 Abirami Devadas
Abirami Devadas Stève Baranton
Stève Baranton Christophe Coutanceau
Christophe Coutanceau- IC2MP, UMR 7285 CNRS-Université de Poitiers, Poitiers cedex, France
Ion exchange method as a green synthesis route is proposed to prepare hydrous ruthenium oxide nanoparticles (H-RuO2). Calcination of H-RuO2 at 350°C resulted in the crystalline rutile RuO2 nanoparticles (C-RuO2). Treatment of H-RuO2 with 20 vol% ammonium hydroxide solution under microwave irradiation and calcination at 350°C resulted in a highly electrocatalytic active crystalline RuO2 nanoparticles (A-C-RuO2). Electrocatalytic performances of H-RuO2, C-RuO2 and A-C-RuO2 for the oxygen evolution reaction in 0.50 mol L−1 H2SO4 medium are evaluated and compared. Improved performances towards the oxygen evolution reaction are observed for A-C-RuO2 when compared to C-RuO2. Based on XRD, TEM, XPS and Raman characterizations performed on all the specimens, it is deduced that the physicochemical properties (crystallinity, mean crystallite size, level of hydrous rutile content) are varied for A-C-RuO2 when compared to C-RuO2. Structure-property correlation has been established to describe the higher electrocatalytic activity of A-C-RuO2.
Introduction
Renewable energy sources such as hydroelectric, wind, solar and tidal powers have been improving constantly, despite these energies have supplied instabilities (International Agency of Energy, 2019; Shariatzadeh et al., 2015). Electrochemical energy storage and conversion technologies are therefore gaining importance due to the growing electric energy demand without impacting the greenhouse gas emission (Badwal et al., 2015; Carmo and Stolten, 2019). Electrodes (anode and cathode) constitute the important aspect of all electrochemical technological processes (water electrolysis, fuel-cell, redox batteries, fuel production from CO2, etc.). More developmental work on electrode materials, also known as electrocatalysts, is monumental because the electrodes determine the efficacy, feasibility, cost-effectiveness and durability of electrochemical processes (Walsh, 2019). In particular, the oxygen evolution reaction (OER) is a challenging reaction with high activation overpotential and high potential conditions, leading to strongly oxidizing operating conditions for the anode materials. Many different materials have been investigated for this reaction (Wang et al., 2018; Laha et al., 2019; Retuerto et al., 2019; Chen et al., 2020).
In this context, ruthenium oxide (RuO2) has become a very important material owing to its potency as supercapacitor (Zheng et al., 1995; Liu et al., 1997; Over, 2012) and as electrocatalyst for the OER (Trasatti, 2000; Carmo et al., 2013). RuO2 is a versatile oxide material used in several electrochemical processes, in the view of its high thermal and chemical stabilities, metallic type conductivity (i.e., low resistivity), highly reversible redox property and long-life cycle. Hence, RuO2 coated materials were used as anodes in many electrochemical industrial processes like cathodic protection, metal recovery, photoelectrochemical water oxidation, electroplating, electrowinning, electroflotation, electrosynthesis, cathodic protection, etc. (Cardarelli et al., 1998; Trasatti, 1999; Bock et al., 2000; Shibli et al., 2004; Katsaounis et al., 2007). Importantly, RuO2 exhibits as a corrosion-resistant electrode at low overpotentials for the OER in water electrolysis cells. The electrocatalytic activity, selectivity and stability of rutile-type RuO2 catalysts for the water splitting reaction depend on the surface and bulk structure of the material, and further on the synthetic method as well as on the synthesis precursors (Pelegrino et al., 2002).
Microwave assisted “instant method” (Devadas et al., 2011; Audichon et al., 2017a) and microwave assisted “polyol method” (Audichon et al., 2017b) were reported by our group for rapid synthesis of RuO2. Synthesized RuO2 was further investigated for its structure to tune the electrocatalytic activity towards the water electrolysis reaction or the capacitance for capacitor application (Devadas et al., 2011; Audichon et al., 2017a; Audichon et al., 2017b). In the case of the “instant method,” it was observed that an impurity phase containing lithium ruthenium oxide or lithium carbonate was formed in the synthesis process (Devadas et al., 2011; Audichon et al., 2017a). Moreover, the use of ethanol-water mixture was needed to centrifuge and to separate precipitant formed. In the case of the polyol method, the reaction was carried out in ethylene glycol as organic solvent, with formation of glycolate and other organic compounds as by-products (Coutanceau et al., 2011; Audichon et al., 2017b).
In the present study, a simple, effective and economic synthesis route for the large-scale synthesis of RuO2 nanoparticles is proposed based on ion-exchange method. In this ion-exchange method, the principles of green chemistry (time, energy and atom savings, no organic solvent, no organic precursor, safe and environmental-benign process) are being realized for the synthesis of materials (Anastas and Eghbali, 2010). The possibility to improve the electrocatalytic activity of RuO2 was also investigated by treating under microwave irradiation the ion-exchange derived product with ammonium hydroxide. All the specimens were evaluated for the OER in acidic medium; their electrocatalytic activity was analyzed and compared in light of their physicochemical characteristics (compositions, structure and crystallinity).
Materials and Methods
Materials
All experiments were carried out using ultrapure water (18.2 MΩ cm) obtained from a Milli-Q Millipore system. The anion exchange resin was obtained from Amberlite IRA910 in the chloride form (15–60 mesh, ion exchange capacity of 3.8 meq/g dry weight). Ruthenium chloride (RuCl3.xH2O, 99.9% purity, Ru 38 wt%) and ammonium hydroxide (NH4OH, 28–30%, ACS reagent grade) were obtained from Alfa Aesar and Sigma Aldrich, respectively. Nafion solution (5 wt% in aliphatic alcohols) from Aldrich was used as received. All the electrochemical measurements were carried out with the electrolyte prepared from suprapur H2SO4 obtained from Merck and electrolyte was de-aerated by bubbling nitrogen of U quality from l'Air Liquide.
Synthesis of RuO2 Nanoparticles Using Anion Exchanger
5 g of as received anion exchange resin were washed using ultrapure water and transformed into hydroxide form by agitating in NaOH solution (2.0 mol L−1, 100 ml) for 18 h followed by repeated washing with ultrapure water till the pH of washing water becomes neutral. Hydroxide form of anion exchanger was then closely packed in a column and an aqueous solution containing RuCl3 (0.010 mol L−1, 50 ml) was passed through the column with flow of 1 ml/min. The contents at the end of the column were collected in a conical flask. A part of the contents in the conical flask was dried at 80°C to obtain hydrous ruthenium oxide nanoparticles (H-RuO2). A part of H-RuO2 was put aside and a part was calcined at 350°C (heating rate of 5°C/min under air atmosphere) to obtain the sample denoted as C-RuO2. The remaining part of the contents in conical flask was transferred to a microwave reactor chamber and treated under microwave irradiation (impulsion power of 400 W) with 20 vol% ammonium hydroxide solution in a microwave oven MARS 5 (CEM Corporation). The reactor chamber in the microwave was attached to a water condenser in order to avoid water evaporation and temperature was maintained at 60°C. After 1 h of microwave irradiation, the mixture in reactor was centrifuged. Finally, the obtained product was calcined at 350°C (heating rate of 5°C/min under air atmosphere) and the obtained sample was denoted as A-C-RuO2. The designed synthesis process is shown in Figure 1.
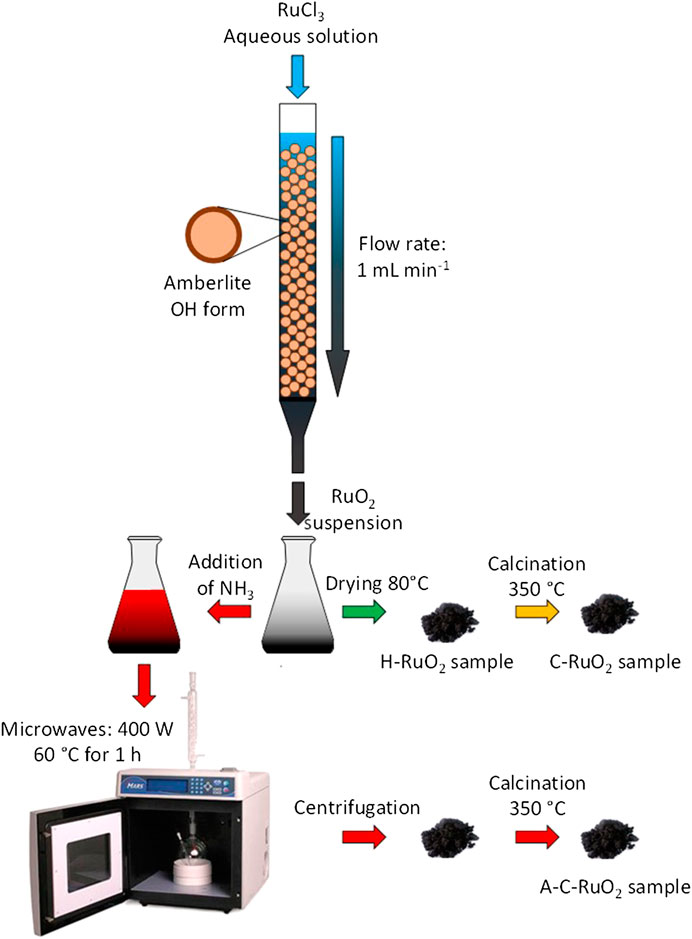
FIGURE 1. Schema of the synthesis processes of C-RuO2 H-RuO2 and A-C-RuO2 samples.
Physicochemical Characterizations
All the XRD Patterns were recorded on a Brucker D8-Advance X-ray diffractometer with a copper anode (Kα = 1.5405 Å) powered at 40 kV and 40 mA. The in situ XRD analysis on H-RuO2 was performed by heating the sample from room temperature to 350°C under air atmosphere. Kanthal holder was used in order to perform the heat treatment with a heating rate of 10°C min-1 between each set temperature, where the XRD patterns were recorded. XRD measurements were carried out in the 2θ range from 10° to 70° in step mode of 0.065° and a fixed acquisition time of 5 s/step. The diffraction peaks were fitted with Lorentzian-Gaussian functions using Fityk software (Fityk, 2010) and the lattice parameters of C-RuO2 were calculated using the linear least square method, considering the rutile structure of RuO2.
Raman spectra (Stokes spectra) were obtained at room temperature using a HR UV 800 confocal scanning spectrometer (Horiba Jobin Yvon) equipped with a Peltier-cooled charge coupled device (1,152 × 298 pixels). The Raman scattering was excited using a 632.8 nm excitation wavelength supplied by an internal, air-cooled, He-Ne laser (Melles Griot) through an Olympus high-stability BXFM microscope coupled confocally. Laser power delivered at the specimen (ca. 20 mW without filtering) could be monitored via a filter wheel with optical densities of 0.3, 0.6, and 1. The backscattered radiation was collected using the same microscope with a ×100 Olympus objective (numerical aperture 0.9). An 1,800 grooves mm-1 holographic grating was used, and the confocal hole aperture was 300 mm, resulting in an instrumental bandwidth of 6.3 cm-1 and a spectral resolution of 0.5 cm-1 in the 50–1,000 cm-1 Raman shift range. The spectrometer was calibrated using the G25 phonon of diamond Si (Fd-3m) resulting in an accuracy of less than 0.5 cm-1. LabSPEC v.5 software was used to perform both acquisition and data processing.
H-RuO2, C-RuO2 and A-C-RuO2 nanoparticle morphologies were examined using a transmission electron microscope (JEOL 2100, UHR, 200 kV) and the qualitative elemental analysis of the specimens was made by EDS analyzer equipped with a LaB6 filament. The mean particle size and size distribution were determined by measuring the féret’s diameter of 250 isolated particles using ImageJ free software (Rasband, 2009).
X-ray photoelectron spectroscopy (XPS) core level spectra were collected using a VG ESCALAB three MKII spectrometer using a monochromatized Mg Kα radiation (1,253.6 eV). The source was operated at 300 W (15 kV and 20 mA). The data were collected at room temperature and the operating pressure in the analysis chamber was set below 8 × 10–9 Torr. The core level spectra were recorded with a resolution of 50 meV and sample analysis covered a surface of 2 × 3 mm.
Electrochemical Measurements
The electrocatalytic activity for the OER was evaluated in a standard three-electrode electrochemical cell fitted with a 3 cm2 glassy carbon plate as auxiliary electrode and a reversible hydrogen electrode (RHE) as reference electrode, which was connected through a Luggin capillary to the working electrode compartment. The anode was made from an ink consisting in a suspension of the prepared materials (10 mg) in a mixture of 1,000 µl ultrapure water and 100 µl Nafion solution. The working electrode was prepared by dipping 5 µl of the ink on a polished glassy-carbon electrode (0.071 cm2 geometric surface area), leading to a catalyst loading of 45.5 µg catalyst on the electrode. The applied potential controlled by a VoltaLab PGZ 402 potentiostat and the outputs were monitored by a computer. The electrolyte used was a 0.50 mol L-1 H2SO4 solution in ultrapure water saturated with nitrogen for 15 min prior to the measurements. Nitrogen atmosphere was maintained in the electrochemical cell throughout the measurements. The linear scan voltammograms were recorded at a scan rate of 0.010 V s-1. Chronoamperometric curves were recorded at +1.600 V vs RHE for 60 min, under the similar experimental conditions of linear voltammograms.
Results
Green Synthesis of Hydrous RuO2
Ion-exchange method adopted in this work leans on a soft hydrolysis route with aid of hydroxyl anions (OH−) from anion exchanger packed in column. Ion exchange method has been used for the batch synthesis of the colloidal precursors of hydroxide and oxide materials of Fe, Al, Cr, Zr and Ti with aid of anion exchangers (Vertegel et al., 1995; Tretyakov et al., 1997; Shafran et al., 2005). To the best of our knowledge the synthesis of ruthenium hydroxide/oxide materials using anion exchanger enabling hydrolysis of a ruthenium salt is not reported. Dynamic/column mode and static/batch mode of operations were used in ion exchange synthesis route (Pelletier et al., 2000). In the column mode, a solution of a metal ion precursor is forced through a column containing a densely packed ion exchange resin. The formed metal hydroxide leads to a stable colloidal phase, which can stabilize the concentration gradient along the column due to variation of the degree of exchange. The slow release of hydroxyl ions from the surface of the resin gives kinetic control of the hydrolysis reaction when compared to hydrolysis of metal ions in conventional solution-based methods. In this study, the contact of RuCl3 solution with OH form of Amberlite IRA910 anion exchanger leads to the formation of Ru(OH)3 precipitate. Van Muylder and Pourbaix (1963) and Music et al. (Music et al., 2002) proposed that Ru(OH)3 was not stable in the presence of air and oxidized into hydrous RuO2. Dmowski et al. (Dmowski et al., 2002) examined the hydrous RuO2 by atomic pair density function (PDF) method and pointed out that hydrous RuO2 is a composite of anhydrous rutile-like RuO2 nanocrystals dispersed by boundaries of structural water associated with Ru-O bonds. The precipitate obtained in the present study is free of any other impurities and no separation step is needed. Moreover, the spent anion exchangers can be reused after treatment with NaOH solution, which suggests the elimination of waste streams during the synthesis process.
Physicochemical Characterization of Hydrous and Crystalline RuO2
Figure 2 illustrates the in-situ XRD patterns of H-RuO2 calcined from room temperature to 350°C. As expected, only broad peaks are present, and no characteristic peak related to the crystalline rutile structure is identified for H-RuO2 specimen. It is observed that H-RuO2 specimen did not show any characteristic peak of crystalline RuO2 even after calcination at 200°C, whereas the rutile type crystalline RuO2 phase is observed at 350°C (JCPDS 88-0322). Similar behavior was observed for the samples prepared by the “instant method” with Li2CO3 as hydrolyzing agent and crystallinity was obtained only after reaching the calcination temperature of 350°C (Devadas et al., 2011; Audichon et al., 2017a). Also, the diffractograms obtained on RuO2 prepared by the “instant method” showed impurity diffraction peaks of lithium ruthenium oxide (Devadas et al., 2011; Audichon et al., 2017a) and/or lithium carbonate (Audichon et al., 2017a). In contrary, the present ion-exchange method (no excess hydrolyzing agents, no purification steps) resulted in the pure rutile phase RuO2 structure. It was reported that well crystalline RuO2 will be obtained only after calcination at high temperatures (Cruz et al., 2011; Tsuji et al., 2011). However, calcination of RuO2 above 350°C may decrease its electrocatalytic activity towards the OER (Devadas et al., 2011; Audichon et al., 2017a). Hence, in the present study, C-RuO2 was obtained after calcination of H-RuO2 at 350°C for 1 h.
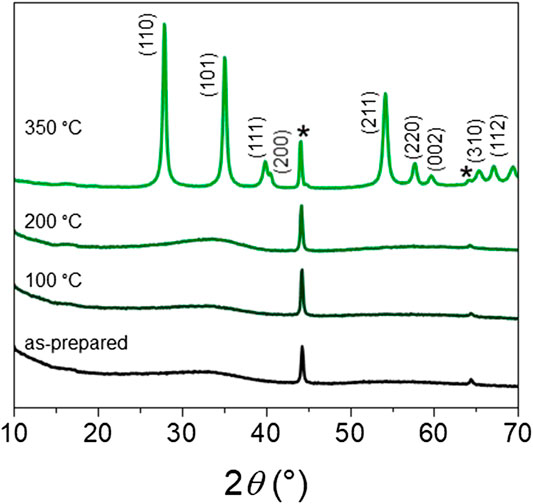
FIGURE 2. In situ XRD pattern of H-RuO2 calcined at different temperature under air atmosphere (*sample holder).
Based on XRD investigation of calcined RuO2 at different temperatures by Jirkovsky et al. (2006), it was mentioned that broadening of the diffraction peaks was directly related to the coherent domain size of crystallites for calcination temperatures lower than 500°C and internal strain/stress effects on broadening of diffracted peaks appeared only for calcination temperatures higher than 600°C. In this study, the calcination temperature is set at 350°C and the mean crystallite size determined using the Scherrer equation is representative of the average coherent domain size. The C-RuO2 is found to have a mean crystallite size close to 23 nm, which is in comparison with the earlier reports (Devadas et al., 2011; Audichon et al., 2017a). The unit cell parameters are calculated using the linear least square method and results indicate that the C-RuO2 crystal lattice has the unit cell values of a = b = 0.4483 nm and c = 0.3102 nm.
The TEM images for the H-RuO2 show the formation of particles with homogeneous size distribution (Figure 3A,B). The inset of Figure 3A depicts the size distribution histogram for the H-RuO2 sample. The average particle size is found to be lower than 2.0 nm. The TEM micrographs of C-RuO2 exemplify the growth/agglomeration of nanoparticles (Figure 3C,D) with well crystallization and particle sizes in the range from 10 to 30 nm, which agrees with results obtained on RuO2 nanostructures prepared by other synthesis methods (Natarajan et al., 2013; Audichon et al., 2014).

FIGURE 3. (A) TEM image of H-RuO2 (inset: related histogram of particle size distribution) and (B) HRTEM image of H-RuO2 (C,D) TEM image and HRTEM image of C-RuO2.
As illustrated in Figure 4, Raman spectra of H-RuO2 and C-RuO2 specimens exhibit three bands; frequencies are obtained accurately by fitting the bands with a Gaussian - Lorentzian function. In the case of H-RuO2, the Eg vibration mode is centered at 507 cm−1; A1g and B2g vibration modes are observed at 652 cm−1 and 685 cm−1, respectively. According to Raman-active phonon frequencies predicted by group theory, the single crystal rutile RuO2 is characterized by Raman bands located at 528, 646 and 716 cm−1 for Eg, A1g, and B2g vibration modes, respectively (Mar et al., 1995). Thus, Raman spectra of H-RuO2 leads to the conclusion that when RuCl3 solution passes through the anion exchange column, the rutile RuO2 phase is formed rather than an amorphous phase, conversely to the conclusion drawn from the XRD pattern. Also, Raman studies on H-RuO2 agree with Dmowski et al. (Dmowski et al., 2002) studies suggesting that hydrous RuO2 is a composite of anhydrous rutile-like RuO2 nanocrystals dispersed by boundaries of structural water. C-RuO2 is characterized by the Raman bands centered at 512, 630 and 690 cm−1 for Eg, A1g, and B2g vibration modes, respectively. The Raman features indicate a decrease of the linewidth for C-RuO2 which can be attributed to well crystallization and structural ordering due to calcination, whereas the red shift of the peak locations compared to those of the single crystal rutile type RuO2 is attributed to the nanoscopic nature of C-RuO2 material (Chen et al., 2004). The observed peak positions of H-RuO2 and C-RuO2 are significantly different from those of characteristic bands of crystalline-RuO2, which is due to the influence of structural water bounded to nanocrystals on the Raman spectrum vibrational frequencies (Mo et al., 2001).
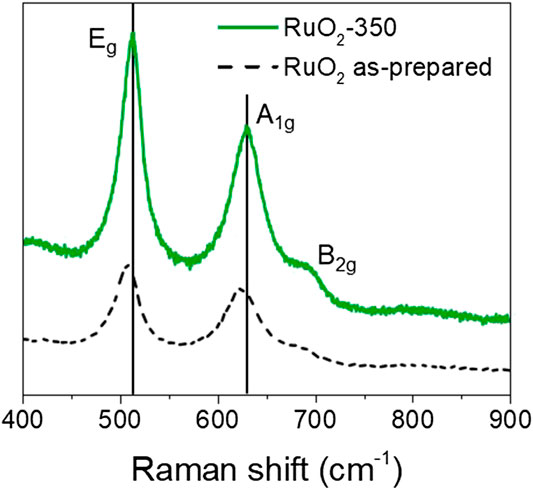
FIGURE 4. Raman spectra of H-RuO2 (dashed line) and C-RuO2 (solid line).
XPS core level spectra for the C-RuO2 sample is presented in Figure 5 and Ru 3d core level spectrum resembles to that of hydrous RuO2•xH2O material reported by Morgan (Mo et al., 2001). Based on the XPS data, different oxidation states for Ru are identified in C-RuO2 specimen and these oxidation states are RuIV (Ru 3d5/2 peaks located at 280.6 eV with the corresponding satellites at 282.5 eV) and RuVI−VIII (Ru 3d5/2 peak located at 284.2 eV). The attribution of both XPS signals at 280.6 and 282.5 eV to RuIV is based on results widely discussed in the literature (Chan et al., 1997; Kim et al., 1997; Mo et al., 2001; Over et al., 2002; Chang and Hu, 2004; Foelske et al., 2006; Morgan, 2015; Walsh, 2019), although the origin of the signal close to 282.5 eV differs depending on the authors. Recently, Näslund et al. (2014) explained that “the strong Coulomb interaction between valence electrons and the core hole produced in the photoionization process” was responsible of this peak (final-state screening effect). The XPS signal observed at 284.2 eV corresponds to RuO4 surface species (Chang and Hu, 2004). These RuO4 species are attributed to highly oxidized surface ruthenium, namely RuVI−VIII, since RuO4 is a volatile compound. The O 1s core level spectrum (Figure 5B) presents three different XPS signals attributed to physisorbed water (H2O), oxygen involved in RuO2 lattice as hydrous ruthenium oxide (OH−) and oxygen in RuO2 lattice (O2-). Table 1 presents the atomic ratios calculated from XPS spectra for C-RuO2.
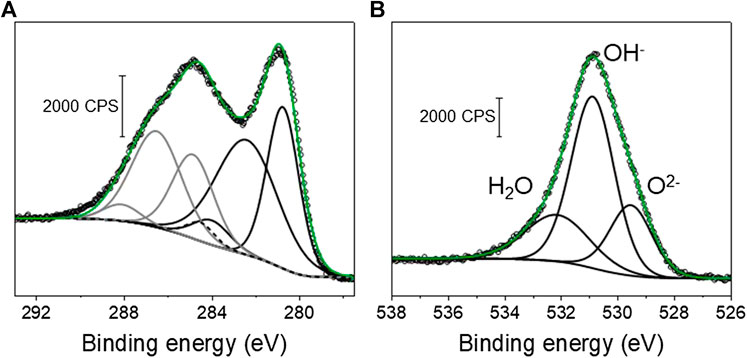
FIGURE 5. (A) XPS Ru 3d core level spectrum of C-RuO2. The XPS signal components represented with black lines are Ru 3d5/2 orbitals and the components represented with grey lines correspond to Ru 3d3/2 orbital signals. The C 1s component of the signal is presented with a dashed line. (B) XPS O 1s core level spectrum of C-RuO2.
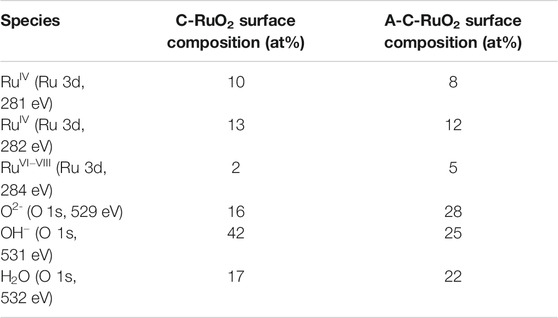
TABLE 1. Atomic percentages of ruthenium and oxygen surface species determined from XPS core level spectra.
Figure 6 presents the characterization data for the A-C-RuO2 specimen. It can be seen from comparison of Figures 3, 6 that structural differences appeared between C-RuO2 and A-C-RuO2. Based on observations of X-ray diffraction (Figure 6A) and Raman scattering measurements (Figure 6B), increased linewidth broadenings and shifts in the peak positions are noticed for A-C-RuO2 when compared to XRD pattern/Raman spectrum of C-RuO2. The shift to higher angles of XRD peaks (Figure 6A), the red shift of Raman peaks (Figure 6B) and the increase of the linewidth broadening for A-C-RuO2 can be correlated to changes in crystallinity, local disorder and mean crystallite size. The unit cell parameters for A-C-RuO2 have values of a = b = 0.4478 nm and c = 0.3097 nm. Contraction of lattice parameters for A-C-RuO2 when compared to C-RuO2 is correlated to the decrease of mean nanocrystallite size in A-C-RuO2 specimen (Leontyev et al., 2014). The mean crystallite size determined using the Scherrer equation for A-C-RuO2 sample was found to be close to 11 nm, which is twofold smaller than the mean crystallite size of C-RuO2. In agreement with XRD results, the micrographs of the A-C-RuO2 sample (Figure 6D) represents the growth of nanoparticles with sizes similar to that of C-RuO2 (i.e. in the range from 10 to 30 nm) (Figure 3D). The smaller mean crystallite size measured on A-C-RuO2 specimen than that measured on C-RuO2 sample does not involve a smaller mean particle size, it only implies that the A-C-RuO2 sample exhibits polycrystalline nanoparticles (Figure 6E). The comparison of XPS data for C-RuO2 (Figure 5) and A-C-RuO2 (Figure 6C) evidences another anomaly in their microstructure. An increase of highly oxidized surface ruthenium species, an increase of oxygen in RuO2 lattice and a decrease of the amount of hydrous ruthenium oxide (OH−) is observed for A-C-RuO2. For comparison, the atomic ratios obtained from XPS analysis for C-RuO2 and A-C-RuO2 are presented in Table 1. Thus, ammonium hydroxide treatment under microwave irradiation on hydrous RuO2 has influenced to limit the growth of nanocrystals of H-RuO2 during calcination process, thereby resulting in lower crystallite sizes/lattice parameters of A-C-RuO2 when compared to C-RuO2 (a blank test of H-RuO2 treatment under microwave irradiation without addition of ammonium hydroxide was performed and the RuO2 structure after calcination at 350°C was the same as that of C-RuO2).
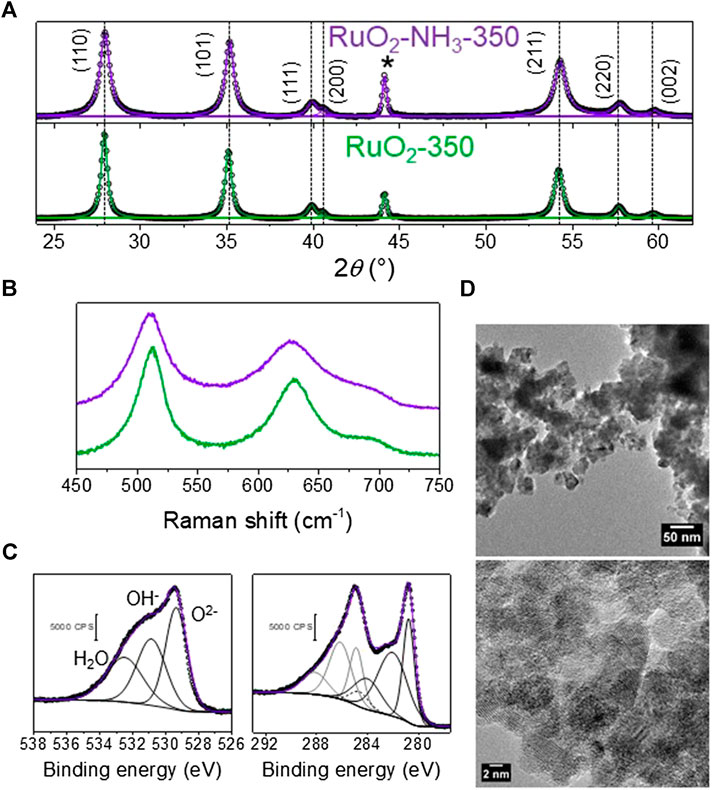
FIGURE 6. (A) XRD pattern of C-RuO2 (green lines) and A-C-RuO2 (purple lines) (*sample holder), peak position of C-RuO2 are shown as dashed lines for comparison to that of A-C-RuO2; (B) Raman spectra C-RuO2 (green line) and A-C-RuO2 (purple line); (C) XPS Ru 3d and O 1s core level spectra of A-C-RuO2 sample (the C 1s component of the signal in the Ru 3d core level spectrum is presented with a dashed line); (D,E) TEM image and HRTEM image of A-C-RuO2.
Electrocatalytic Activity
The electrocatalytic activity of the H-RuO2 and C-RuO2 samples was evaluated by recording the current-potential curves of the OER in 0.50 mol L−1 H2SO4 electrolyte at the quasi-stationary potential scan rate of 0.010 V s−1. It is very difficult to give an accurate evaluation of the real surface area of the catalyst due to the absence of faradaic process in the supporting electrolyte and to the non-proportional increase of the capacitance with the real surface area (Devadas et al., 2011). Moreover, it was previously observed that the capacitive current measured could vary independently on the surface area for RuO2 materials with particle sizes from two to almost 100 nm (Devadas et al., 2011; Audichon et al., 2017a). Hence, considering the large difference of OH species present at the surface of the C-RuO2 and A-C-RuO2 samples, as shown by XPS measurements, the determination of capacitance may not reflect accurately the difference of electrochemical active surface area. The determination of the BET porosity and surface area suffers the same problem. The confined water amount in the materials has a huge influence on adsorption of N2 on internal RuO2 domains (Yoshida et al., 2013) However, according to XPS measurements, C-RuO2 and A-C-RuO2 catalysts display significantly different amounts of water, which will therefore lead to inaccuracies in the determination of porosity and surface area by BET, and even to discrepancies in the evaluation of the active surface areas by both these different methods (Siviglia et al., 1983). Hence, because all electrode are loaded with the same mass of catalytic material, the mass specific activity is used for the representation of polarization curves. As shown in Figure 7, H-RuO2 is found to exhibit the oxygen evolution with an onset potential at ca. +1.40 V vs RHE. After +1.500 V vs RHE, a steep decrease of current values is noticed for H-RuO2. Such behavior was also observed with hydrous RuO2 nanomaterials synthesized in our previous investigations (Devadas et al., 2011; Audichon et al., 2017a; Audichon et al., 2017b), which is due to structural instability at higher potentials. C-RuO2 shows the onset potential of oxygen evolution at ca. +1.42 V vs RHE and is found to be active at higher potentials (Figure 7). The chronoamperometric study of the OER on C-RuO2 at E = +1.600 V vs RHE is shown in Figure 7 inset and a stable mass specific activity plateau of 22 A g−1 is reached after 10 min. These results infer the stability of C-RuO2 for the OER in 0.50 mol L−1 H2SO4 solution for the applied time period in comparison with H-RuO2 material.
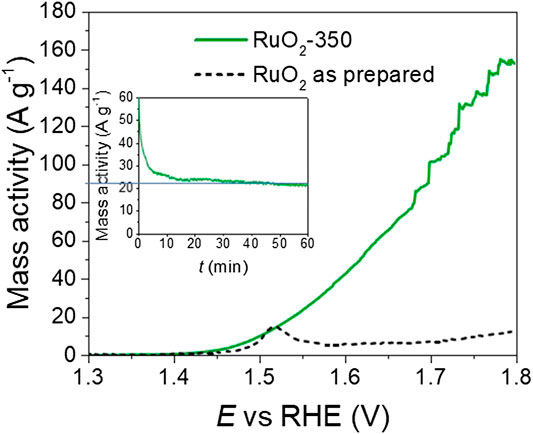
FIGURE 7. Linear scan voltammograms for oxygen evolution reaction (OER) on H-RuO2 (dashed lines) and C-RuO2 sample (green line) recorded in 0.50 mol L−1 H2SO4 at a scan rate s = 0.010 V s−1 and T = 25°C. Inset: chronoamperometric curves for the C-RuO2 recorded at E = +1.600 V vs. RHE.
A-C-RuO2 exhibits improved performance towards the OER when compared to C-RuO2 (Figure 8A). At E = +1.600 V vs RHE, the mass specific activities of C-RuO2 and A-C-RuO2 were found to be 41 and 57 A g−1, respectively. Table 2 compares the mass activities for these two catalysts with recent results in literature. C-RuO2 and A-C-RuO2 catalysts match well with rutile RuO2 nanoparticles (Laha et al., 2019). The addition of transition group metals helps enhancing the catalytic activity (Tariq et al., 2020; Wu et al., 2020). Note that such binary or ternary materials could be achieved with the ion exchange method on resin discussed in the present work. Figure 8B illustrates the Tafel plots for the OER on C-RuO2 and A-C-RuO2. The curves exhibit linear and parallel parts at lower potentials or overpotentials with slopes of ca. 69 mV decade−1. The Tafel slope of 60 mV decade−1 was reported for (100) and (110) RuO2 surfaces (Castelli et al., 1986; Stoerzinger et al., 2014). This indicates that the mechanism of water splitting is likely the same on both catalysts at low overpotentials. According to Antolini (Antolini, 2014), a Tafel slope of 60 mV decade−1 corresponds to the following mechanism, with step 1 as rate determining step:
where, S represents an active site, S-O
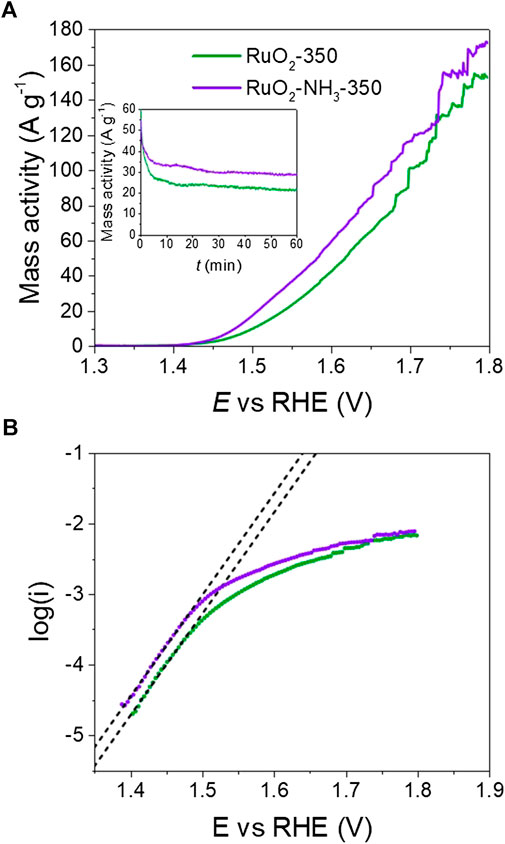
FIGURE 8. (A) Polarization curves for oxygen evolution reaction (OER) on C-RuO2 (green line) and A-C-RuO2 (purple line) recorded in 0.50 mol L−1 H2SO4 at a scan rate s = 0.010 V s−1 and T = 25°C. Inset: corresponding chronoamperometric curves for the OER recorded at E = +1.600 V vs. RHE. (B) Tafel plots of C-RuO2 (green circles) and A-C-RuO2 (purple circles).

TABLE 2. Comparison of mass activities measured on C-RuO2 and A-C-RuO2 with that of ruthenium-based catalysts from the literature.
At higher overpotentials, the Tafel slope values for the OER on C-RuO2 and A-C-RuO2 are found to be ca. 166 and ca. 190 mV decade−1, respectively. Based on the mechanistic studies for the OER on RuO2, IrO2 and RuO2@IrO2 core-shell nanocatalysts, it was reported that different Tafel slopes at high overpotentials were characteristic of the electrocatalytic material, whereas similar Tafel slopes (60 mV decade−1 for both RuO2 and IrO2) were found at low overpotentials (Ma et al., 2018). Thus, in the present study, varying Tafel slope value of C-RuO2 and A-C-RuO2 at high overpotentials corroborates the differences in their physicochemical characteristics.
At last, chronoamperometric curves of OER on C-RuO2 and A-C-RuO2 at E = +1.600 V vs RHE are compared in Figure 8 (inset). A higher mass activity for A-C-RuO2 when compared to C-RuO2 was recorded, confirming the linear scan voltammetry measurements. The mass specific activity reaches a plateau of 29 A g−1 (activity gain of 32% compared with C-RuO2) which also demonstrates the high stability of A-C-RuO2 for the OER in 0.50 mol L−1 H2SO4 solution for the applied time period.
Discussion
DFT simulations have pointed out that under electrochemical conditions for the OER, two types of adsorption sites exist for RuO2 electrocatalyst (Rossmeisl et al., 2007; Hansen et al., 2010). The first one forming a bridge between two adjacent Ru atoms along the [001] direction is electrocatalytically inactive. The second one denoted as coordinatively unsaturated site, involving one of the Ru-O bonds, is found to be electrocatalytically active (Rossmeisl et al., 2007; Hansen et al., 2010). DFT calculations have also identified a peroxo group formed by two adjacent octahedral oxygen sites that favors the rate determining intermediates for the OER (Rossmeisl et al., 2007; Hansen et al., 2010) and such peroxo group formation can be minimized by doping of RuO2 (Petrykin et al., 2010). Ma et al. proposed that OER rate on RuO2 was limited by OOH formation on the Oads and OER activity of RuO2@IrO2 was limited by Oads (Ma et al., 2018). In the present study, A-C-RuO2 material appears less ordered and possibly a part of the hydroxyl group present in the catalyst was converted into oxygen group that are coordinated to Ru atom. Plausibly, nitrogen doping in RuO2 can be anticipated and its effect may be the microstructural changes of A-C-RuO2 specimen, thereby exhibiting the enhanced electrocatalytic activity for the OER when compared to C-RuO2. However, EDX and XPS analyses of A-C-RuO2 did not show any evidence of the presence of nitrogen either because of its low concentration or of its absence. Indeed, heat treatment at 350°C under pure N2 atmosphere of as-prepared RuO2 from the “instant method” also induced changes in crystallinity, crystallite size and morphology of the final material compared with the same materials heat treated under air, while nitrogen was not detected (Audichon et al., 2017b). The treatment by ammonium hydroxide under microwave irradiation of the H-RuO2 from the anion exchange method also affects the microstructure of the final material, which highlights the importance of the role of nitrogen atoms on RuO2 microstructure even if not detected.
The increase of active surface area could also be proposed to explain the improved activity of the A-C-RuO2 catalyst in comparison to that of C-RuO2 catalyst. But if XRD measurements indicated a smaller mean crystallite size for A-C-RuO2 than for C-RuO2, TEM characterizations showed same particle size as for C-RuO2 and polycrystallinity of A-C-RuO2 nanoparticles. Therefore, both catalysts should display same active surface area. The hydroxyl content in the A-C-RuO2 compound has decreased and that of lattice oxygen has increased as shown by XPS measurements, but the material is made less ordered than C-RuO2, as evidenced by Raman measurements. In the case of RuO2 material, it was highlighted that the electronic conductivity is supported by the rutile-like nanocrystals, while proton conduction is facilitated by the structural water along the RuO2 grain boundaries (Dmowski et al., 2002). Based on the electrocatalytic activity of RuO2 for various applications, it was reported that the right combination of electronic and protonic conduction of RuO2 is the most obvious parameter to tune the electrochemical activity of RuO2 (Walsh, 2019). In another study, it was proposed that the better electrochemical activity for the OER required more interconnected RuO6 octahedra (which is the structure of anhydrous rutile) as well as optimized structural water content (Trasatti and Lodi, 1981). Since the calcination temperatures of both samples are the same, the ammonium hydroxide treatment might lead to right balance between the number of available oxygen and proton sites (OH− species) on the catalytic surface of A-C-RuO2, also balancing the electronic and protonic conduction of the catalyst. Moreover, XPS measurements indicated that the Ru oxidation state was predominantly +IV, which makes the compound extremely electrochemically stable (Sugimoto et al., 2006).
But finally, the role of nitrogen on A-C-RuO2 specimen is still not well established and the physicochemical properties of nitrogen treated RuO2 need to be further investigated for the better understanding of the electrocatalytic activity enhancement of ammonium hydroxide treated RuO2.
Conclusion
Anion exchanger assisted synthesis of hydrous RuO2 nanocrystals (H-RuO2) was reported. Ion-exchange method proposed in this study offers a promising way respecting most of the twelve green chemistry principles (Anastas and Warner, 2000) for synthesis of RuO2 electrocatalysts. The crystalline RuO2 was obtained by calcination of hydrous RuO2 at 350°C (C-RuO2), which is evidenced by XRD and Raman characterizations. All the calcined specimens in this work resulted in rutile structure of RuO2 without any impurities. The ammonia treated H-RuO2 under microwave irradiation (A-C-RuO2) showed improved performances towards the OER when compared to C-RuO2. Tafel slope value of A-C-RuO2 was found to be 60 mV/decade, which represents the hydroxyl radical adsorption on catalytic active sites as the rate-limiting step for the OER at lower overpotentials. XRD, TEM measurements indicated the decrease in crystallite sizes of A-C-RuO2 (11 nm) when compared to C-RuO2 (23 nm) specimen. XPS analysis of A-C-RuO2 pointed out the increase of highly oxidized surface ruthenium species and decrease of the amount of hydrous ruthenium oxide. Raman spectral investigations of A-C-RuO2 indicated the local structural disorder. Structure-property correlation was established to describe the higher electrocatalytic activity of ammonium hydroxide treated specimen (A-C-RuO2) when compared to C-RuO2.
Overall, ion-exchange method as green synthesis route facilitates the formation of highly pure rutile phase RuO2 electrocatalysts. Further improvement of RuO2 electrocatalytic activity was realized by microwave assisted ammonium hydroxide treatment of ion-exchange derived hydrous RuO2. The approach proposed in this work is simple, environmental-friendly and more cost effective than traditional ones for obtaining RuO2 nanoparticles with the same activity. The ease to proceed the scale up to produce metal oxides and mixed oxides (RuIrO2, for example) could make this synthesis method technologically and industrially interesting.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors greatly acknowledge the CNRS Research Federation (FRH2 no. 2044), the European Union (ERDF) and the "Région Nouvelle Aquitaine" for supporting this work.
References
Anastas, P., and Eghbali, N. (2010). Green Chemistry: principles and practice. Chem. Soc. Rev. 39 (1), 301–312. doi:10.1039/B918763B
Anastas, P., and Warner, J. C. (2000). Green chemistry: theory and practice. Oxford, UK: Oxford University Press.
Antolini, E. (2014). Iridium as catalyst and cocatalyst for oxygen evolution/reduction in acidic polymer electrolyte membrane electrolyzers and fuel cells. ACS Catal. 4, 1426–1440. doi:10.1021/cs4011875
Audichon, T., Guenot, B., Baranton, S., Cretin, M., Lamy, C., and Coutanceau, C. (2017a). Effect of the annealing atmosphere on the electrochemical properties of RuO2 nano-oxides synthesized by the Instant Method. Appl. Catal. B-Environ. 218, 385–397. doi:10.1016/j.apcatb.2017.06.081
Audichon, T., Guenot, B., Baranton, S., Cretin, M., Lamy, C., and Coutanceau, C. (2017b). Preparation and characterization of supported RuxIr(1-x)O2 nano-oxides using a modified polyol synthesis assisted by microwave activation for energy storage applications. Appl. Catal. B-Environ. 200, 493–502. doi:10.1016/j.apcatb.2016.07.048
Audichon, T., Mayousse, E., Napporn, T. W., Morais, C., Comminges, C., and Kokoh, K. B. (2014). Elaboration and characterization of ruthenium nano-oxides for the oxygen evolution reaction in a proton exchange membrane water electrolyzer supplied by a solar profile. Electrochim. Acta. 132, 284–291. doi:10.1016/j.electacta.2014.03.141
Badwal, S. P. S., Giddey, S. S., Munnings, C., Bhatt, A. I., and Hollenkamp, A. F. (2015). Emerging electrochemical energy conversion and storage technologies, Front. Chem. 2, 79. doi:10.3389/fchem.2014.00079
Bock, C., Spinney, H., and MacDougall, B. (2000). A study of the deactivation and service life of Ir oxide anodes supported on Al substrates. J. Appl. Electrochem. 30, 523–532. doi:10.1023/A:1003961701335
Cardarelli, F., Taxil, P., Savall, A., Comninellis, C., Manoli, G., and Leclerc, O. (1998). Preparation of oxygen evolving electrodes with long service life under extreme conditions. J. Appl. Electrochem. 28, 245–250. doi:10.1023/A:1003251329958
Carmo, M., Fritz, D. L., Mergel, J., and Stolten, D. (2013). A comprehensive review on PEM water electrolysis. Int. J. Hydrogen Energy. 38, 4901–4934. doi:10.1016/j.ijhydene.2013.01.151
Carmo, M., and Stolten, D. (2019). “Energy storage using hydrogen produced from excess renewable electricity: power to hydrogen,” in Science and engineering of hydrogen-based energy technologies: hydrogen production and practical applications in energy generation. Editors P. E. V. de Miranda (Amsterdam, Netherlands: Elsevier BV), 165–199
Castelli, P., Trasatti, S., Pollak, F. H., and O’Grady, W. E. (1986). Single crystals as model electrocatalysts: oxygen evolution on RuO2 (110). J. Electroanal. Chem. 210, 189–194. doi:10.1016/0022-0728(86)90325-6
Chan, H. Y. H., Takoudis, C. G., and Weaver, M. J. (1997). High-pressure oxidation of ruthenium as probed by surface-enhanced Raman and X-ray photoelectron spectroscopies. J. Catal. 172, 336–345. doi:10.1006/jcat.1997.1841
Chang, K., and Hu, C. (2004). Oxidative synthesis of RuOx⋅n H2O with ideal capacitive characteristics for supercapacitors. J. Electrochem. Soc. 151, A958. doi:10.1149/1.1755591
Chen, D., Pu, Z. H., Lu, R. H., Ji, P. X., Wang, P. Y., Zhu, J. W., et al. (2020). Ultralow Ru loading transition metal phosphides as high-efficient bifunctional electrocatalyst for a solar-to-hydrogen generation system. Adv. Energy Mater. 10, 2000814. doi:10.1002/aenm.202000814
Chen, R. S., Chen, C. C., Huang, Y. S., Chia, C. T., Chen, H. P., Tsai, D. S., et al. (2004). A comparative study of microstructure of RuO2 nanorods via Raman scattering and field emission scanning electron microscopy. Solid State Commun. 131, 349–353. doi:10.1016/j.ssc.2004.06.002
Coutanceau, C., Baranton, S., and Napport, T. W. (2011). “Platinum fuel cell nanoparticle syntheses: effect on Morphology, Structure and Electrocatalytic Behavior,” in The delivery of nanoparticles. Editor A. A. Hashim (Rijeka, Croatia: InTech Publisher), 403–430.
Cruz, J. C., Baglio, V., Siracusano, S., Antonucci, V., Aricò, A. S., Ornelas, R., et al. (2011). Preparation and characterization of RuO2 catalysts for oxygen evolution in a solid polymer electrolyte. Int J. Electrochem. Sci. 6, 6607–6619.
Devadas, A., Baranton, S., Napporn, T. W., and Coutanceau, C. (2011). Tailoring of RuO2 nanoparticles by microwave assisted “Instant method” for energy storage applications. J. Power Sources. 196, 4044–4053. doi:10.1016/j.jpowsour.2010.11.149
Dmowski, W., Egami, T. K. E., Swider-Lyons, K. E., Love, C. T., and Rolison, D. R. (2002). Local atomic structure and conduction mechanism of nanocrystalline hydrous RuO2 from X-ray scattering. J. Phys. Chem. B. 106, 12677–12683. doi:10.1021/jp503960q
Fityk, W. M. (2010). A general-purpose peak fitting program. J. Appl. Crystallogr. 43, 1126–1128. doi:10.1107/S0021889810021199
Foelske, A., Barbieri, O., Hahn, M., and Kötz, R. (2006). An X-ray photoelectron spectroscopy study of hydrous ruthenium oxide powders with various water contents for supercapacitors. Electrochem. Solid State Lett. 9, A268. doi:10.1149/1.2188078
Hansen, H. A., Man, I. C., Studt, F., Abild-Pederson, F., Bligaard, T., and Rossmeisl, J. (2010). Electrochemical chlorine evolution at rutile oxide (110) surfaces. Phys. Chem. Chem. Phys. 12, 283–290. doi:10.1039/B917459A
International Agency of Energy (2019). Renewables 2019 Market analysis and forecast from 2019 to 2024. Available at: https://www.iea.org/reports/renewables-2019/power (Accessed on May 11, 2020).
Jirkovsky, J., Hoffmannova, H., Klementova, M., and Krtil, P. (2006). Particle size dependence of the electrocatalytic activity of nanocrystalline RuO2 electrodes. J. Electrochem. Soc. 153, E111–E118. doi:10.1149/1.2189953
Katsaounis, A., Brosda, S., and Vayenas, C. G. (2007). “Electrocatalysis,” in Encyclopedia of electrochemistry. Editors A. J. Bard and M. Stratmann (Weinheim, Germany: Wiley-VCH), 1.
Kim, Y. J., Gao, Y., and Chambers, S. A. (1997). Core-level X-ray photoelectron spectra and X-ray photoelectron diffraction of RuO2 (110) grown by molecular beam epitaxy on TiO2 (110). Colloid. Surface. 120, 250–260. doi:10.1016/S0169-4332(97)00233-X
Laha, S., Lee, Y., Podjaski, F., Weber, D., Duppel, V., Schoop, L. M., et al. (2019). Ruthenium oxide nanosheets for enhanced oxygen evolution catalysis in acidic medium. Adv. Energy Mater. 9, 1803795. doi:10.1002/aenm.201803795
Leontyev, I. N., Kuriganova, A. B., Leontyev, N. G., Hennet, L., Rakhmatullin, A., Smirnova, N. V., et al. (2014). Size dependence of the lattice parameters of carbon supported platinum nanoparticles: X-ray diffraction analysis and theoretical considerations. RSC Adv. 4, 35959–35965. doi:10.1039/C4RA04809A
Liu, T., Pell, W. G., and Conway, B. E. (1997). Self-discharge and potential recovery phenomena at thermally and electrochemically prepared RuO2 supercapacitor electrodes. Electrochim. Acta. 42, 3541–3552. doi:10.1016/S0013-4686(97)81190-5
Ma, Z., Zhang, Y., Liu, S., Xu, W., Wu, L., Hsieh, Y.-C., et al. (2018). Reaction mechanism for oxygen evolution on RuO2, IrO2, and RuO2@IrO2 core-shell nanocatalysts. J. Electroanal. Chem. 819, 296–305. doi:10.1016/j.jelechem.2017.10.062
Mar, S. Y., Chen, C. S., Huang, Y. S., and Tiong, K. K. (1995). Characterization of RuO2 thin films by Raman spectroscopy. Appl. Surf. Sci. 90, 497–504. doi:10.1016/0169-4332(95)00177-8
Mo, Y. B., Cai, W. B., Dong, J., Carey, P. R., and Scherson, D. A. (2001). In situ surface enhanced Raman scattering of ruthenium dioxide films in acid electrolytes. Electrochem. Solid State Lett. 4, E37–E38. doi:10.1149/1.1387226
Morgan, D. J. (2015). Resolving ruthenium: XPS studies of common ruthenium materials. Surf. Interface Anal. 47, 1072–1079. doi:10.1002/sia.5852
Music, S., Popović, S., Maljković, M., Furić, K., and Gajović, A. (2002). Influence of synthesis procedure on the formation of RuO2. Mater. Lett. 56, 806–811. doi:10.1016/S0167-577X(02)00618-3
Näslund, L. Å., Ingason, Á. S., Holmin, S., and Rosen, J. (2014). Formation of RuO(OH)2 on RuO2-based electrodes for hydrogen production. J. Phys. Chem. C. 118, 15315–15323. doi:10.1021/jp503960q
Natarajan, V., Basu, S., and Scott, K. (2013). Effect of treatment temperature on the performance of RuO2 anode electrocatalyst for high temperature proton exchange membrane water electrolysers. Int. J. Hydrogen Energy. 38, 16623–16630. doi:10.1016/j.ijhydene.2013.05.133
Over, H. (2012). Surface chemistry of ruthenium dioxide in heterogeneous catalysis and electrocatalysis: from Fundamental to Applied Research. Chem. Rev. 112, 3356–3426. doi:10.1021/cr200247n
Over, H., Seitsonen, A. P., Lundgran, E., Smedh, M., and Andersen, J. N. (2002). On the origin of the Ru-3d5/2 satellite feature from RuO2(110). Surf. Sci. 504, L196–L200. doi:10.1016/S0039-6028(01)01979-3
Pelegrino, R. R. L., Vicentin, L. C., De Andrade, A. R., and Betazzoli, R. (2002). Thirty minutes laser calcination method for the preparation of DSA® type oxide electrodes. Electrochem Comm. 4, 139–142. doi:10.1016/S1388-2481(01)00295-8
Pelletier, O., Davidson, P., Bourgaux, C., Coulon, C., Regnault, S., and Livage, J. (2000). A detailed study of the synthesis of aqueous vanadium pentoxide nematic gels. Langmuir. 16, 5295–5303. doi:10.1021/la991444f
Petrykin, V., Macounova, K., Shlyakhtin, O. A., and Krtil, P. (2010). Tailoring the selectivity for electrocatalytic oxygen evolution on ruthenium oxides by zinc substitution. Angew. Chem. Int. Ed. 49, 4813–4815. doi:10.1002/anie.200907128
Rasband, W. S. (2009). Image J. US National Institutes of Health Bethesda, MD. Available at: http://rsbweb.nih.gov/ij/ (Accessed May 11, 2020).
Retuerto, M., Pascual, L., Calle-Vallejo, F., Ferrer, P., Gianolio, D., González Pereira, A., et al. (2019). Na-doped ruthenium perovskite electrocatalysts with improved oxygen evolution activity and durability in acidic media. Nat. Commun. 10, 2041. doi:10.1038/s41467-019-09791-w
Rossmeisl, J., Qu, Z.-W., Zhu, H., Kroes, G.-J., and Nørskov, J. K. (2007). Electrolysis of water on oxide surfaces. J. Electroanal. Chem. 607, 83–89. doi:10.1016/j.jelechem.2006.11.008
Shafran, K. L., Deschaume, O., and Perry, C. C. (2005). The static anion exchange method for generation of high purity aluminium polyoxocations and monodisperse aluminium hydroxide nanoparticles. J. Mater. Chem. 15, 3415–3423. doi:10.1039/B505466D
Shariatzadeh, F., Mandal, P., and Srivastava, A. K. (2015). Demand response for sustainable energy systems: a review, application and implementation strategy. Renew. Sustain. Energy Rev. 45, 343–350. doi:10.1016/j.rser.2015.01.06
Shibli, S. M. A., Gireesh, V. S., and George, S. (2004). Surface catalysis based on ruthenium dioxide for effective activation of aluminium sacrificial anodes. Corrosion Sci. 46, 819–830. doi:10.1016/S0010-938X(03)00038-6
Siviglia, P., Daghetti, A., and Trasatti, S. (1983). Influence of the preparation temperature of ruthenium dioxide on its point of zero charge. Colloids Surf. A. 7, 15–27. doi:10.1016/0166-6622(83)80038-9
Stoerzinger, K. A., Qiao, L., Biegalski, M. D., and Shao-Horn, Y. (2014). Orientation-dependent oxygen evolution activities of rutile IrO2 and RuO2. J. Phys. Chem. Lett. 5, 1636–1641. doi:10.1021/jz500610u
Sugimoto, W., Yokoshima, K., Murakami, Y., and Takasu, Y. (2006). Charge storage mechanism of nanostructured anhydrous and hydrous ruthenium-based oxides. Electrochim. Acta. 52, 1742–1748. doi:10.1016/j.electacta.2006.02.054
Tariq, M., Wu, Y. Y., Ma, C. L., Ali, M., Zaman, W. Q., Abbas, Z., et al. (2020). Boosted up stability and activity of oxygen vacancy enriched RuO2/MoO3 mixed oxide composite for oxygen evolution reaction. Int. J. Hydrogen Energy. 45, 17287–17298. doi:10.1016/j.ijhydene.2020.04.101
Trasatti, S. (1999). “Interfacial electrochemistry of conductive oxides for electrocatalysis,” in Interfacial electrochemistry – theory, experiment and applications. Editor A. Wieckowski (New York, NY: Marcel Dekker Inc.), 769–792.
Trasatti, S. (2000). Electrocatalysis: understanding the success of DSA®. Electrochim. Acta. 45, 2377–2385. doi:10.1016/S0013-4686(00)00338-8
Trasatti, S., and Lodi, G. (1981). “Oxygen and chlorine evolution at conductive metallic oxides,” in Electrodes of conductive metal Oxides. Editor S. Trasatti (Amsterdam, NL: Elsevier), 521–626. doi:10.1002/bbpc.19810851141
Tretyakov, Y. D., Oleynikov, N. N., and Shlyakhtin, O. A. (1997). Cryochemical technology of advanced materials. London, England: Chapman & Hall.
Tsuji, E., Imanishi, A., Fukui, K.-I., and Nakato, Y. (2011). Electrocatalytic activity of amorphous RuO2 electrode for oxygen evolution in an aqueous solution. Electrochim. Acta. 56, 2009–2016. doi:10.1016/j.electacta.2010.11.062
Van Muylder, J., and Pourbaix, M. (1963). Atlas d’équilibres électrochimiques à 25 °C. Paris, France: Gauthier-Villars & Cie.
Vertegel, A. A., Kalinin, S. V., Oleynikov, N. N., and Tretyakov, Y. D. (1995). The fractal particles of iron (III) hydroxonitrate: from solution to solid state. J. Non-Cryst. Solids. 181, 146–150. doi:10.1016/0022-3093(94)00495-1
Walsh, F. C. (2019). Modern developments in electrodes for electrochemical technology and the role of surface finishing. Transactions of the IMF. 97, 28–42. doi:10.1080/00202967.2019.1551277
Wang, L., Zhou, Q., Pu, Z. H., Zhang, Q., Mu, X. Q., Jing, H. Y., et al. (2018). Surface reconstruction engineering of cobalt phosphides by Ru inducement to form hollow Ru-RuPx-CoxP pre-electrocatalysts with accelerated oxygen evolution reaction. Nanomater. Energy. 53, 270–276. doi:10.1016/j.nanoen.2018.08.061
Wu, Y. Y., Tariq, M., Zaman, W. Q., Sun, W., Zhou, Z., and Yang, J. (2020). Bimetallic doped RuO2 with manganese and iron as electrocatalysts for favorable oxygen evolution reaction performance. ACS Omega. 5, 7342–7347. doi:10.1021/acsomega.9b04237
Yoshida, N., Yamada, Y., Nishimura, S., Oba, Y., Ohnuma, M., and Yamada, A. (2013). Unveiling the origin of unusual pseudocapacitance of RuO2·nH2O from its hierarchical nanostructure by small-angle X-ray scattering. J. Phys. Chem. C. 117, 12003–12009. doi:10.1021/jp403402k
Keywords: green synthesis, ion exchange method, hydrous RuO2, ammonia treated RuO2, oxygen evolution reaction
Citation: Devadas A, Baranton S and Coutanceau C (2020) Green Synthesis and Modification of RuO2 Materials for the Oxygen Evolution Reaction. Front. Energy Res. 8:571704. doi: 10.3389/fenrg.2020.571704
Received: 11 June 2020; Accepted: 19 August 2020;
Published: 28 October 2020.
Edited by:
Cesar Augusto Correia De Sequeira, University of Lisbon, PortugalReviewed by:
Shichun Mu, Wuhan University of Technology, ChinaSenthil Kumar Sakkarapalayam Murugesan, Central Electrochemical Research Institute (CSIR), India
Copyright © 2020 Devadas, Baranton and Coutanceau. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christophe Coutanceau, Q2hyaXN0b3BoZS5jb3V0YW5jZWF1QHVuaXYtcG9pdGllcnMuZnI=