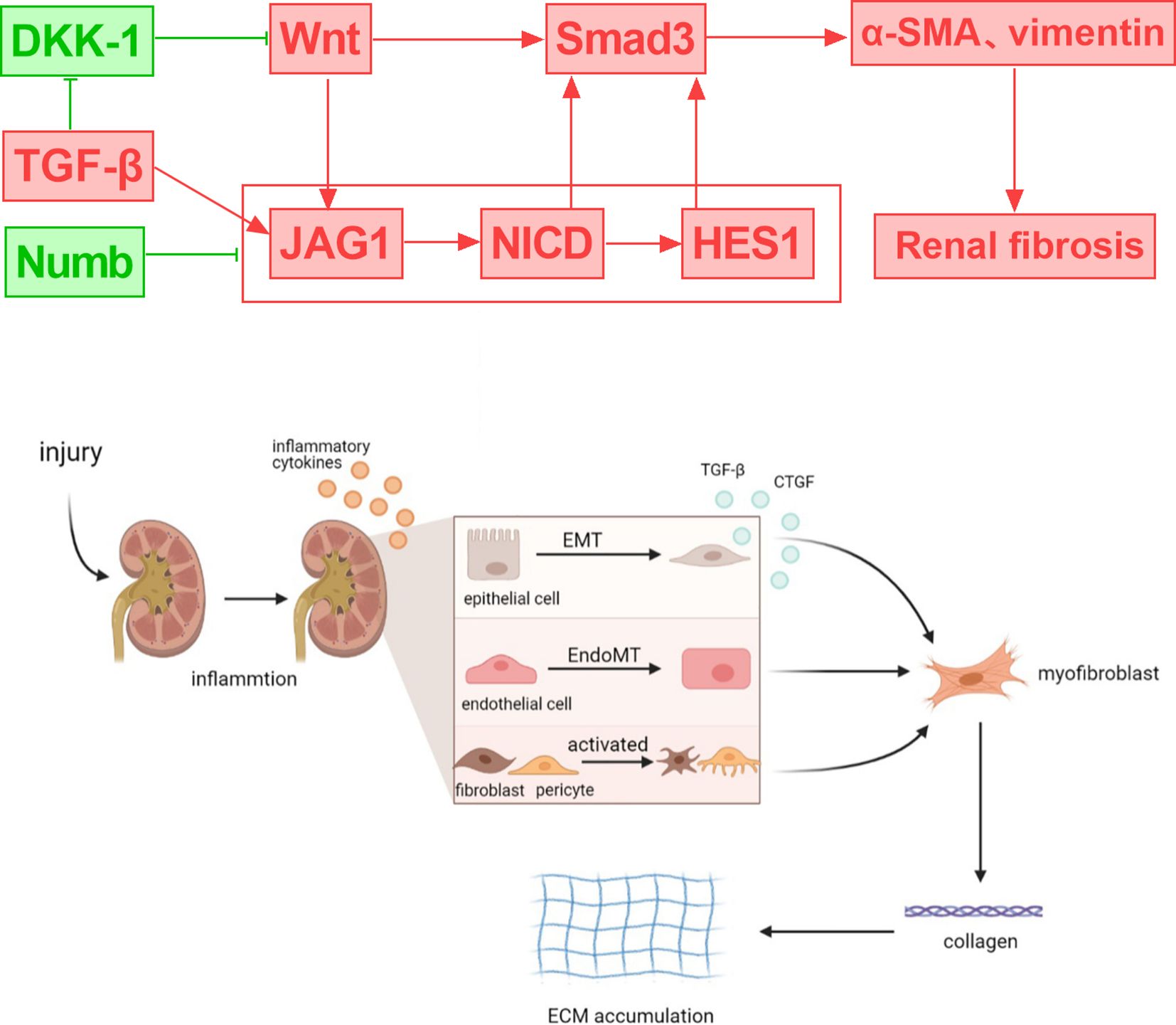
Figure 3. Crosstalk between notch signaling and DKD-related signals and their role in regulating renal inflammation and fibrosis.
In the development of diabetic nephropathy, a variety of inflammatory cells participate and play an important role, especially in the process of renal fibrosis (75). Damaged intrinsic renal cells in diabetic nephropathy attract monocytes and macrophages to the site of tissue injury in order to prevent and remove cell damage. Polarization of macrophages in diabetic nephropathy was caused by interaction between the Notch system and NF-κB signaling in macrophages. Numerous inflammatory cytokines are secreted by M1 phenotype macrophages, which further worsened the intrinsic kidney cells’ fibrosis, necroptosis, extracellular matrix production, and inflammatory response (8). Inhibition of the macrophage Notch pathway can alleviate the pathological changes of renal cells. In addition to being a key regulator of kidney development and being silenced afterwards, Notch signaling is reactivated in kidney injury and is involved in acute and chronic kidney injury. In vivo studies have shown that Notch activation is associated with interstitial fibrosis and glomerulosclerosis (76). In the glomeruli and tubules of patients with chronic kidney disease, expression of both Notch receptors and ligands increases renal injury.
5 Roles of Notch-related miRNA in DKD
miR-124a can promote the differentiation of bone marrow mesenchymal stem cells into islet-like cells. Bone marrow mesenchymal stem cells combine with miR-124a to regulate specific transcription factors and genes and inhibit the activity of Notch signaling, exerting significant anti-fibrosis and repair effects, thereby effectively alleviating the progression of diabetic nephropathy (77). By blocking the Notch pathway, miR-135a inhibition can lessen renal fibrosis in DKD rats (78). Through its targeting of MAML1 and inactivation of the Notch signaling pathway, miR-133a-3p reduces the damage caused by high hyperglycemia to human renal tubular epithelial cells and prevents the progression of DKD (79). In podocytes exposed to high hyperglycemia, overexpression of miR-34c suppresses Notch signaling by specifically targeting Notch1 and Jaggged1 (80). Increased expression of miR-34a reduced podocyte damage and NICD, Hes1, Hey1, Jagged1, and Notch 1 protein expression under high glucose circumstances (81). MiR34a further enhances the effect of mesenchymal stem cell microvesicles on mouse renal fibrosis by regulating EMT and Notch pathways (82). miR-34a may be a candidate molecular therapeutic target for the treatment of renal fibrosis. Loss of miR-146a in renal tubular cells is associated with an increased risk of DKD. miR-146a normally inhibits the expression of Notch1 and ErbB4 mRNA by binding to their 3’ UTR region. When miR-146a expression is reduced or absent, this inhibitory effect is lost, resulting in increased transcription levels of Notch1 and ErbB4. Upregulation of Notch1 and ErbB4 leads to activation of the EGFR pathway, which further promotes tubular cell damage and disease progression (83).
Exosomal miR-30a-5p significantly promoted the proliferation and migration, and reduced apoptosis of glomerular endothelial cells under high glucose conditions and reduced the mRNA and protein expression levels of Notch1 and VEGF. Exosomal miR-30a-5p inhibits EMT and abnormal angiogenesis of glomerular endothelial cells by regulating the Notch1/VEGF signaling pathway. One possible therapeutic approach for DKD treatment might be miR-30a-5p (84). Targeted administration of miRNA-30a via engineered nanoplexes to save dying podocytes in DKD. miRNA-30a is primarily responsible for podocyte homeostasis. In DKD, miRNA-30a is directly and predominantly inhibited by hyperglycemic kidney-induced Notch signaling, leading to podocyte damage and apoptosis. The nanocomplex can upregulate the expression level of miRNA-30a in high glucose-exposed podocytes, significantly inhibit Notch1 signaling in diabetic C57BL/6 mice, and reduce glomerular expansion and glomerular fibrosis (85).
6 Clinical use of Notch signaling in DKD
In diabetic nephropathy, high concentrations of glucose promote fibrosis through the Notch1 pathway. Natural flavonoid ombuin significantly down-regulated the expression of TGF-β1, Notch 1, and Hes 1 and up-regulated the expression of peroxisome proliferator-activated receptor γ (PPAR γ), significantly improved renal function and pathological damage in DKD rats, and improved renal interstitial fibrosis. Ombuin may exert anti-inflammatory and anti-fibrotic effects by inhibiting Notch 1 activity and activating PPARγ (86). Under the Danggui-Shaoyao-San (DSS) treatment group, there were substantial decreases in the protein and mRNA levels of Jagged1, Notch1, Hes5, and NICD and significant increases in the protein and mRNA levels of E-cadherin. By blocking Notch signaling, DSS inhibits renal tubular EMT and prevents diabetic nephropathy (87). Trichostatin A (TSA) does not affect the phosphorylation levels of Smad2, Smad3, p38, and ERK but significantly reduces the phosphorylation of JNK, thereby inhibiting the activation of the Notch-2 signaling pathway. This effect leads to the downregulation of fibrosis markers such as α-SMA and fibronectin stimulated by TGF-β1, thereby alleviating the occurrence and development of renal fibrosis. By interfering with the JNK/Notch-2 signaling pathway, TSA shows potential therapeutic effect in the treatment of renal fibrosis (–).-Epigallocatechin gallate (EGCG), as a natural antioxidant, can inhibit the expression of Notch1 and reduce the activation of the TGF-β/Smad3 signaling pathway, thereby alleviating renal fibrosis in diabetic mice (88). In addition, EGCG treatment can significantly reduce the levels of Notch1 and TGF-βRII in HEK293 cells stimulated by high glucose, further supporting the potential of EGCG in the treatment of diabetic nephropathy. Therefore, the Notch signaling pathway is an important target for the treatment of fibrosis, and EGCG may alleviate related diseases by regulating the Notch pathway (67).
Baicalin inhibits the activation of the Notch1-Snail axis in podocytes, alleviates glomerular structural destruction and dysfunction, and reduces proteinuria. Baicalin is a new renal protective agent against podocyte EMT (89). Under a high glucose environment, C-peptide significantly reduces EMT and renal fibrosis by reducing the expression of Snail, Vimentin, α-SMA, and CTGF. C-peptide also inhibits the activation of Notch1 and Jagged1 in the Notch signaling pathway and TGF-β1 in the TGF-β signaling pathway, thereby alleviating the progression of diabetic nephropathy (90). Gliquidone inhibits the expression of proteins associated with the Notch signaling system, including Jagged1, Notch1, Hes1, and Snail1, hence delaying the EMT process of renal tubular epithelial cells. Gliquidone has high therapeutic promise and slows the evolution of diabetic nephropathy by inhibiting the activation of the Notch/Snail signaling pathway (91). Glucagon-like peptide-1 agonist combined with crocin treatment can inhibit Notch signaling and mesangial growth in animal models of diabetic kidney disease and significantly improve renal function (92). In the kidney tissue of model rats, the Traditional Chinese medicine (TCM) capsule for qi replenishment, yin nourishment, and blood activation can lower Hes1, CD34, and CD144, safeguard kidney function, and postpone the onset of DKD (93). By triggering autophagy and blocking the Notch pathway to reduce podocyte dedifferentiation, dasatinib and quercetin prevent DKD (94).
High glucose environments activate the Notch pathway, leading to oxidative damage of renal tubular epithelial cells and renal interstitial fibrosis. Studies have shown that the Notch pathway affects cell apoptosis by regulating mitochondrial function and related genes (such as Drp1 and PGC-1α) under high glucose conditions. Inhibitors of the Notch pathway, such as DAPT, can reduce these negative effects. GH treatment will trigger an increase in the expression of EMT markers (vimentin, SMA, etc.), while DAPT effectively prevents these changes induced by GH (95). DAPT treatment not only reduced the release of cytokines but also prevented the thickening of the renal tubular basement membrane, proteinuria, and decreased renal function caused by GH (96). Therefore, DAPT significantly improved GH-induced EMT and its related renal injury by blocking the Notch1 signaling pathway. Following therapy with DAPT, blood urea nitrogen and creatinine levels were dramatically lowered. By inhibiting the Notch signaling system, DAPT dramatically lowers the expression of Notch signaling components in renal tissue, including Jagged1, Notch1-3, and Hes1. This lessens kidney damage in diabetic rats. With its ability to suppress the Notch signaling system, DAPT offers fresh promise as a therapy for kidney damage caused by diabetes. Trichosanthes kirilowii lectin (TKL) can prevent macrophage polarization from the M2 (anti-inflammatory) to the M1 (pro-inflammatory) phenotype by inhibiting the Notch signaling pathway, reducing the expression of Notch1, NICD1, and Hes1 to inhibit Notch signaling activity, and reducing kidney damage in rats with diabetic nephropathy (97). SIRT1 promotes macrophage polarization toward the M2 phenotype by inhibiting the NOTCH signaling pathway (98). Regulating macrophage polarization through Notch signaling is an important direction for treating diabetic nephropathy.
According to research on animals, the intrauterine diabetes environment hinders the differentiation of progenitor cells into nephrons, potentially through interference with the Notch and Wnt/β-catenin signaling pathways (99). Research has shown that p66Shc activates the Notch-PTEN-PI3K/Akt/mTOR signaling pathway to cause apoptosis and block podocyte autophagy, two actions that may be useful in the management of diabetic kidney disease (DKD) (100). The human renal tubular epithelial cells (HK-2 cells) exhibit a considerable activation of their Notch1 and Hes-1 signaling pathways during high glucose stimulation, which therefore results in a reduction in autophagy levels. Autophagy activity was restored when Sirt3 overexpression was achieved using pCMV-Sirt3 transfection. This resulted in a considerable inhibition of the Notch1/Hes-1 pathway activation (101). Based on this, it appears that Sirt3 protects autophagy in HK-2 cells and might be a target for diabetic nephropathy therapy (Table 1).
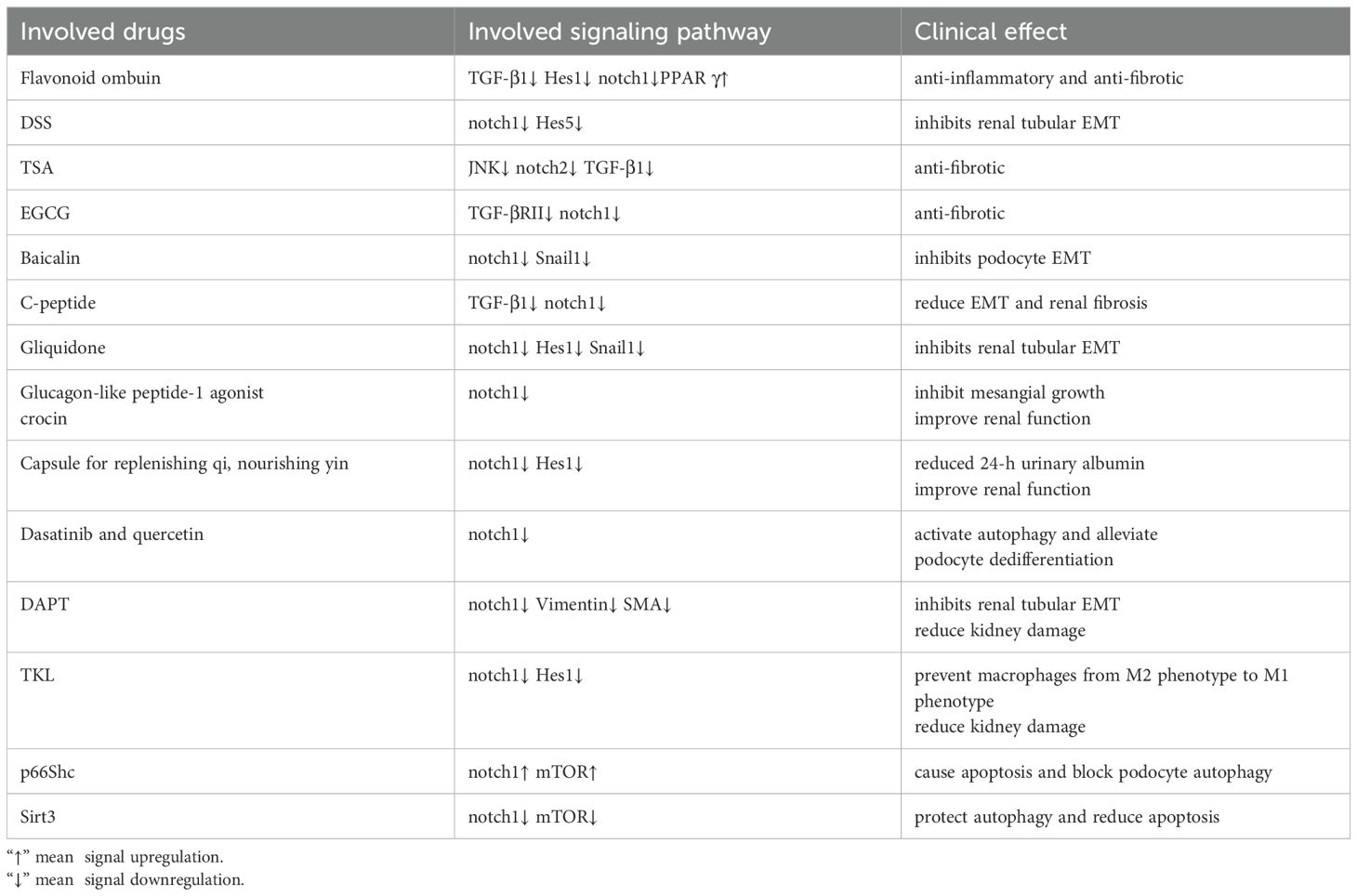
Table 1. Clinical use of Notch signaling in DKD.
7 Discussion
DKD is the main driver of death in diabetic patients. Controlling blood sugar alone is not enough to eliminate diabetic complications and improve survival (102). Drug treatment should focus on preventing complications rather than simply lowering blood sugar. SGLT2 is only expressed by PT cells, and SGLT2 inhibitors (SGLT2i), drugs targeting PT cells, have achieved good results. In addition to reducing heart failure mortality and mortality, they can also reduce comprehensive renal outcomes by 40% (103–105). Combination therapy such as SGLT2i and angiotensin-converting enzyme inhibitors has non-overlapping synergistic effects and can reduce PT cell damage, which suggests that combination therapy is the trend of the future (106). However, there are currently no approved drugs targeting podocytes. Current drugs primarily target renal PT cells and have been associated with GFR preservation. Future research will examine the potential therapeutic benefits of targeting different cell types, such as fibroblasts, immune cells, or podocytes.
The Notch signaling pathway regulates kidney development, angiogenesis, glomeruli, tubules, renal interstitium, macrophage function, etc. It is involved in multiple processes such as glomerular endothelial dysfunction, filtration barrier damage, podocyte EMT and dedifferentiation, tubulointerstitial fibrosis, PT cell dedifferentiation, macrophage polarization, etc. in DKD and is closely related to the onset of DKD. Notch signaling is involved in the entire process of DKD. It may become a reality to develop drugs targeting different types of kidney cells using NOTCH signaling, and combined treatment of multiple cell targets may be expected to improve the future of DKD patients. In summary, although there are still some issues to be resolved, we believe that correcting notch signaling abnormalities will become a new therapeutic strategy for DKD.
Author contributions
ZW: Writing – original draft, Writing – review & editing. WT: Writing – review & editing, Writing – original draft. YL: Writing – review & editing. PL: Writing – review & editing. KH: Writing – review & editing. JW: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Cho NH, Shaw JE, Karuranga S, Huang Y, da Rocha Fernandes JD, Ohlrogge AW, et al. Idf diabetes atlas: global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res Clin Pract. (2018) 138:271–81. doi: 10.1016/j.diabres.2018.02.023
2. Sun H, Saeedi P, Karuranga S, Pinkepank M, Ogurtsova K, Duncan BB, et al. Idf diabetes atlas: global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract. (2022) 183:109119. doi: 10.1016/j.diabres.2021.109119
3. Sagoo MK and Gnudi L. Diabetic nephropathy: an overview. Methods Mol Biol. (2020) 2067:3–7. doi: 10.1007/978-1-4939-9841-8_1
4. Jung CY and Yoo TH. Pathophysiologic mechanisms and potential biomarkers in diabetic kidney disease. Diabetes Metab J. (2022) 46:181–97. doi: 10.4093/dmj.2021.0329
5. Gupta S, Dominguez M, and Golestaneh L. Diabetic kidney disease: an update. Med Clin North Am. (2023) 107:689–705. doi: 10.1016/j.mcna.2023.03.004
6. Yang J and Liu Z. Mechanistic pathogenesis of endothelial dysfunction in diabetic nephropathy and retinopathy. Front Endocrinol (Lausanne). (2022) 13:816400. doi: 10.3389/fendo.2022.816400
7. Lavoz C, Poveda J, Marquez-Exposito L, Rayego-Mateos S, Rodrigues-Diez RR, Ortiz A, et al. Gremlin activates the notch pathway linked to renal inflammation. Clin Sci (Lond). (2018) 132:1097–115. doi: 10.1042/CS20171553
8. Ma T, Li X, Zhu Y, Yu S, Liu T, Zhang X, et al. Excessive activation of notch signaling in macrophages promote kidney inflammation, fibrosis, and necroptosis. Front Immunol. (2022) 13:835879. doi: 10.3389/fimmu.2022.835879
9. Tung CW, Hsu YC, Shih YH, Chang PJ, and Lin CL. Glomerular mesangial cell and podocyte injuries in diabetic nephropathy. Nephrol (Carlton). (2018) 23 Suppl 4:32–7. doi: 10.1111/nep.13451
10. Zhou B, Lin W, Long Y, Yang Y, Zhang H, Wu K, et al. Notch signaling pathway: architecture, disease, and therapeutics. Signal Transduct Target Ther. (2022) 7:95. doi: 10.1038/s41392-022-00934-y
11. Lemmon MA and Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. (2010) 141:1117–34. doi: 10.1016/j.cell.2010.06.011
12. Dorsam RT and Gutkind JS. G-protein-coupled receptors and cancer. Nat Rev Cancer. (2007) 7:79–94. doi: 10.1038/nrc2069
13. Meurette O and Mehlen P. Notch signaling in the tumor microenvironment. Cancer Cell. (2018) 34:536–48. doi: 10.1016/j.ccell.2018.07.009
14. Kopan R and Ilagan MX. The canonical notch signaling pathway: unfolding the activation mechanism. Cell. (2009) 137:216–33. doi: 10.1016/j.cell.2009.03.045
15. Yuan X, Wu H, Xu H, Xiong H, Chu Q, Yu S, et al. Notch signaling: an emerging therapeutic target for cancer treatment. Cancer Lett. (2015) 369:20–7. doi: 10.1016/j.canlet.2015.07.048
16. Capaccione KM and Pine SR. The notch signaling pathway as a mediator of tumor survival. Carcinogenesis. (2013) 34:1420–30. doi: 10.1093/carcin/bgt127
17. Owen DH, Giffin MJ, Bailis JM, Smit MD, Carbone DP, and He K. Dll3: an emerging target in small cell lung cancer. J Hematol Oncol. (2019) 12:61. doi: 10.1186/s13045-019-0745-2
18. Pitulescu ME, Schmidt I, Giaimo BD, Antoine T, Berkenfeld F, Ferrante F, et al. Dll4 and notch signalling couples sprouting angiogenesis and artery formation. Nat Cell Biol. (2017) 19:915–27. doi: 10.1038/ncb3555
19. Langridge PD and Struhl G. Epsin-dependent ligand endocytosis activates notch by force. Cell. (2017) 171:1383–96.e12. doi: 10.1016/j.cell.2017.10.048
20. Lambrecht BN, Vanderkerken M, and Hammad H. The emerging role of Adam metalloproteinases in immunity. Nat Rev Immunol. (2018) 18:745–58. doi: 10.1038/s41577-018-0068-5
21. Seegar TCM, Killingsworth LB, Saha N, Meyer PA, Patra D, Zimmerman B, et al. Structural basis for regulated proteolysis by the alpha-secretase adam10. Cell. (2017) 171:1638–48.e7. doi: 10.1016/j.cell.2017.11.014
22. Du H, Shih CH, Wosczyna MN, Mueller AA, Cho J, Aggarwal A, et al. Macrophage-released adamts1 promotes muscle stem cell activation. Nat Commun. (2017) 8:669. doi: 10.1038/s41467-017-00522-7
23. Siebel C and Lendahl U. Notch signaling in development, tissue homeostasis, and disease. Physiol Rev. (2017) 97:1235–94. doi: 10.1152/physrev.00005.2017
24. Aster JC, Pear WS, and Blacklow SC. The varied roles of notch in cancer. Annu Rev Pathol. (2017) 12:245–75. doi: 10.1146/annurev-pathol-052016-100127
25. Bray SJ. Notch signalling in context. Nat Rev Mol Cell Biol. (2016) 17:722–35. doi: 10.1038/nrm.2016.94
26. Liu Z, Zhou L, Zhao W, Jin L, Zhang J, Zhang Y, et al. Predict and prevent microvascular complications of type 2 diabetes: A cross-sectional and longitudinal study in Chinese communities. Front Endocrinol (Lausanne). (2025) 16:1541663. doi: 10.3389/fendo.2025.1541663
27. Zhang X, Zhao S, Huang Y, Ma M, Li B, Li C, et al. Diabetes-related macrovascular complications are associated with an increased risk of diabetic microvascular complications: A prospective study of 1518 patients with type 1 diabetes and 20–802 patients with type 2 diabetes in the UK biobank. J Am Heart Assoc. (2024) 13:e032626. doi: 10.1161/JAHA.123.032626
28. Salmon AH, Ferguson JK, Burford JL, Gevorgyan H, Nakano D, Harper SJ, et al. Loss of the endothelial glycocalyx links albuminuria and vascular dysfunction. J Am Soc Nephrol. (2012) 23:1339–50. doi: 10.1681/ASN.2012010017
29. Li L, Liu Q, Shang T, Song W, Xu D, Allen TD, et al. Aberrant activation of notch1 signaling in glomerular endothelium induces albuminuria. Circ Res. (2021) 128:602–18. doi: 10.1161/CIRCRESAHA.120.316970
30. Belyea BC, Xu F, Wiltsie M, Fountain H, Charlton J, Fogo AB, et al. Overexpression of notch signaling in renin cells leads to a polycystic kidney phenotype. Clin Sci (Lond). (2023) 137:35–45. doi: 10.1042/CS20220496
31. Farber G, Hurtado R, Loh S, Monette S, Mtui J, Kopan R, et al. Glomerular endothelial cell maturation depends on adam10, a key regulator of notch signaling. Angiogenesis. (2018) 21:335–47. doi: 10.1007/s10456-018-9599-4
32. Lin JS and Susztak K. Podocytes: the weakest link in diabetic kidney disease? Curr Diabetes Rep. (2016) 16:45. doi: 10.1007/s11892-016-0735-5
33. Ai X, Yu P, Luo L, Sun J, Tao H, Wang X, et al. Berberis dictyophylla F. Inhibits angiogenesis and apoptosis of diabetic retinopathy via suppressing Hif-1alpha/Vegf/Dll-4/Notch-1 pathway. J Ethnopharmacol. (2022) 296:115453. doi: 10.1016/j.jep.2022.115453
34. Lin CL, Wang FS, Hsu YC, Chen CN, Tseng MJ, Saleem MA, et al. Modulation of notch-1 signaling alleviates vascular endothelial growth factor-mediated diabetic nephropathy. Diabetes. (2010) 59:1915–25. doi: 10.2337/db09-0663
35. Fortini F, Vieceli Dalla Sega F, Marracino L, Severi P, Rapezzi C, Rizzo P, et al. Well-known and novel players in endothelial dysfunction: updates on a notch(Ed) landscape. Biomedicines. (2021) 9(8):997. doi: 10.3390/biomedicines9080997
36. Akil A, Gutierrez-Garcia AK, Guenter R, Rose JB, Beck AW, Chen H, et al. Notch signaling in vascular endothelial cells, angiogenesis, and tumor progression: an update and prospective. Front Cell Dev Biol. (2021) 9:642352. doi: 10.3389/fcell.2021.642352
37. Certelli A, Valente P, Uccelli A, Grosso A, Di Maggio N, D’Amico R, et al. Robust angiogenesis and arteriogenesis in the skin of diabetic mice by transient delivery of engineered Vegf and Pdgf-Bb proteins in fibrin hydrogels. Front Bioeng Biotechnol. (2021) 9:688467. doi: 10.3389/fbioe.2021.688467
38. Wang Y, Zhao S, Ni N, Chen H, Zhao W, Xing K, et al. Nanoparticles induced glomerular endothelial leakiness promoting albuminuria level. NanoImpact. (2025) 37:100548. doi: 10.1016/j.impact.2025.100548
39. Huangfu Q, Zhang J, Xu J, Xu J, Yang Z, Wei J, et al. Mechanosensitive Ca(2+) channel Trpv1 activated by low-intensity pulsed ultrasound ameliorates acute kidney injury through Notch1-Akt-Enos signaling. FASEB J. (2025) 39:e70304. doi: 10.1096/fj.202401142RR
40. Pandya Thakkar N, Pereira BMV, Katakia YT, Ramakrishnan SK, Thakar S, Sakhuja A, et al. Elevated H3k4me3 through Mll2-Wdr82 upon hyperglycemia causes jagged ligand dependent notch activation to interplay with differentiation state of endothelial cells. Front Cell Dev Biol. (2022) 10:839109. doi: 10.3389/fcell.2022.839109
41. Zhao Y, Qiao X, Wang L, Tan TK, Zhao H, Zhang Y, et al. Correction to: matrix metalloproteinase 9 induces endothelial-mesenchymal transition via notch activation in human kidney glomerular endothelial cells. BMC Mol Cell Biol. (2020) 21:72. doi: 10.1186/s12860-020-00318-6
42. Mukhi D and Susztak K. The transcriptomic signature of the aging podocyte. Kidney Int. (2020) 98:1079–81. doi: 10.1016/j.kint.2020.08.004
43. Rutkowski JM, Wang ZV, Park AS, Zhang J, Zhang D, Hu MC, et al. Adiponectin promotes functional recovery after podocyte ablation. J Am Soc Nephrol. (2013) 24:268–82. doi: 10.1681/ASN.2012040414
44. Raij L, Tian R, Wong JS, He JC, and Campbell KN. Podocyte injury: the role of proteinuria, urinary plasminogen, and oxidative stress. Am J Physiol Renal Physiol. (2016) 311:F1308–F17. doi: 10.1152/ajprenal.00162.2016
45. Niranjan T, Bielesz B, Gruenwald A, Ponda MP, Kopp JB, Thomas DB, et al. The notch pathway in podocytes plays a role in the development of glomerular disease. Nat Med. (2008) 14:290–8. doi: 10.1038/nm1731
46. He Y, Zhang M, Wu Y, Jiang H, Fu H, Cai Y, et al. Aberrant activation of notch-1 signaling inhibits podocyte restoration after islet transplantation in a rat model of diabetic nephropathy. Cell Death Dis. (2018) 9:950. doi: 10.1038/s41419-018-0985-z
47. Yuan J, Lin F, Chen L, Huang H, Ni X, Pan X, et al. Microencapsulated islet transplantation alleviates podocyte injury in diabetic nephropathy via inhibiting notch-1 signaling. Transpl Immunol. (2022) 72:101579. doi: 10.1016/j.trim.2022.101579
48. Liu M, Liang K, Zhen J, Zhou M, Wang X, Wang Z, et al. Sirt6 deficiency exacerbates podocyte injury and proteinuria through targeting notch signaling. Nat Commun. (2017) 8:413. doi: 10.1038/s41467-017-00498-4
49. Li MR, Lei CT, Tang H, Yin XJ, Hao Z, Qiu Y, et al. Mad2b promotes podocyte injury through regulating numb-dependent notch 1 pathway in diabetic nephropathy. Int J Biol Sci. (2022) 18:1896–911. doi: 10.7150/ijbs.68977
50. Xiao M, Bai S, Chen J, Li Y, Zhang S, and Hu Z. Cdkn2b-As1 participates in high glucose-induced apoptosis and fibrosis via Notch2 through functioning as a Mir-98-5p decoy in human podocytes and renal tubular cells. Diabetol Metab Syndr. (2021) 13:107. doi: 10.1186/s13098-021-00725-5
51. Majumder S, Thieme K, Batchu SN, Alghamdi TA, Bowskill BB, Kabir MG, et al. Shifts in podocyte histone H3k27me3 regulate mouse and human glomerular disease. J Clin Invest. (2018) 128:483–99. doi: 10.1172/JCI95946
52. Jiang L, Liu X, Hu X, Gao L, Zeng H, Wang X, et al. Mettl3-mediated M(6)a modification of Timp2 Mrna promotes podocyte injury in diabetic nephropathy. Mol Ther. (2022) 30:1721–40. doi: 10.1016/j.ymthe.2022.01.002
53. Kim D, Lim S, Park M, Choi J, Kim J, Han H, et al. Ubiquitination-dependent carm1 degradation facilitates notch1-mediated podocyte apoptosis in diabetic nephropathy. Cell Signal. (2014) 26:1774–82. doi: 10.1016/j.cellsig.2014.04.008
54. Nishad R, Meshram P, Singh AK, Reddy GB, and Pasupulati AK. Activation of notch1 signaling in podocytes by glucose-derived ages contributes to proteinuria. BMJ Open Diabetes Res Care. (2020) 8(1):e001203. doi: 10.1136/bmjdrc-2020-001203
55. Ying Q and Wu G. Molecular mechanisms involved in podocyte emt and concomitant diabetic kidney diseases: an update. Ren Fail. (2017) 39:474–83. doi: 10.1080/0886022X.2017.1313164
56. Nishad R, Mukhi D, Singh AK, Motrapu M, Chintala K, Tammineni P, et al. Growth hormone induces mitotic catastrophe of glomerular podocytes and contributes to proteinuria. Cell Death Dis. (2021) 12:342. doi: 10.1038/s41419-021-03643-6
57. Quinn GZ, Dhillon P, and Susztak K. It takes two to tango: the role of dysregulated metabolism and inflammation in kidney disease development. Semin Nephrol. (2020) 40:199–205. doi: 10.1016/j.semnephrol.2020.01.010
58. Vallon V and Thomson SC. The tubular hypothesis of nephron filtration and diabetic kidney disease. Nat Rev Nephrol. (2020) 16:317–36. doi: 10.1038/s41581-020-0256-y
59. Luo J, Xu H, Su C, Dong W, Xiao M, Xiao N, et al. Polo-like kinase2 regulates renal tubulointerstitial fibrosis via notch signaling pathway in diabetic kidney disease. FASEB J. (2025) 39:e70455. doi: 10.1096/fj.202402793R
60. Antar SA, Ashour NA, Marawan ME, and Al-Karmalawy AA. Fibrosis: types, effects, markers, mechanisms for disease progression, and its relation with oxidative stress, immunity, and inflammation. Int J Mol Sci. (2023) 24(4):4004. doi: 10.3390/ijms24044004
61. Zhang Y, Jin D, Kang X, Zhou R, Sun Y, Lian F, et al. Signaling pathways involved in diabetic renal fibrosis. Front Cell Dev Biol. (2021) 9:696542. doi: 10.3389/fcell.2021.696542
62. Meyer-Schwesinger C, Seipold L, and Saftig P. Ectodomain shedding by Adam proteases as a central regulator in kidney physiology and disease. Biochim Biophys Acta Mol Cell Res. (2022) 1869:119165. doi: 10.1016/j.bbamcr.2021.119165
63. Murea M, Park JK, Sharma S, Kato H, Gruenwald A, Niranjan T, et al. Expression of notch pathway proteins correlates with albuminuria, glomerulosclerosis, and renal function. Kidney Int. (2010) 78:514–22. doi: 10.1038/ki.2010.172
64. Yu C, Xiong C, Tang J, Hou X, Liu N, Bayliss G, et al. Histone demethylase Jmjd3 protects against renal fibrosis by suppressing Tgfbeta and notch signaling and preserving Pten expression. Theranostics. (2021) 11:2706–21. doi: 10.7150/thno.48679
65. Wang Y, Shen RW, Han B, Li Z, Xiong L, Zhang FY, et al. Notch signaling mediated by Tgf-Beta/Smad pathway in concanavalin a-induced liver fibrosis in rats. World J Gastroenterol. (2017) 23:2330–6. doi: 10.3748/wjg.v23.i13.2330
66. Liu L, Gao C, Chen G, Li X, Li J, Wan Q, et al. Notch signaling molecules activate Tgf- Beta in rat mesangial cells under high glucose conitions. J Diabetes Res. (2013) 2013:979702. doi: 10.1155/2013/979702
67. Zhu QQ, Yang XY, Zhang XJ, Yu CJ, Pang QQ, Huang YW, et al. Egcg targeting notch to attenuate renal fibrosis via inhibition of Tgfbeta/Smad3 signaling pathway activation in streptozotocin-induced diabetic mice. Food Funct. (2020) 11:9686–95. doi: 10.1039/d0fo01542c
68. Xiao Z, Zhang J, Peng X, Dong Y, Jia L, Li H, et al. The notch gamma-secretase inhibitor ameliorates kidney fibrosis via inhibition of Tgf-Beta/Smad2/3 signaling pathway activation. Int J Biochem Cell Biol. (2014) 55:65–71. doi: 10.1016/j.biocel.2014.08.009
69. Liu W, Wu Y, Yu F, Hu W, Fang X, and Hao W. The implication of numb-induced notch signaling in endothelial-mesenchymal transition of diabetic nephropathy. J Diabetes Complications. (2018) 32:889–99. doi: 10.1016/j.jdiacomp.2018.06.011
70. Perkins TN, Dentener MA, Stassen FR, Rohde GG, Mossman BT, Wouters EF, et al. Alteration of canonical and non-canonical Wnt-signaling by crystalline silica in human lung epithelial cells. Toxicol Appl Pharmacol. (2016) 301:61–70. doi: 10.1016/j.taap.2016.04.003
71. Bertrand FE, Angus CW, Partis WJ, and Sigounas G. Developmental pathways in colon cancer: crosstalk between Wnt, Bmp, Hedgehog and Notch. Cell Cycle. (2012) 11:4344–51. doi: 10.4161/cc.22134
72. Modder UI, Oursler MJ, Khosla S, and Monroe DG. Wnt10b activates the Wnt, Notch, and Nfkappab pathways in U2os osteosarcoma cells. J Cell Biochem. (2011) 112:1392–402. doi: 10.1002/jcb.23048
73. Chen X, Stoeck A, Lee SJ, Shih Ie M, Wang MM, and Wang TL. Jagged1 expression regulated by Notch3 and Wnt/Beta-catenin signaling pathways in ovarian cancer. Oncotarget. (2010) 1:210–8. doi: 10.18632/oncotarget.127
74. Xiao X, Wang W, Guo C, Wu J, Zhang S, Shi H, et al. Hypermethylation leads to the loss of Hoxa5, resulting in Jag1 expression and notch signaling contributing to kidney fibrosis. Kidney Int. (2024) 106:98–114. doi: 10.1016/j.kint.2024.02.023
75. Kanasaki K, Taduri G, and Koya D. Diabetic nephropathy: the role of inflammation in fibroblast activation and kidney fibrosis. Front Endocrinol (Lausanne). (2013) 4:7. doi: 10.3389/fendo.2013.00007
76. Sweetwyne MT, Tao J, and Susztak K. Kick it up a notch: notch signaling and kidney fibrosis. Kidney Int Suppl (2011). (2014) 4:91–6. doi: 10.1038/kisup.2014.17
77. Sun J, Zhao F, Zhang W, Lv J, Lv J, and Yin A. Bmscs and Mir-124a ameliorated diabetic nephropathy via inhibiting notch signalling pathway. J Cell Mol Med. (2018) 22:4840–55. doi: 10.1111/jcmm.13747
78. Zhang LY, Wang Y, Yang YR, Shao JJ, and Liang B. Mir-135a regulates renal fibrosis in rats with diabetic kidney disease through the notch pathway. Eur Rev Med Pharmacol Sci. (2020) 24:1979–87. doi: 10.26355/eurrev_202002_20375
79. Li Y, Tan P, Liu Q, Liu M, Wang Y, Kong W, et al. Mirna-133a-3p attenuates renal tubular epithelial cell injury via targeting Malm1 and suppressing the notch signaling pathway in diabetic nephropathy. Cell Biochem Biophys. (2024) 82:2401–11. doi: 10.1007/s12013-024-01351-4
80. Liu XD, Zhang LY, Zhu TC, Zhang RF, Wang SL, and Bao Y. Overexpression of mir-34c inhibits high glucose-induced apoptosis in podocytes by targeting notch signaling pathways. Int J Clin Exp Pathol. (2015) 8:4525–34. Available at: online https://www.ncbi.nlm.nih.gov/pubmed/26191142.
81. Zhang X, Song S, and Luo H. Regulation of podocyte lesions in diabetic nephropathy via mir-34a in the Notch signaling pathway. Med (Baltimore). (2016) 95:e5050. doi: 10.1097/MD.0000000000005050
82. Wang Y, Hu P, Li Y, Dong R, and He J. Microvesicles from mesenchymal stem cells overexpressing Mir-34a ameliorate renal fibrosis in vivo. Iran J Kidney Dis. (2024) 18:99–107. doi: 10.5254/s9bdqs74
83. Lee HW, Khan SQ, Khaliqdina S, Altintas MM, Grahammer F, Zhao JL, et al. Absence of Mir-146a in podocytes increases risk of diabetic glomerulopathy via up-regulation of Erbb4 and Notch-1. J Biol Chem. (2017) 292:732–47. doi: 10.1074/jbc.M116.753822
84. Ning Y, Zhou X, Wang G, Zhang L, and Wang J. Exosome Mir-30a-5p regulates glomerular endothelial cells’ Endmt and angiogenesis by modulating Notch1/Vegf signaling pathway. Curr Gene Ther. (2024) 24:159–77. doi: 10.2174/0115665232258527230919071328
85. Raval N, Gondaliya P, Tambe V, Kalia K, and Tekade RK. Engineered nanoplex mediated targeted Mirna delivery to rescue dying podocytes in diabetic nephropathy. Int J Pharm. (2021) 605:120842. doi: 10.1016/j.ijpharm.2021.120842
86. Liu B, Deng C, and Tan P. Ombuin ameliorates diabetic nephropathy in rats by anti-inflammation and antifibrosis involving Notch 1 and Ppar gamma signaling pathways. Drug Dev Res. (2022) 83:1270–80. doi: 10.1002/ddr.21956
87. Xiaobing L, Chunling N, Wenyu C, Yan C, and Zhenzhen L. Effect of danggui-shaoyao-san-containing serum on the renal tubular epithelial-mesenchymal transition of diabetic nephropathy. Curr Pharm Biotechnol. (2020) 21:1204–12. doi: 10.2174/1389201021666200416094318
88. Tung CW, Hsu YC, Cai CJ, Shih YH, Wang CJ, Chang PJ, et al. Trichostatin a ameliorates renal tubulointerstitial fibrosis through modulation of the Jnk-dependent Notch-2 signaling pathway. Sci Rep. (2017) 7:14495. doi: 10.1038/s41598-017-15162-6
89. Dou Y, Shang Y, Shen Y, Qu J, Liu C, and Cao J. Baicalin alleviates adriamycin-induced focal segmental glomerulosclerosis and proteinuria by inhibiting the notch1-snail axis mediated podocyte emt. Life Sci. (2020) 257:118010. doi: 10.1016/j.lfs.2020.118010
90. Luo J, Jiang J, Huang H, Jiang F, Xu Z, Zhou Z, et al. C-peptide ameliorates high glucose-induced podocyte dysfunction through the regulation of the notch and Tgf-Beta signaling pathways. Peptides. (2021) 142:170557. doi: 10.1016/j.peptides.2021.170557
91. Tian H, Yang J, Xie Z, and Liu J. Gliquidone alleviates diabetic nephropathy by inhibiting notch/snail signaling pathway. Cell Physiol Biochem. (2018) 51:2085–97. doi: 10.1159/000495827
92. Amin SN, El-Gamal EM, Rashed LA, Kamar SS, and Haroun MA. Inhibition of notch signalling and mesangial expansion by combined glucagon like peptide-1 agonist and Crocin therapy in animal model of diabetic nephropathy. Arch Physiol Biochem. (2023) 129:544–54. doi: 10.1080/13813455.2020.1846203
93. Zhou X, Xu C, Wang K, Chu Q, Dong C, Wu C, et al. Effect of traditional chinese medicine for replenishing qi, nourishing yin and activating blood on renal Notch/Hes1 signaling in rats with diabetic nephropathy. Nan Fang Yi Ke Da Xue Xue Bao. (2019) 39:855–60. doi: 10.12122/j.issn.1673-4254.2019.07.17
94. Zhu X, Zhang C, Liu L, Xu L, and Yao L. Senolytic combination of dasatinib and quercetin protects against diabetic kidney disease by activating autophagy to alleviate podocyte dedifferentiation via the notch pathway. Int J Mol Med. (2024) 53(3):26. doi: 10.3892/ijmm.2024.5350
95. Nishad R, Mukhi D, Tahaseen SV, Mungamuri SK, and Pasupulati AK. Growth hormone induces Notch1 signaling in podocytes and contributes to proteinuria in diabetic nephropathy. J Biol Chem. (2019) 294:16109–22. doi: 10.1074/jbc.RA119.008966
96. Duan X and Qin G. Notch inhibitor mitigates renal ischemia−Reperfusion injury in diabetic rats. Mol Med Rep. (2020) 21:583–8. doi: 10.3892/mmr.2019.10857
97. Jiandong L, Yang Y, Peng J, Xiang M, Wang D, Xiong G, et al. Trichosanthes kirilowii lectin ameliorates streptozocin-induced kidney injury via modulation of the balance between M1/M2 phenotype macrophage. BioMed Pharmacother. (2019) 109:93–102. doi: 10.1016/j.biopha.2018.10.060
98. Song BF, Li BJ, Ning JZ, Xia YQ, Ye ZH, Yuan TH, et al. Overexpression of sirtuin 1 attenuates calcium oxalate-induced kidney injury by promoting macrophage polarization. Int Immunopharmacol. (2023) 121:110398. doi: 10.1016/j.intimp.2023.110398
99. Cerqueira DM, Hemker SL, Bodnar AJ, Ortiz DM, Oladipupo FO, Mukherjee E, et al. In utero exposure to maternal diabetes impairs nephron progenitor differentiation. Am J Physiol Renal Physiol. (2019) 317:F1318–F30. doi: 10.1152/ajprenal.00204.2019
100. Zheng D, Tao M, Liang X, Li Y, Jin J, and He Q. P66shc regulates podocyte autophagy in high glucose environment through the notch-pten-Pi3k/Akt/Mtor pathway. Histol Histopathol. (2020) 35:405–15. doi: 10.14670/HH-18-178
101. Wang Y, Chang J, Wang ZQ, and Li Y. Sirt3 promotes the autophagy of Hk−2 human proximal tubular epithelial cells via the inhibition of Notch−1/Hes−1 signaling. Mol Med Rep. (2021) 24(3):634. doi: 10.3892/mmr.2021.12273
102. Skyler JS, Bergenstal R, Bonow RO, Buse J, Deedwania P, Gale EA, et al. Intensive glycemic control and the prevention of cardiovascular events: implications of the accord, advance, and Va diabetes trials: A position statement of the American diabetes association and a scientific statement of the American college of cardiology foundation and the American heart association. Circulation. (2009) 119:351–7. doi: 10.1161/CIRCULATIONAHA.108.191305
103. Chertow GM, Vart P, Jongs N, Toto RD, Gorriz JL, Hou FF, et al. Effects of dapagliflozin in stage 4 chronic kidney disease. J Am Soc Nephrol. (2021) 32:2352–61. doi: 10.1681/ASN.2021020167
104. Heerspink HJL, Stefansson BV, Correa-Rotter R, Chertow GM, Greene T, Hou FF, et al. Dapagliflozin in patients with chronic kidney disease. N Engl J Med. (2020) 383:1436–46. doi: 10.1056/NEJMoa2024816
105. Perkovic V, Jardine MJ, Neal B, Bompoint S, Heerspink HJL, Charytan DM, et al. Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. N Engl J Med. (2019) 380:2295–306. doi: 10.1056/NEJMoa1811744
Keywords: Notch signaling, diabetic kidney disease, renal fibrosis, podocyte, therapy
Citation: Wang Z-H, Tu W, Long Y-N, Li P-F, He K-Y and Wu J (2025) Notch signaling in diabetic kidney disease: recent progress. Front. Endocrinol. 16:1537769. doi: 10.3389/fendo.2025.1537769
Received: 01 December 2024; Accepted: 18 July 2025;
Published: 31 July 2025.
Edited by:
Yong Fan, Institute of Cellular Therapeutics, United StatesCopyright © 2025 Wang, Tu, Long, Li, He and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Tu, MzI2MzE5NjI5QHFxLmNvbQ==; Jing Wu, Zno0MjEyQHllYWgubmV0