 Amit A. Lugade1
Amit A. Lugade1
- 1 Department of Immunology, Roswell Park Cancer Institute, Buffalo, NY, USA
- 2 Department of Pathology, Roswell Park Cancer Institute, Buffalo, NY, USA
- 3 Department of Medicine and New York State Center of Excellence in Bioinformatics and Health Sciences, Division of Infectious Diseases, University at Buffalo, State University of New York, Buffalo, NY, USA
Non-typeable Haemophilus influenzae (NTHI) colonizes the lower respiratory tract of patients with chronic obstructive pulmonary disease and also causes exacerbations of the disease. The 16-kDa lipoprotein P6 has been widely studied as a potential vaccine antigen due to its highly conserved expression amongst NTHI strains. Although P6 is known to induce potent inflammatory responses, its role in the pathogenesis of NTHI infection in vivo has not been examined. Additionally, the presence of an amino-terminal lipid motif on P6 serves to activate host Toll-like receptor 2 (TLR2) signaling. The role of host TLR2 and NTHI expression of the lipoprotein P6 on the induction of airway inflammation and generation of adaptive immune responses following chronic NTHI stimulation was evaluated with TLR2-deficient mice and a P6-deficient NTHI strain. Absence of either host TLR2 or bacterial P6 resulted in diminished levels of immune cell infiltration within lungs of mice exposed to NTHI. Pro-inflammatory cytokine secretion was also reduced in lungs that did not express TLR2 or were exposed to NTHI devoid of P6. Induction of specific antibodies to P6 was severely limited in TLR2-deficient mice. Although mice exposed to the P6-deficient NTHI strain were capable of generating antibodies to other surface antigens of NTHI, these levels were lower compared to those observed in mice exposed to P6-expressing NTHI. Therefore, cognate interaction between host TLR2 and bacterial P6 serves to enhance lung inflammation and elicit robust adaptive immune responses during NTHI exposure. Strategies to limit NTHI inflammation while simultaneously promoting robust immune responses may benefit from targeting the TLR2:P6 signaling axis.
Introduction
Within the respiratory mucosa, there exists a delicate balance between induction of inflammation to limit pathogen spread and bystander lung tissue destruction. Ligation of pathogen recognition receptors, such as Toll-like receptors (TLRs), during infection is necessary to initiate the protective inflammatory processes and signaling from these receptors is critical for the generation of adaptive immune responses (Manicassamy and Pulendran, 2009). Therefore, protective immune responses to respiratory pathogens induce and require inflammation to activate antigen-specific lymphocytes. We have previously shown that chronic exposure to a gram-negative bacteria, non-typeable Haemophilus influenzae (NTHI), in the lung results in high levels of bronchovascular inflammation and the generation of specific T cells and circulating antibodies (Lugade et al., 2011).
Non-typeable Haemophilus influenzae is a gram-negative bacterium that resides in the nasopharynx of adults and children. Introduction of NTHI into the middle ear of children causes otitis media. NTHI also causes lower respiratory tract infections, called exacerbations, in adults with chronic obstructive pulmonary disease (COPD; Sethi and Murphy, 2001). The outer membrane of the bacterium contains several TLR ligands that have been evaluated as potential vaccine antigens. Included within the outer membrane is the 16-kDa lipoprotein P6, which comprises approximately 1–5% of total outer membrane proteins and is highly conserved among strains of NTHI (Munson et al., 1985; Sethi and Murphy, 2001). P6 plays a critical role in the structural integrity of the outer membrane as previous work has demonstrated that its absence increases sensitivity of the mutant NTHI strain to a panel of antimicrobial agents (Murphy et al., 2006). Although several studies have evaluated the potential of P6 as a vaccine antigen (Hotomi et al., 1998; Bertot et al., 2004; McMahon et al., 2005; Wu et al., 2005; Ishida et al., 2006; Nomura et al., 2008; Noda et al., 2010), there are no studies detailing its role in the initiation of inflammation during infection in vivo.
P6 is a key mediator in the interaction of NTHI with host immune cells (Chen et al., 2004). The lipoprotein expresses an amino-terminal tripalmitoyl residue that is required for signaling via TLR2 (Lugade et al., submitted). Other investigators have shown that P6-mediated induction of the pro-inflammatory cytokines IL-8 and TNF-α from human epithelial cells is dependent on TLR2 signaling via an NF-κB pathway (Shuto et al., 2001; Berenson et al., 2005). Using TLR2-deficient mice and a P6-deficient NTHI strain, we have evaluated whether the cognate interaction between host TLR2 and bacterial P6 influences the extent of lung inflammation and the generation of antigen-specific lymphocytes. Such information will provide valuable data regarding the pathogenesis of NTHI infection in the lower respiratory tract and the potential of protective responses from vaccination to P6.
Materials and Methods
Mice
Six-week old C57BL/6NCr (WT) female mice were purchased from NCI. Female B6.129-Tlr2tm1Kir/J (TLR2−/−) mice were purchased from Jackson Laboratories (Bar Harbor, ME, USA). Mice were maintained under specific pathogen free conditions. All procedures performed on animals were IACUC-approved, and complied with all state, federal, and NIH regulations. Groups of five mice per genotype were used in all experiments.
Preparation and Oropharyngeal Administration of NTHI
Frozen glycerol stock of P6-expressing NTHI strains 1479 and 49P5H1 (P6+ NTHI) and P6-deficient NTHI strain 49P5H1 (P6− NTHI) were streaked on chocolate-agar plates. Generation of the P6-deficient strain has been described elsewhere (Murphy et al., 2006). Single colonies of both strains were grown in a liquid culture of brain–heart infusion (BHI) media supplemented with 10 μg/ml hemin and 10 μg/ml β-nicotinamide adenine dinucleotide (β-NAD). After 3–4 h of culture in a 37°C shaking incubator, OD600 was determined in order to dilute the required number of colony forming units (CFU) to 2 × 108 CFU/ml in PBS. Bacteria was pelleted in microcentrifuge tubes at 13000 × g for 10 min and washed twice in PBS. NTHI was introduced to mice by oropharyngeal instillation via the trachea as previously described (Lugade et al., 2011). Mice received bi-weekly instillations of live NTHI for 16 consecutive weeks before analysis.
Bronchoalveolar Lavage
On the day of sacrifice, mice were injected intraperitoneally with 1 ml of warmed 2.5% Avertin (2,2,2-tribromoethanol). The trachea exposed for cannulation with a 22-gage i.v. catheter. PBS (750 μl) was injected and withdrawn from the lung two times using a tuberculin syringe. White blood cell count of bronchoalveolar lavage (BAL) fluid was assessed using a hemocytometer. Cells were cytocentrifuged onto clean glass slides and stained with Hema 3® (Fisher Scientific) to obtain cell differential counts of macrophages, lymphocytes, and neutrophils. Cytokine levels in BAL supernatants were measured by sandwich ELISA.
Lung Histology
Lungs were excised and fixed in 10% formaldehyde (Polysciences, Inc.) in PBS, embedded in paraffin, sectioned, and stained with H&E by the Roswell Park Cancer Institute histopathology core facility. Images were obtained on an Olympus light microscope equipped with a CCD camera and Spot image analysis software (v25.4, Diagnostics Instruments). A scoring schema (outlined in Table 1) was developed to quantify the extent of inflammation and immune cell infiltration in the lungs of mice exposed to NTHI. Identity of the slides was blinded during two independent scoring sessions by the pathologist (Paul N. Bogner) and a consensus score of 0–3 was given for each of the parameters evaluated.
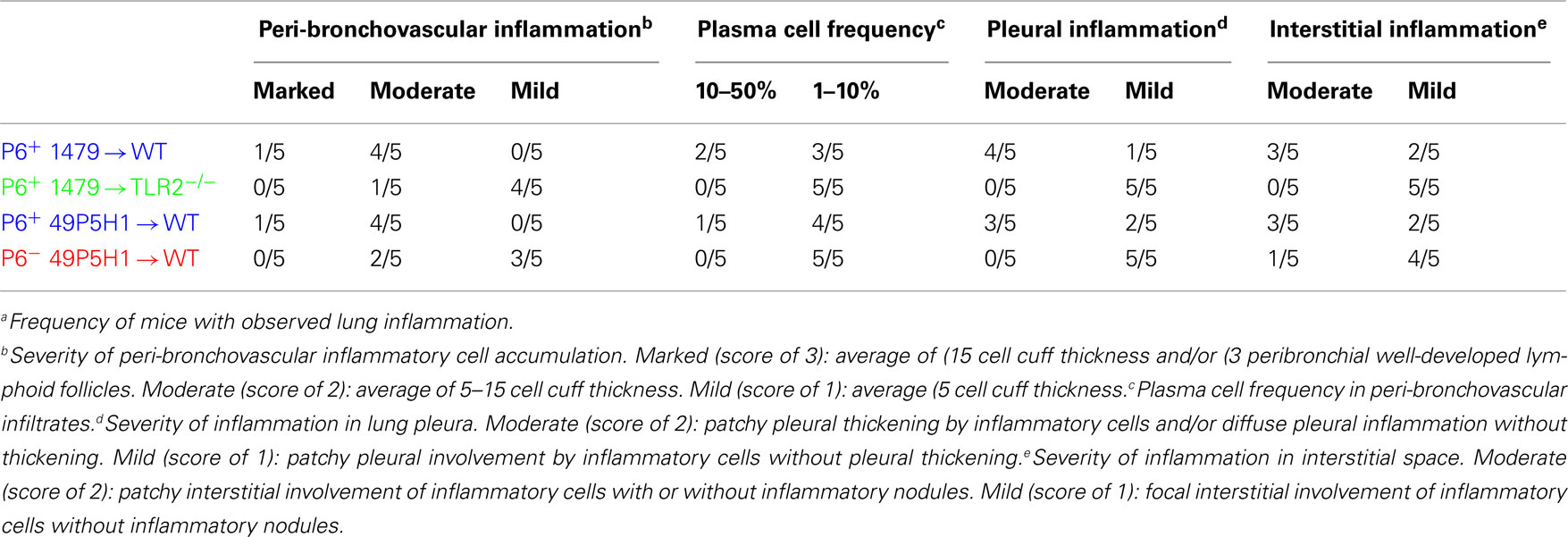
Table 1. Pathological evaluation of lung inflammationa.
Cytokine ELISPOTs
Frequency of cytokine-secreting antigen-specific T cells from the spleen of NTHI-challenged mice was evaluated by ELISPOT. Multiscreen Immobilon-P plates (Millipore) were coated overnight at 4°C with 3 μg/ml of rat anti-mouse IFN-γ (clone AN-18), anti-mouse IL-4 (clone 11B11), or anti-mouse IL-17A (clone eBio17CK15A5). Lymphocytes were co-cultured with syngeneic antigen presenting cells (APCs) pulsed with 1 μM P641–55 peptide or lysates of the P6+ NTHI 49P5H1 strain. After an 18h culture, the plates were washed extensively and cytokines detected with biotinylated antibodies (IFN-γ, R4-6A2; IL-4, BVD6-24G2; IL-17A, eBio17B7) followed by addition of streptavidin-HRP. Spots were developed with tetramethylbenzidine (TMB) substrate and enumerated microscopically.
B Cell ELISPOTs
Multiscreen Immobilon-P plates (Millipore) were coated overnight at 4°C with 3 μg/ml of P6 protein or 4 × 106 CFU of intact heat-killed P6− NTHI. Cells were added to plates and cultured for 18 h. Rabbit anti-mouse IgG-subclass specific polyclonal antibodies (Jackson ImmunoResearch) was added to wells followed by HRP-conjugated mouse anti-rabbit polyclonal antibodies (Jackson ImmunoResearch). Spots were developed and enumerated as described for cytokine ELISPOTs.
Serum Anti-P6 and Anti-NTHI Ig
Titers of antigen-specific antibodies were measured using an indirect ELISA as described previously (Badr et al., 1999). Briefly, ELISA plates were coated overnight at 4°C with 3 μg/ml of P6 protein or 4 × 106 CFU of intact heat-killed P6+ NTHI. Weekly serum samples from individual mice were added to blocked plates and detected with AP-conjugated goat anti-mouse Ig (Sigma). Plates were developed with p-nitrophenylphosphate (pNPP) and absorbance measured at 405 nm.
Results
Cognate Interaction of Host TLR2 and NTHI Lipoprotein Promotes Inflammation
Our previous work has demonstrated that chronic exposure to NTHI in the lower respiratory tract induces an inflammatory microenvironment characterized by immediate neutrophil accumulation and long-term lymphocytic accumulation (Lugade et al., 2011). The role and requirement of host pattern recognition receptors for the initiation of NTHI-mediated lung inflammation has not been evaluated. WT and TLR2−/− mice were exposed to NTHI strain 1479 by oropharyngeal instillation for 16 consecutive weeks in order to determine whether host expression of TLR2 is required for the generation of lung inflammation. Characteristic lymphocytic accumulation surrounding airways and bronchovasculature was observed in lungs of WT mice exposed to P6-expressing NTHI strain 1479 (P6+ 1479 → WT; Figure 1, top row). In contrast, the extent of inflammation was not as evident histologically in the lungs of TLR2−/− mice exposed to NTHI strain 1479 (P6+ 1479 → TLR2−/−; Figure 1, second row). Although the localization pattern of infiltrating cells was similar between WT and TLR2−/− mice, the extent of infiltration was substantially lower in TLR2−/− mice, suggesting that TLR2 plays an important role in the generation of robust chronic NTHI-mediated lung inflammation.
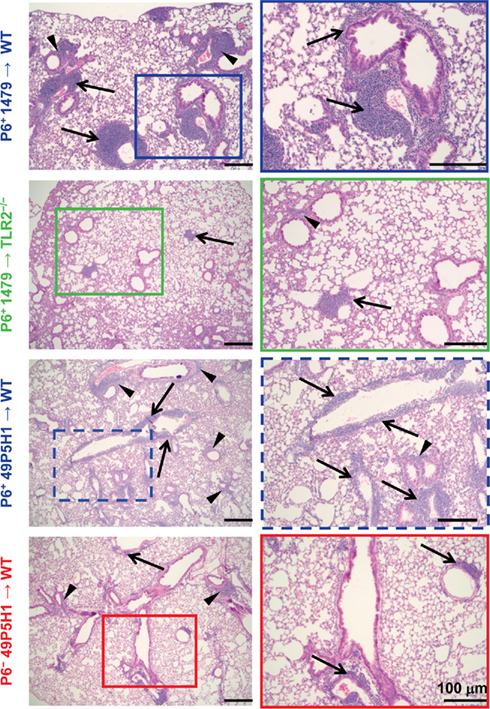
Figure 1. Absence of host TLR2 or bacterial P6 limits immune cell infiltration. Mice were exposed to NTHI for 16 consecutive weeks. H&E stained lung sections were analyzed for inflammation by light microscopy. Representative images of H&E sections taken from five mice per group. Images in the right column are enlarged magnification of the outlined region. Immune cell infiltrates surrounding perivascular (arrows) and peribronchial (arrowheads) regions are indicated.
Like many gram-negative bacteria, NTHI expresses a variety of TLR2 ligands such as Pam3Cys-expressing lipoproteins and peptidoglycan (Li, 2003). One of the components of the NTHI outer membrane is P6, an immunogenic lipoprotein that expresses an amino-terminal tripalmitoyl residue and initiates TLR2 signaling. P6 is highly conserved among NTHI strains and despite its potential as a promising vaccine candidate, has not been directly tested for its role in NTHI-mediated inflammation. The chronic inflammation model was initially established using the well characterized NTHI strain 1479. However, we were unable to construct a P6-deficient mutant in this strain, perhaps because of the well described variability in the transformability observed among strains of NTHI (Murphy et al., 2006). Thus, the P6− mutant was constructed in strain 49P5H1 which was isolated from the sputum of an adult with COPD (Murphy et al., 2006). In order to precisely define the role of P6 in the elicitation of chronic inflammation, WT mice were exposed to 49P5H1 strains that either expressed P6 (P6+ 49P5H1 → WT) or were devoid of P6 (P6− 49P5H1 → WT). The P6-deficient strain (P6− 49P5H1) was instilled into WT mice in order to determine whether the presence of P6 in the outer membrane of NTHI is required for the induction of lung inflammation in TLR2-expressing hosts. Lymphocytic accumulation was observed in WT mice exposed to P6+ 49P5H1 in a pattern similar to that observed in WT mice exposed to strain P6+ 1479 (Figure 1, third row compared to top row). Exposure to P6− 49P5H1 in WT mice did not result in the extensive lymphocytic accumulation that was observed with P6+ 49P5H1 (Figure 1, bottom row compared to third row). Clearly, the presence of other TLR ligands expressed by the P6− 49P5H1 mutant strain did not appreciably contribute to the induction of lung inflammation in the WT host, suggesting that P6 plays a critical role in NTHI-mediated inflammation.
The level of lung inflammation generated was scored using a semi-quantitative system by a pathologist to determine whether any differences exist due to the absence of host TLR2 or bacterial P6 (Table 1). All WT mice receiving P6-expressing NTHI strains (1479 or 49P5H1) were scored for bronchovascular inflammation at “moderate” or “marked” levels, whereas the majority of TLR2−/− mice receiving P6+ 1479 or WT mice receiving P6− 49P5H1 scored primarily as “mild.” Additionally, pleural and interstitial inflammation was very different in TLR2-deficient hosts or mice exposed to P6− 49P5H1. Thus, histopathological evidence of inflammation is greatest in a host that expresses TLR2 and when NTHI expresses the P6 lipoprotein in the outer membrane.
The role and contribution of cognate TLR2 and P6 to the immune cell composition (Table 2) and cytokine production (Figure 2) from BAL fluid was also assessed. Total immune cell infiltration was greatest in WT mice receiving either strain of P6-expressing NTHI (i.e., 1479 or 49P5H1). There was a statistically significant difference in the total number of leukocytes in TLR2−/− mice receiving P6+ 1479 (346 vs 98, p < 0.01) compared to WT mice receiving the same strain (Table 2). Similarly, WT mice receiving P6− 49P5H1 also exhibited a statistically significant difference in total immune cell infiltration when compared WT mice that were instilled with P6+ 49P5H1 (285 vs 154, p < 0.01). Macrophages comprised the majority of immune cells in the BAL fluid in TLR2−/− mice and WT mice receiving P6− 49P5H1, accounting for greater than 70% whereas macrophages accounted for roughly 60% in WT mice receiving either strain of P6-expressing NTHI, a difference that was statistically significant (p < 0.01). In conjunction with an increase in macrophages, there was a statistically significant decrease in the frequency of lymphocytes present in TLR2−/− mice receiving NTHI strain 1479 and WT mice receiving P6− NTHI strain 49P5H1 as compared to their counterpart WT mice receiving P6-expressing NTHI. This altered leukocyte frequency suggests that a lung microenvironment in which host TLR2 or P6 is absent during NTHI exposure results in a limited cell infiltrate and altered inflammatory phenotype.
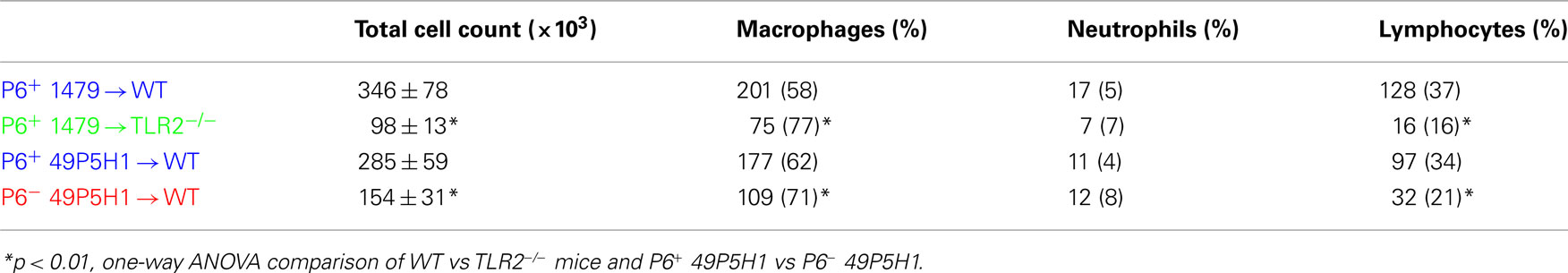
Table 2. Immune cell composition in bronchoalveolar lavage fluid.
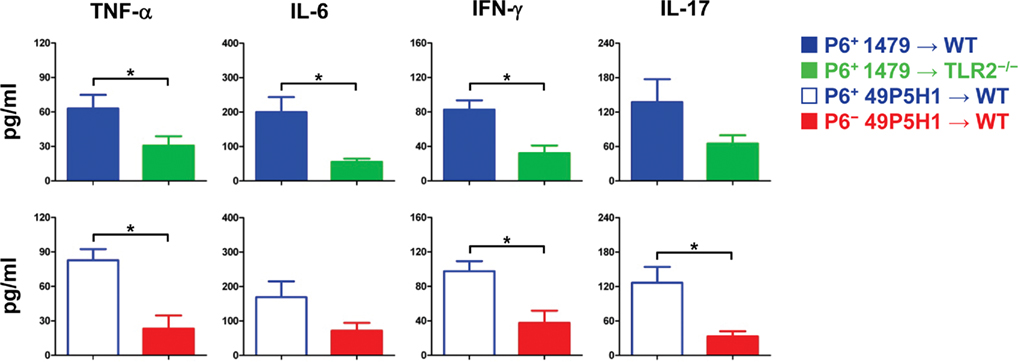
Figure 2. Pro-inflammatory cytokine levels in BAL fluid. Concentration of TNF-α, IL-6, IFN-γ, IL-17 in BAL fluid of mice exposed to either P6+ 1479  , P6+ 49P5H1 (
, P6+ 49P5H1 ( ), or P6− 49P5H1 (
), or P6− 49P5H1 ( ) NTHI were analyzed by sandwich ELISA. *p < 0.01, one-way ANOVA. Average ± SEM.
) NTHI were analyzed by sandwich ELISA. *p < 0.01, one-way ANOVA. Average ± SEM.
Differences in BAL fluid pro-inflammatory cytokines were assessed by cytokine-specific ELISA (Figure 2). TNF-α, IL-6, IL-17, and IFN-γ were detected in the BAL fluid from WT mice receiving P6-expressing NTHI, indicative of chronic pulmonary inflammation. Levels of IL-6 and TNF-α were decreased by almost 50% (p < 0.01) in TLR2-deficient mice and WT mice exposed to NTHI devoid of P6. In addition, the levels of cytokines associated with inflammatory T cell activation, IL-17 and IFN-γ, were also diminished in TLR2−/− mice receiving NTHI strain 1479 and WT mice receiving P6− NTHI strain 49P5H1. Collectively our results demonstrate that the ability of the host to respond via surface TLR2 expression to NTHI P6 on its outer membrane is critical toward the generation of inflammatory mediators that contribute to chronic lung inflammation.
Adaptive Immune Responses to Chronic NTHI are Dependent on Host TLR2 and Bacterial P6 Expression
We next assessed whether the absence of TLR2 or bacterial P6 impacts on the generation and function of NTHI-specific T cells and B cells. An immunodominant peptide we had previously identified from P6 (McMahon et al., 2005) was utilized to measure the frequency of cytokine-secreting P6-specific T cells from the spleens of WT and TLR2−/− mice exposed to NTHI strain 1479 (Figure 3A). Although P6-specific T cells were detected in TLR2−/− mice, the frequency of these cells that secreted IFN-γ, IL-17, or IL-4 exhibited a statistically significant decrease compared to WT mice receiving NTHI strain 1479. Thus, the optimal generation of P6-specific T cells is reduced in the absence of TLR2.
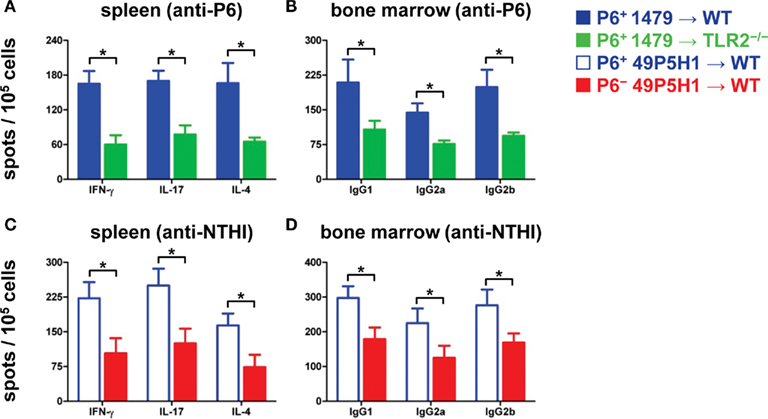
Figure 3. Frequency of antigen-specific immune cells in lymphoid organs. ELISPOT to measure frequency of IFN-γ-, IL-17-, IL-4-secreting T cells (A,C) and frequency of IgG1-, IgG2a-, IgG2b-secreting B cells (B,D) from mice exposed to NTHI. Responses to P6 are measured in (A,B); responses to NTHI strain 49P5H1 are measured in (C,D). *p < 0.01, one-way ANOVA. Average ± SEM.
T cell responses to total NTHI strain 49P5H1 antigens were measured in WT mice that had received NTHI strain 49P5H1, both the P6+ and P6− strain. The frequency of NTHI-specific T cells was significantly lower in WT mice receiving P6− 49P5H1 as compared to WT mice receiving P6+ 49P5H1 (Figure 3C). Interestingly and unexpectedly, we observed that the absence of P6 in the outer membrane of NTHI hampers the generation of T cells to the other NTHI antigens suggesting that the presence of P6 during bacterial exposure is critical for the optimal generation of NTH-specific T cells.
The frequency of antigen-specific Ig-secreting B cells from the bone marrow was measured by B cell ELISPOT. WT and TLR2−/− mice receiving P6-expressing NTHI strain 1479 were evaluated for P6-specific cells (Figure 3B). A statistically significant decrease in the frequency of anti-P6 Ig-secreting cells for all three IgG subclasses tested was observed in TLR2−/− mice as compared to similarly treated WT mice. The presence of all three IgG subclasses tested in TLR2−/− mice suggests that the absence of this receptor did not hamper isotype switching but only the magnitude of P6-specific B cells generated.
B cell frequency in WT mice receiving NTHI strain 49P5H1 was measured using intact heat-killed 49P5H1 (Figure 3D). Similar to the T cell ELISPOT, frequency of total NTHI antigen-specific B cells secreting the three IgG subclasses tested was lower in WT mice receiving P6− 49P5H1 compared to the levels of antigen-specific B cells in mice receiving P6+ 49P5H1. Thus, the presence of P6 in the outer membrane of NTHI is critical also for the optimal generation of B cells that secrete NTHI-specific antibodies.
The titers of P6-specific antibodies in serum was measured by indirect ELISA in WT and TLR2−/− mice. TLR2−/− mice exhibited diminished levels of P6-specific Ig compared to WT mice (Figure 4A). Although, the magnitude of the anti-P6 Ig was lower, the kinetics of anti-P6 Ig generation and maintenance was not altered in TLR2−/− mice. Therefore, the ability of the host to respond to lipoprotein P6 via TLR2 is important for the robust generation of anti-P6 antibodies. To determine whether the absence of P6 from the outer membrane of NTHI impacted the ability of WT mice to elicit antibodies against other NTHI surface antigens, ELISA plates were coated with intact 49P5H1 (Figure 4B). WT mice receiving P6+ NTHI strain 49P5H1 exhibited endpoint titers of NTHI-specific Ig significantly greater than that observed in mice receiving P6− NTHI strain 49P5H1. Thus, overall, the ability of the host to mount a robust adaptive immune response to NTHI is dependent on host TLR2 expression and the presence of the P6 lipoprotein in the outer membrane of NTHI during chronic exposure.
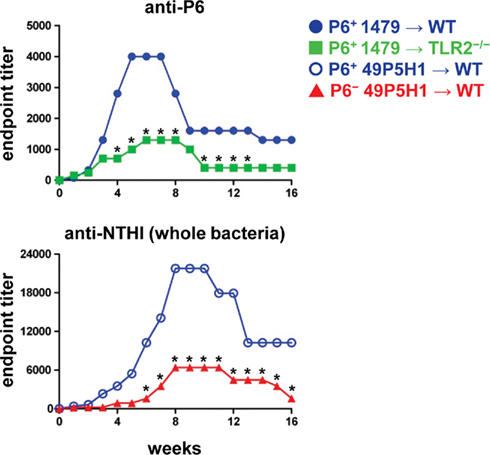
Figure 4. Titers of anti-P6 and anti-NTHI Ig in serum. Indirect ELISA was used to measure Ig levels in mice exposed to NTHI. Purified P6 was used to measure anti-P6 Ig in mice exposed to P6+ 1479 ( ,
, ) and intact NTHI 49P5H1 was used to measure anti-NTHI levels in mice exposed to P6+ 49P5H1 (
) and intact NTHI 49P5H1 was used to measure anti-NTHI levels in mice exposed to P6+ 49P5H1 ( ), or P6− 49P5H1 (
), or P6− 49P5H1 ( ). *p < 0.01, two-way ANOVA with Bonferroni post-test comparison between groups. Average ± SEM.
). *p < 0.01, two-way ANOVA with Bonferroni post-test comparison between groups. Average ± SEM.
Discussion
Toll-like receptors and their cognate ligands expressed in pathogens exert a potent response on the immune system during infection. The pathogenesis of bacterial infections includes tissue inflammation, which is initiated by TLR signaling. Inflammatory responses to pathogens are regulated to minimize overt damage, especially at mucosal surfaces. Mucosal tissues are populated with commensal bacteria and therefore they must tightly regulate the response to pathogens via TLR activation (Alexopoulou and Kontoyiannis, 2005). Conversely, the absence of inflammation or insufficient inflammation can also be deleterious by allowing unrestrained pathogen growth and abrogating immune cell activation. In this study, we have demonstrated that absence of the TLR2:P6 signaling axis severely limits the extent of NTHI-mediated airway inflammation and the optimal generation of adaptive immune responses.
Exposure to outer membrane protein P6 results in production of prototypical inflammatory signals. In one such example, P6 stimulation of primary human epithelial cells upregulates MUC5AC mucin gene transcription (Chen et al., 2004) and the upregulation of this gene is an important contributor to airway obstruction in COPD patients (Caramori et al., 2009). Overproduction of mucin by epithelial cells increases bacterial adherence and colonization (Knowles and Boucher, 2002) and the absence of this signaling pathway may also play a role in the diminished inflammatory response observed with NTHI devoid of P6. In fact, Moghaddam et al. (2008) have demonstrated increased mucin gene transcription in mice chronically exposed to NTHI lysates. In our model, there was no evidence of mucus overproduction when mice were exposed to live, intact P6+ NTHI, therefore it was not surprising that exposure to P6− NTHI would also prevent accumulation of mucus in the airways. Monoclonal antibody inhibition of TLR2 prevented MUC5AC mucin gene transcription from Mycoplasma pneumoniae treated epithelial cells (Kraft et al., 2008), supporting the importance of this signaling axis for the induction of NTHI-mediated airway obstruction in COPD patients.
Non-typeable Haemophilus influenzae is present in the airways under stable conditions, in adults with (COPD;Sethi and Murphy, 2001). The degeneration of normal mucociliary processes in the COPD airways due to accumulated lifetime cigarette-smoke exposure contributes to the colonization of NTHI in the lower respiratory tract. It is now well accepted that cigarette-smoke alters the immune response in the airways and even impacts adversely on systemic immunity (Domagala-Kulawik, 2008; Mehta et al., 2008; Dhillon et al., 2009; Stämpfli and Anderson, 2009). This alteration negatively influences the innate immune response to bacterial infection, which further dysregulates inflammatory cascades in the respiratory mucosa. Using a model of acute intranasal NTHI exposure (4 h), Gaschler et al. (2009) demonstrated that short-term NTHI exposure induces airway TNF-α production similar to that observed in our model involving exposure to NTHI for up to 16 weeks. Interestingly, the production of inflammatory cytokines, most notably MCP-1, increased in the respiratory mucosa of mice exposed to 8 weeks of cigarette-smoke prior to the acute NTHI exposure (Gaschler et al., 2009). Therefore, the inflammatory profile is additionally exaggerated and has the potential to cause further respiratory damage in lungs that have been assaulted by cigarette-smoke. Innate immune responses to NTHI are further impaired by cigarette-smoke exposure through the inactivation of alveolar macrophage clearance of the colonizing bacteria (Martí-Lliteras et al., 2009). This double hit, of increased inflammatory cytokine production and diminished bacterial clearance, likely contributes to exacerbations that characterize the course of COPD.
In addition to alterations to the innate immune response, cigarette-smoke exposure also reduces the generation of IFN-γ producing Th1 cells (Phaybouth et al., 2006); although there is some question whether both Th1 and Th17 skewed cells are present in the airways of chronic smokers (Harrison et al., 2008). Exposure to P6+ NTHI-induced antigen-specific T cells capable of secreting IFN-γ (Th1) and IL-17 (Th17; Lugade et al., 2011). Both types of cells are likely required for clearance of the infiltrating bacteria and maintenance of the chronic inflammatory milieu. Attenuation of NTHI-specific Th1 responses by cigarette-smoke exposure in the airways of COPD patients therefore serves to exacerbate pulmonary inflammation at the expense of bacterial clearance. Generation of the protective IFN-γ producing cells is dependent on the presence of the TLR2:P6 signaling axis, but TLR2 signaling can be negatively modulated by cigarette-smoke exposure (Droemann et al., 2005; Bagaitkar et al., 2010). Thus, vaccination strategies that induce the generation of P6-specific Th1 cells will be essential for clearance of NTHI from airways during disease exacerbations (McMahon et al., 2005; Lugade et al., submitted). Our previous work has revealed the requirement of TLR2 for the vaccination efficacy of P6 (Lugade et al., submitted). Similarly, the current study has demonstrated the role of TLR2 in driving the immunopathology of NTHI infection and generation of adaptive immune responses. Thus, TLR2 plays a critical role in the setting of NTHI vaccination and immune response following NTHI infection. Protective immune responses are required to limit the extent of pathogen-mediated inflammation during infection. It is therefore critical to overcome TLR2 hyposensitivity observed as a result of cigarette-smoke exposure (Bagaitkar et al., 2010). Although the absence of TLR2-mediated signaling upon NTHI infection is beneficial for limiting airway inflammation, its absence prevents stimulation of protective immune responses that can limit pathogen growth and through this mechanism reduce pathogen-mediated inflammation. Additionally, the presence of Th1 and Th17 effector cells serves to initiate rapid secondary immune responses upon pathogen re-infection.
Signaling from TLRs has the dual function of initiating inflammation and serving as an adjuvant for the induction of adaptive immune responses. Immune-mediated pathology is commonly observed in mucosal tissues upon pathogen infection and the mechanism behind these processes is a result of TLR ligation (Heimesaat et al., 2007; Layland et al., 2007; Sayi et al., 2011). However, suppression of TLR function in order to limit immunopathology is detrimental, as these same receptors are required to initiate immune responses to mucosal pathogens, such as respiratory syncytial virus and Toxoplasma gondii (Cyr et al., 2009; Murawski et al., 2009; Yarovinsky et al., 2008). In our model of NTHI-mediated airway inflammation, TLR2 is essential for the induction of IL-6 and TNF-α in the BAL fluid and for the generation of IFN-γ- and IL-17-secreting T cells. The requirement of TLR2 for the optimal generation of these cells suggests that COPD patients with frequent disease exacerbations may have a defect in the ability to generate these cells. This defect may be attributed to a lifetime of cigarette-smoke exposure that has diminished TLR2 signaling (Droemann et al., 2005).
In the present study, we used non-isogenic NTHI strains 1479 and 49P5H1. In view of this, we have been cautious to confine our comparisons and conclusions to immune responses to the P6 molecule because comparisons of immune responses to antigens other than P6 could be confounded by strain differences. Importantly, the amino acid sequences of P6 from strains 1479 and 49P5H1 are identical in all 153 amino acids based on the gene sequences (Murphy et al., 2006).
Of particular interest is the finding that anti-P6 antibodies are an important proportion of the total antibody response elicited against NTHI during respiratory mucosal exposure, despite the lipoprotein being a relatively minor constituent of the total outer membrane protein on this bacterium. The absence of P6 from the outer membrane resulted in lower titers of anti-NTHI antibodies suggesting that responses to P6 are critical for generating effective immune responses to the bacteria during exacerbations (Abe et al., 2002). We are currently investigating whether bacterial colonization in the respiratory mucosa and airway inflammation upon NTHI exposure can be limited by prophylactic vaccination with P6.
The results of the current study establish an essential role for TLR2 and P6 in calibrating the inflammatory response to NTHI in the respiratory mucosa. Signaling via the TLR2:P6 axis has broad regulatory effects on both innate and T and B cell effector functions. Our work identifies the dual function of this cognate interaction on the induction of inflammation and adaptive immune responses, thereby serving as a potential therapeutic target for NTHI-induced exacerbations of COPD.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Galina Elkin for assisting in serum collection, Charmaine Kirkham for assistance propagating the P6− NTHI strain, and Mehrab Nasirikenari for assisting in BAL collection. The work was supported by AI069379 from NIAID to Yasmin Thanavala.
References
Abe, Y., Murphy, T. F., Sethi, S., Faden, H. S., Dmochowski, J., Harabuchi, Y., and Thanavala, Y. M. (2002). Lymphocyte proliferative response to P6 of Haemophilus influenzae is associated with relative protection from exacerbations of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 165, 967–971.
Alexopoulou, L., and Kontoyiannis, D. (2005). Contribution of microbial-associated molecules in innate mucosal responses. Cell. Mol. Life Sci. 62, 1349–1358.
Badr, W. H., Loghmanee, D., Karalus, R. J., Murphy, T. F., and Thanavala, Y. (1999). Immunization of mice with P6 of nontypeable Haemophilus influenzae: kinetics of the antibody response and IgG subclasses. Vaccine 18, 29–37.
Bagaitkar, J., Demuth, D. R., Daep, C. A., Renaud, D. E., Pierce, D. L., and Scott, D. A. (2010). Tobacco upregulates P. gingivalis fimbrial proteins which induce TLR2 hyposensitivity. PLoS ONE 5, e9323. doi: 10.1371/journal.pone.0009323
Berenson, C. S., Murphy, T. F., Wrona, C. T., and Sethi, S. (2005). Outer membrane protein P6 of nontypeable Haemophilus influenzae is a potent and selective inducer of human macrophage proinflammatory cytokines. Infect. Immun. 73, 2728–2735.
Bertot, G. M., Becker, P. D., Guzmán, C. A., and Grinstein, S. (2004). Intranasal vaccination with recombinant P6 protein and adamantylamide dipeptide as mucosal adjuvant confers efficient protection against otitis media and lung infection by nontypeable Haemophilus influenzae. J. Infect. Dis. 189, 1304–1312.
Caramori, G., Casolari, P., Di Gregorio, C., Saetta, M., Baraldo, S., Boschetto, P., Ito, K., Fabbri, L. M., Barnes, P. J., Adcock, I. M., Cavallesco, G., Chung, K. F., and Papi, A. (2009). MUC5AC expression is increased in bronchial submucosal glands of stable COPD patients. Histopathology 55, 321–331.
Chen, R., Lim, J. H., Jono, H., Gu, X. X., Kim, Y. S., Basbaum, C. B., Murphy, T. F., and Li, J. D. (2004). Nontypeable Haemophilus influenzae lipoprotein P6 induces MUC5AC mucin transcription via TLR2-TAK1-dependent p38 MAPK-AP1 and IKKbeta-IkappaBalpha-NF-kappaB signaling pathways. Biochem. Biophys. Res. Commun. 324, 1087–1094.
Cyr, S. L., Angers, I., Guillot, L., Stoica-Popescu, I., Lussier, M., Qureshi, S., Burt, D. S., and Ward, B. J. (2009). TLR4 and MyD88 control protection and pulmonary granulocytic recruitment in a murine intranasal RSV immunization and challenge model. Vaccine 27, 421–430.
Dhillon, N. K., Murphy, W. J., Filla, M. B., Crespo, A. J., Latham, H. A., and O’Brien-Ladner, A. (2009). Down modulation of IFN-gamma signaling in alveolar macrophages isolated from smokers. Toxicol. Appl. Pharmacol. 237, 22–28.
Domagala-Kulawik, J. (2008). Effects of cigarette smoke on the lung and systemic immunity. J. Physiol. Pharmacol. 59(Suppl. 6), 19–34.
Droemann, D., Goldmann, T., Tiedje, T., Zabel, P., Dalhoff, K., and Schaaf, B. (2005). Toll-like receptor 2 expression is decreased on alveolar macrophages in cigarette smokers and COPD patients. Respir. Res. 6, 68.
Gaschler, G. J., Skrtic, M., Zavitz, C. C., Lindahl, M., Onnervik, P. O., Murphy, T. F., Sethi, S., and Stämpfli, M. R. (2009). Bacteria challenge in smoke-exposed mice exacerbates inflammation and skews the inflammatory profile. Am. J. Respir. Crit. Care Med. 179, 666–675.
Harrison, O. J., Foley, J., Bolognese, B. J., Long, E. III, Podolin, P. L., and Walsh, P. T. (2008). Airway infiltration of CD4+ CCR6+ Th17 type cells associated with chronic cigarette smoke induced airspace enlargement. Immunol. Lett. 121, 13–21.
Heimesaat, M. M., Fischer, A., Jahn, H. K., Niebergall, J., Freudenberg, M., Blaut, M., Liesenfeld, O., Schumann, R. R., Göbel, U. B., and Bereswill, S. (2007). Exacerbation of murine ileitis by Toll-like receptor 4 mediated sensing of lipopolysaccharide from commensal Escherichia coli. Gut 56, 941–948.
Hotomi, M., Saito, T., and Yamanaka, N. (1998). Specific mucosal immunity and enhanced nasopharyngeal clearance of nontypeable Haemophilus influenzae after intranasal immunization with outer membrane protein P6 and cholera toxin. Vaccine 16, 1950–1956.
Ishida, Y., Abe, Y., Yanai, M., Kobayashi, H., and Harabuchi, Y. (2006). Identification of human T-cell epitopes and highly immunogenic analog peptides on the non-typeable Haemophilus influenzae P6 outer membrane protein. Clin. Immunol. 121, 90–99.
Knowles, M. R., and Boucher, R. C. (2002). Mucus clearance as a primary innate defense mechanism for mammalian airways. J. Clin. Invest. 109, 571–577.
Kraft, M., Adler, K. B., Ingram, J. L., Crews, A. L., Atkinson, T. P., Cairns, C. B., Krause, D. C., and Chu, H. W. (2008). Mycoplasma pneumoniae induces airway epithelial cell expression of MUC5AC in asthma. Eur. Respir. J. 31, 43–46.
Layland, L. E., Rad, R., Wagner, H., and da Costa, C. U. (2007). Immunopathology in schistosomiasis is controlled by antigen-specific regulatory T cells primed in the presence of TLR2. Eur. J. Immunol. 37, 2174–2184.
Li, J. D. (2003). Exploitation of host epithelial signaling networks by respiratory bacterial pathogens. J. Pharmacol. Sci. 91, 1–7.
Lugade, A. A., Vethanayagam, R. R., Nasirikenari, M., Bogner, P., Segal, B. H., and Thanavala, Y. (2011). Nrf2 regulates chronic lung inflammation and B cell responses to non-typeable Haemophilus influenzae. Am. J. Respir. Cell Mol. Biol. doi: 10.1165/rcmb.2010-0321OC [Epub ahead of print].
Manicassamy, S., and Pulendran, B. (2009). Modulation of adaptive immunity with Toll-like receptors. Semin. Immunol. 21, 185–193.
Martí-Lliteras, P., Regueiro, V., Morey, P., Hood, D. W., Saus, C., Sauleda, J., Agustí, A. G., Bengoechea, J. A., and Garmendia, J. (2009). Nontypeable Haemophilus influenzae clearance by alveolar macrophages is impaired by exposure to cigarette smoke. Infect. Immun. 77, 4232–4242.
McMahon, M., Murphy, T. F., Kyd, J., and Thanavala, Y. (2005). Role of an immunodominant T cell epitope of the P6 protein of nontypeable Haemophilus influenzae in murine protective immunity. Vaccine 23, 3590–3596.
Mehta, H., Nazzal, K., and Sadikot, R. T. (2008). Cigarette smoking and innate immunity. Inflamm. Res. 57, 497–503.
Moghaddam, S. J., Clement, C. G., De la Garza, M. M., Zou, X., Travis, E. L., Young, H. W., Evans, C. M., Tuvim, M. J., and Dickey, B. F. (2008). Haemophilus influenzae lysate induces aspects of the chronic obstructive pulmonary disease phenotype. Am. J. Respir. Cell Mol. Biol. 38, 629–638.
Munson, R. S. Jr., and Granoff, D. M. (1985). Purification and partial characterization of outer membrane proteins P5 and P6 from Haemophilus influenzae type b. Infect. Immun. 49, 544–549.
Murawski, M. R., Bowen, G. N., Cerny, A. M., Anderson, L. J., Haynes, L. M., Tripp, R. A., Kurt-Jones, E. A., and Finberg, R. W. (2009). Respiratory syncytial virus activates innate immunity through Toll-like receptor 2. J. Virol. 83, 1492–1500.
Murphy, T. F., Kirkham, C., and Lesse, A. J. (2006). Construction of a mutant and characterization of the role of the vaccine antigen P6 in outer membrane integrity of nontypeable Haemophilus influenzae. Infect. Immun. 74, 5169–5176.
Noda, K., Kodama, S., Umemoto, S., Abe, N., Hirano, T., and Suzuki, M. (2010). Nasal vaccination with P6 outer membrane protein and alpha-galactosylceramide induces nontypeable Haemophilus influenzae-specific protective immunity associated with NKT cell activation and dendritic cell expansion in nasopharynx. Vaccine 28, 5068–5074.
Nomura, Y., Abe, Y., Ishida, Y., Kobayashi, H., and Harabuchi, Y. (2008). Promiscuous peptides on the nontypeable Haemophilus influenzae P6 outer membrane protein. J. Clin. Immunol. 28, 361–369.
Phaybouth, V., Wang, S. Z., Hutt, J. A., McDonald, J. D., Harrod, K. S., and Barrett, E. G. (2006). Cigarette smoke suppresses Th1 cytokine production and increases RSV expression in a neonatal model. Am. J. Physiol. Lung Cell Mol. Physiol. 290, L222-31.
Sayi, A., Kohler, E., Toller, I. M., Flavell, R. A., Müller, W., Roers, A., and Müller, A. (2011). TLR-2-activated B cells suppress Helicobacter-induced preneoplastic gastric immunopathology by inducing T regulatory-1 cells. J. Immunol. 186, 878–890.
Sethi, S., and Murphy, T. F. (2001). Bacterial infection in chronic obstructive pulmonary disease in 2000: a state-of-the-art review. Clin. Microbiol. Rev. 14, 336–363.
Shuto, T., Xu, H., Wang, B., Han, J., Kai, H., Gu, X. X., Murphy, T. F., Lim, D. J., and Li, J. D. (2001). Activation of NF-kappa B by nontypeable Haemophilus influenzae is mediated by toll-like receptor 2-TAK1-dependent NIK-IKK alpha/beta-I kappa B alpha and MKK3/6-p38 MAP kinase signaling pathways in epithelial cells. Proc. Natl. Acad. Sci. U.S.A. 98, 8774–8779.
Stämpfli, M. R., and Anderson, G. P. (2009). How cigarette smoke skews immune responses to promote infection, lung disease and cancer. Nat. Rev. Immunol. 9, 377–384.
Wu, T., Chen, J., Murphy, T. F., Green, B. A., and Gu, X. X. (2005). Investigation of non-typeable Haemophilus influenzae outer membrane protein P6 as a new carrier for lipooligosaccharide conjugate vaccines. Vaccine 23, 5177–5185.
Keywords: NTHI, P6, inflammation, T cells, B cells
Citation: Lugade AA, Bogner PN, Murphy TF and Thanavala Y (2011) The role of TLR2 and bacterial lipoprotein in enhancing airway inflammation and immunity. Front. Immun. 2:10. doi: 10.3389/fimmu.2011.00010
Received: 28 February 2011; Paper pending published: 30 March 2011;
Accepted: 12 April 2011; Published online: 27 April 2011.
Edited by:
Paul W. Bland, University of Gothenburg, SwedenCopyright: © 2011 Lugade, Bogner, Murphy and Thanavala. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Yasmin Thanavala, Department of Immunology, Roswell Park Cancer Institute, Elm and Carlton Streets, Buffalo, NY 14263, USA. e-mail:eWFzbWluLnRoYW5hdmFsYUByb3N3ZWxscGFyay5vcmc=