


- Department of Immunology, Erasmus MC, University Medical Center, Rotterdam, Netherlands
The vast majority of patients suffering from a primary immunodeficiency (PID) have defects in their T- and/or B-cell compartments. Despite advances in molecular diagnostics, in many patients no underlying genetic defect has been identified. B- and T-lymphocytes are unique in their ability to create a receptor by genomic rearrangement of their antigen receptor genes via V(D)J recombination. During this process, stable circular excision products are formed that do not replicate when the cell proliferates. Excision circles can be reliably quantified using real-time quantitative (RQ-)PCR techniques. Frequently occurring δREC–ψJα T-cell receptor excision circles (TRECs) have been used to assess thymic output and intronRSS–Kde recombination excision circles (KREC) to quantify B-cell replication history. In this perspective, we describe how TRECs and KRECs are formed during precursor – T- and B-cell differentiation, respectively. Furthermore, we discuss new insights obtained with TRECs and KRECs and specifically how these excision circles can be applied to support therapy monitoring, patient classification and newborn screening of PID.
Primary Immunodeficiencies of the Lymphoid System
Primary immunodeficiencies (PID) are rare inherited disorders of the immune system. Mutations in a single gene can affect one or more immunological components of the individual, resulting in increased susceptibility to infections, often accompanied by immunoregulatory defects (Notarangelo, 2010).
The vast majority of PID patients have impaired T- and/or B-cell function. T- and B-cells have the ability to create a unique receptor to recognize antigen. Furthermore, upon encounter with their specific antigen, they generate immunological memory by means of long-lived antigen-specific memory cells and antibody-producing plasma cells. Thus, defects in these cells grossly impair immunity. T-cell defects result in combined immunodeficiency (CID), affecting both cellular and humoral immunity. These patients suffer from opportunistic infections. The most severe CID variant (SCID) is fatal in the first months of life unless stem cell transplantation is provided. These patients lack T-cells, often combined with lack of B-cells, due to early arrests in precursor lymphocyte differentiation (Figure 1). B-cell defects lead to antibody deficiency. This defect in humoral immunity results in increased susceptibility to encapsulated bacteria. A subgroup of patients has agammaglobulinemia, due to a B-cell differentiation arrest in bone marrow and the consequent absence of mature B-cells and serum immunoglobulins (Figure 1). Over 30% of patients develop irreversible organ damage, mainly in the lungs, in childhood or early adulthood (Quinti et al., 2007). Early treatment with intravenous immunoglobulins and prophylactic antibiotics may prevent this (Lucas et al., 2010).
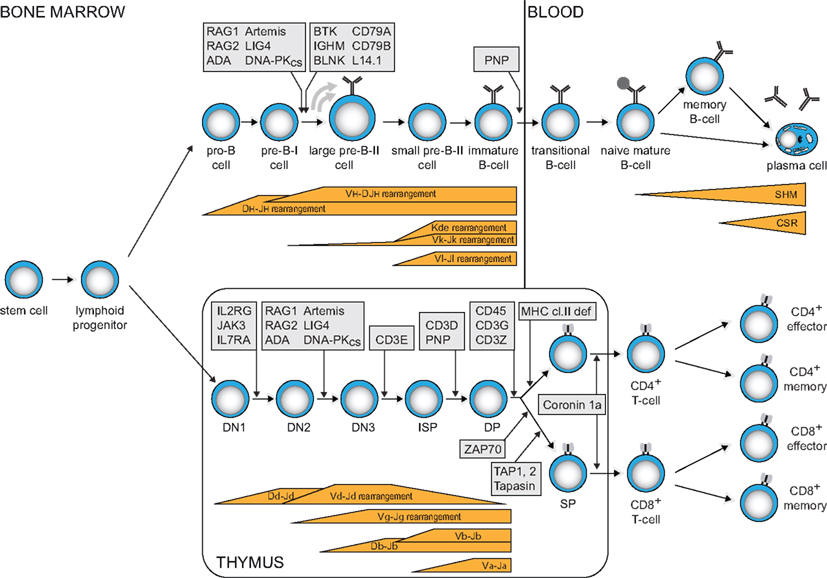
Figure 1. Human B- and T-cell differentiation stages including V(D)J recombination bars (Dik et al., 2005; van Zelm et al., 2005) and genetic defects underlying PID that result in impaired precursor differentiation. Blocks in B-cell development are derived from immunophenotyping studies on bone marrow samples from agammaglobulinemia and SCID patients. Developmental blocks in the T-cell lineage largely rely on data from targeted mutation studies in the mouse.
Whereas many genetic defects have been identified (Notarangelo et al., 2009), a molecular diagnosis has not been made in the vast majority of patients with a primary antibody deficiency (van der Burg et al., 2011). Furthermore, mild genetic variants in known disease-causing genes can give rise to a broad clinical spectrum of diseases (Schuetz et al., 2008; Conley et al., 2009; IJspeert et al., 2011). Thus, new assays are required to establish an early identification of disease and proper diagnosis with estimation of future complication risks. Over the past 10 years, many studies have addressed immunological defects to define subgroups of CID or antibody deficient patients. One of the newly explored methods involves quantification of circular DNA products that are generated in early lymphocyte development during the creation of unique B- and T-cell receptors. In this perspective, we will first address how recombination excision circles are formed and how these products can be used to study the mature B- and T-cell compartments. Subsequently, we will discuss recent insights obtained by quantification of these excision circles and potential applications to support early diagnosis, classification, therapy monitoring and newborn screening of PID.
V(D)J Recombination and Formation of Excision Circles
The gene complexes encoding the T- and B-cell receptor components do not contain a functional first exon. Instead, they contain multiple variable (V), diversity (D), and joining (J) genes. During antigen-independent differentiation in bone marrow (B-cells) and thymus (T-cells), stepwise rearrangements are introduced in the genome to couple one of each segment together to form a functional first exon (Figure 1).
Precursor-T cells in thymus first start to rearrange their T-cell receptor (TCR) D and TCRG genes (Figure 1). When this leads to a functional receptor, the cell exits the thymus as TCRγδ+ T-cell. Most cells, however, do not form a functional γδ TcR and start rearranging their TCRB and TCRA genes. Upon successful completion, these cells exit the thymus with an αβ TcR. In TCRD and TCRB loci, D to J gene rearrangements are induced prior to V to DJ rearrangements. TCRA and TCRG lack D gene segments and directly couple a V to a J gene segment.
Precursor B-cells start with D to J gene rearrangements in the Ig heavy chain locus (IGH), followed by V to DJ gene rearrangements (Figure 1). Upon successful completion, Ig light chain rearrangements are induced in the IGK locus, leading to direct V to J coupling. When IGK light chain rearrangements are not successful, V to J gene rearrangements are induced in the IGL light chain locus. Thus, mature B-cells can exit the bone marrow expressing an antigen receptor with either an Igκ or an Igλ isotype.
Double strand DNA breaks are introduced by the recombination activating gene products RAG1 and RAG2 on the borders of two gene segments and the flanking recombination signal sequences (RSS). The resulting coding ends at the side of the gene segments are ligated together forming a coding joint. The intervening DNA is excised and circularized by ligating the blunt DNA signal ends, thereby forming a signal joint. While multiple of these episomal recombination excision circles are formed in every developing B- and T-cell, they seem to have no specific function. They are stable products, but cannot replicate in the cell, do not duplicate during mitosis, and consequently are diluted during cell division, while the corresponding rearrangement on genomic DNA (coding joint) is inherited by all daughter cells (Livak and Schatz, 1996; Breit et al., 1997; van Zelm et al., 2007a). Thus, quantification of coding joints and signal joints from a specific gene rearrangement can be used to study cellular turnover and proliferation (van Zelm et al., 2007a).
Generation of TRECs and KRECs in Lymphoid Cells
When a B- or T-cell has completed a gene rearrangement and starts to proliferate, only one of the daughter cells will contain the corresponding excision circle. Thus, it is not possible to study turnover or proliferation of single cells: each cell either contains a circle or not. In fact, measurement of turnover or proliferation via excision circles is performed at the level of a cell population. By comparing the number of detected signal joints on excision circles to the total number of coding joints in the genome, the average proliferation is established. The condition to detect signal joints at the population level requires the need to select a common rearrangement. However, V(D)J recombination of Ig and TCR loci is optimized to generate a large repertoire of antigen receptors: in each B- and T-cell unique combinations of V, D, and J gene segments are formed. Therefore, most rearrangements are formed to generate a functional B- or T-cell receptor and will consequently not be useful as targets to study proliferation.
Fortunately, most B- and T-cells initiate a single-step gene rearrangement to render a specific locus non-functional. This concerns the δREC–ψJα rearrangement that deletes the entire TCRD locus in T-cells and the intronRSS–Kde rearrangement that deletes the constant region of IGK in B-cells (Figure 2). The δREC–ψJα rearrangement is initiated following unsuccessful generation of a γδ TcR and is estimated to have occurred on 70–80% of TCRD alleles in mature αβ+ T-cells (Verschuren et al., 1997). Nearly all newly formed αβ+ T-cells will have a T-cell receptor excision circle (TREC) with the δREC–ψJα signal joint (Figure 2A). However, the coding joint of this rearrangement is not stably present in the genome. As the TCRD locus is located within the TCRA locus, subsequent Vα–Jα rearrangements will remove the δREC–ψJα coding joint from the locus and place it on a novel circular excision product (Breit et al., 1997; Hazenberg et al., 2000).
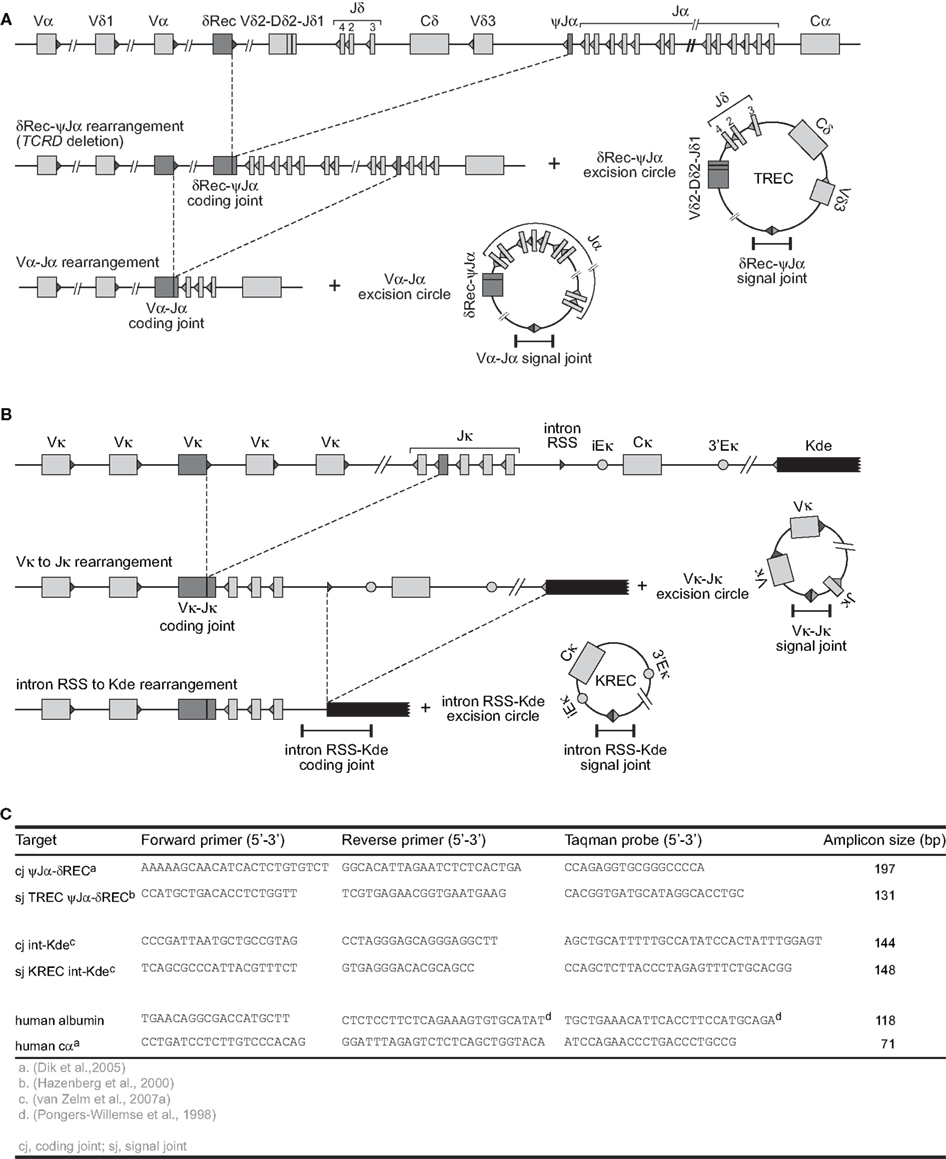
Figure 2. V(D)J recombination and the formation of excision circles. (A) Sequential rearrangements in the TCRAD locus. Following V(D)J recombination of TCRD, the whole locus is then preferentially deleted by δREC–ψJα rearrangements in the αβ+ T-cell lineage. This gives rise to a δREC–ψJα signal joint on an excision circle (TREC) and a δREC–ψJα coding joint in the genome. The coding joint is deleted from the genome by TCRA (Vα-Jα) rearrangements and is then located on a TREC as well. (B) V(D)J recombination on the IGK locus results in a VJκ coding joint. Subsequent rearrangement between the intronRSS and the Kde elements can render the IGK allele non-functional by deleting the Cκ exons and the enhancers. Consequently, the coding joint precludes any further rearrangements in the IGK locus and therefore remains present in the genome, whereas an intronRSS–Kde signal joint is formed on an excision circle (KREC). (C) Oligonucleotide characteristics for TREC, KREC, and control gene quantification (Pongers-Willemse et al., 1998; Hazenberg et al., 2000; Dik et al., 2005; van Zelm et al., 2007a).
In contrast, the intronRSS–Kde rearrangement does generate a stable coding joint in the IGK locus of B-cells and cannot be removed by subsequent gene rearrangements, because all enhancers are removed from the allele (Figure 2B). Moreover, this rearrangement is one of the last to occur on 35–40% of IGK alleles prior to migration of B-cells to the periphery and is not followed by proliferation in bone marrow (van Zelm et al., 2005). Thus, most B-cells will have a kappa-deletion recombination excision circle (KREC) with the intronRSS–Kde signal joint. The coding joint to signal joint ratio of this rearrangement can be used to quantify B-cell proliferation in secondary lymphoid organs (van Zelm et al., 2007a). In fact, the intronRSS–Kde fulfills all criteria to be a robust target for replication history studies. (a) it is a frequently occurring gene rearrangement; (b) it is one of the last Ig gene rearrangements in bone marrow–derived B cells before obtaining a functional Ig molecule, ensuring that the corresponding KRECs are abundantly present in naive B lymphocytes; (c) it is a single-step rearrangement, which allows easy design of RQ-PCR primers and probes for accurate detection of the coding joints and signal joints; and (d) it is an end-stage rearrangement, precluding further rearrangements.
The δREC–ψJα rearrangement fulfills all but the last criterion. Therefore, the δREC–ψJα TRECs cannot be straightforwardly used to quantify T-cell proliferation. Still, TREC quantification is suitable for many applications without need for the corresponding coding joint.
How to Quantify and Interpret Recombination Excision Circles
V(D)J recombination excision circles are formed by blunt end ligation of signal ends after RAG cleavage. Thus, a signal joint is formed between two pieces of DNA that were not in close proximity before and is an ideal target for a specific PCR for excision circle detection. δREC–ψJα TRECs can be reliably detected with TaqMan-based real-time quantitative (RQ-) PCR (Figure 2; Table 1). For exact quantification, several additional steps need to be undertaken. First, a standard curve for quantification is required. This is commonly done on serial dilutions of a signal joint construct cloned in a bacterial plasmid. Thus, the exact number of TRECs can be quantified for a given DNA sample (TRECs/μg DNA). Furthermore, the number of TRECs per 106 cells can be estimated based on the theoretical recovery of 1 μg DNA from approximately 150,000 cells (Hazenberg et al., 2002; Bains et al., 2009). Alternatively, more accurate TREC contents can be obtained when quantification is performed relative to a control gene, such as albumin or the TCRA constant region (Hazenberg et al., 2000; Zubakov et al., 2010). This would require a parallel RQ-PCR on the same sample and quantification using a dilution series of a bacterial plasmid containing the target gene. Because such a control gene is biallelically present in the genome, the number of TRECs per 106 cells can be calculated as follows (Sottini et al., 2010):

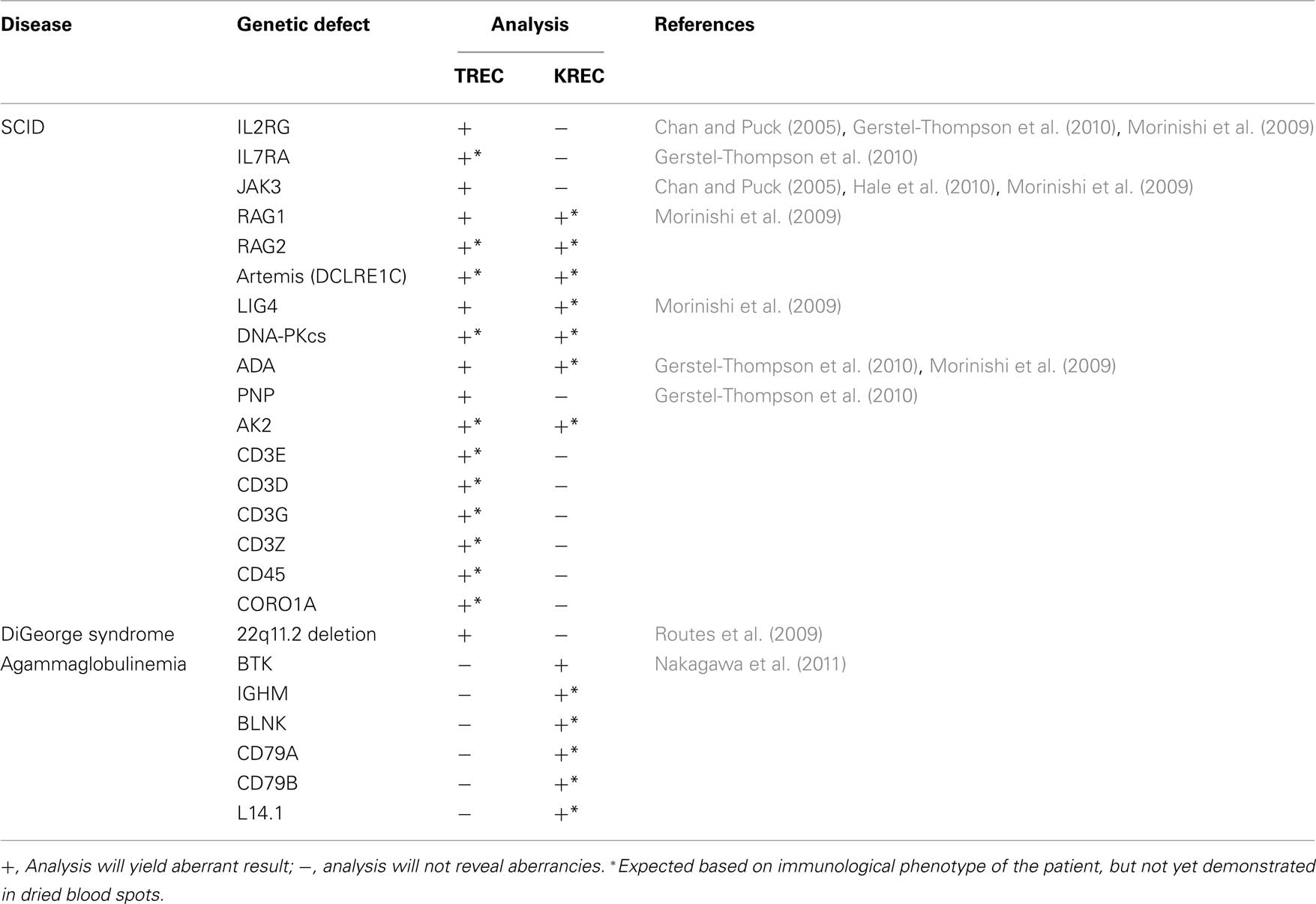
Table 1. Application of TREC and/or KREC analysis in newborn screening to identify immunodeficient patients with various types of genetic defects that result in extremely low total T- and/or B-cell numbers.
δREC–ψJα rearrangements are formed in nearly all αβ+ T-cells and subsequently, most thymic emigrants contain δREC–ψJα TRECs. Since TRECs cannot replicate, the TREC content is diluted when T-cells undergo homeostatic or antigen-induced proliferation. Still, quantification of mature T-cell proliferation is complicated, since the exact frequency of TRECs per cell of new thymic emigrants is unknown. Unfortunately, the δREC–ψJα coding joint cannot be used as marker, since it is also removed from the genome by subsequent Vα–Jα rearrangements (Figure 2). Furthermore, TREC measurements do not allow direct quantitative statements on thymic output. TRECs are highly abundant in recent thymic emigrants, but they are stable structures and they can remain present in “old” thymic emigrants (Verschuren et al., 1997; Douek et al., 1998; Hazenberg et al., 2000, 2001; Sodora et al., 2000). Therefore TRECs are not by definition markers of recent thymic emigrants.
Recently, we have introduced the recombination excision circle of the intronRSS–Kde rearrangement (KREC) to study the replication history of B-cells (van Zelm et al., 2007a). Since the intronRSS–Kde rearrangement is one of the last rearrangements to occur in bone marrow and the coding joint remains stably present in the genome, it allowed us to reliably quantify cell divisions. To further enable straightforward quantification of proliferation, we created a cell line that contains one intronRSS–Kde coding joint and one signal joint per genome. DNA from this cell line, U698-DB01, can be used to correct for differences in signal joint and coding joint PCR efficiencies. When PCRs are performed on similar DNA quantities of both the cell line and the samples of interest, the average number of cell divisions of the sample can be quantified as follows:
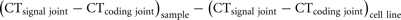
Furthermore, the frequency of B-cells that contain an intronRSS–Kde coding joint can be determined when a control gene is quantified in both the sample and the control cell line. The frequency of IGK alleles contain the intronRSS–Kde coding joint can be calculated by:
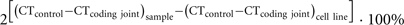
The above described approaches for quantification of excision circles are very important to take into account for correct interpretation of the obtained data. Still, in a few settings, straightforward quantification of excision circles with respect to a control gene can be sufficient to address a specific question. First, since TRECs and KRECs are specifically formed in developing lymphocytes, they can be used to determine the presence of T- and B-cells, respectively. This has been exploited in recent years to develop newborn screening tests for SCID and agammaglobulinemia (see below). Furthermore, very recent studies demonstrated a linear correlation between the frequency of TRECs in blood with human individual age (Zubakov et al., 2010). These analyses only require the difference in CT value of the TREC signal joint PCR as compared with the CT value of the control gene PCR (Zubakov et al., 2010). The strong linear decline in TRECs with age is very intriguing. Since TRECs were determined from full blood, the decrease is likely to result from both a decrease in blood T-cell numbers in young children and increased proliferation of mature T-cells in later childhood and adulthood (Comans-Bitter et al., 1997). The TREC decline in an aging immune system in normal individuals can be used as a reference for studies on premature or early immunosenescence under particular disease conditions (Betjes et al., 2011). Interestingly, the decline in KRECs was much less pronounced (Menno C. van Zelm, unpublished observations). A potential explanation is the more stable generation of new B-cells from bone marrow in older individuals as compared with new T-cell from thymus. Thus, the KREC content remains much higher since the dilution from additional proliferation is limited.
Newborn Screening of SCID and Agammaglobulinemia
Early diagnosis is critical for patients suffering from PID to prevent irreversible organ damage or early death. This is especially true for SCID patients, in whom infections become quickly fatal in the first months of life. Ideally, patients should be identified in a newborn screening protocol prior to developing disease symptoms to allow successful stem cell replacement therapy (Buckley, 2004; Puck, 2007). This would require a straightforward screening method that can be applied on dried heel-stick blood spots on Guthrie cards from neonates.
In 2005, Chan and Puck (2005) introduced a newborn screening method based on RQ-PCR detection of δREC–ψJα TRECs. This method proved to be very reliable in detecting T-cells in healthy neonates. Furthermore, TRECs were consistently very low to absent in blood from SCID patients suffering from various gene defects Table 1; Chan and Puck, 2005; Morinishi et al., 2009; Routes et al., 2009; Gerstel-Thompson et al., 2010; Hale et al., 2010). Based on these promising results, state-wide newborn screening is implemented in Wisconsin and assay validation has started in Massachusetts (Routes et al., 2009; Gerstel-Thompson et al., 2010).
Besides the easy detection with RQ-PCR and the specific generation of TRECs in developing T-cells, the success of SCID newborn screening with TRECs benefited from the additional phenomena of TREC dilution in adults. Young children have high TREC numbers, which provide a good signal to noise ratio. Furthermore, the presence of maternal T-cells or residual T-cells in “leaky” SCID patients thus hardly affects TREC numbers due to their extensive replication history.
Analogous to SCID, efforts have been initiated to set-up KREC-based screening for B-cell maturation defects (Nakagawa et al., 2011). These initial results revealed the presence of >200 KREC copies/μg DNA in healthy children, whereas no KRECs were detected in 30 XLA patients and 5 non-BTK agammaglobulinemia patients.
Recent Insights and New Directions for Analysis of TRECs and KRECs in PID
Hematopoietic Stem Cell Treatment Evaluation
The only corrective treatment for patients with PID used to be hematopoietic stem cell replacement therapies. Stem cell replacement therapy is the first treatment choice for patients with a severe T-cell defect. Over the past decades, enormous advances have been made with stem cell transplantation and the success rate is very high in young patients and with good donor matches. Still, the limited availability of properly matched donors requires the need for alternatives. Supported by the advances in molecular diagnostics of PID, gene therapy protocols have been developed, in which the patients’ own hematopoietic stem cells are corrected with a wild type copy of the defective gene (“gene therapy”). Despite setbacks due to adverse insertional mutagenesis (Hacein-Bey-Abina et al., 2003), many gene therapy protocols have been restarted in the last years (Aiuti et al., 2009).
Quantification of δREC–ψJα TRECs has been informative for assessment of thymic T-cell neogenesis (Douek et al., 1998; Hazenberg et al., 2000). Initial insights have been obtained from HIV patients, in whom a low TREC content was demonstrated that could be reverted upon antiretroviral treatment (Douek et al., 1998; Lewin et al., 2002; Di Mascio et al., 2006). Furthermore, low TREC levels following hematopoietic stem cell transplantation have been shown to correlate with clinical morbidity and mortality from infection (Douek et al., 2000; Hazenberg et al., 2002; Lewin et al., 2002; Brown et al., 2010).
Recently, B- and T-cell recovery of PID patients following stem cell transplantation have been studied in a multiplex setting for KREC and TREC detection (Sottini et al., 2010). Such evaluations can be very informative to obtain early insight into B- and T-cell neogenesis, and a valuable addition to flow cytometric and donor/acceptor chimerism analysis.
Classification of CVID Patients
Despite recent advances in genetics of PID, in many PID patients no molecular diagnosis is made. The number of patients with a humoral immunodeficiency without molecular diagnosis is especially high. Most of these patients are diagnosed as having common variable immunodeficiency (CVID). As apparent from the name, these patients have a highly diverse clinical phenotype, likely in part resulting from genetic variation. In the past decade the “Freiburg” and “Paris” CVID classifications have been developed based on the composition of the peripheral B-cell compartment (Warnatz et al., 2002; Piqueras et al., 2003). Several patient subgroups display distinct abnormalities in B-cell subsets of which the cause remains unknown. Quantification of the replication history (with KRECs) of sorted B-cell subsets on top of flow cytometric analysis could help to explain aberrancies in the B-cell compartments of these patients. In absence of a molecular analysis, such immunological parameters will be important to define subgroups of patients and prediction of future complications.
Studies on Normal and Aberrant Lymphoid Maturation
Memory B cells in blood have been classically divided into CD27+IgD+ and CD27+IgD− subsets. The presence of CD27+IgD+ cells in patients with CD40L deficiency who lack functional germinal centers has lead to the conclusion that these can be generated independently of T cell help (Agematsu et al., 1998; Weller et al., 2001, 2004). Furthermore, they strongly resemble splenic marginal zone B cells (Weller et al., 2004). However, these conclusions have been challenged by the facts that natural effector cells showed molecular footprints of the GC reaction and that similar IGH gene rearrangements were found in natural effector B cells and in CD27+IgG+ B cells (Seifert and Kuppers, 2009). Furthermore, the absolute numbers of CD27+IgD+ B cells are clearly reduced in CD40L-deficient patients (Agematsu et al., 1998; Weller et al., 2001, 2004). Thus, this B-cell subset in healthy individuals is likely to be a mixed population of GC-derived and splenic marginal zone-derived “natural effector” memory B cells. By using the intronRSS–Kde coding joint / signal joint ratio, we quantified homeostatic and antigen-induced B-cell proliferation in purified B-cell subsets of healthy individuals (van Zelm et al., 2007a,b). The replication history of CD27+IgD+ B-cells was consistently lower than CD27+IgD− B-cells, both in childhood tonsil (4 vs 8 cell cycles) and in adult blood (6 vs 11 cell cycles; van Zelm et al., 2007a). Interestingly, germinal center B cells from childhood tonsil already had a replication history of eight cell cycles (van Zelm et al., 2007a). The replication history of the total subset of CD27+IgD+ B cells was clearly less than B cells undergoing a T-cell dependent response in the germinal center and supports the idea that at least part of the CD27+IgD+ B cells are derived from T-cell dependent responses (Weller et al., 2001). Thus, the use of KRECs to quantify the replication history can be of added value to identify the origin and maturation pathways of memory B cells. This is especially interesting, since CD27+IgD− memory B cells are in fact a mixture of IgM-only, IgG+ and IgA+ B cells. Furthermore, recent studies have identified CD27−IgA+ and CD27−IgG+ memory B cells (Fecteau et al., 2006; Wei et al., 2007; Cagigi et al., 2009). Quantification of their replication history in combination with additional molecular and phenotypic analysis could help to delineate their origin and maturation pathways.
The replication history of normal B-cell subsets has provided a template for analysis of the aberrant population in B-cells of patients with immunological disease (Moir et al., 2008; Rakhmanov et al., 2009; van der Burg et al., 2010). Interestingly, both in patients suffering from HIV and in a subgroup of CVID patients, an expanded CD21low B-cell population has been observed (Warnatz et al., 2002; Moir et al., 2008). These cells showed a replication history of ∼4 cell cycles, which was increased as compared with naive mature B-cells (two divisions), but decreased as compared with memory B-cells (6–10 cell cycles). It is currently debated whether these cells are naive anergic B-cells or tissue-like memory-B cells (Moir et al., 2008; Isnardi et al., 2010; Rakhmanov et al., 2009). Further analysis of CD27 and Ig isotype expression could help to resolve this discussion.
Recently, we analyzed the replication history of B-cells in patients with Nijmegen breakage syndrome (NBS; repair syndrome; van der Burg et al., 2010). NBS patients have a DNA repair defect and reduced B- and T-cell numbers resulting from inefficient V(D)J recombination. Still, both coding joints and signal joints can be robustly formed in absence of functional NBN protein and show normal characteristics (Saidi et al., 2010; van der Burg et al., 2010). Interestingly, the replication history of NBS patients’ naive mature B-cells was ∼4 cell cycles, which was similar to the CD21low B-cells in HIV and CVID patients that were described above. The increased proliferation in NBS most likely resulted from decreased output of new B-cell emigrants from bone marrow and increased homeostatic proliferation to compensate the total peripheral B-cell number (van der Burg et al., 2010). The decreased KREC levels thus reflect a form of premature senescence of the immune system in NBS patients.
The above described observations clearly illustrate how KRECs can be used to support studies on the B-cell compartment of patients suffering from a PID. Unfortunately, analogous analysis of the T-cell compartment is hampered by the lack of a good internal control for quantification with δREC–ψJα TRECs. The corresponding coding joint is removed from the genome by subsequent Vα–Jα rearrangements. Therefore, it remains unclear how many TRECs are formed in thymus. Furthermore, following δREC–ψJα rearrangements, thymocytes undergo several rounds of proliferation. Still, these disadvantages as compared to the B-cell compartment can be overcome. First, the frequency of δREC–ψJα coding joints and signal joints can be quantified in thymocyte subsets and the proliferation in thymus can be calculated for CD4+ and CD8+ single positive thymocytes. Based on these data, the TREC frequencies in peripheral T-cell subsets can be corrected to calculate peripheral T-cell proliferation (K. van der Weerd et al. submitted for publication). Additionally, TRECs from other TCR rearrangements can be used to calculate T-cell proliferation. These would have to be single-step rearrangements, relatively frequent, and the coding joint should remain stably present in the genome. We are currently evaluating the potential of such TRECs in other TCR loci.
Newborn Screening for SCID and Agammaglobulinemia
With the introduction of TRECs to detect patients with SCID, all major criteria were met for inclusion in newborn screening protocols. The estimated incidence of SCID is >1:100,000 live births (Chan and Puck, 2005) and life-saving therapies exist. Furthermore, it has become clear that early diagnosis and early treatment initiation increases successful outcome rates. Using RQ-PCR techniques, the absence of blood T-cells can be robustly determined from DNA of dried blood spots. It should be possible to detect all SCID patients, independently of the underlying genetic defect. This is supported by multiple studies in which patients with various identified and unidentified genetic defects have been detected (Table 1).
The successful detection of agammaglobulinemia patients with KRECs now opens the way to inclusion of this technique for detection of agammaglobulinemia in newborn screening. The clinical phenotype of agammaglobulinemia is milder than SCID. Still, the disease is underdiagnosed in the first years of life and patients can develop serious organ damage before a diagnosis of immunodeficiency has been made. Thus, early identification by newborn screening can contribute to quality of life and reduction of health care costs.
Finally, RQ-PCR-based newborn screening has the advantage that tests can be combined in a multiplex setting. This has been successfully tested for detection of TRECs and an internal control gene (Gerstel-Thompson et al., 2010). Theoretically, it would be possible to also include KREC detection in this assay. Thus, a single assay could be developed to screen for both agammaglobulinemia and SCID with a combined estimated incidence of 1:30,000–1:50,000 births. Furthermore, T-cell deficient cases can be further separated in B-cell positive and B-cell negative subgroups, guiding additional diagnostics toward either T−B+ or T−B− SCID (Table 1). Consequently, such a combined test should keep the costs low, while increasing the benefits with regard to disease incidence and guiding of further diagnostics.
In conclusion, we here provide an overview of how V(D)J recombination excision circles can be used to obtain insights into the normal and aberrant lymphoid immune system. We are convinced that KREC and TREC analysis in combination with flow cytometric evaluation of T- and B-cell subpopulations of PID patients will provide new insights into their disease and may contribute to the identification of new genetic defects. Especially, combined quantification of TRECs and KRECs could support early diagnosis in newborn screening protocols, patient subgroup classification and evaluation of stem cell replacement therapy.
Conflict of Interest Statement
Jacques J. M. van Dongen is inventor of the KREC assay, which has been patented (PCT/NL 2005/000761; priority date 25 Oct 2004) and licensed to InVivoScribe Technologies, San Diego, CA; revenues of the patent go to Erasmus MC. The other authors have declared that no conflict of interest exists.
Acknowledgments
We thank Dr. K. Imai (National Defense Medical College, Saitama, Japan), and K. van der Weerd (Erasmus MC, Rotterdam, Netherlands) for sharing unpublished data, and Mrs. S. de Bruin-Versteeg for assistance in preparing the figures. Menno C. van Zelm is supported by fellowships from the Erasmus University Rotterdam (EUR-Fellowship) and the Erasmus MC, and by Veni grant 916.110.90 from ZonMW/NWO. Mirjam van der Burg is supported by a grant from the foundation “Sophia Kinderziekenhuis Fonds” (SKF; grant 589).
References
Agematsu, K., Nagumo, H., Shinozaki, K., Hokibara, S., Yasui, K., Terada, K., Kawamura, N., Toba, T., Nonoyama, S., Ochs, H. D., and Komiyama, A. (1998). Absence of IgD−CD27(+) memory B cell population in X-linked hyper-IgM syndrome. J. Clin. Invest. 102, 853–860.
Aiuti, A., Cattaneo, F., Galimberti, S., Benninghoff, U., Cassani, B., Callegaro, L., Scaramuzza, S., Andolfi, G., Mirolo, M., Brigida, I., Tabucchi, A., Carlucci, F., Eibl, M., Aker, M., Slavin, S., Al-Mousa, H., Al Ghonaium, A., Ferster, A., Duppenthaler, A., Notarangelo, L., Wintergerst, U., Buckley, R. H., Bregni, M., Marktel, S., Valsecchi, M. G., Rossi, P., Ciceri, F., Miniero, R., Bordignon, C., and Roncarolo, M. G. (2009). Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N. Engl. J. Med. 360, 447–458.
Bains, I., Thiebaut, R., Yates, A. J., and Callard, R. (2009). Quantifying thymic export: combining models of naive T cell proliferation and TCR excision circle dynamics gives an explicit measure of thymic output. J. Immunol. 183, 4329–4336.
Betjes, M. G. H., Langerak, A. W., Van der Spek, A., De Wit, L. E. A., and Litjens, N. H. R. (2011). Excessive premature ageing of circulating T cells in ESRD patients. Kidney Int. doi: 10.1038/ki.2011.110. [Epub ahead ofprint].
Breit, T. M., Verschuren, M. C., Wolvers-Tettero, I. L., Van Gastel-Mol, E. J., Hahlen, K., and van Dongen, J. J. (1997). Human T cell leukemias with continuous V(D)J recombinase activity for TCR-delta gene deletion. J. Immunol. 159, 4341–4349.
Brown, J. A., Stevenson, K., Kim, H. T., Cutler, C., Ballen, K., McDonough, S., Reynolds, C., Herrera, M., Liney, D., Ho, V., Kao, G., Armand, P., Koreth, J., Alyea, E., McAfee, S., Attar, E., Dey, B., Spitzer, T., Soiffer, R., Ritz, J., Antin, J. H., and Boussiotis, V. A. (2010). Clearance of CMV viremia and survival after double umbilical cord blood transplantation in adults depends on reconstitution of thymopoiesis. Blood 115, 4111–4119.
Buckley, R. H. (2004). Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution. Annu. Rev. Immunol. 22, 625–655.
Cagigi, A., Du, L., Dang, L. V., Grutzmeier, S., Atlas, A., Chiodi, F., Pan-Hammarstrom, Q., and Nilsson, A. (2009). CD27(−) B-cells produce class switched and somatically hyper-mutated antibodies during chronic HIV-1 infection. PLoS ONE 4, e5427. doi: 10.1371/journal.pone.0005427
Chan, K., and Puck, J. M. (2005). Development of population-based newborn screening for severe combined immunodeficiency. J. Allergy Clin. Immunol. 115, 391–398.
Comans-Bitter, W. M., de Groot, R., van den Beemd, R., Neijens, H. J., Hop, W. C., Groeneveld, K., Hooijkaas, H., and van Dongen, J. J. (1997). Immunophenotyping of blood lymphocytes in childhood. Reference values for lymphocyte subpopulations. J. Pediatr. 130, 388–393.
Conley, M. E., Dobbs, A. K., Farmer, D. M., Kilic, S., Paris, K., Grigoriadou, S., Coustan-Smith, E., Howard, V., and Campana, D. (2009). Primary B cell immunodeficiencies: comparisons and contrasts. Annu. Rev. Immunol. 27, 199–227.
Di Mascio, M., Sereti, I., Matthews, L. T., Natarajan, V., Adelsberger, J., Lempicki, R., Yoder, C., Jones, E., Chow, C., Metcalf, J. A., Sidorov, I. A., Dimitrov, D. S., Polis, M. A., and Kovacs, J. A. (2006). Naive T-cell dynamics in human immunodeficiency virus type 1 infection: effects of highly active antiretroviral therapy provide insights into the mechanisms of naive T-cell depletion. J. Virol. 80, 2665–2674.
Dik, W. A., Pike-Overzet, K., Weerkamp, F., de Ridder, D., de Haas, E. F., Baert, M. R., van der Spek, P., Koster, E. E., Reinders, M. J., van Dongen, J. J., Langerak, A. W., and Staal, F. J. (2005). New insights on human T cell development by quantitative T cell receptor gene rearrangement studies and gene expression profiling. J. Exp. Med. 201, 1715–1723.
Douek, D. C., McFarland, R. D., Keiser, P. H., Gage, E. A., Massey, J. M., Haynes, B. F., Polis, M. A., Haase, A. T., Feinberg, M. B., Sullivan, J. L., Jamieson, B. D., Zack, J. A., Picker, L. J., and Koup, R. A. (1998). Changes in thymic function with age and during the treatment of HIV infection. Nature 396, 690–695.
Douek, D. C., Vescio, R. A., Betts, M. R., Brenchley, J. M., Hill, B. J., Zhang, L., Berenson, J. R., Collins, R. H., and Koup, R. A. (2000). Assessment of thymic output in adults after haematopoietic stem-cell transplantation and prediction of T-cell reconstitution. Lancet 355, 1875–1881.
Fecteau, J. F., Cote, G., and Neron, S. (2006). A new memory CD27−IgG+ B cell population in peripheral blood expressing VH genes with low frequency of somatic mutation. J. Immunol. 177, 3728–3736.
Gerstel-Thompson, J. L., Wilkey, J. F., Baptiste, J. C., Navas, J. S., Pai, S. Y., Pass, K. A., Eaton, R. B., and Comeau, A. M. (2010). High-throughput multiplexed T-cell-receptor excision circle quantitative PCR assay with internal controls for detection of severe combined immunodeficiency in population-based newborn screening. Clin. Chem. 56, 1466–1474.
Hacein-Bey-Abina, S., Von Kalle, C., Schmidt, M., McCormack, M. P., Wulffraat, N., Leboulch, P., Lim, A., Osborne, C. S., Pawliuk, R., Morillon, E., Sorensen, R., Forster, A., Fraser, P., Cohen, J. I., de Saint Basile, G., Alexander, I., Wintergerst, U., Frebourg, T., Aurias, A., Stoppa-Lyonnet, D., Romana, S., Radford-Weiss, I., Gross, F., Valensi, F., Delabesse, E., Macintyre, E., Sigaux, F., Soulier, J., Leiva, L. E., Wissler, M., Prinz, C., Rabbitts, T. H., Le Deist, F., Fischer, A., and Cavazzana-Calvo, M. (2003). LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 302, 415–419.
Hale, J. E., Bonilla, F. A., Pai, S. Y., Gerstel-Thompson, J. L., Notarangelo, L. D., Eaton, R. B., and Comeau, A. M. (2010). Identification of an infant with severe combined immunodeficiency by newborn screening. J. Allergy Clin. Immunol. 126, 1073–1074.
Hazenberg, M. D., Otto, S. A., Cohen Stuart, J. W., Verschuren, M. C., Borleffs, J. C., Boucher, C. A., Coutinho, R. A., Lange, J. M., Rinke de Wit, T. F., Tsegaye, A., van Dongen, J. J., Hamann, D., de Boer, R. J., and Miedema, F. (2000). Increased cell division but not thymic dysfunction rapidly affects the T-cell receptor excision circle content of the naive T cell population in HIV-1 infection. Nat. Med. 6, 1036–1042.
Hazenberg, M. D., Otto, S. A., de Pauw, E. S., Roelofs, H., Fibbe, W. E., Hamann, D., and Miedema, F. (2002). T-cell receptor excision circle and T-cell dynamics after allogeneic stem cell transplantation are related to clinical events. Blood 99, 3449–3453.
Hazenberg, M. D., Verschuren, M. C., Hamann, D., Miedema, F., and van Dongen, J. J. (2001). T cell receptor excision circles as markers for recent thymic emigrants: basic aspects, technical approach, and guidelines for interpretation. J. Mol. Med. 79, 631–640.
IJspeert, H., Lankester, A. C., van den Berg, J. M., Wiegant, W., van Zelm, M. C., Weemaes, C. M., Warris, A., Pan-Hammarstrom, Q., Pastink, A., van Tol, M. J., van Dongen, J. J., van Gent, D. C., and van der Burg, M. (2011). Artemis splice defects cause atypical SCID and can be restored in vitro by an antisense oligonucleotide. Genes Immun. doi: 10.1038/gene.2011.16.
Isnardi, I., Ng, Y. S., Menard, L., Meyers, G., Saadoun, D., Srdanovic, I., Samuels, J., Berman, J., Buckner, J. H., Cunningham-Rundles, C., and Meffre, E. (2010). Complement receptor 2/CD21− human naive B cells contain mostly autoreactive unresponsive clones. Blood 115, 5026–5036.
Lewin, S. R., Ribeiro, R. M., Kaufmann, G. R., Smith, D., Zaunders, J., Law, M., Solomon, A., Cameron, P. U., Cooper, D., and Perelson, A. S. (2002). Dynamics of T cells and TCR excision circles differ after treatment of acute and chronic HIV infection. J. Immunol. 169, 4657–4666.
Livak, F., and Schatz, D. G. (1996). T-cell receptor alpha locus V(D)J recombination by-products are abundant in thymocytes and mature T cells. Mol. Cell. Biol. 16, 609–618.
Lucas, M., Lee, M., Lortan, J., Lopez-Granados, E., Misbah, S., and Chapel, H. (2010). Infection outcomes in patients with common variable immunodeficiency disorders: relationship to immunoglobulin therapy over 22 years. J. Allergy Clin. Immunol. 125, 1354–1360 e1354.
Moir, S., Ho, J., Malaspina, A., Wang, W., DiPoto, A. C., O’Shea, M. A., Roby, G., Kottilil, S., Arthos, J., Proschan, M. A., Chun, T. W., and Fauci, A. S. (2008). Evidence for HIV-associated B cell exhaustion in a dysfunctional memory B cell compartment in HIV-infected viremic individuals. J. Exp. Med. 205, 1797–1805.
Morinishi, Y., Imai, K., Nakagawa, N., Sato, H., Horiuchi, K., Ohtsuka, Y., Kaneda, Y., Taga, T., Hisakawa, H., Miyaji, R., Endo, M., Oh-Ishi, T., Kamachi, Y., Akahane, K., Kobayashi, C., Tsuchida, M., Morio, T., Sasahara, Y., Kumaki, S., Ishigaki, K., Yoshida, M., Urabe, T., Kobayashi, N., Okimoto, Y., Reichenbach, J., Hashii, Y., Tsuji, Y., Kogawa, K., Yamaguchi, S., Kanegane, H., Miyawaki, T., Yamada, M., Ariga, T., and Nonoyama, S. (2009). Identification of severe combined immunodeficiency by T-cell receptor excision circles quantification using neonatal guthrie cards. J. Pediatr. 155, 829–833.
Nakagawa, N., Imai, K., Kanegane, H., Sato, H., Yamada, M., Kondoh, K., Okada, S., Kobayashi, M., Agematsu, K., Takada, H., Mitsuiki, N., Oshima, K., Ohara, O., Suri, D., Rawat, A., Singh, S., Pan-Hammarstrom, Q., Hammarstrom, L., Reichenbach, J., Seger, R., Ariga, T., Hara, T., Miyawaki, T., and Nonoyama, S. (2011). Quantification of kappa-deleting recombination excision circles in Guthrie cards for the identification of early B-cell maturation defects. J. Allergy Clin. Immunol. doi: 10.1016/J.Jaci.2011.01.052. [Epub ahead of print].
Notarangelo, L. D., Fischer, A., Geha, R. S., Casanova, J. L., Chapel, H., Conley, M. E., Cunningham-Rundles, C., Etzioni, A., Hammartrom, L., Nonoyama, S., Ochs, H. D., Puck, J., Roifman, C., Seger, R., and Wedgwood, J. (2009). Primary immunodeficiencies: 2009 update. J. Allergy Clin. Immunol. 124, 1161–1178.
Piqueras, B., Lavenu-Bombled, C., Galicier, L., Bergeron-van der Cruyssen, F., Mouthon, L., Chevret, S., Debre, P., Schmitt, C., and Oksenhendler, E. (2003). Common variable immunodeficiency patient classification based on impaired B cell memory differentiation correlates with clinical aspects. J. Clin. Immunol. 23, 385–400.
Pongers-Willemse, M. J., Verhagen, O. J., Tibbe, G. J., Wijkhuijs, A. J., de Haas, V., Roovers, E., van der Schoot, C. E., and van Dongen, J. J. (1998). Real-time quantitative PCR for the detection of minimal residual disease in acute lymphoblastic leukemia using junctional region specific TaqMan probes. Leukemia 12, 2006–2014.
Puck, J. M. (2007). Neonatal screening for severe combined immune deficiency. Curr. Opin. Allergy Clin. Immunol. 7, 522–527.
Quinti, I., Soresina, A., Spadaro, G., Martino, S., Donnanno, S., Agostini, C., Claudio, P., Franco, D., Maria Pesce, A., Borghese, F., Guerra, A., Rondelli, R., Plebani, A., and Italian Primary Immunodeficiency, N. (2007). Long-term follow-up and outcome of a large cohort of patients with common variable immunodeficiency. J. Clin. Immunol. 27, 308–316.
Rakhmanov, M., Keller, B., Gutenberger, S., Foerster, C., Hoenig, M., Driessen, G., van der Burg, M., van Dongen, J. J., Wiech, E., Visentini, M., Quinti, I., Prasse, A., Voelxen, N., Salzer, U., Goldacker, S., Fisch, P., Eibel, H., Schwarz, K., Peter, H. H., and Warnatz, K. (2009). Circulating CD21low B cells in common variable immunodeficiency resemble tissue homing, innate-like B cells. Proc. Natl. Acad. Sci. U.S.A. 106, 13451–13456.
Routes, J. M., Grossman, W. J., Verbsky, J., Laessig, R. H., Hoffman, G. L., Brokopp, C. D., and Baker, M. W. (2009). Statewide newborn screening for severe T-cell lymphopenia. JAMA 302, 2465–2470.
Saidi, A., Li, T., Weih, F., Concannon, P., and Wang, Z. Q. (2010). Dual functions of Nbs1 in the repair of DNA breaks and proliferation ensure proper V(D)J recombination and T-cell development. Mol. Cell. Biol. 30, 5572–5581.
Schuetz, C., Huck, K., Gudowius, S., Megahed, M., Feyen, O., Hubner, B., Schneider, D. T., Manfras, B., Pannicke, U., Willemze, R., Knuchel, R., Gobel, U., Schulz, A., Borkhardt, A., Friedrich, W., Schwarz, K., and Niehues, T. (2008). An immunodeficiency disease with RAG mutations and granulomas. N. Engl. J. Med. 358, 2030–2038.
Seifert, M., and Kuppers, R. (2009). Molecular footprints of a germinal center derivation of human IgM+ (IgD+)CD27+ B cells and the dynamics of memory B cell generation. J. Exp. Med. 206, 2659–2669.
Sodora, D. L., Douek, D. C., Silvestri, G., Montgomery, L., Rosenzweig, M., Igarashi, T., Bernacky, B., Johnson, R. P., Feinberg, M. B., Martin, M. A., and Koup, R. A. (2000). Quantification of thymic function by measuring T cell receptor excision circles within peripheral blood and lymphoid tissues in monkeys. Eur. J. Immunol. 30, 1145–1153.
Sottini, A., Ghidini, C., Zanotti, C., Chiarini, M., Caimi, L., Lanfranchi, A., Moratto, D., Porta, F., and Imberti, L. (2010). Simultaneous quantification of recent thymic T-cell and bone marrow B-cell emigrants in patients with primary immunodeficiency undergone to stem cell transplantation. Clin. Immunol. 136, 217–227.
van der Burg, M., Pac, M., Berkowska, M. A., Goryluk-Kozakiewicz, B., Wakulinska, A., Dembowska-Baginska, B., Gregorek, H., Barendregt, B. H., Krajewska-Walasek, M., Bernatowska, E., van Dongen, J. J., Chrzanowska, K. H., and Langerak, A. W. (2010). Loss of juxtaposition of RAG-induced immunoglobulin DNA ends is implicated in the precursor B-cell differentiation defect in NBS patients. Blood 115, 4770–4777.
van der Burg, M., van Zelm, M. C., Driessen, G. J., and van Dongen, J. J. (2011). Dissection of B-cell development to unravel defects in patients with a primary antibody deficiency. Adv. Exp. Med. Biol. 697, 183–196.
van Zelm, M. C., Szczepanski, T., van der Burg, M., and van Dongen, J. J. (2007a). Replication history of B lymphocytes reveals homeostatic proliferation and extensive antigen-induced B cell expansion. J. Exp. Med. 204, 645–655.
van Zelm, M. C., van der Burg, M., and van Dongen, J. J. (2007b). Homeostatic and maturation-associated proliferation in the peripheral B-cell compartment. Cell Cycle 6, 2890–2895.
van Zelm, M. C., van der Burg, M., de Ridder, D., Barendregt, B. H., de Haas, E. F., Reinders, M. J., Lankester, A. C., Revesz, T., Staal, F. J., and van Dongen, J. J. (2005). Ig gene rearrangement steps are initiated in early human precursor B cell subsets and correlate with specific transcription factor expression. J. Immunol. 175, 5912–5922.
Verschuren, M. C., Wolvers-Tettero, I. L., Breit, T. M., Noordzij, J., van Wering, E. R., and van Dongen, J. J. (1997). Preferential rearrangements of the T cell receptor-delta-deleting elements in human T cells. J. Immunol. 158, 1208–1216.
Warnatz, K., Denz, A., Drager, R., Braun, M., Groth, C., Wolff-Vorbeck, G., Eibel, H., Schlesier, M., and Peter, H. H. (2002). Severe deficiency of switched memory B cells (CD27(+)IgM(−)IgD(−)) in subgroups of patients with common variable immunodeficiency: a new approach to classify a heterogeneous disease. Blood 99, 1544–1551.
Wei, C., Anolik, J., Cappione, A., Zheng, B., Pugh-Bernard, A., Brooks, J., Lee, E. H., Milner, E. C., and Sanz, I. (2007). A new population of cells lacking expression of CD27 represents a notable component of the B cell memory compartment in systemic lupus erythematosus. J. Immunol. 178, 6624–6633.
Weller, S., Braun, M. C., Tan, B. K., Rosenwald, A., Cordier, C., Conley, M. E., Plebani, A., Kumararatne, D. S., Bonnet, D., Tournilhac, O., Tchernia, G., Steiniger, B., Staudt, L. M., Casanova, J. L., Reynaud, C. A., and Weill, J. C. (2004). Human blood IgM “memory” B cells are circulating splenic marginal zone B cells harboring a prediversified immunoglobulin repertoire. Blood 104, 3647–3654.
Weller, S., Faili, A., Garcia, C., Braun, M. C., Le Deist, F. F., de Saint Basile, G. G., Hermine, O., Fischer, A., Reynaud, C. A., and Weill, J. C. (2001). CD40-CD40L independent Ig gene hypermutation suggests a second B cell diversification pathway in humans. Proc. Natl. Acad. Sci. U.S.A. 98, 1166–1170.
Keywords: V(D)J recombination, KREC, TREC, T-cell, B-cell, primary immunodeficiencies, SCID, argammaglobulinemia
Citation: van Zelm MC, van der Burg M, Langerak AW and van Dongen JJM (2011) PID comes full circle: applications of V(D)J recombination excision circles in research, diagnostics and newborn screening of primary immunodeficiency disorders. Front. Immun. 2:12. doi: 10.3389/fimmu.2011.00012
Received: 04 February 2011; Accepted: 20 April 2011;
Published online: 04 May 2011.
Edited by:
Stuart G. Tangye, Garvan Institute of Medical Research, AustraliaReviewed by:
Klaus Warnatz, University of Freiburg, GermanyStuart G. Tangye, Garvan Institute of Medical Research, Australia
Tim Niehues, HELIOS, Germany
Copyright: © 2011 Zelm, Burg, Langerak and Dongen. This is an open-access article subject to an on-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Menno C. van Zelm, Molecular Immunology Unit, Department of Immunology, Erasmus MC, University Medical Center Rotterdam, Dr. Molewaterplein 50, 3015 GE Rotterdam, Netherlands. e-mail:bS52YW56ZWxtQGVyYXNtdXNtYy5ubA==