



- 1 Centre for Inflammation Research, Centre for Multiple Sclerosis Research and Centre for Immunity, Infection and Evolution, Queen’s Medical Research Institute, University of Edinburgh, Edinburgh, UK
- 2 Inserm, U1043, Toulouse, France
- 3 CNRS, U5282, Toulouse, France
- 4 Centre de Physiopathologie de Toulouse Purpan, Université de Toulouse, Université Paul Sabatier, Toulouse, France
CD4+ T cells have a well-defined pathogenic role in experimental autoimmune encephalomyelitis, the rodent model of multiple sclerosis (MS), yet CD8+ T cells are commonly found in MS lesions. To determine whether immunological tolerance might impact differently on CD4+ versus CD8+ T cells, we studied T cell responses in mice genetically deficient for the central nervous system (CNS) autoantigen myelin oligodendrocyte glycoprotein (MOG) versus wild type (WT) C57BL/6 mice. We show that MOG−/− mice have enhanced sensitivity to immunization with the immunodominant peptide of MOG (35–55), as evidenced by increased expansion of both CD4+ and CD8+ T cell subsets. Most strikingly, CD8+ T cells from MOG−/− mice responded to a novel T cell epitope which binds to MHC class I with high affinity. Despite this, MOG-responsive CD8+ T cells sourced from either WT or MOG−/− mice failed to initiate CNS inflammation upon transfer to MOG-sufficient mice. In our hands, this capacity was only found in CD4+ T cells. However, MOG−/− CD4+ cells did not show greater pathogenic activity than their WT counterparts. Our data indicate that, in the presence of endogenous MOG, CD8+ T cells capable of responding to a MHC class I-restricted epitope that can be stably expressed are subject to rigorous control through central and/or peripheral tolerance.
Introduction
Given their importance in orchestrating immune responses, both quantitatively and qualitatively, it is not surprising that much work has focused on the actions of CD4+ T helper cell responses to myelin autoantigens. The clear importance of HLA class II alleles in genetic susceptibility to multiple sclerosis (MS) supports this (Holmes et al., 2005) as do studies in experimental autoimmune encephalomyelitis (EAE) which have overwhelmingly described this disease being absolutely dependent on the CD4+ T cell compartment (Zamvil et al., 1985; Flugel et al., 1999). Furthermore, a series of elegant studies have identified means by which immunological tolerance to MHC class II-restricted T cell epitopes of myelin can fail, providing for the immunodominance of certain encephalitogenic peptide antigens (reviewed in Goverman, 1999; Anderton and Wraith, 2002).
There are some discrepancies to this paradigm, however, suggesting pathogenic roles for CD8+ T cells. CD8+ cells appear at high frequencies within CNS lesions (Babbe et al., 2000) and parenchyma and are also enriched within the CSF of patients with relapsing–remitting MS (Jilek et al., 2007; Malmestrom et al., 2008). Although depleting all lymphocytes with the anti-CD52 mAb Campath 1-H can reduce clinical signs in patients with relapsing–remitting and secondary progressive stages of MS (Coles et al., 2006; Hirst et al., 2008), trials that selectively targeted only CD4+ T cells showed limited benefit (Racadot et al., 1993; Rumbach et al., 1996; van Oosten et al., 1997), suggesting that CD8+ T cells might also be an important target. Others have suggested that transfer of myelin oligodendrocyte glycoprotein (MOG)-reactive CD8+ T cells can also induce EAE (Sun et al., 2003; Ford and Evavold, 2005). The pathogenic contribution of CD8+ T cells to MS and its animal models is therefore of increasing interest (Mars et al., 2011).
An elegant way to assess the impact of the endogenous expression of an autoantigen upon the mature T cell repertoire is to compare T cell responsiveness of autoantigen-deficient mice with that of their WT counterparts. In C57BL/6 mice, the immunodominant epitope of MOG lies within the 35–55 peptide (pMOG; Mendel et al., 1995). An initial study of MOG−/− mice generated on the C57BL/6 background revealed that their T cell response remained focused on MOG(35–55). No novel responses to peptides outside this region were identified and that T cell populations sourced from MOG−/− did not have enhanced encephalitogenic activity when transferred into MOG-sufficient mice (Delarasse et al., 2003).
In the present study we sought to determine the relative influence of immunological tolerance on the CD4+ and CD8+ autoreactive T cell repertoires, by a new comparison of WT and MOG−/−. We were able to detect both subtle differences in the CD4+ response and, particularly, enhanced sensitivity in the CD8+ response to MOG in T cell populations that had matured in absence of MOG. The effect in the CD8+ compartment was most pronounced. As previously reported (Ford and Evavold, 2005) we found that pMOG-responsive CD8+ T cells from WT mice responded to an epitope contained with MOG(37–46) which poorly binds to the Db MHC class I molecule. In contrast, the CD8+ pMOG-responsive repertoire from MOG−/− mice concentrated on an epitope within MOG(42–50) which binds to the Db with greater affinity. This indicates that, in the absence of endogenous MOG, these MOG(42–50)-responsive CD8+ T cells escape the normal constraints of immunological tolerance. Although this did not manifest in enhanced pathological activity in MOG−/− CD8+ T cells (in our hands the ability to initiate CNS inflammation is retained only by pMOG-responsive CD4+ cells, not CD8+ cells), it does suggest that for this region of MOG CD8+ T cells are subject to more rigorous control through central and/or peripheral tolerance than their CD4+ counterparts.
Materials and methods
Mice and Antigens
C57BL/6J and MOG−/− mice (Delarasse et al., 2003) were bred under specific pathogen-free conditions at the University of Edinburgh. MOG−/− mice were also crossed with the 2D2 transgenic line that expresses a pMOG-responsive TCR (Bettelli et al., 2003), kindly provided by Dr V. Kuchroo, Harvard. Six- to twelve-week-old, sex-matched mice were used for all experiments. All experiments were approved by the University of Edinburgh ethical review panel and were conducted under United Kingdom legislation.
The MOG(35–55; pMOG) and OVA(323–339) peptides were obtained from the Advanced Biotechnology Centre, Imperial College (London, UK). A panel of overlapping 15-mer peptides with five residue shifts covering the 1–123 extracellular domain of mouse MOG were synthesized by the laboratory of Professor D. Wraith, University of Bristol, UK. Peptides corresponding to 37–46, 41–50, and 42–50 of MOG were synthesized by GL Biochem (Shanghai, China). The LCMV gp(33–41) was obtained from Proimmune Ltd (Oxford, UK).
Recombinant expression and purification of the extracellular domain of murine MOG (rMOG), has been described previously (Fillatreau et al., 2002).
Immunization and Assessment of Lymphoid Recall Responses
Mice were immunized subcutaneously in each hind leg as indicated either with 20 or 100 μg of MOG(35–55), with 100 μg of rMOG, or with 20 μg OVA(323–339), emulsified in complete Freund’s adjuvant (CFA) containing a total of 50 μg of heat-killed mycobacterium tuberculosis H37Ra (Sigma, UK). Draining inguinal and para-aortic lymph nodes (LN) and splenocytes were sampled 10 days later. Cell suspensions from individual mice were cultured in 96-well flat-bottomed plates (BD, Oxford, UK) at 6 × 105 LN cells/well, or 8 × 105 splenocytes/well, using x-Vivo 15™ serum-free medium (BioWhittaker, Maidenhead, UK) supplemented with 2 mM L-glutamine and 50 μM 2-ME (Invitrogen Life Technologies, Paisley, UK). Cells were stimulated in triplicate with overlapping 15-mer peptides, or with a dose range of pMOG for 48 h prior to addition of [3H] thymidine at 0.5 μCi/well. Thymidine incorporation was measured 18 h later using a liquid scintillation β counter (LKB Wallac, Turku, Finland). Results are expressed as the mean counts per minute (CPM) for each group. Supernatants from 72 h cultures were tested for IFN-γ and IL-17 by ELISA.
For CFSE dilution assays, cells were labeled with 5 μM CFSE and cultured with or without 20 μM pMOG for 3 days. Cells were counterstained with anti-CD4 and anti-CD8 prior to flow cytometric analysis.
Induction and Assessment of EAE
Active EAE was induced by immunization with 100 μg pMOG as above. Mice also received 200 ng pertussis toxin (Health Protection Agency, Dorset, UK) i.p. in 500 μl PBS on the day of immunization and 2 days later.
Passive EAE was induced using a previously described protocol (O’Connor et al., 2007). Donor mice were immunized with 100 μg pMOG as above. Eight days later draining LN cells were prepared and cultured at 4 × 106 cells/ml with 10 μg/ml pMOG, 0.5 ng/ml IL-2, 25 ng/ml rIL-12 (R&D Systems), and 25 ng/ml rIL-18 (MBP International). After 48 h, the final concentration of IL-2 was increased to 2 ng/ml for a further 24 h. In some experiments CD4+ cells were selected using magnetic anti-CD4 microbeads. CD8+ cells were selected by removal of CD19+ and CD4+ cells from the cell suspension by magnetic anti-CD4 and anti-CD19 microbeads (Miltenyi Biotec, Germany). Both CD4+ and CD8+ populations were further enriched by FACS sorting to ensure high purity (96 ± 2%). Host mice received an intravenous injection of cells (unsorted cells, sorted CD4+ or CD8+ cells, or mixed populations, as indicated). Mice also received 200 ng pertussis toxin i.p. in 500 μl PBS on the same day as cell transfer.
Clinical signs of EAE were assessed daily with the following scoring system: 0, no signs; 1, flaccid tail; 2, impaired righting reflex and/or gait; 3, partial hind limb paralysis; 4, complete hind limb paralysis; 5, hind limb paralysis and partial front limb paralysis; 6, moribund or dead.
Flow Cytometric Analysis
For phenotypic analysis of T cell populations, cells were stained with anti-CD3, anti-CD4, and anti-CD8 (all from eBioscience, USA). T cells were gated on live, single CD3+ T cells and the frequency of CD4+ or CD8+ T cells assessed. Data was acquired on a BD LSR II or LSR Fortessa (BD Biosciences, USA) and analyzed with Flowjo analysis software (Treestar, USA).
MOG-Reactive T Cell Lines
Lymph node cells were harvested from mice immunized 10 days previously as indicated. CD4+ T cell lines were generated as previously described by repeated rounds of restimulation with pMOG (Anderton et al., 1998; Sweenie et al., 2007). To generate pMOG-responsive CD8+ T cell lines, CD4+ T cells were first depleted from the starting LN population using magnetic anti-CD4 microbeads (Miltenyi Biotec). The remaining cells were then cultured in the presence of 5 μM pMOG, 10 U/ml IL-2, and 10 ng/ml IL-7 for 3 days. Viable cells were recovered by density gradient centrifugation and rested in 20 U/ml IL-2 and 10 ng/ml IL-7 for 4 days. Cells were subjected to three to five rounds of stimulation prior to testing in recall assays to MOG peptides.
RMA-S Db Stabilization Assay
The RMA-S cell line was kindly provided by Professor R. Zamoyska (University of Edinburgh). Cells were grown at 37°C in RPMI 1640 medium supplemented with 10% FCS, 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 50 μM 2-ME (Invitrogen Life Technologies). Cells were incubated at 1.2 × 106/ml with 100 μM of the indicated peptide for 3 h at 37°C prior to counterstaining first with purified anti-Db, then anti-mouse biotinylated IgG2a and then streptavidin–allophycocyanin (BD Pharmingen). Data are expressed as the geometric MFI of Db+ cells.
Results
In the Absence of MOG, the MOG-Responsive T Cell Repertoire Remains Focused of the 35–55 Region
In H-2b mice, the immunodominant region of the extracellular domain of MOG capable of stimulating T cells is contained within the pMOG peptide (Mendel et al., 1995; Sweenie et al., 2007). Within this peptide, the core T cell epitope has been described as 40–48 (Mendel et al., 1996). Our own previous studies using immunization with the recombinant extracellular domain of mouse MOG (rMOG) had not identified additional naturally processed epitopes, out with the 35–55 region, that were recognized by T cells from WT C57BL/6 mice (Sweenie et al., 2007). Other studies have probed whether MOG-deficient mice are able to mount T cell responses to additional epitopes, but those studies used immunization with synthetic peptides, rather than intact protein (Delarasse et al., 2003). We made the assumption that, in the presence of MOG (i.e., in WT mice), immunological tolerance to any additional potential T cell epitopes would involve those epitopes that can be generated after antigen processing of the intact MOG protein (i.e., naturally processed, rather than cryptic T cell epitopes; Sweenie et al., 2007). With this in mind, we decided to revisit the question of whether T cells from MOG-deficient mice can respond to additional epitopes within MOG. We immunized MOG−/− mice with rMOG and subsequently rechallenged their primed LN cells with overlapping peptides covering the MOG 1–123 sequence. As shown in Figure 1, these recall responses remained absolutely focused on peptide 36–50, the only peptide to contain the 40–48 core epitope. These data were consistent with the previous reports on MOG−/− T cell responses using peptide immunization protocols (Delarasse et al., 2003). We therefore have found no evidence that T cells recognizing additional epitopes contained in the extracellular domain of MOG (i.e., not within 35–55) are either purged from the repertoire or rendered unresponsive in the presence of MOG.
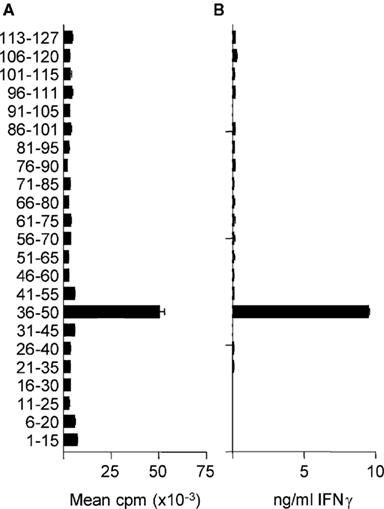
Figure 1. MOG−/− T cells remain focused on MOG(35–55). MOG−/− mice were immunized with 100 μg rMOG in CFA. Ten days later, draining LN were tested for recall responses to individual overlapping peptides as measured by proliferation (A) or IFN-y secretion (B). Data shown are from a representative experiment of two.
MOG−/− Mice are More Sensitive to Immune Challenge with MOG(35–55)
The above data indicated that in WT and MOG−/− mice, the T cell response to rMOG was focused on the 35–55 region. To test whether there was greater sensitivity to this region in the absence of endogenous MOG, we immunized mice with differing doses of the pMOG peptide. As shown in Figures 2A–C, when using an immunizing dose of 100 μg of pMOG, there was the suggestion that the resulting primed T cells from MOG−/− mice had a greater capacity to respond to pMOG, particularly in terms of IFN-γ release. This difference was accentuated in those mice immunized with a lower dose of pMOG (20 μg; Figures 2D–F). In this setting, T cells from WT mice failed to proliferate and did not produce IFN-γ or IL-17 in response to in vitro rechallenge with pMOG. In contrast, T cells from MOG−/− mounted a robust response, not discernibly weaker than the recall response seen after immunization with 100 μg pMOG, particularly in terms of IL-17 release. This effect could not be attributed to a general, intrinsically greater capacity of MOG−/− mice to respond to antigen immunization, because responses to immunization with the OVA(323–339) peptide were equivalent in MOG−/− and WT mice (Figures 2G–I).
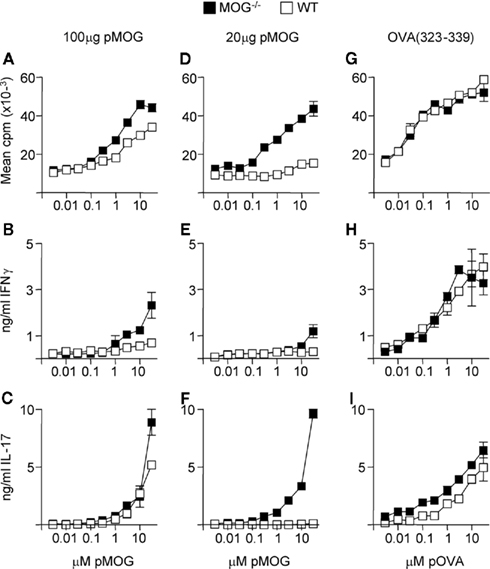
Figure 2. MOG−/− mice are highly sensitive to MOG(35–55). Mice were immunized with either 100 μg pMOG (A–C), 20 μg pMOG (D–F) or 20 μg OVA(323–339) (G–I) in CFA. Ten days later, splenic recall responses to pMOG or OVA(323–339) were tested as measured by proliferation (A,D,G), IFN-y production (B,E,H) or IL-17 production (C,F,I). Each experiment shown in one of two experiments giving consistent results; three mice per group were used.
These data led us to conclude that the presence of MOG in WT mice does, in fact, impact subtly on the ability of the T cell repertoire to respond to the immunodominant region of MOG.
In the Absence of Endogenous MOG, Both CD4+ and CD8+ T Cells are More Responsive to pMOG
The increased sensitivity that we observed in MOG−/− mice might reflect the presence of more pMOG-reactive precursors than in their WT counterparts, or that the same repertoire was present, but in a fitter condition to respond to pMOG. There have been previous reports that immunization with pMOG is also capable of activating Db-restricted CD8+ T cells (Sun et al., 2003). We therefore loaded lymphoid cells from immunized mice with CFSE prior to culture with pMOG and subsequent assessment of the proliferation of CD4+ and CD8+ T cells by flow cytometry (Figure 3A). These assays revealed that MOG−/− samples had greater numbers of cells that had undergone division in both the CD4+ and the CD8+ compartments.
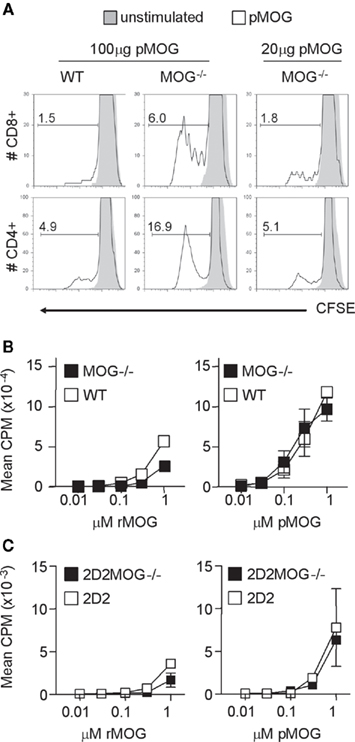
Figure 3. Both CD4+ and CD8+ T cells respond to MOG(35–55). Mice were immunized with either 100 μg or 20 μg pMOG in CFA. Ten days later, draining LN cells were stained with CFSE and cultured with or without pMOG. CFSE dilution was analyzed on day 3 of culture in CD4+ and CD8+ T cells (A), percentages represent the frequency of gated cells that had diluted CFSE. Results shown are one of three experiments giving consistent results; three mice per group were used. CD4+ T cell lines developed from pMOG-immunized mice were tested for recall responses to rMOG or pMOG (B). Naive CD4+ T cells from 2D2 and 2D2MOG−/− mice were tested for primary in vitro responses to rMOG and pMOG (C).
To test for increased responsiveness on per cell basis, we first generated CD4+ pMOG-responsive T cell lines from WT and MOG−/− mice. The dose response profiles of these lines did not show a greater sensitivity for T cells obtained from MOG−/− mice (Figure 3B). Furthermore, we crossed MOG−/− mice with the 2D2 line that is transgenic for a pMOG-responsive TCR (Bettelli et al., 2003) Naive splenocytes from this MOG−/− line showed no greater sensitivity to either pMOG or rMOG than their counterparts derived form MOG-sufficient 2D2 mice (Figure 3C).
In the Absence of Endogenous MOG, a Functional Cohort of MOG(42–50)-Responsive CD8+ T Cells Persists
No gross differences in pMOG-responsiveness were evident in the CD4+ T cell lines derived from WT versus MOG−/− mice. We therefore decided to pursue potential differences in the CD8+ T cell repertoire for a number of reasons. Firstly, although dependent on CD4+ T cells, the CNS lesions of mice with pMOG-induced EAE clearly contain CD8+ cells (Figure 4). Secondly, CD8+ T cells have been reported to have some pathogenic activity in this model (Sun et al., 2003; Ford and Evavold, 2005; Bettini et al., 2009). Thirdly, although the T cell epitope recognized by CD8+ T cells from WT mice has been defined as 37–46 (Ford and Evavold, 2005), this peptide has relatively poor binding affinity for the Db MHC class-I molecule. In contrast, the adjacent 41–50 peptide was found to have greater binding affinity for Db (Ford and Evavold, 2005). We therefore developed the hypothesis that the enhanced CD8+ T cell response to pMOG seen in MOG−/− mice reflected the survival of T cells capable of responding to the more stable Db/MOG(41–50) complex, and that these cells would be purged from, or rendered unresponsive in WT mice due to the presence of endogenous MOG.
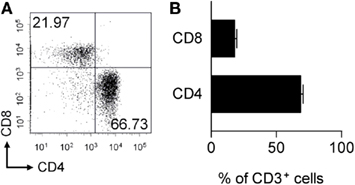
Figure 4. The CNS infiltrate in pMOG-induced EAE includes CD4+ and CD8+ T cells. EAE was induced in WT mice by immunization with pMOG. Spinal cord and brain tissue was collected 13 days later and analyzed for the presence of CD4+ and CD8+ T cell subsets within the live, singlet, CD3+ T cell gate (A). (B) Shows the frequency of CD4+ or CD8+ T cells.
To test this hypothesis, we first used a Db stabilization assay to confirm the observation that the MOG(37–46) peptide is a relatively poor Db binder (Figures 5A,B) RMA-S cells cultured with this peptide, showed no elevation in their surface expression of Db, compared with cells cultured in medium alone. Importantly, culture with the MOG(41–50) peptide induced Db expression to a similar level as seen with the strong Db-binding peptide, gp33–41 of LCMV (Klavinskis et al., 1990; Hudrisier et al., 1997; Ford and Evavold, 2005). The MOG(42–50) peptide stabilized Db to a lesser extent than MOG(41–50) and LCMVgp(33–41), whereas MOG(35–55) induced only a marginal shift in Db expression.
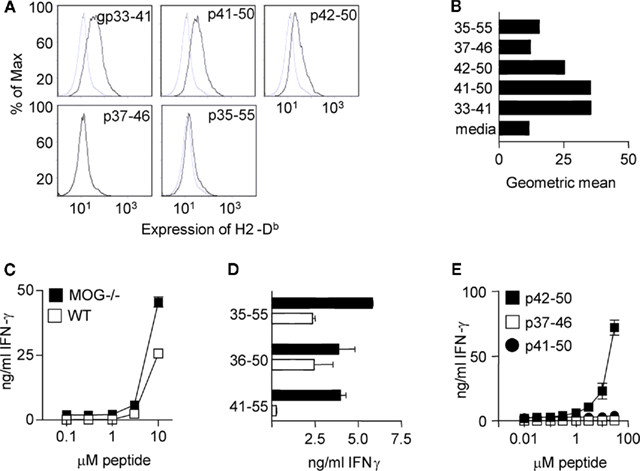
Figure 5. The CD8+ T cell response in MOG−/− mice focuses on an epitope with high affinity for Db.(A) RMA-s cells were cultured with the indicated peptides and expression of Db assessed by flow cytometry (B) shows the geometric mean fluorescence (MFI). Mice were immunized with 100 μg pMOG in CFA. Ten days later, CD8+ T cell lines were derived by restimulation of CD4-depleted LN cells with pMOG. After five rounds of stimulation, (C) T cells were tested for recall responses to pMOG or (D) to the indicated peptides. (E) A MOG−/− CD8+ T cell line was cultured with dose ranges of the indicated peptides. Each experiment shown is one of two/three experiments giving consistent results.
We next generated pMOG-responsive CD8+ T cell lines from WT versus MOG−/− mice that had been immunized with pMOG. These lines responded in a similar fashion to restimulation with pMOG; i.e., there was no greater sensitivity apparent in the MOG−/− T cell lines (Figure 5C). However, there was a clear difference in which of the overlapping 15-mer peptides these CD8+ T cell lines would respond to. Lines from WT mice responded to MOG(36–50) but not MOG(41–55), indicative of a response to the previously described 37–46 epitope. In contrast, lines generated from MOG−/− mice responded to both the MOG(36–50) and the MOG(41–55) peptides (Figure 5D). These data could indicate MOG−/− responses either to both 37–46 and 41–50, or 41–50 alone. We therefore pursued the response profiles of these CD8+ TCL generated from pMOG-primed MOG−/− mice. Using shorter peptides for in vitro stimulation, with found that these CD8+ T cells responded to the MOG(42–50) peptide, but not to the MOG(37–46) peptide (Figure 5E). Interestingly these T cells responded to this alternative epitope as the MOG(42–50) 9-mer rather than the MOG(41–50) 10-mer (Figure 5E) even though the latter seemed optimal in the RMA-S Db stabilization assay (Figures 5A,B).
Our conclusion from these studies is that MOG−/−, but not WT mice harbor CD8+ T cells that are capable of responding to the MOG(42–50) peptide, which binds well to the Db MHC class I molecule. The inference therefore is that, in the presence of endogenous MOG (in WT mice), such CD8+ T cells are either deleted, or functionally silenced by the stable expression of their cognate peptide-MHC complex under steady state, tolerogenic conditions.
Neither WT Nor MOG−/− CD8+ T Cells Show EAE Inducing, or Enhancing Properties
Previous studies have reported that pMOG-responsive CD8+ T cells generated from WT mice can transfer EAE (Sun et al., 2003; Ford and Evavold, 2005). Our observation that MOG−/− mice harbor CD8+ T cells recognizing a T cell epitope that has the potential to be stably expressed in MOG-sufficient mice led us to predict that such CD8+ T cells from MOG−/− would be highly pathogenic. We have previously reported the passive transfer of EAE using pMOG-primed LN cells from WT mice. We find the most robust way of triggering pathogenic activity in these LN populations is by exposing them to a 72-h in vitro restimulation with pMOG in the presence of a Th1-polarizing cytokine cocktail (IL-12 + IL-18 + IL-2; O’Connor et al., 2007). Transferring such populations from either WT or MOG−/− mice into WT recipients produced indistinguishable clinical EAE courses (Figure 6B). These LN populations contain CD4+ cells, CD8+ cells and also a large non-T cell population (Figure 6A). Using traceable populations of donor cells we have always recovered a predominantly CD4+ donor population from the inflamed CNS of diseased recipients (O’Connor et al., 2007, 2008). However, to test (a) whether CD8+ T cells present in these transferred populations could be pathogenic on their own and (b) whether their pathogenic activity might be elevated if sourced from MOG−/− mice, we transferred CD4+ and CD8+ T populations that had been sorted (>95% purity) following the in vitro restimulation culture. We found that the transfer of CD4+ cells, but not CD8+, was able to reliably achieve clinical EAE. Again, no difference in clinical picture was apparent when using cells sourced from WT versus MOG−/− mice (Figure 6C). We also transferred purified T cells into RAG2−/− H-2b mice. In these lymphopenic recipients, either WT or MOG−/− CD4+ cells produced aggressive, fatal EAE within 10 days of transfer, but again CD8+ cells showed no pathogenic activity (data not shown).
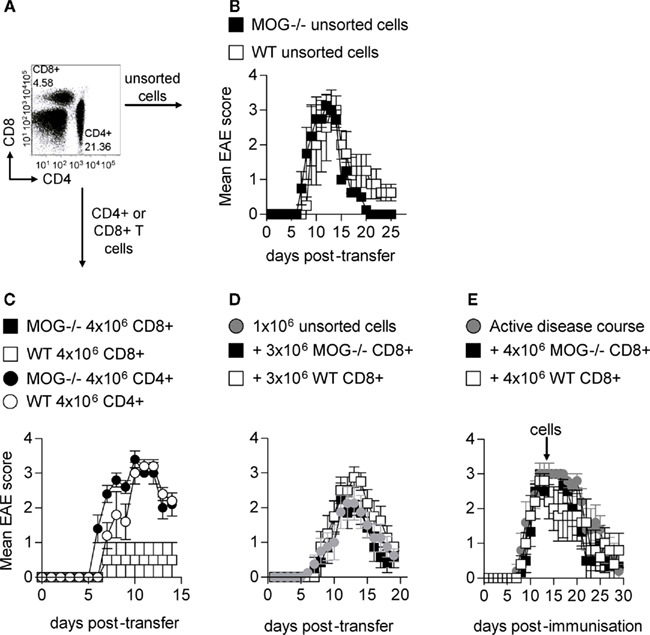
Figure 6. CD8+ T cells are unable to induce EAE. Donor mice were immunized with 100 μg pMOG in CFA. Ten days later, LN cells were restimulated with pMOG for 3 days in the presence of IL-12 and IL-18. Cells were analyzed pre-transfer for CD4+ and CD8+ T cells within the donor population (A). Unsorted cells were injected i.v. into WT host mice (B). CD4+ and CD8+ T cells were purified by FACS and injected into WT hosts (C). CD8+ T cells (3 × 106) were transferred alongside 1 × 106 unsorted cells (D) or 4 × l06 injected into mice with active disease (immunized 13 days previously) (E). Each experiment shown in one of two/three experiments giving consistent results; three–five mice per group were used. Figure (E) is a pool of three experiments with eight–nine mice per group.
These data led us to conclude that CD4+ T cells were indispensable for the induction of EAE in these passive transfer models. As an aside, this is the first time that we definitively report that the pathogenic capacity of these stimulated LN cultures is truly retained by the CD4+ compartment (Figure 6C).
It has been suggested previously that CD4+ T cells enter the CNS prior to the arrival of CD8+ T cells in MOG-induced active EAE (Sonobe et al., 2007). Thus the inability of CD8+ T cells to induce passive disease may be related to their ability to enter the CNS only once inflammation has been established by pMOG-responsive CD4+ cells. We therefore tested whether transfer of increased CD8+ cell numbers could exacerbate EAE induced by unpurified “Th1” LN preparations. This was attempted in two ways; (a) by co-transfer of additional CD8+ T cells at the time of Th1 transfer, (b) by transfer of CD8+ T cells at the peak of clinical EAE (we have recently shown that the transfer of a Th1 preparation at this time can exacerbate the course of EAE; Carrillo-Vico et al., 2010). Under either protocol, transferring increased numbers of pMOG-stimulated CD8+ T cells, sourced from either WT or MOG−/− mice, showed no ability to enhance EAE pathology (Figures 6D,E).
Our conclusion from these data is that, despite our exhaustive efforts, pMOG-responsive CD8+ T cells cannot initiate CNS inflammation upon passive transfer and cannot exacerbate EAE once it is underway.
Discussion
The extent to which the T cell repertoire against a particular autoantigen is molded by the presence of that autoantigen remains an issue that is central both to immunological tolerance and to the risk of developing an autoaggressive T cell response to that antigen. The current paradigms for how autoreactive T cells can avoid negative selection during thymic development have been informed by studies of neuroantigen-responsive T cells in particular, largely because of the array of well-characterized T cell epitopes capable of provoking EAE (Anderton et al., 1998; Goverman, 1999; Huseby and Goverman, 2000; Anderton and Wraith, 2002). For example, low level thymic expression of the immunodominant 139–151 peptide of proteolipid protein (PLP) in H-2S mice occurs because the exon containing 139–151 is not included in the DM20 splice variant of PLP which is dominantly expressed in the thymus (Anderson et al., 2000; Klein et al., 2000). Although myelin basic protein (MBP) is expressed in the thymus, the immunodominant encephalitogenic epitope of MBP in H-2U mice (the Ac1-9 peptide) binds very poorly to AU, generating an insufficient TCR-signal to trigger clonal deletion of Ac1-9-responsive T cells (Liu and Wraith, 1995; Liu et al., 1995; Seamons et al., 2003). The 85–99 peptide of MBP is commonly recognized by T cells from MS patients (this peptide can also induce EAE in H-2s mice). The antigen processing enzyme asparaginyl endopeptidase can cleave MBP after the 94Asn residue, thereby destroying the 85–99 epitope, and has been shown to be expressed in the thymus, at least providing the opportunity for destructive antigen processing (Manoury et al., 2002).
These EAE-relevant examples highlight three ways in which developing thymocytes can avoid encountering their cognate antigen with sufficient avidity to trigger their clonal deletion. Of note, such effects would be active irrespective of the potential for AIRE-driven ectopic expression of the autoantigen in the thymus (Mathis and Benoist, 2009). Of course, negative selection does stringently remove some neuroantigen-responsive T cells. For example, in MBP-deficient H-2u mice, the immunodominant T cell epitope of MBP lies within 121–150, not Ac1-9 as found in their MBP-sufficient counterparts. This is because the 121–150-responsive population is purged from the T cell repertoire when MBP is endogenously expressed (Harrington et al., 1998).
An important point is that all the above examples involve CD4+ T cell responses to neuroantigen; with good reason because, in general, EAE is a CD4+ T cell-dependent disease. Only a few similar studies have analyzed how autoantigen impacts on the myelin-reactive CD8+ T cell compartment. Using MBP-deficient H-2k mice, the Goverman lab initially identified a MBP(79–87)-responsive KK-restricted CD8+ population and showed that such cells could be highly pathogenic when transferred to a WT H-2k mice (Huseby et al., 2001). Evidence that CD8+ T cells have the capacity to provoke profound CNS inflammation has come from two transgenic models in which the expression of “neo-self” antigens [OVA, or influenza hemagglutinin (HA)] was driven by oligodendrocyte-specific promoters (Na et al., 2008; Saxena et al., 2008). In the OVA model, crossing the antigen transgenic with the OT-I transgenic that expresses Kb-restricted TCR recognizing OVA led to a spontaneous and fatal disease that was IFN-γ-dependent (Na et al., 2008). In the HA model, disease expression required transfer of pre-activated HA-responsive CD8+ TCR transgenic cells, which led to oligodendrocyte loss, demyelination, and axonal damage (Saxena et al., 2008).
In agreement with the initial published observations made using MOG−/− mice (Delarasse et al., 2003), we observed that both WT and MOG−/− mice respond only to the same immunodominant region within recombinant mouse MOG, MOG(35–55). Given the profound differences seen in some earlier comparisons of myelin autoantigen-deficient and -sufficient mice (Huseby et al., 2001), it was somewhat surprising when Delarasse et al. (2003) did not find gross differences in the T cell response to MOG in MOG−/− versus WT mice. This was born out by subsequent TCR usage analyses of pMOG-responsive CD4+ T cells from the two strains (Fazilleau et al., 2006).
Nevertheless, our data described here do indicate some subtle differences that have so far gone unappreciated. Firstly, the finding that only MOG−/− mice can mount a productive T cell response when immunized with a low dose of pMOG suggests that these mice do respond to MOG as a foreign antigen and, by inference, that the T cell repertoire of WT mice has been influenced by exposure to endogenous autoantigen. However, although CD4+ T cells within the lymphoid organs of MOG−/− mice responded to low dose immunization with pMOG, we were unable to confirm that these cells had any meaningful change in their function. pMOG-responsive CD4+ T cell lines were not more sensitive in vitro if they were originally sourced from MOG−/− mice. Again consistent with the Delarasse et al. (2003) study, the transfer of purified pMOG-responsive CD4+ T cells provoked equivalent degrees of clinical disease regardless of which strain the T cells were sourced from. Freshly isolated “naïve” pMOG-responsive T cells from 2D2 TCR transgenic mice responded to pMOG in a similar fashion, regardless of whether they had developed in a mouse that did, or did not express MOG. This latter point may be weakened by the fact that the 2D2 TCR can cross-react with a peptide from the neuronal autoantigen neurofilament-M (Krishnamoorthy et al., 2009). It is therefore possible that 2D2 T cell sensitivity may be controlled by exposure to this autoantigen, irrespective of MOG expression. A further caveat on these data is that the T cell clone from which the 2D2 TCR was originally derived came from a WT mouse that did express MOG (Bettelli et al., 2003). Therefore, it remains possible that this provenance imposes an “intrinsic” sensitivity limit upon T cells that bear this TCR, which cannot be increased even when MOG is absent throughout T cell development as was the case for the 2D2 × MOG−/− line.
The most striking feature of the MOG−/− T cell response to pMOG was found in the CD8+ population. In WT mice, the pMOG-responsive repertoire focuses on MOG(37–46), consistent with previous reports (Ford and Evavold, 2005). In contrast, the CD8+ response of MOG−/− mice was focused on the MOG(42–50) epitope, which has a higher binding affinity for Db. This would be consistent with an avidity-based negative selection of these cells in WT mice, although we should stress that our data do not address whether the lack of a response on WT mice was the result of physical deletion or functional inactivation. A previous report suggested the presence of T cells in the repertoire of WT C57BL/6 mice that could bind to dimeric Db loaded with the MOG(44–54) peptide and that immunization with that peptide could produce mild clinical signs of EAE with low incidence (Sun et al., 2003). However, the nature (CD4+ versus CD8+) of the CNS infiltrate in those mice was not explored. Immunization with the MOG(40–54) peptide gave a higher incidence of EAE, but that peptide contained the core CD4+ epitope (40–48) so, overall, no definitive conclusions can be drawn from that study (Sun et al., 2003).
The ultimate evidence for the importance of a suitably restrained MOG-responsive CD8+ T cell repertoire would have been to show enhanced pathogenic activity when transferring CD8+ T cells from pMOG-primed MOG−/− mice, when compared to their WT counterparts. However, despite extensive attempts using a range of strategies, we came to the conclusion that, in our hands, pMOG-responsive CD8+ T cells do not transfer disease, even in lymphopenic hosts. Early studies reported CD8+ T cells to have suppressive function, whereby genetic ablation or antibody-mediated depletion of CD8+ T cells resulted in exacerbated EAE (Koh et al., 1992; Montero et al., 2004). More recent papers show a potential regulatory function of CD8+ T cells with transfer of cells at the peak of disease resulting in early resolution (Lee et al., 2008; York et al., 2010). These contrast with the reported pathogenic activity for CD8+ T cells as already discussed. The failure of CD8+ T cells to induce EAE in our study, despite numerous attempts with alterations in cell numbers and method of cell isolation, is in agreement with York et al. (2010). However, in contrast to York et al. (2010), we also found no evidence for CD8+ regulatory function.
The CD8+ T cells we transferred secreted high quantities of IFN-γ and TNF-α in response to pMOG (data not shown). The populations were of high purity (>95%). In contrast highly pure pMOG-responsive CD4+ T cells were fully pathogenic. Of note, we were able to induce disease with as few as 1 × 106 unpurified, pMOG-stimulated LN preparations, which contained only ∼2 × 105 CD4+ T cells. Previous reports of pathogenic CD8+ T cell transfer have not achieved the level of purity we have used here and at least had the potential to include a contaminating CD4+ population of that order (Sun et al., 2003; Ford and Evavold, 2005). A further complicating factor is the use of lymphopenic hosts, which might allow the rapid expansion of a small contaminating CD4+ population. Importantly in the current study, we found purified CD4+ cells, but not highly pure CD8+ cells to be highly encephalitogenic.
Our observations led us to the conclusion that, at least in the system studied here, the processes of immunological tolerance meld the autoreactive CD8+ T cell repertoire in a more obvious manner than the CD4+ repertoire. Whether this is a general rule is unclear, but CD8+ T cells do have certain qualities that make them particularly dangerous as agents of autoimmune attack. Firstly, once activated, they can act more easily as autonomous effector cells (by killing antigen presenting stromal cells) than their CD4+ counterparts, which tend to require the destructive assistance of intermediary innate immune cells (as highlighted by the importance of the macrophage compartment to EAE pathology; Tran et al., 1998). Secondly, CD8+ effector cells can be exquisitely sensitive to TCR stimulation by very few peptide-MHC complexes, whereas CD4+ cell may require a higher level of antigen presentation for their reactivation in inflammatory sites. Thirdly, as evidenced from peptide-MHC tetramer-based studies of humans with viral infections, CD8+ T cells can undergo a remarkable clonal expansion (Klenerman et al., 2002). These features all make the need to control the degree of autoreactivity in the CD8+ T cell compartment acute.
In summary, this study has revisited whether the endogenous expression of MOG has an impact on immunological tolerance and indicates that there are indeed some subtle effects not previously identified, most notably upon the pMOG-responsive CD8+ T cell repertoire. Our data show very clearly that, at least in our hands, the ability to initiate autoimmune inflammation in the CNS is entirely contained within the CD4+ T cell population. They also highlight, however, that the mechanisms of immunological tolerance that silence CD8+ T cells capable of responding to a peptide-MHC complex that can be stably expressed are at least as robust as those controlling their CD4+ T cells counterparts. Understanding how these might break down during MS remains a major challenge.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the Medical Research Council, UK (Stephen M. Anderton). Antonio Carrillo-Vico was supported by a long-term fellowship from the Federation of European Biochemical Societies. Roland S. Liblau is supported by grants from ARSEP and FRM Team 2009.
References
Anderson, A. C., Nicholson, L. B., Legge, K. L., Turchin, V., Zaghouani, H., and Kuchroo, V. K. (2000). High frequency of autoreactive myelin proteolipid protein-specific T cells in the periphery of naive mice: mechanisms of selection of the self-reactive repertoire. J. Exp. Med. 191, 761–770.
Anderton, S. M., Manickasingham, S. P., Burkhart, C., Luckcuck, T. A., Holland, S. J., Lamont, A. G., and Wraith, D. C. (1998). Fine specificity of the myelin-reactive T cell repertoire: implications for TCR antagonism in autoimmunity. J. Immunol. 161, 3357–3364.
Anderton, S. M., and Wraith, D. C. (2002). Selection and fine-tuning of the autoimmune T-cell repertoire. Nat. Rev. Immunol. 2, 487–498.
Babbe, H., Roers, A., Waisman, A., Lassmann, H., Goebels, N., Hohlfeld, R., Friese, M., Schroder, R., Deckert, M., Schmidt, S., Ravid, R., and Rajewsky, K. (2000). Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J. Exp. Med. 192, 393–404.
Bettelli, E., Pagany, M., Weiner, H. L., Linington, C., Sobel, R. A., and Kuchroo, V. K. (2003). Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J. Exp. Med. 197, 1073–1081.
Bettini, M., Rosenthal, K., and Evavold, B. D. (2009). Pathogenic MOG-reactive CD8+ T cells require MOG-reactive CD4+ T cells for sustained CNS inflammation during chronic EAE. J. Neuroimmunol. 213, 60–68.
Carrillo-Vico, A., Leech, M. D., and Anderton, S. M. (2010). Contribution of myelin autoantigen citrullination to T cell autoaggression in the central nervous system. J. Immunol. 184, 2839–2846.
Coles, A. J., Cox, A., Le Page, E., Jones, J., Trip, S. A., Deans, J., Seaman, S., Miller, D. H., Hale, G., Waldmann, H., and Compston, D. A. (2006). The window of therapeutic opportunity in multiple sclerosis: evidence from monoclonal antibody therapy. J. Neurol. 253, 98–108.
Delarasse, C., Daubas, P., Mars, L. T., Vizler, C., Litzenburger, T., Iglesias, A., Bauer, J., Della Gaspera, B., Schubart, A., Decker, L., Dimitri, D., Roussel, G., Dierich, A., Amor, S., Dautigny, A., Liblau, R., and Pham-Dinh, D. (2003). Myelin/oligodendrocyte glycoprotein-deficient (MOG-deficient) mice reveal lack of immune tolerance to MOG in wild-type mice. J. Clin. Invest. 112, 544–553.
Fazilleau, N., Delarasse, C., Sweenie, C. H., Anderton, S. M., Fillatreau, S., Lemonnier, F. A., Pham-Dinh, D., and Kanellopoulos, J. M. (2006). Persistence of autoreactive myelin oligodendrocyte glycoprotein (MOG)-specific T cell repertoires in MOG-expressing mice. Eur. J. Immunol. 36, 533–543.
Fillatreau, S., Sweenie, C. H., McGeachy, M. J., Gray, D., and Anderton, S. M. (2002). B cells regulate autoimmunity by provision of IL-10. Nat. Immunol. 3, 944–950.
Flugel, A., Willem, M., Berkowicz, T., and Wekerle, H. (1999). Gene transfer into CD4+ T lymphocytes: green fluorescent protein-engineered, encephalitogenic T cells illuminate brain autoimmune responses. Nat. Med. 5, 843–847.
Ford, M. L., and Evavold, B. D. (2005). Specificity, magnitude, and kinetics of MOG-specific CD8+ T cell responses during experimental autoimmune encephalomyelitis. Eur. J. Immunol. 35, 76–85.
Goverman, J. (1999). Tolerance and autoimmunity in TCR transgenic mice specific for myelin basic protein. Immunol. Rev. 169, 147–159.
Harrington, C. J., Paez, A., Hunkapiller, T., Mannikko, V., Brabb, T., Ahearn, M., Beeson, C., and Goverman, J. (1998). Differential tolerance is induced in T cells recognizing distinct epitopes of myelin basic protein. Immunity 8, 571–580.
Hirst, C. L., Pace, A., Pickersgill, T. P., Jones, R., McLean, B. N., Zajicek, J. P., Scolding, N. J., and Robertson, N. P. (2008). Campath 1-H treatment in patients with aggressive relapsing remitting multiple sclerosis. J. Neurol. 255, 231–238.
Holmes, S., Friese, M. A., Siebold, C., Jones, E. Y., Bell, J., and Fugger, L. (2005). Multiple sclerosis: MHC associations and therapeutic implications. Expert Rev. Mol. Med. 7, 1–17.
Hudrisier, D., Oldstone, M. B., and Gairin, J. E. (1997). The signal sequence of lymphocytic choriomeningitis virus contains an immunodominant cytotoxic T cell epitope that is restricted by both H-2D(b) and H-2K(b) molecules. Virology 234, 62–73.
Huseby, E. S., and Goverman, J. (2000). Tolerating the nervous system: a delicate balance. J. Exp. Med. 191, 757–760.
Huseby, E. S., Liggitt, D., Brabb, T., Schnabel, B., Ohlen, C., and Goverman, J. (2001). A pathogenic role for myelin-specific CD8(+) T cells in a model for multiple sclerosis. J. Exp. Med. 194, 669–676.
Jilek, S., Schluep, M., Rossetti, A. O., Guignard, L., Le Goff, G., Pantaleo, G., and Du Pasquier, R. A. (2007). CSF enrichment of highly differentiated CD8+ T cells in early multiple sclerosis. Clin. Immunol. 123, 105–113.
Klavinskis, L. S., Whitton, J. L., Joly, E., and Oldstone, M. B. (1990). Vaccination and protection from a lethal viral infection: identification, incorporation, and use of a cytotoxic T lymphocyte glycoprotein epitope. Virology 178, 393–400.
Klein, L., Klugmann, M., Nave, K. A., Tuohy, V. K., and Kyewski, B. (2000). Shaping of the autoreactive T-cell repertoire by a splice variant of self protein expressed in thymic epithelial cells. Nat. Med. 6, 56–61.
Klenerman, P., Cerundolo, V., and Dunbar, P. R. (2002). Tracking T cells with tetramers: new tales from new tools. Nat. Rev. Immunol. 2, 263–272.
Koh, D. R., Fung-Leung, W. P., Ho, A., Gray, D., Acha-Orbea, H., and Mak, T. W. (1992). Less mortality but more relapses in experimental allergic encephalomyelitis in CD8(/( mice. Science 256, 1210–1213.
Krishnamoorthy, G., Saxena, A., Mars, L. T., Domingues, H. S., Mentele, R., Ben-Nun, A., Lassmann, H., Dornmair, K., Kurschus, F. C., Liblau, R. S., and Wekerle, H. (2009). Myelin-specific T cells also recognize neuronal autoantigen in a transgenic mouse model of multiple sclerosis. Nat. Med. 15, 626–632.
Lee, Y. H., Ishida, Y., Rifa’i, M., Shi, Z., Isobe, K., and Suzuki, H. (2008). Essential role of CD8+ CD122+ regulatory T cells in the recovery from experimental autoimmune encephalomyelitis. J. Immunol. 180, 825–832.
Liu, G. Y., Fairchild, P. J., Smith, R. M., Prowle, J. R., Kioussis, D., and Wraith, D. C. (1995). Low avidity recognition of self-antigen by T cells permits escape from central tolerance. Immunity 3, 407–415.
Liu, G. Y., and Wraith, D. C. (1995). Affinity for class II MHC determines the extent to which soluble peptides tolerize autoreactive T cells in naive and primed adult mice – implications for autoimmunity. Int. Immunol. 7, 1255–1263.
Malmestrom, C., Lycke, J., Haghighi, S., Andersen, O., Carlsson, L., Wadenvik, H., and Olsson, B. (2008). Relapses in multiple sclerosis are associated with increased CD8+ T-cell mediated cytotoxicity in CSF. J. Neuroimmunol. 196, 159–165.
Manoury, B., Mazzeo, D., Fugger, L., Viner, N., Ponsford, M., Streeter, H., Mazza, G., Wraith, D. C., and Watts, C. (2002). Destructive processing by asparagine endopeptidase limits presentation of a dominant T cell epitope in MBP. Nat. Immunol. 3, 169–174.
Mars, L. T., Saikali, P., Liblau, R. S., and Arbour, N. (2011). Contribution of CD8 T lymphocytes to the immuno-pathogenesis of multiple sclerosis and its animal models. Biochim. Biophys. Acta 1812, 151–161.
Mendel, I., Kerlero de Rosbo, N., and Ben-Nun, A. (1995). A myelin oligodendrocyte glycoprotein peptide induces typical chronic experimental autoimmune encephalomyelitis in H-2b mice: fine specificity and T cell receptor V beta expression of encephalitogenic T cells. Eur. J. Immunol. 25, 1951–1959.
Mendel, I., Kerlero de Rosbo, N., and Ben-Nun, A. (1996). Delineation of the minimal encephalitogenic epitope within the immunodominant region of myelin oligodendrocyte glycoprotein: diverse Vb gene usage by T cells recognising the core epitope encephalitogenic for T cell receptor Vbeta-b and T cell receptor Vbeta-a H-2b mice. Eur. J. Immunol. 26, 2470–2479.
Montero, E., Nussbaum, G., Kaye, J. F., Perez, R., Lage, A., Ben-Nun, A., and Cohen, I. R. (2004). Regulation of experimental autoimmune encephalomyelitis by CD4+, CD25+ and CD8+ T cells: analysis using depleting antibodies. J. Autoimmun. 23, 1–7.
Na, S. Y., Cao, Y., Toben, C., Nitschke, L., Stadelmann, C., Gold, R., Schimpl, A., and Hunig, T. (2008). Naive CD8 T-cells initiate spontaneous autoimmunity to a sequestered model antigen of the central nervous system. Brain 131, 2353–2365.
O’Connor, R. A., Malpass, K. H., and Anderton, S. M. (2007). The inflamed central nervous system drives the activation and rapid proliferation of Foxp3+ regulatory T cells. J. Immunol. 179, 958–966.
O’Connor, R. A., Prendergast, C. T., Sabatos, C. A., Lau, C. W., Leech, M. D., Wraith, D. C., and Anderton, S. M. (2008). Cutting edge: Th1 cells facilitate the entry of Th17 cells to the central nervous system during experimental autoimmune encephalomyelitis. J. Immunol. 181, 3750–3754.
Racadot, E., Rumbach, L., Bataillard, M., Galmiche, J., Henlin, J. L., Truttmann, M., Herve, P., and Wijdenes, J. (1993). Treatment of multiple sclerosis with anti-CD4 monoclonal antibody. A preliminary report on B-F5 in 21 patients. J. Autoimmun. 6, 771–786.
Rumbach, L., Racadot, E., Armspach, J. P., Namer, I. J., Bonneville, J. F., Wijdenes, J., Marescaux, C., Herve, P., and Chambron, J. (1996). Biological assessment and MRI monitoring of the therapeutic efficacy of a monoclonal anti-T CD4 antibody in multiple sclerosis patients. Mult. Scler. 1, 207–212.
Saxena, A., Bauer, J., Scheikl, T., Zappulla, J., Audebert, M., Desbois, S., Waisman, A., Lassmann, H., Liblau, R. S., and Mars, L. T. (2008). Cutting edge: multiple sclerosis-like lesions induced by effector CD8 T cells recognizing a sequestered antigen on oligodendrocytes. J. Immunol. 181, 1617–1621.
Seamons, A., Sutton, J., Bai, D., Baird, E., Bonn, N., Kafsack, B. F., Shabanowitz, J., Hunt, D. F., Beeson, C., and Goverman, J. (2003). Competition between two MHC binding registers in a single peptide processed from myelin basic protein influences tolerance and susceptibility to autoimmunity. J. Exp. Med. 197, 1391–1397.
Sonobe, Y., Jin, S., Wang, J., Kawanokuchi, J., Takeuchi, H., Mizuno, T., and Suzumura, A. (2007). Chronological changes of CD4(+) and CD8(+) T cell subsets in the experimental autoimmune encephalomyelitis, a mouse model of multiple sclerosis. Tohoku J. Exp. Med. 213, 329–339.
Sun, D., Zhang, Y., Wei, B., Peiper, S. C., Shao, H., and Kaplan, H. J. (2003). Encephalitogenic activity of truncated myelin oligodendrocyte glycoprotein (MOG) peptides and their recognition by CD8+ MOG-specific T cells on oligomeric MHC class I molecules. Int. Immunol. 15, 261–268.
Sweenie, C. H., Mackenzie, K. J., Rone-Orugboh, A., Liu, M., and Anderton, S. M. (2007). Distinct T cell recognition of naturally processed and cryptic epitopes within the immunodominant 35–55 region of myelin oligodendrocyte glycoprotein. J. Neuroimmunol. 183, 7–16.
Tran, E. H., Hoekstra, K., van Rooijen, N., Dijkstra, C. D., and Owens, T. (1998). Immune invasion of the central nervous system parenchyma and experimental allergic encephalomyelitis, but not leukocyte extravasation from blood, are prevented in macrophage-depleted mice. J. Immunol. 161, 3767–3775.
van Oosten, B. W., Lai, M., Hodgkinson, S., Barkhof, F., Miller, D. H., Moseley, I. F., Thompson, A. J., Rudge, P., McDougall, A., McLeod, J. G., Adèr, H. J., and Polman, C. H. (1997). Treatment of multiple sclerosis with the monoclonal anti-CD4 antibody cM-T412: results of a randomized, double-blind, placebo-controlled, MR-monitored phase II trial. Neurology 49, 351–357.
York, N. R., Mendoza, J. P., Ortega, S. B., Benagh, A., Tyler, A. F., Firan, M., and Karandikar, N. J. (2010). Immune regulatory CNS-reactive CD8( T cells in experimental autoimmune encephalomyelitis. J. Autoimmun. 35, 33–44.
Keywords: CD8+ T cells, EAE, multiple sclerosis
Citation: Leech MD, Carrillo-Vico A, Liblau RS and Anderton SM (2011) Recognition of a high affinity MHC class I-restricted epitope of myelin oligodendrocyte glycoprotein by CD8+ T cells derived from autoantigen-deficient mice. Front. Immun. 2:17. doi: 10.3389/fimmu.2011.00017
Received: 23 February 2011; Paper pending published: 29 March 2011;
Accepted: 13 May 2011; Published online: 24 May 2011.
Edited by:
Bruno Kyewski, German Cancer Research Center, GermanyReviewed by:
Ari Waisman, University Medical Center of the Johannes-Gutenberg University of Mainz, GermanyLudger Klein, Ludwig-Maximilians-University, Germany
Hartmut Wekerle, Max-Planck-Society, Germany
Copyright: © 2011 Leech, Carrillo-Vico, Liblau and Anderton. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Stephen M. Anderton, Centre for Inflammation Research, Queen’s Medical Research Institute, University of Edinburgh, 47 Little France Crescent, Edinburgh EH16 4TJ, UK. e-mail:c3RldmUuYW5kZXJ0b25AZWQuYWMudWs=