 Nhat-Long L. Pham1
Nhat-Long L. Pham1

- 1 Interdisciplinary Graduate Program in Immunology, University of Iowa, Iowa City, IA, USA
- 2 Department of Pathology, University of Iowa, Iowa City, IA, USA
- 3 Department of Microbiology, University of Iowa, Iowa City, IA, USA
Following an infection, naïve CD8 T cells are stimulated by dendritic cells (DC) displaying pathogen-derived peptides on MHC class I molecules (signal 1) and costimulatory molecules (signal 2). Additionally, pathogen-induced inflammatory cytokines also act directly on the responding CD8 T cells to regulate their expansion and differentiation. In particular, both type I interferons (IFNs) and IL-12 have been described as critical survival signals (signal 3) for optimal CD8 T cell accumulation during the expansion phase. Furthermore, expansion in numbers of antigen-specific CD8 T cells is coupled with their acquisition of effector functions to combat the infection. However, it still remains unclear whether these same cytokines also regulate the effector/memory differentiation program of the CD8 T cell response in vivo. Here, we demonstrate that defective signaling by either type I IFNs or IL-12 to the responding CD8 T cells impairs maximal expansion in response to DC immunization + CpG ODN, but neither of these cytokines is essential to regulate the effector/memory differentiation program. In addition, lack of direct IL-12 signaling to CD8 T cells accelerates the development of central memory phenotype in both primary and secondary antigen-specific memory CD8 T cells.
Introduction
The naive CD8 T cell repertoire for any specific pathogen–peptide ranges between 10 and 1000 cells per mouse (Blattman et al., 2002). In order to combat infection, the pathogen specific CD8 T cells must expand in number and acquire effector functions. Following an infection, pathogen-induced inflammatory cytokines directly modulate all phases of the CD8 T cell response including proliferation, survival, effector, and memory differentiation (Mescher et al., 2006; Xiao et al., 2009). In particular, type I interferons (IFN, i.e., IFN-α, -β) and IL-12 have been demonstrated to serve as “signal 3” for optimal CD8 T cell expansion (Haring et al., 2006) and have also been implicated in the differentiation of effector CD8 T cells (Mescher et al., 2006).
Signal 3 inflammatory cytokines regulate multiple aspects of the CD8 T cell response. Interestingly, the specific cytokine serving as the signal 3 depends on the pathogen under investigation (Harty and Badovinac, 2008). For example, type I IFN signaling in CD8 T cells is most critical for numerical expansion in the lymphocytic choriomeningitis virus infection model, whereas IL-12 serves as the critical signal 3 for CD8 T cells responding to Listeria monocytogenes (LM) and vaccinia infection (Kolumam et al., 2005; Thompson et al., 2006; Xiao et al., 2009). However, it remains unclear if these same critical signal 3 inflammatory cytokines also directly regulate the commitment of responding CD8 T cells to effector or memory differentiation in vivo. While experimental infection models have provided crucial insights into the differentiation of antigen-specific CD8 T cells into effector and memory lineages, it has been difficult to dissociate the contribution of antigen and costimulation signals (signal 1 and 2) from inflammatory cytokines (signal 3) to this differentiation process in vivo. Several reports from Mescher’s lab demonstrated a role for type I IFNs and IL-12 as the third signal required for the enhancement of cytolytic activity and IFN-γ production by the naïve OT-I TcR transgenic CD8 T cells responding to in vitro stimulation with antigen/MHC class I complexes and B7 ligands coupled microspheres (Curtsinger et al., 1999, 2003, 2005). However, it is unclear how in vitro system relates to complex in vivo sensing of infection. For example, CD8 T cells could potentially receive multiple inflammatory signals (signal 3) directly from the priming dendritic cells (DC) that present the antigen (signal 1) and costimulation (signal 2) or they could receive the signal 3 from the pathogen-induced systemic inflammatory cytokines after priming or both. Thus, dissociating the inflammatory signals from the activating signal 1 and 2 would provide a better understanding of how specific cytokines regulate the commitment of the responding CD8 T cells to effector or memory differentiation.
Recent studies suggest that IL-12 signaling directly to CD8 T cells enhances expansion, promotes terminal differentiation (Pearce and Shen, 2007; Cui et al., 2009), and regulates memory formation through a gradient of T-bet expression (Joshi et al., 2007). In addition, inflammatory cytokines play an important role in regulating the rate at which responding CD8 T cells acquire memory phenotype and function (Harty and Badovinac, 2008). Priming of CD8 T cells in a low inflammatory environment (i.e., DC immunization) accelerates memory CD8 T cell development (Badovinac et al., 2005). As early as 6 or 7 days after priming, the antigen-specific CD8 T cells exhibit memory characteristics defined by IL-7Rα (CD127) and KLRG-1 expression profiles (CD127hiKLRG-1lo), and the ability to vigorously expand in response to booster immunization (Badovinac et al., 2005). In contrast, the acquisition of phenotypic and functional memory characteristics by CD8 T cells takes several months after clearance of acute infection (Kaech et al., 2002a). Consistent with this, DC immunization in the presence of CpG-induced inflammation also delayed memory CD8 T cell development (Badovinac et al., 2005). Therefore, inflammatory cytokines promote and sustain the effector differentiation program and prevent the early acquisition of memory characteristics by the responding CD8 T cells (Pham et al., 2009). Thus, the DC immunization model in the presence or absence of inflammation allows us to specifically address whether the specific inflammatory cytokines, type I IFNs, or IL-12, signaling to the responding antigen-specific CD8 T cells, not only enhance effector cell commitment but also directly regulate the effector/memory differentiation program.
Here, we show that both type I IFN and IL-12 serve as signal 3 cytokines to promote optimal CD8 T cell expansion after DC immunization in the presence of CpG-induced systemic inflammation. However, neither cytokine receptor on CD8 T cells is essential for inflammation-induced effector commitment. Thus, these results show that the role of Type I IFN and IL-12 as signal 3 during expansion can be dissociated from the signal(s) that regulate the CD8 T cell effector/memory differentiation program.
Materials and Methods
Mice
C57BL/6 (CD45.1) mice were obtained from the National Cancer Institute (Frederick, MD, USA). B6.PL-Thy1a/CyJ (Thy1.1) and IL-12Rβ2−/− mice were obtained from the Jackson Laboratory (JAX). OT-I and IFNABR−/− mice were previously described (Hogquist et al., 1994; Kolumam et al., 2005). IFNABR−/− and IL-12Rβ1−/− OT-I cells were kindly provided by Dr. Matthew Mescher (University of Minnesota, Minneapolis, MN, USA). Experiments were conducted according to federal and institutional guidelines and approved by the University of Iowa Animal Care and Use Committee (Iowa City, IA, USA).
Antibodies, Peptides, and MHC Class I Tetramers
Antibodies of the indicated specificity with appropriate fluorochromes were used. The following antibodies were from eBioscience (San Diego, CA, USA): IFN-γ (XMG1.2), CD8 (53–6.7), CD127 (A7R34), KLRG-1 (2F1), CD11c (N418), CD86 (GL-1), MHC II (I-A/I-E; M5/114.15.2), (CD45.1 (A20), CD45.2 (104) isotype control rat IgG2a (eBR2a), rat IgG2b (KLH/G2b-1-2), Armenian Hamster IgG, and Golden Syrian Hamster IgG. The following antibodies were from BD Pharmingen (San Diego, CA, USA): CD11c (HL3), Thy1.1 (OX-7). The following antibodies were from Caltag (San Diego, CA, USA): anti-human Granzyme B and isotype control mouse IgG1. MHC class I tetramers (Kb) specific for Ova257–264 prepared using published protocols.
Dendritic Cells
Splenic DC were prepared as previously described (Pham et al., 2009). In brief, splenic DC were isolated after subcutaneous injection of B6 mice with 5 × 106 B16 cells expressing Flt3L (provided by M. Prlic and M. Bevan, University of Washington). When tumors were palpable (5 mm × 5 mm), mice were injected with 2 μg LPS (Sigma) i.v. to mature the DC. Spleens were harvested 16 h later and were digested with DNase and Collagenase for 20 min at 37°C/7% CO2 with shaking (120 RPM). Spleen pieces were smashed through a nylon cell strainer (70 μm) to generate a single cell suspension, red blood cells (RBC) were lysed and splenocytes were resuspended in two parts of 10% FCS RPMI-1640 to one part B16-Flt3L conditioned medium + rGM − CSF (1000 μ/ml) + 2 μM Ova257–264 and incubated 2 h at 37°C/7% CO2 with shaking (100 RPM). Spleen cells were washed three times and CD11c+ cells were isolated using anti-CD11c microbeads (Miltenyi Biotec, Auburn, CA, USA). The purity and activation status of DC were determined by staining for CD11c, CD86, and MHC class II. Routinely, greater than 90% pure CD11c+ DC were obtained and the yield was approximately 15–20 × 106 DC per mouse. DC were resuspended in saline and injected i.v.
Bacteria
actA-deficient L. monocytogenes expressing Ova257–264 (attLM-Ova) were grown, injected ∼5 × 106 cfu i.v. per mouse for boosting and quantified as described (Badovinac et al., 2005).
Adoptive-Transfer Experiments
Wild-type (WT) (Thy1.1/1.2) and receptor-deficient OT-I cells (Thy1.2/1.2) both (CD45.2) were obtained from the spleen of naïve donors. IFNABR−/− and IL-12Rβ1−/− OT-I cells were kindly provided by Dr. Matthew Mescher (University of Minnesota, Minneapolis, MN, USA). Spleens of transgenic donor mice were obtained fresh within 24 h of the day of adoptive-transfer experiment and RBC were removed with lysis buffer. To ensure the naïve status, OT-I transgenic cells were isolated fresh and were not in in vitro culture. Frequency of OT-I transgenic cells were determined by staining for CD8, Vα2, Vβ5, and analyzed with FACS. The number of input OT-I was calculated from the total number of splenocytes and the frequency of OT-I transgenic cells determined via FACS. Based on the number of input OT-I calculated, the WT (Thy1.1/1.2) and receptor-deficient OT-I cells (Thy1.2/1.2) both (CD45.2) were mixed and the ratio (WT:receptor-deficient OT-I) was checked via FACS prior to transfer into naive CD45.1 B6 mice (Badovinac et al., 2007). Approximately 600 WT OT-I and 600 receptor-deficient OT-I were co-transferred to each recipient CD45.1 mouse. All mice were immunized with ∼1 × 106 Ova257-coated matured DC at day 0 with or without CpG 1826 (IDT, Iowa City, IA, USA; 100 μg i.p.).
Generation of Radiation Bone Marrow Chimeric Mice
Bone marrow cells were obtained from tibia and femurs from naïve WT B6 (Thy1.1, CD45.2) and receptor-deficient B6 (Thy1.2, CD45.2) mice. Following removal of RBC, approximately 5 × 106 WT cells and 5 × 106 receptor-deficient cells were mixed and transplanted to each lethally irradiated (90 Gy) CD45.1 recipient mouse. Reconstitution of donor lymphocytes was verified by flow cytometry at 8 weeks following transplantation. All experiments were carried out after 8 weeks of reconstitution.
Quantification and Phenotypic Analysis of Antigen-Specific T Cells
The magnitude of the epitope-specific CD8 T cell response was determined either by tetramer staining as described (Badovinac et al., 2002) or by staining for the CD45.2 marker that is exclusively expressed on transferred TCR-tg cells (Badovinac et al., 2007). In the experiments with bone marrow chimeric mice, staining for CD45.1 was used to distinguish between donor and recipient cells. Differential expression of Thy1.1 and Thy1.2 allowed separation of WT and receptor-deficient TCR-tg cells or transplanted cells. For analysis of circulating TCR-tg T cells, ∼50 μl of blood was obtained from each mouse. The number of Tg OT-I cells or antigen-specific CD8 T cells was presented as frequency of total PBL. For phenotypic analysis, blood was pooled from all mice in either DC only or DC + CpG immunization groups for OT-I cells or analyzed individually for bone marrow chimeric mice.
Results
Direct Signaling by Signal 3 Inflammatory Cytokines, Type I IFN, or IL-12, to CD8 T Cells Promotes Their Expansion After DC + CpG DNA Immunization
The requirement of direct type I IFN or IL-12 signaling for optimal CD8 T cell expansion depends critically on the inflammatory cytokines produced by the specific pathogen in the study (Aichele et al., 2006; Thompson et al., 2006; Xiao et al., 2009). The role of these cytokines in supporting CD8 T cell expansion in vivo in response to DC immunization in the presence of CpG-induced inflammation has not been well established. To control for the potential variations in antigen load and inflammatory environment, we examined receptor-sufficient and receptor-deficient Tg OT-I cell responses in the same host (Figure 1A). Prior to adoptive transfer, the ratio of WT and receptor-deficient OT-I was monitored by flow cytometry to ensure approximately equal numbers of each type were being transferred (Figures 1B,C). WT (WT)- and IFNABR−/−- and IL-12Rβ1−/− (not shown)-OT-I T cells exhibit similar naïve phenotypes (CD127hi and KLRG-1lo; Figure 1D). To approximate endogenous precursor frequencies (Blattman et al., 2002; Badovinac et al., 2007), 600 WT (CD45.2; Thy1.1/1.2) and 600 of either IFNABR−/− or IL-12Rβ1−/− (CD45.2; Thy1.2/1.2) naïve OT-I cells were co-transferred into congenic (CD45.1) hosts (Figure 1A). Mice were then immunized with Ova257-coated DC and CpG 1826 (a TLR9 agonist) to induce systemic inflammatory cytokines. We then tracked the frequency of OT-I cells in blood during the primary expansion from day 5–7 after DC immunization by analyzing the CD8+ CD45.2+ cell populations and distinguishing WT and receptor-deficient OT-I cells by Thy1.1 staining (Figure 1E).
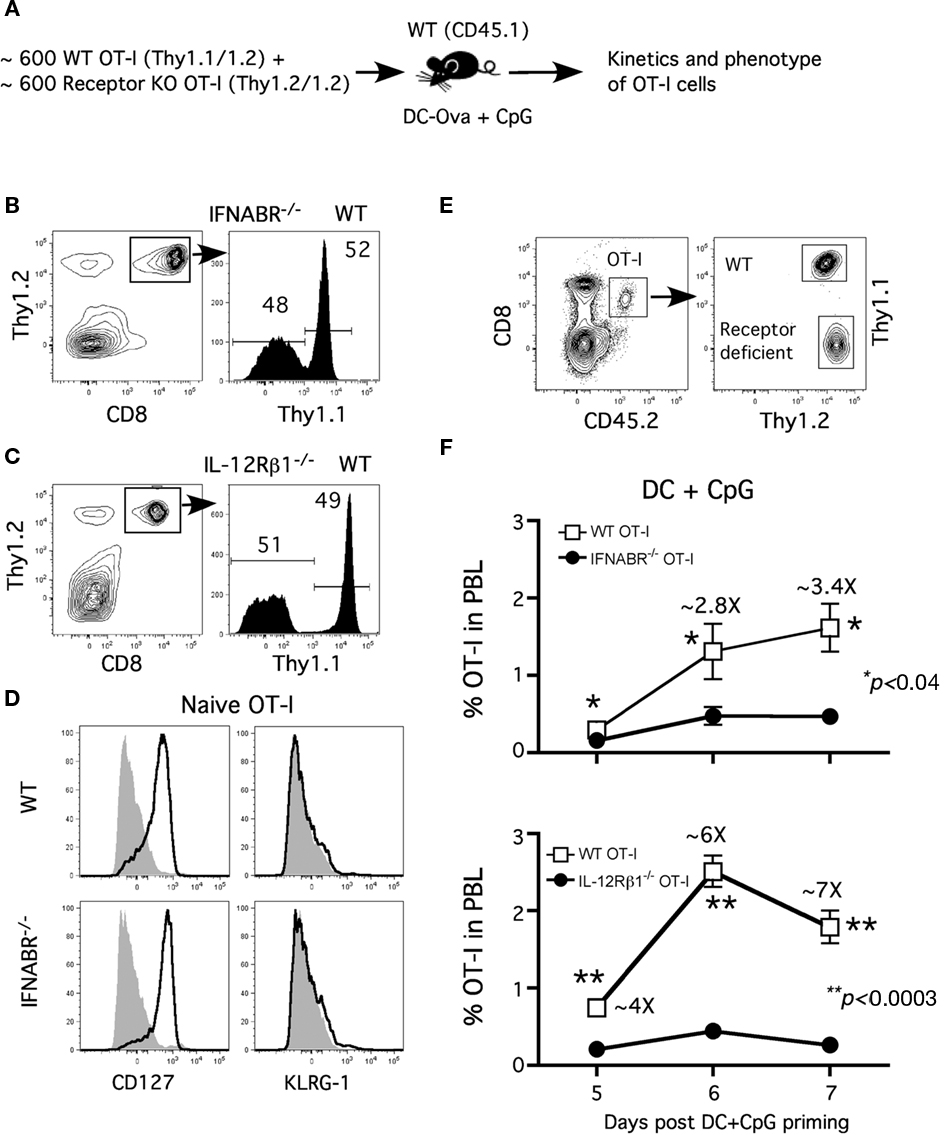
Figure 1. Type I IFN and IL-12 serve as “signal 3” to promote optimal CD8 T cell expansion. (A) Experimental design: Naïve CD45.2 mice received 600 naïve WT OT-I (Thy1.1)+ ∼600 receptor-deficient OT-I (CD45.1, Thy1.2) cells and were immunized with ∼1 × 106 Ova257-264-coated DC (i.v.) + CpG (100 μg i.p.) 1 day later. The ratio of WT and (B) IFNABR−/− or (C) IL-12Rβ1−/− OT-I cells prior to transfer is shown. (D) Representative profiles of CD127 and KLRG-1 expression on naïve WT or IFNABR−/− OT-I cells prior to adoptive transfer. (E) Representative gating strategy to distinguish between WT and receptor-deficient OT-I cells. (F) WT and receptor-deficient OT-I responses from day 5 to day 7 after DC + CpG immunization expressed as frequency (mean ± SEM, n = 4) of OT-I cells in PBL. Data are representative of three independent experiments.
OT-I cells deficient for either type I IFN- or IL-12-receptor did not expand as robustly as their WT counterparts following DC immunization in the presence of CpG-induced inflammation (Figure 1F). Furthermore, the differences in CD8 T cell numbers between WT and receptor-deficient OT-I were more pronounced at day 6 and 7 compared to day 5 following immunization. These results are consistent with the notion that direct type I IFN or IL-12 signaling to CD8 T cells both provide critical survival signals for their accumulation during the proliferative expansion phase after DC immunization in the presence of CpG-induced inflammation (Curtsinger et al., 2005; Aichele et al., 2006; Pearce and Shen, 2007). Interestingly, the expansion defect was apparent earlier and was slightly more severe for IL-12β1−/− OT-I cells than IFNABR−/− OT-I cells when compared to WT OT-I cells (Figure 1F), suggesting a more prominent role of IL-12 as the survival signal in this model (Cui et al., 2009). These results demonstrate an important role for both type I IFN and IL-12 as the signal 3 for optimal CD8 T cell expansion after DC immunization in the presence of CpG-induced inflammatory cytokines.
Neither Type I IFN Nor IL-12 Direct Signaling in CD8 T Cells is Required for Effector Differentiation
Dendritic cells immunization in the absence of systemic inflammation accelerates the memory differentiation in antigen-specific CD8 T cells (Badovinac et al., 2005). However, co-injection of CpG 1826 induces systemic inflammatory cytokines that sustain effector CD8 T cell differentiation and prevent the accelerated progression to memory after DC immunization (Badovinac et al., 2005). These findings strongly suggest that inflammatory cytokines regulate memory CD8 T cell development. In addition, the ability of CpG to induce both IL-12p70 and IFN-αβ has been described (Krieg, 2002). Thus, DC immunization in the presence or absence of CpG-induced systemic inflammatory cytokines offers a unique opportunity to determine the direct role of type I IFN or IL-12 signaling in regulating effector/memory CD8 T cell differentiation in vivo.
To address this question, we examined the phenotype of WT and receptor-deficient Tg OT-I cells from the peripheral blood at day 7 following Ova257-coated DC immunization ± CpG. Consistent with previous findings, a substantial fraction of OT-I cells responding to DC immunization exhibited a memory-like phenotype (CD127hiKLRG-1lo) at day 7 after immunization (Badovinac et al., 2005; Pham et al., 2009). This was true for both WT and IFNABR−/− OT-I or WT and IL-12Rβ1−/− OT-I cells responding in the same host (Figures 2A–C). As previously shown, most WT OT-I cells responding to DC immunization in the presence of systemic inflammation (DC + CpG) displayed an effector phenotype (CD127loKLRG-1hi) at day 7 after immunization. Interestingly and in contrast to the clear impact that the absence of IFNABR or IL-12Rβ1 had on OT-I expansion in this model, IFNABR−/− OT-I, or IL-12Rβ1−/− OT-I cells clearly down-regulate CD127 and up-regulate KLRG-1 expression in the presence of CpG-induced inflammation (Figures 2A–C). To ensure that the phenotypic differences between the DC and DC + CpG groups that we observed truly reflect the effector/memory differentiation process, we analyzed the kinetics of CD127 and KLRG-1 expression from day 5 to day 7 following DC immunization (Figures 2D,E). In the absence of CpG-induced inflammation, both WT and IFNABR−/− OT-I cells (Figure 2D, left panels) or WT and IL-12Rβ1−/− OT-I cells. (Figure 2E, left panels) quickly re-acquire CD127 expression and only a small fraction of these cells expresses KLRG-1 from day 5 to day 7 following DC immunization. In sharp contrast, both WT and IFNABR−/− OT-I cells (Figure 2D, right panels) or WT and IL-12Rβ1−/− OT-I cells. (Figure 2E, right panels) do not up-regulate CD127 expression and increasing frequencies of these cells express KLRG-1 from day 5 to day 7 following DC immunization in the presence CpG-induced inflammatory cytokines. Thus, OT-I cells that could not receive type I IFN or IL-12 signals during expansion still acquired an effector phenotype (CD127loKLRG-1hi) that was indistinguishable from WT OT-I cells in the same host. Hence, although both are required as signal 3 for optimal expansion in this model, neither type I IFN nor IL-12 serves as an essential signal to promote and sustain effector differentiation by the responding CD8 T cells.
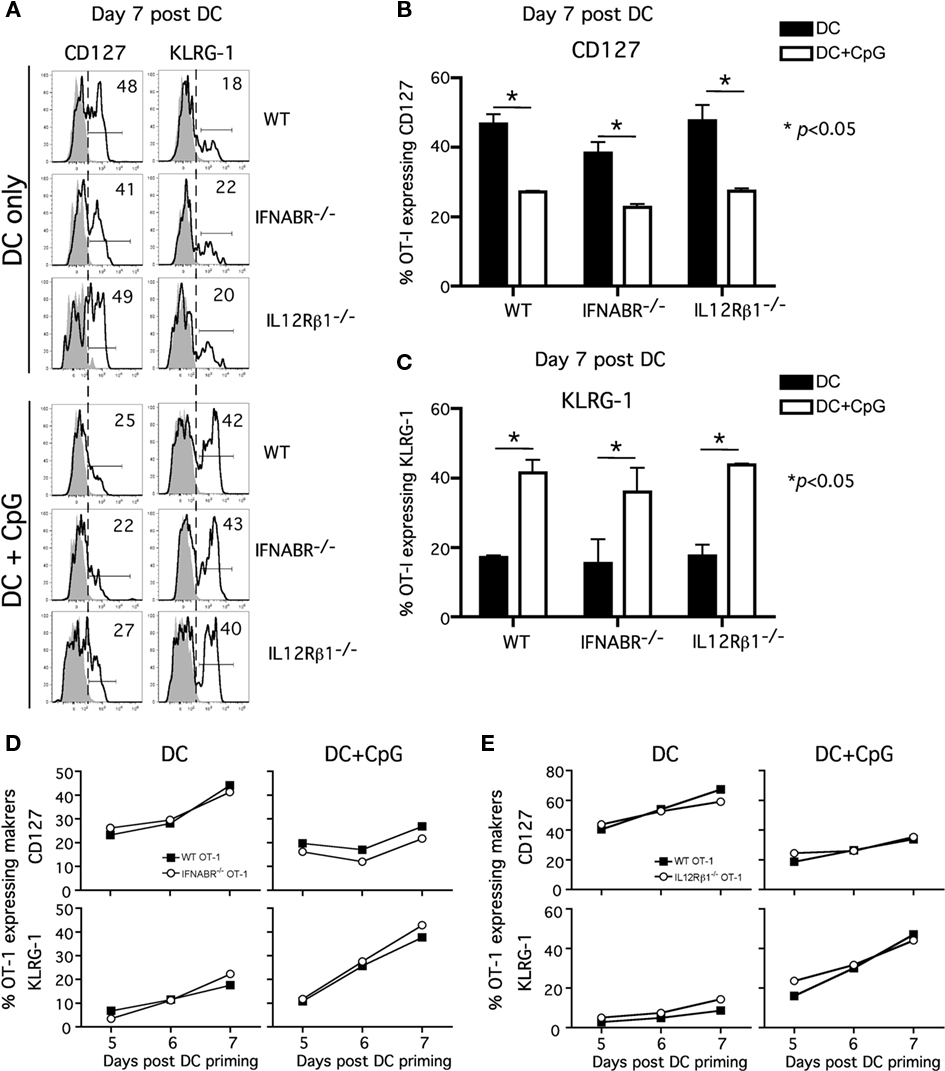
Figure 2. Neither type I IFN nor IL-12 direct signaling to CD8 T cells is required for acquisition of effector phenotype. Experiment design is in Figure 1. PBL were pooled from mice in either DC or DC + CpG immunization groups for analysis. (A) Representative histograms showing CD127 and KLRG-1 expression on WT, IFNABR-/-, and IL-12Rβ1-/- OT-I cells at day 7 following priming. Gray histograms represent isotype controls. Numbers indicate percentage of OT-I positive for the indicated marker. Cumulative data from two independent experiments showing frequency of WT, IFNABR-/-, and IL-12Rβ1-/- OT-I expressing (B) CD127 and (C) KLRG-1 at day 7 following DC immunization in the presence or absence of CpG. Kinetics of CD127 and KLRG-1 expression by (D) WT and IFNABR-/- OT-I cells and (E) WT and IL-12Rβ1-/- OT-I cells from day 5 to day 7 following DC immunization in the presence or absence of CpG. Data are from two independent experiments.
Our results differ from a recent study showing that the lack of direct IL-12 signaling promotes CD127hiKLRG-1lo phenotype on the responding CD8 T cells in the presence of CpG-induced inflammation (Cui et al., 2009). This discrepancy may be explained by the use of different transgenic CD8 T cells (OT-I vs P14), experimental approach, or the quality and quantity of inflammatory cytokines induced by CpG 1826 in the studies. We have observed that varying doses of CpG 1826 had differential effect on CD127 and KLRG-1 expression on the responding CD8 T cells (data not shown). In addition, the different results may arise from evaluating WT and receptor-deficient TCR-tg cells in the same (our study) or in different hosts (Cui et al., 2009). To address this issue, we investigated the polyclonal antigen-specific CD8 T cell response in chimeric mice containing both cytokine receptor-sufficient and receptor-deficient CD8 T cells (Figure 3A).
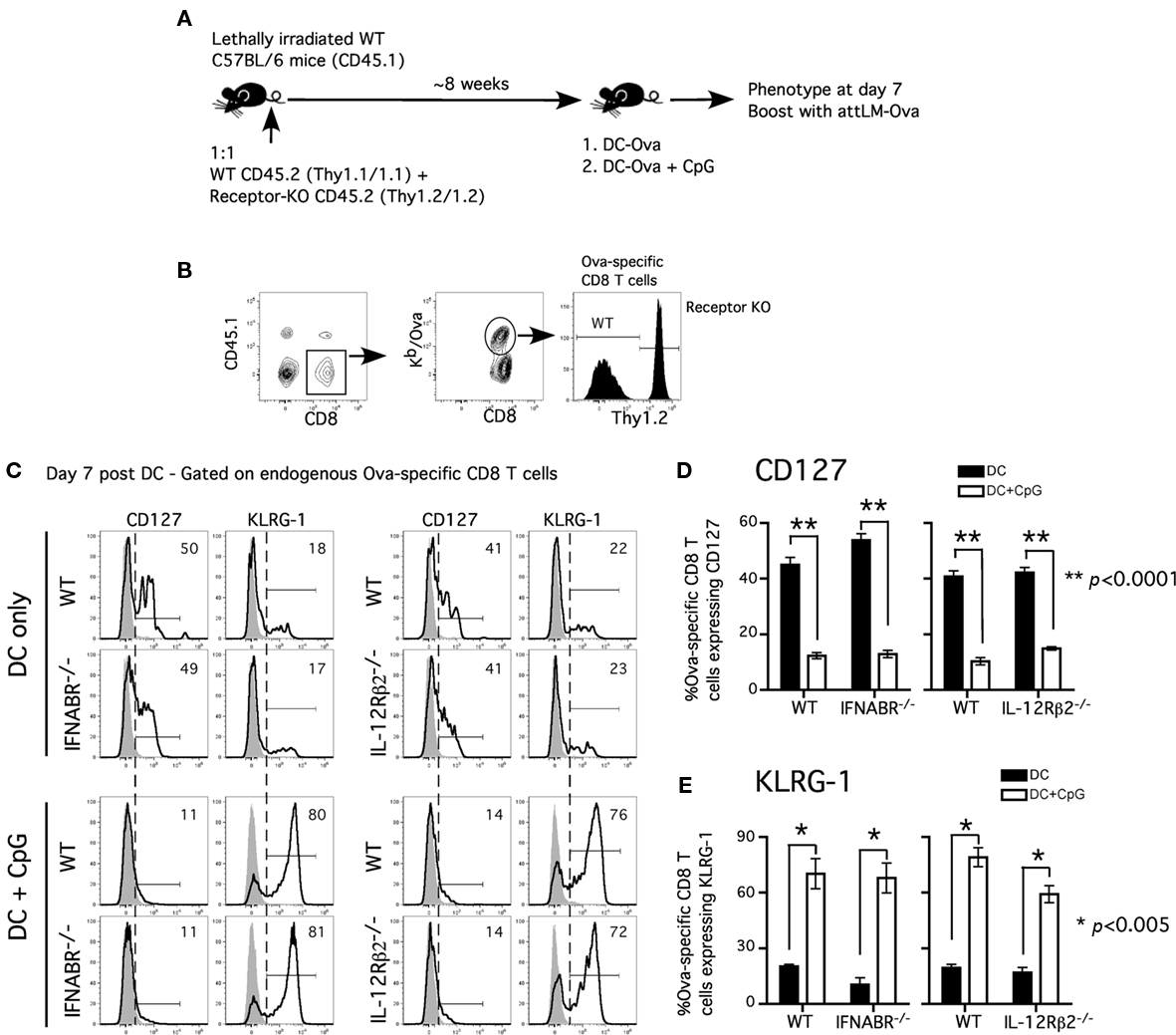
Figure 3. Acquisition of effector phenotype is independent of type I IFN receptor or IL-12 receptor signaling in polyclonal antigen-specific CD8 T cell response. (A) Experimental design: WT CD45.1 C57BL/6 mice were lethally irradiated and reconstituted with 1:1 mixture of either WT (CD45.2, Thy1.1) and IFNABR−/− (CD45.2, Thy1.2) or WT (CD45.2, Thy1.1) and IL-12Rβ2−/− (CD45.2, Thy1.2). Mixed bone marrow chimeric mice were allowed to reconstitute for at least 8 weeks before experiments were performed. (B) Representative gating strategy to distinguish between WT and receptor-deficient Ova257-specific CD8 T cells in bone marrow chimeric mice. Representative histograms (C) and cumulative data (D,E) showing CD127 and KLRG-1 expression on WT, IFNABR−/−, and IL-12Rβ2−/− Ova257-specific CD8 T cells at day 7 following DC or DC + CpG immunization. Gray histograms represent isotype controls. Statistical analysis was performed with student’s t-test between DC and DC + CpG groups. Data are representative of two independent experiments.
Chimeric mice were immunized with Ova257-coated DC in the presence or absence of CpG-induced inflammation. Following DC immunization, Ova257-specific, receptor-deficient, and -sufficient CD8 T cell populations can be easily distinguished by Thy1.2 staining on the CD45.1-negative, Ova-tetramer positive CD8 T cells in the same mouse (Figure 3B). Both WT and IFNABR−/− Ova257-specific CD8 T cells responding to DC immunization exhibited an accelerated memory phenotype (CD127hiKLRG-1lo) at day 7 (Figure 3C). In contrast, both WT and IFNABR−/− Ova257-specific CD8 T cells displayed an effector phenotype (CD127loKLRG-1hi) when primed in the presence of CpG-induced inflammation and similar results were obtained in WT/IL-12Rβ2−/− chimeric mice (Figure 3C). DC immunization alone promoted early acquisition of a memory phenotype (CD127hiKLRG-1lo) while CpG-induced inflammation resulted in effector phenotype (CD127loKLRG-1hi) by both WT and IL-12Rβ2−/− Ova257-specific CD8 T cells at day 7 (Figure 3C). Moreover, the difference in inflammation-induced CD127 downregulation and KLRG-1 upregulation on both receptor-sufficient and receptor-deficient Ova257-specific CD8 T cells was statistically significant when compared with DC immunization alone (Figures 3D,E). Importantly, these data with polyclonal, endogenous Ova257-specific CD8 T cells recapitulate the earlier data obtained for TCR-tg OT-I CD8 T cells. Thus, we conclude that direct signaling by either type I IFN or IL-12 in the responding CD8 T cells was not an absolute requirement for the acquisition of effector phenotype in the presence of CpG-induced inflammation. Taken together, these results suggested that other yet to be identified inflammatory cytokine(s) could fulfill this function or that there is a redundancy in inflammatory cytokine signaling to promote and sustain effector differentiation of the responding CD8 T cells.
Functional Effector/Memory Differentiation of CD8 T Cells is Independent of Type I Ifn or IL-12
Previous work in our lab demonstrated that the (CD127hiKLRG-1lo) phenotype correlated well with the ability of the DC-stimulated memory-like CD8 T cells to expand vigorously to short-interval booster immunization (Badovinac et al., 2005). Therefore, we investigated the secondary response potential in chimeric mice following DC immunization with or without CpG co-injection. A booster immunization with actA-deficient L. monocytogenes expressing Ova257-264 epitope (attLM-Ova) was administered to the chimeric mice at day 7 following DC-Ova ± CpG DNA immunization. We reasoned that if either type I IFN or IL-12 signaling in the antigen-specific CD8 T cells is critical in promoting their effector differentiation, CD8 T cells lacking either receptor would function more like accelerated memory cells when primed in the presence of CpG-induced inflammation and undergo more robust secondary expansion compared to the receptor-sufficient CD8 T cells in response to day-7 booster immunization with attLM-Ova.
Consistent with their memory phenotype (CD127hiKLRG-1lo), total Ova257-specific CD8 T cells (including both WT and receptor-deficient cells) in both WT/IL-12Rβ2−/− and WT/IFNABR−/− chimeric mice groups that received DC immunization alone underwent substantial responses to booster infection with attLM-Ova at day 7 after DC immunization (Figures 4A,B, filled squares). In contrast, total Ova257-specific CD8 T cells in both chimeric mice groups receiving DC + CpG failed to further expand in response to day-7 booster attLM-Ova infection, reflecting their effector phenotype (CD127loKLRG-1hi; Figures 4A,B, open circles). Importantly, this short-interval booster infection resulted in higher frequency of total Ova257-specific memory CD8 T cells 43 days later in both sets of chimeric mice when they were primed in the absence of inflammation compared to priming in the presence of CpG-induced inflammation (Figures 4A,B).
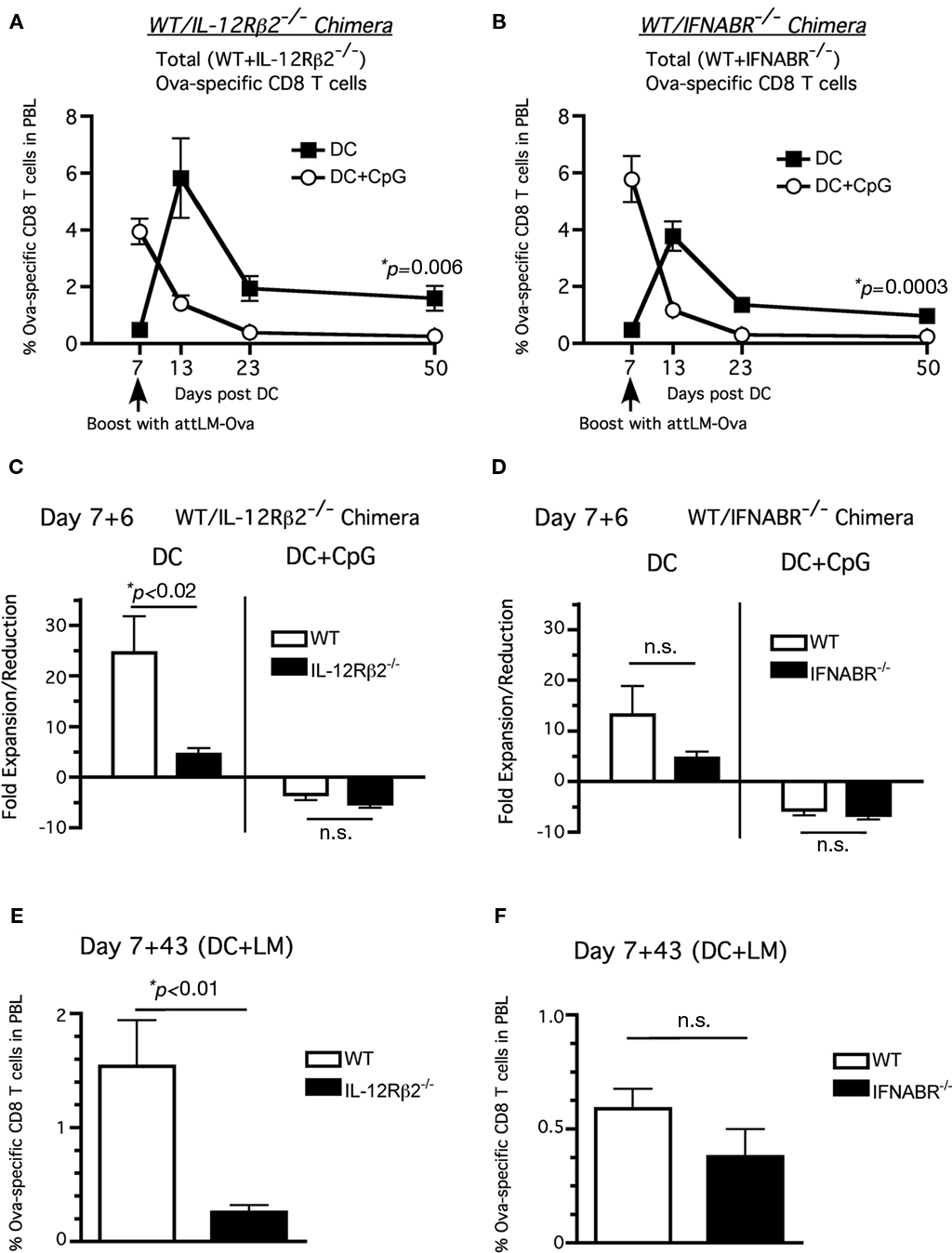
Figure 4. Neither type I IFN nor IL-12 signaling is required to inhibit accelerated memory differentiation in the presence of CpG-induced inflammation. WT/IFNABR-/- or WT/IL-12Rβ2-/- chimeric mice were immunized with ∼1 × 106 Ova257-264-coated DC ± CpG (100 μg i.p.) and were boosted with attLM-Ova 7 days later. Ova257-specific CD8 T cell response expressed as frequency (mean ± SEM, n = 6–8, data are posted from two independent experiments) in total PBL from (A) DC or (B) DC + CpG groups. Fold expansion/reduction = frequency of Ova257-specific CD8 T cells in PBL at day 6 post booster infection/frequency prior to booster infection (day 7 post DC immunization) for (C) WT/IFNABR−/− and (D) WT/IL-12Rβ2−/− chimeric mice. Frequency of Ova-specific CD8 T cells in the PBL at d43 after attLM-Ova boost in (E) WT/IL-12R2−/− and (F) WT/IFNABR−/− chimeric mice.
The day-7 booster infection following DC priming resulted in increased secondary expansion for both WT and IL-12Rβ2−/− Ova257-specific CD8 T cells following DC immunization only in the absence of CpG (Figure 4C). Similar results were obtained for WT and IFNABR−/− Ova257-specific CD8 T cells (Figure 4D). Interestingly, DC primed IL-12Rβ2−/− Ova257-specific CD8 T cells did not expand as robustly as their WT counterparts following booster infection 25× to 5×, respectively (Figure 4C, left side) while there was no statistically significant difference in fold expansion between WT and IL-12Rβ2−/− Ova257-specific CD8 T cells in the DC + CpG group (Figure 4C, right side). The difference in fold expansion between WT and IL-12Rβ2−/− Ova257-specific CD8 T cells resulted in significantly higher number of WT memory CD8 T cells at day 43 post boosting compared to the IL-12Rβ2−/− memory CD8 T cells. This result possibly reflects the diminished capacity of IL-12Rβ2−/− Ova257-specific CD8 T cells to accumulate following booster immunization, suggesting for the first time an important role of direct IL-12 signaling to promote optimal CD8 T cell expansion during secondary responses. On the other hand, we did not observe any statistically significant differences in fold expansion, fold reduction, or frequency of memory cells between WT and IFNABR−/− Ova257-specific CD8 T cells at day 6 and day 43 post booster infection (Figures 4D–F). Taken together, these results suggest that neither type I IFN nor IL-12 signaling to the CD8 T cells was required for secondary memory development. However, IL-12 appears to play a more critical role for optimal secondary expansion of memory CD8 T cells after short-interval attLM-Ova boosting.
Lack of IL-12 Signaling Accelerates the Development of Central Memory Phenotype in both Primary and Secondary Antigen-Specific Memory CD8 T Cells
Up to this point, our results suggested that the absence of IL-12 signaling did not prevent the Ova257-specific CD8 T cells from acquiring effector phenotype (CD127loKLRG-1hi) or function (failing to boost) when primed in the presence of CpG-induced systemic inflammation. However, the impact of IL-12 signaling on number and phenotype of antigen-specific memory CD8 T cells between primary and secondary response was not entirely clear. Following booster immunization, we observed significant differences in both the fold expansion (day 6) and memory frequency (day 43) between WT and IL-12Rβ2−/− Ova257-specific CD8 T cells (Figures 4C–E). We, thus, decided to analyze in detail the Ova257-specific memory CD8 T cells and their phenotype following attLM-Ova infection only (primary response) or DC priming followed by day-7 attLM-Ova booster infection (secondary response) at a long-term memory time point in WT/IL-12Rβ2−/− chimeric mice. Consistent with our earlier results, the number of IL-12Rβ2−/− Ova257-specific memory CD8 T cells was significantly lower than their WT counterparts in both primary (attLM-Ova infection only) and secondary (DC + attLM-Ova) responses (Figure 5A). The lower number of IL-12Rβ2−/− Ova257-specific memory CD8 T cells reflected the critical role of IL-12 as a survival signal for antigen-specific CD8 T cells during expansion following priming with attLM-Ova. In addition, our results indicated that IL-12 signaling in antigen-specific CD8 T cells is not required for either primary or secondary memory generation. We could readily detect IL-12Rβ2−/− Ova257-specific memory CD8 T cells at more than 100 days following either primary or secondary responses, albeit at a lower number compared to the WT cells (Figure 5A).
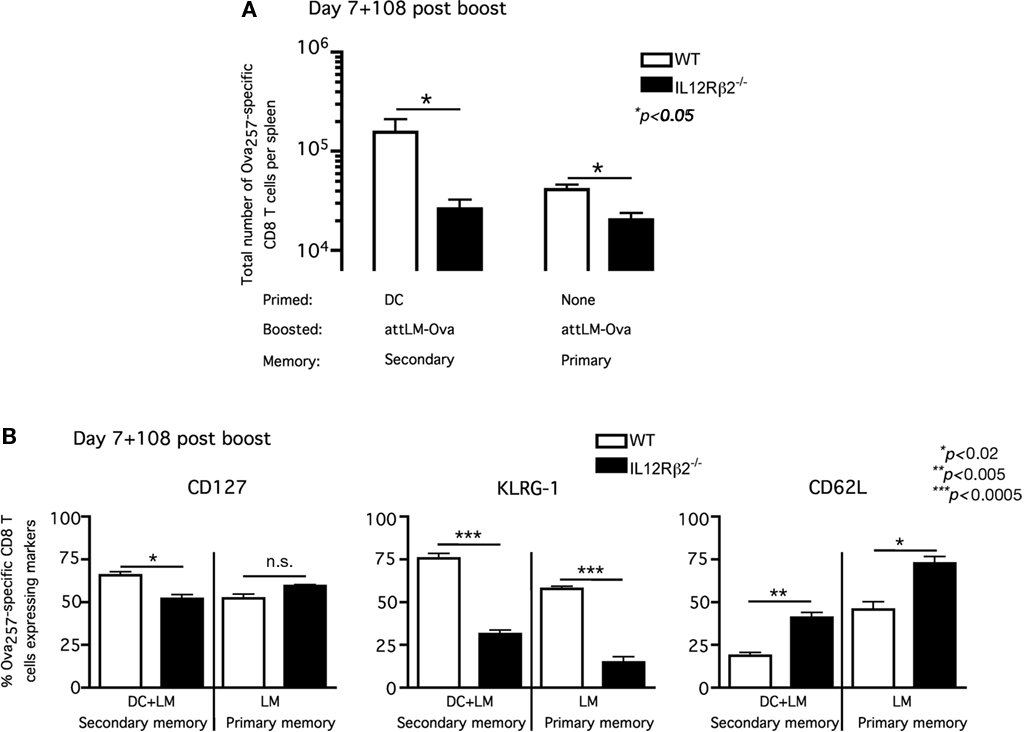
Figure 5. Lack of IL-12 signaling accelerates the development of central memory phenotype in both primary and secondary antigen-specific memory CD8 T cells. WT/IL-12Rβ2−/− chimeric mice were either immunized with ∼1 × 106 Ova257-264-coated DC or nothing and they all received ∼5 × 106 cfu attLM-Ova 7 days later. (A) Total number of WT and IL-12Rβ2−/− Ova257-specific CD8 T cells per spleen was determined at day 108 after attLM-Ova challenge. (B) Frequency of WT and IL-12Rβ2−/− Ova257-specific CD8 T cells expressing CD127, KLRG-1, and CD62L at day 108 following attLM-Ova in both DC + LM or LM only groups.
Phenotypic analysis of these stable WT and IL-12Rβ2−/− Ova257-specific memory CD8 T cells in the WT/IL-12Rβ2−/− chimeric mice revealed interesting differences. In both primary memory populations (attLM-Ova only) and secondary memory populations (DC + attLM-Ova) at more than 100 days post priming, IL-12Rβ2−/− Ova257-specific CD8 T cells exhibited markedly different CD127, KLRG-1, and CD62L expression patterns. In primary memory population (attLM-Ova only), CD127 expression was not statistically different between WT and IL-12Rβ2−/− Ova257-specific CD8 T cells (Figure 5B, left panel). However, in the absence of IL-12 signaling, a higher frequency of the memory CD8 T cells expressed lymph node homing molecule CD62L and a lower frequency of these cells expressed KLRG-1. Thus, the lack of IL-12 signaling in these antigen-specific CD8 T cells resulted in them possessing a more central memory phenotype (CD127hiCD62LhiKLRG-1lo) when compared directly to the corresponding WT cells in the same host (Figure 5B). These results were consistent with previous publication showing that lack of IL-12 signaling on CD8 T cells favors the development of primary memory CD8 T cells with central memory phenotype (Pearce and Shen, 2007). Furthermore, IL-12 signaling on responding CD8 T cells has been shown to positively regulate T-bet expression (Takemoto et al., 2006) and T-bet-deficient mice enhanced the generation of antigen-specific CD8 T cells with central memory phenotype (Intlekofer et al., 2007).
In addition and consistent with published reports (Badovinac et al., 2003; Wherry et al., 2003; Jabbari and Harty, 2006), secondary memory CD8 T cells were slower to acquire central memory phenotype compared to primary memory CD8 T cells (Figure 5B). Lower frequencies of secondary memory CD8 T cells expressed CD62L and higher frequencies expressed KLRG-1. This was true for both WT and IL-12Rβ2−/− Ova257-specific memory CD8 T cells, although IL-12Rβ2−/− cells consistently had higher frequency expressing CD62L and lower frequency expressing KLRG-1 compared to WT cells in either primary or secondary memory populations (Figure 5B). Taken together, these results suggested that direct IL-12 signaling to antigen-specific CD8 T cells clearly influences, but is not required for optimal generation of stable, long-term memory CD8 T cell populations. In addition, the lack of IL-12 signaling accelerates the development of central memory phenotype by antigen-specific CD8 T cells. However, IL-12 signaling does not significantly impact the memory phenotype between primary and secondary memory populations.
Discussion
Following activation by professional antigen presenting cells, antigen-specific CD8 T cells undergo a programmed response of proliferation, acquisition of effector functions, and formation of memory cells (Bevan and Fink, 2001; Kaech and Ahmed, 2001; van Stipdonk et al., 2001; Badovinac and Harty, 2006). Furthermore, the inflammatory environment in which the CD8 T cells are primed regulates multiple aspects of their response including expansion and effector and memory differentiation (Kaech et al., 2002b; Mescher et al., 2006; Harty and Badovinac, 2008). However, the “inflammatory signature” is often pathogen specific. The same is likely true for different immunostimulatory agents (i.e., adjuvants) used in vaccination protocols to generate a strong immune response. Thus, understanding the inflammatory mediator(s) that potentially regulate the effector and memory CD8 T cell differentiation would benefit vaccine design. Among the inflammatory cytokines, IL-12, type I IFNs, and IFN-γ have been studied most extensively in the CD8 T cell differentiation in various infection models and in vitro stimulation (Mescher et al., 2006).
The critical role of direct signaling by IL-12 and/or type I IFNs in the responding CD8 T cells to optimize their survival during the expansion phase has been well documented (Curtsinger et al., 2005; Kolumam et al., 2005; Pearce and Shen, 2007; Cui et al., 2009; Xiao et al., 2009). However, the role of these cytokines in the regulating of the balance between CD8 T cell effector and memory differentiation and maintenance in vivo still remains less well characterized. Here we analyzed with DC immunization ± CpG DNA to dissociate the role of type I IFN and IL-12 receptor signaling as the survival “signal 3” during expansion from the signal(s) that are required to regulate the CD8 T cell effector/memory differentiation program. Both type I IFN and IL-12 signaling to antigen-specific CD8 T cells are required for the optimal expansion in the presence of CpG-induced systemic inflammation since the lack of either receptor on CD8 T cells resulted in diminished expansion following DC + CpG immunization. However, neither type I IFN nor IL-12 signaling is required for CD8 T cell effector differentiation in the same immunization model. Using both adoptive transfer of transgenic CD8 T cells in the same host and mixed bone marrow chimeric mice, we demonstrated that direct signaling by either IL-12 or type I IFNs in the responding CD8 T cells was not required for their acquisition of effector phenotype and function.
A Recent publication by Xiao et al. (2009) showed that memory CD8 T cells lacking IL-12 receptors or type I IFN receptors are significantly reduced following vaccinia or LM infection compared to their WT counterparts. It is still not entirely clear whether CD8 T cells lacking either IL-12 receptors or type I IFN receptors fail to differentiate into memory population or their survival is compromised. Interestingly, however, the IL-12 requirement for memory development is not absolute since WT and type I IFN receptor-deficient CD8 T cells developed memory population following vaccinia infection in both IL-12p40- and IL-12p35-deficient mice (Xiao et al., 2009). These results suggested that the cytokine milieu of the host might play an important role in supporting memory development. Consistent with this notion, our results showed that the lack of IL-12 or type I IFN direct signaling on CD8 T cells did not promote accelerated memory differentiation in the presence of CpG-induced systemic inflammation. Since CpG induces various pro-inflammatory cytokines in addition to type I IFN and IL-12 (Krieg, 2002), it is possible that a yet to be identified pro-inflammatory cytokine serves as the critical signal that regulates the rate of CD8 T cell effector/memory differentiation. In addition, our study does not rule out the possibility that type I IFN and/or IL-12 are important in regulating the expression of other pro-inflammatory molecules that signal directly in CD8 T cells and control their differentiation. Alternatively, multiple inflammatory cytokines potentially serve a redundant role to enforce the effector CD8 T cell differentiation program. Thus, elucidating the signal(s) that regulate CD8 T cell memory differentiation remains a potential exciting strategy to manipulate memory generation and enhance protective immunity.
The role of direct IL-12 signaling in CD8 T cells has been extensively studied in primary responses (Mescher et al., 2006). However, less is known about its role in secondary responses. Our model of DC priming followed by short-interval booster immunization allowed us to examine the secondary response post booster infection. Our results show that lack of direct IL-12 signaling in responding CD8 T cells impairs their expansion but did not prevent their acquisition of effector phenotype and function during the primary response. However, antigen-specific CD8 T cells that do not receive IL-12 signals acquire central memory phenotype (CD127hi, KLRG-1lo, and CD62Lhi) earlier than their WT counterparts in the same host. In particular, this is true for both primary response (LM infection) and secondary response (DC prime followed by LM boost). A recent publication from Mescher’s laboratory demonstrated that direct IL-12 signaling in CD8 T cells regulates chromatin remodeling and epigenetic modifications at multiple loci after in vitro stimulation (Agarwal et al., 2009). These results raise the possibility that IL-12-induced epigenetic modifications potentially regulate the rate of central memory formation. However, it is still unclear at this point whether accelerated acquisition of central memory phenotype in the absence of direct IL-12 signaling results from the suboptimal expansion of CD8 T cells both in the primary and secondary response (Schluns and Lefrancois, 2003) or epigenetic modifications or both. Additional studies remain warranted to understand the complex regulation of CD8 T cell memory differentiation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank members of the Harty laboratory for helpful discussion and Dr. Steven Varga for critical comments. This workis supported by NIH grants AI46653, AI150073 and AI42767 (John T. Harty), and AI83286 (Vladimir P. Badovinac).
References
Agarwal, P., Raghavan, A., Nandiwada, S. L., Curtsinger, J. M., Bohjanen, P. R., Mueller, D. L., and Mescher, M. F. (2009). Gene regulation and chromatin remodeling by IL-12 and type I IFN in programming for CD8 T cell effector function and memory. J. Immunol. 183, 1695–1704.
Aichele, P., Unsoeld, H., Koschella, M., Schweier, O., Kalinke, U., and Vucikuja, S. (2006). CD8 T cells specific for lymphocytic choriomeningitis virus require type I IFN receptor for clonal expansion. J. Immunol. 176, 4525–4529.
Badovinac, V. P., Haring, J. S., and Harty, J. T. (2007). Initial T cell receptor transgenic cell precursor frequency dictates critical aspects of the CD8(+) T cell response to infection. Immunity 26, 827–841.
Badovinac, V. P., and Harty, J. T. (2006). Programming, demarcating, and manipulating CD8+T-cell memory. Immunol. Rev. 211, 67–80.
Badovinac, V. P., Messingham, K. A., Hamilton, S. E., and Harty, J. T. (2003). Regulation of CD8+T cells undergoing primary and secondary responses to infection in the same host. J. Immunol. 170, 4933–4942.
Badovinac, V. P., Messingham, K. A., Jabbari, A., Haring, J. S., and Harty, J. T. (2005). Accelerated CD8+ T-cell memory and prime-boost response after dendritic-cell vaccination. Nat. Med. 11, 748–756.
Badovinac, V. P., Porter, B. B., and Harty, J. T. (2002). Programmed contraction of CD8(+) T cells after infection. Nat. Immunol. 3, 619–626.
Blattman, J. N., Antia, R., Sourdive, D. J., Wang, X., Kaech, S. M., Murali-Krishna, K., Altman, J. D., and Ahmed, R. (2002). Estimating the precursor frequency of naive antigen-specific CD8 T cells. J. Exp. Med. 195, 657–664.
Cui, W., Joshi, N. S., Jiang, A., and Kaech, S. M. (2009). Effects of signal 3 during CD8 T cell priming: bystander production of IL-12 enhances effector T cell expansion but promotes terminal differentiation. Vaccine 27, 2177–2187.
Curtsinger, J. M., Lins, D. C., and Mescher, M. F. (2003). Signal 3 determines tolerance versus full activation of naive CD8 T cells: dissociating proliferation and development of effector function. J. Exp. Med. 197, 1141–1151.
Curtsinger, J. M., Schmidt, C. S., Mondino, A., Lins, D. C., Kedl, R. M., Jenkins, M. K., and Mescher, M. F. (1999). Inflammatory cytokines provide a third signal for activation of naive CD4+ and CD8+ T cells. J. Immunol. 162, 3256–3262.
Curtsinger, J. M., Valenzuela, J. O., Agarwal, P., Lins, D., and Mescher, M. F. (2005). Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J. Immunol. 174, 4465–4469.
Haring, J. S., Badovinac, V. P., and Harty, J. T. (2006). Inflaming the CD8+ T cell response. Immunity 25, 19–29.
Harty, J. T., and Badovinac, V. P. (2008). Shaping and reshaping CD8+ T-cell memory. Nat. Rev. Immunol. 8, 107–119.
Hogquist, K. A., Jameson, S. C., Heath, W. R., Howard, J. L., Bevan, M. J., and Carbone, F. R. (1994). T cell receptor antagonist peptides induce positive selection. Cell 76, 17–27.
Intlekofer, A. M., Takemoto, N., Kao, C., Banerjee, A., Schambach, F., Northrop, J. K., Shen, H., Wherry, E. J., and Reiner, S. L. (2007). Requirement for T-bet in the aberrant differentiation of unhelped memory CD8+T cells. J. Exp. Med. 204, 2015–2021.
Jabbari, A., and Harty, J. T. (2006). Secondary memory CD8+T cells are more protective but slower to acquire a central-memory phenotype. J. Exp. Med. 203, 919–932.
Joshi, N. S., Cui, W., Chandele, A., Lee, H. K., Urso, D. R., Hagman, J., Gapin, L., and Kaech, S. M. (2007). Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity 27, 281–295.
Kaech, S. M., and Ahmed, R. (2001). Memory CD8+T cell differentiation: initial antigen encounter triggers a developmental program in naive cells. Nat. Immunol. 2, 415–422.
Kaech, S. M., Hemby, S., Kersh, E., and Ahmed, R. (2002a). Molecular and functional profiling of memory CD8 T cell differentiation. Cell 111, 837–851.
Kaech, S. M., Wherry, E. J., and Ahmed, R. (2002b). Effector and memory T-cell differentiation: implications for vaccine development. Nat. Rev. Immunol. 2, 251–262.
Kolumam, G. A., Thomas, S., Thompson, L. J., Sprent, J., and Murali-Krishna, K. (2005). Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J. Exp. Med. 202, 637–650.
Krieg, A. M. (2002). CpG motifs in bacterial DNA and their immune effects. Annu. Rev. Immunol. 20, 709–760.
Mescher, M. F., Curtsinger, J. M., Agarwal, P., Casey, K. A., Gerner, M., Hammerbeck, C. D., Popescu, F., and Xiao, Z. (2006). Signals required for programming effector and memory development by CD8+ T cells. Immunol. Rev. 211, 81–92.
Pearce, E. L., and Shen, H. (2007). Generation of CD8 T cell memory is regulated by IL-12. J. Immunol. 179, 2074–2081.
Pham, N. L., Badovinac, V. P., and Harty, J. T. (2009). A default pathway of memory CD8 T cell differentiation after dendritic cell immunization is deflected by encounter with inflammatory cytokines during antigen-driven proliferation. J. Immunol. 183, 2337–2348.
Schluns, K. S., and Lefrancois, L. (2003). Cytokine control of memory T-cell development and survival. Nat. Rev. Immunol. 3, 269–279.
Takemoto, N., Intlekofer, A. M., Northrup, J. T., Wherry, E. J., and Reiner, S. L. (2006). Cutting edge: IL-12 inversely regulates T-bet and eomesodermin expression during pathogen-induced CD8+T cell differentiation. J. Immunol. 177, 7515–7519.
Thompson, L. J., Kolumam, G. A., Thomas, S., and Murali-Krishna, K. (2006). Innate inflammatory signals induced by various pathogens differentially dictate the IFN-I dependence of CD8 T cells for clonal expansion and memory formation. J. Immunol. 177, 1746–1754.
van Stipdonk, M. J., Lemmens, E. E., and Schoenberger, S. P. (2001). Naive CTLs require a single brief period of antigenic stimulation for clonal expansion and differentiation. Nat. Immunol. 2, 423–429.
Wherry, E. J., Teichgraber, V., Becker, T. C., Masopust, D., Kaech, S. M., Antia, R., von Andrian, U. H., and Ahmed, R. (2003). Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat. Immunol. 4, 225–234.
Keywords: memory CD8 T cells, inflammatory cytokines
Citation: Pham N-LL, Badovinac VP and Harty JT (2011) Differential role of “signal 3” inflammatory cytokines in regulating CD8 T cell expansion and differentiation in vivo. Front. Immun. 2:4. doi: 10.3389/fimmu.2011.00004
Received: 09 November 2010;
Paper pending published: 06 December 2010;
Accepted: 28 January 2011;
Published online: 11 February 2011.
Edited by:
Bruce Reinhold, Dana-Farber Cancer Institute, USAReviewed by:
Leo Lefrancois, University of Connecticut Health Center, USAJonathan Duke-Cohan, Dana-Farber Cancer Institute, USA
Bo Zhou, Dana-Farber Cancer Institute, USA
Copyright: © 2011 Pham, Badovinac and Harty. This is an open-access article subject to an exclusive license agreement between the authors and Frontiers Media SA, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: John T. Harty, Department of Microbiology, Department of Pathology, and Interdisciplinary Program in Immunology, University of Iowa, 3-530 Bowen Science Building, 51 Newton Road, Iowa City, IA 52242, USA.e-mail:am9obi1oYXJ0eUB1aW93YS5lZHU=