
- 1 Berwin Laboratory, Department of Microbiology and Immunology, Dartmouth Medical Center, Lebanon, NH, USA
- 2 Turk Laboratory, Department of Microbiology and Immunology, Dartmouth Medical Center, >Lebanon, NH, USA
- 3 Usherwood Laboratory, Department of Microbiology and Immunology, Dartmouth Medical Center, Lebanon, NH, USA
Elevated levels of IL-10 in the microenvironment of human ovarian cancer and murine models of ovarian cancer are well established and correlate with poor clinical prognosis. However, amongst a myriad of immunosuppressive factors, the actual contribution of IL-10 to the ovarian tumor microenvironment, the mechanisms by which it acts, and its possible functional redundancy are unknown. We previously demonstrated that elimination of the myeloid-derived suppressor cell (MDSC) compartment within the ovarian tumor ascites inhibited tumor progression and, intriguingly, significantly decreased local IL-10 levels. Here we identify a novel pathway in which the tumor-infiltrating MDSC are the predominant producers of IL-10 and, importantly, require it to develop their immunosuppressive function in vivo. Importantly, we demonstrate that the role of IL-10 is critical, and not redundant with other immunosuppressive molecules, to in vivo tumor progression: blockade of the IL-10 signaling network results in alleviation of MDSC-mediated immunosuppression, altered T cell phenotype and activity, and improved survival. These studies define IL-10 as a fundamental modulator of both MDSC and T cells within the ovarian tumor microenvironment. Importantly, IL-10 signaling is shown to be necessary to the development and maintenance of a permissive tumor microenvironment and represents a viable target for anti-tumor strategies.
Introduction
Ovarian cancer is characterized by a progressive peritoneal ascites and a highly immunosuppressive tumor microenvironment infiltrated by massive numbers of leukocytes. Amongst a plethora of known immunosuppressive factors within this tumor microenvironment, including arginase, TGF-β, and PD-L1, IL-10 has generated a great deal of interest. IL-10 has numerous suppressive functions involved in dampening inflammatory responses of the immune system, including inhibition of myeloid cell maturation and reduction of expression of co-stimulatory molecules on dendritic cells (Moore et al., 2001). High levels of IL-10 are found in the serum and ascites of ovarian cancer patients (Gotlieb et al., 1992; Santin et al., 2001; Mustea et al., 2006, 2009; Giuntoli et al., 2009; Nowak et al., 2010) and, importantly, IL-10 levels consistently correlate with advanced disease and poor patient prognosis in both ovarian cancer and other types of cancers (Lech-Maranda et al., 2006; Mustea et al., 2006; Zeni et al., 2007; Nowak et al., 2010). However, the exact roles IL-10 plays in the ovarian tumor microenvironment and which of these directly contribute to the support of tumor progression are unknown. Additionally, given the multitude of potentially redundant immunosuppressive factors present, how the IL-10 signaling networks function and contribute within the peritoneal ovarian tumor microenvironment is currently unclear.
We and others have shown that the substantial CD115+CD11b+ CD11c+ leukocyte compartment within the ovarian tumor environment functions as myeloid-derived suppressor cells (MDSCs). Of direct relevance, work from our group has shown that targeted elimination of MDSCs resulted in a significant decrease in IL-10 within the ovarian tumor ascites and, concomitantly, inhibited tumor progression (Bak et al., 2007; Peter et al., 2009). These findings, combined with the aforementioned clinical correlations between IL-10 and disease prognosis, led us to hypothesize that IL-10 is a critical factor in ovarian tumor progression, that the MDSCs within the tumor microenvironment are the predominant source of IL-10, and that the IL-10 network may provide a viable therapeutic target.
Here we use the murine ID8 ovarian tumor model, the best available transplantable model of ovarian cancer which recapitulates critical characteristics of human epithelial ovarian cancer (Roby et al., 2000). These features include a progressive accumulation of ascites, elevated levels of IL-10, the recruitment of massive numbers of leukocytes including a substantial population of monocytic-like MDSC (MO-MDSC; Hart et al., 2009), and allow for the generation of chimeric mice to directly test for differences only attributable to specific genetic manipulations within the same host and tumor. With this system we provide the first in vivo analysis of IL-10 signaling partners in the tumor and we identify novel IL-10 pathways that define a critical role for IL-10 in tumor progression. These studies identify the CD11b+CD11c+ MDSCs as the predominant IL-10-producing cells in the ovarian tumor microenvironment and, importantly, with the use of mixed chimeric mice, we demonstrate that IL-10 directly and independently alters both MDSC and T cell phenotype and function. Moreover, we demonstrate that inhibition of the IL-10 signaling network results in dramatically reduced tumor burden and enhanced survival through a mechanism requiring T cells. These studies identify a critical and non-redundant role for IL-10 in the development and maintenance of a hospitable microenvironment for the progression of ovarian tumors, and specifically identify IL-10 signaling as a target for efficacious therapeutic strategies against the MDSCs that are known to potently inhibit the efficacy of other treatment modalities.
Materials and Methods
Reagents
Female C57Bl/6 mice were purchased from the National Cancer Institute (Fredricksburg, MD, USA). This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Dartmouth IACUC Committee (Permit Number: A3259-01). No surgery was performed, and all efforts were made to minimize suffering. IL-10Rα−/− (Friedline et al., 2009) and IL-10 reporter mice (Maynard et al., 2007) were obtained from the Kang (UMASS – Worcester) and Weaver (UAB) labs, respectively. MAFIA mice (Burnett et al., 2004), under agreement with ARIAD Pharmaceuticals (Cambridge, MA, USA), and TCRα mice were purchased from Jackson Laboratories (Bar Harbor, ME, USA). FOXP3-reporter mice (Fontenot et al., 2003) were used in collaboration with Dr. Mary Jo Turk at Dartmouth. All animal experiments were approved by the Dartmouth Medical School Institutional Animal Care and Use Committee. ID8 cells (Roby et al., 2000) transduced with Vegf-A and Defb29 (referred to as ID8 within this manuscript) were maintained as previously described (Conejo-Garcia et al., 2004). Anti-mouse Fc Block, Phosflow phospho-STAT3 (pY705), Rat IgG1 isotype control, and anti-CD62L (MEL-14) were purchased from BD Biosciences (San Jose, CA, USA); anti-mouse CD3 (145-2C11), Gr-1 (RB6-8C5), CD45 (30-F11), CD11b (M1/70), CD8 (53–6.7), CD4 (L3T4), MHC-II (M5/114.15.2), and CD45.1 (A20) antibodies from eBioscience (San Diego, CA, USA); and anti-mouse Thy1.1 (OX-7), CD11c (N418), and IFN-γ (XMG1.2) antibodies from Biolegend (San Diego, CA, USA). IL-10 was assessed using murine DuoSet ELISAs (R&D Systems, Minneapolis, MN, USA). Anti-mouse STAT3 (79D7) and anti-phospho-STAT3 (3E2) were purchased from Cell Signaling Technology (Danvers, MA, USA).
Tumors and Leukocyte Isolation
As indicated, ascites and blood were harvested from mice; red blood cells were removed using ACK lysis buffer (0.15 M NH4Cl, 1.0 mM KHCO3, 0.1 mM EDTA). Cells were resuspended in 0.5% BSA in PBS, or media, for further analysis or sorting. Cells were isolated using human or mouse anti-CD11b microbeads or mouse anti-Ly6G microbead kit (Miltenyi Biotec, Auburn, CA, USA). Immunosuppression assays using isolated cell populations from tumor-bearing mice were performed by our previously described methodology (Bak et al., 2008) and analyzed for IFN-γ production by ELISA (R&D Systems, Minneapolis, MN, USA).
FACS Analyses
Cells were pre-incubated with Fc-blocking antibody (clone 2.4G2) prior to antibody staining. Flow cytometry and cell sorting was done on the Accuri C6 and FACS Aria, and analyzed using CFlow and FlowJo 8.8.2 software. Phospho-STAT3 staining was performed by stimulating 106 cells with 100 ng/ml recombinant IL-10 (R&D Systems, Minneapolis, MN, USA) for 10 min at 37°C. Cells were fixed with pre-warmed 4% paraformaldehyde, and permeabilized in ice cold methanol for 60 min prior to staining. Intracellular IFN-γ staining was performed on splenocytes in 10 mg/ml brefeldin A in complete medium at 37°C for 5 h in the absence or presence of 1 μg/ml anti-CD3 stimulation. Cells were stained with anti-CD8, fixed with paraformaldehyde, and permeabilized with saponin before staining with APC-conjugated anti-IFN-γ. CD62L analyses was performed on bead-selected CD8 splenocytes cultured in the presence or not of 10 ng/ml of IL-10 for 72 h.
Luminex Analysis
Centrifuged supernatants of 4 ml PBS peritoneal lavages were analyzed in the Norris Cotton Cancer Center Immune Monitoring Laboratory using the mouse 12-plex cytokine panel (Bio-Rad, Hercules, CA, USA). Cytokine concentrations were calculated as lavage dilutions from each mouse based on a 250-μl peritoneal volume for mice with no ascites, and the recovered volume for mice with ascites. Samples that were below the level of Luminex detection were set as the minimum detectable concentration.
Western Blot
Phospho-STAT3 western was performed on one million plated Ly6G or CD11b selected Ly6G-depleted cells treated with 10 ng/ml of IL-10 for 30 min, spun down, and resuspended in sample buffer with beta-mercaptoethanol, heated for 5 min at 90°C, then run on a 12% SDS-PAGE gel. After transfer to a PVDF membrane and blocking, membranes were incubated overnight with primary antibody, washed, blocked, and incubated with an HRP-secondary antibody. Bands were then detected with ECL Plus Western Blotting Detection Reagent (Amersham Biosciences, Buckinghamshire, UK).
Chimeric Mice
For chimeric mouse studies, C57Bl/6 and Ly5.1 IL-10R−/− bone marrow was collected from femurs, the red blood cells lysed, and washed. For the 20:80 ratio mixed bone marrow chimeras, IL-10R knockout and TCRα knockout bone marrow was mixed at the appropriate ratio. Mixed bone marrow was injected intravenously by periorbital injection into previously irradiated (1000 rads) naïve C57Bl/6 recipients.
Antibody Treatment and Survival
Mice were injected with ID8 tumor cells and treated with 100 μg of anti-IL-10 receptor antibody (clone 1b1.3A; BioXCell, West Lebanon, NH, USA), PBS, or Rat IgG control antibody (Sigma, St. Louis, MO, USA). Mice were sacrificed at the indicated times following the treatment regimen, or monitored for survival. For depletion experiments mice were injected once a week with anti-CD8 monoclonal antibody (clone 2.43), generously provided by Dr. Mary Jo Turk (Dartmouth Medical School, Lebanon, NH, USA).
Results
Myeloid Cells are the Predominant Producers of IL-10 in the Ascites of Ovarian Tumor-Bearing Mice
A number of studies have reported high levels of IL-10 in the ovarian cancer tumor microenvironment (Gotlieb et al., 1992; Murray, 2006; Mustea et al., 2009). To elucidate the role of IL-10 within the ovarian tumor microenvironment, we analyzed the origin of IL-10 in the ascites of tumor-bearing mice. Compared to cultured CD11b+ cells isolated from the ascites, which secrete substantial amounts of IL-10 into the supernatant, equivalent numbers of ID8 tumor cells secrete undetectable levels of IL-10 as measured by ELISA (Figure 1A). This is consistent with analyses showing human ovarian tumor cell production of IL-10 to be limited (Nash et al., 1998; Berger et al., 2001; Carr et al., 2008).
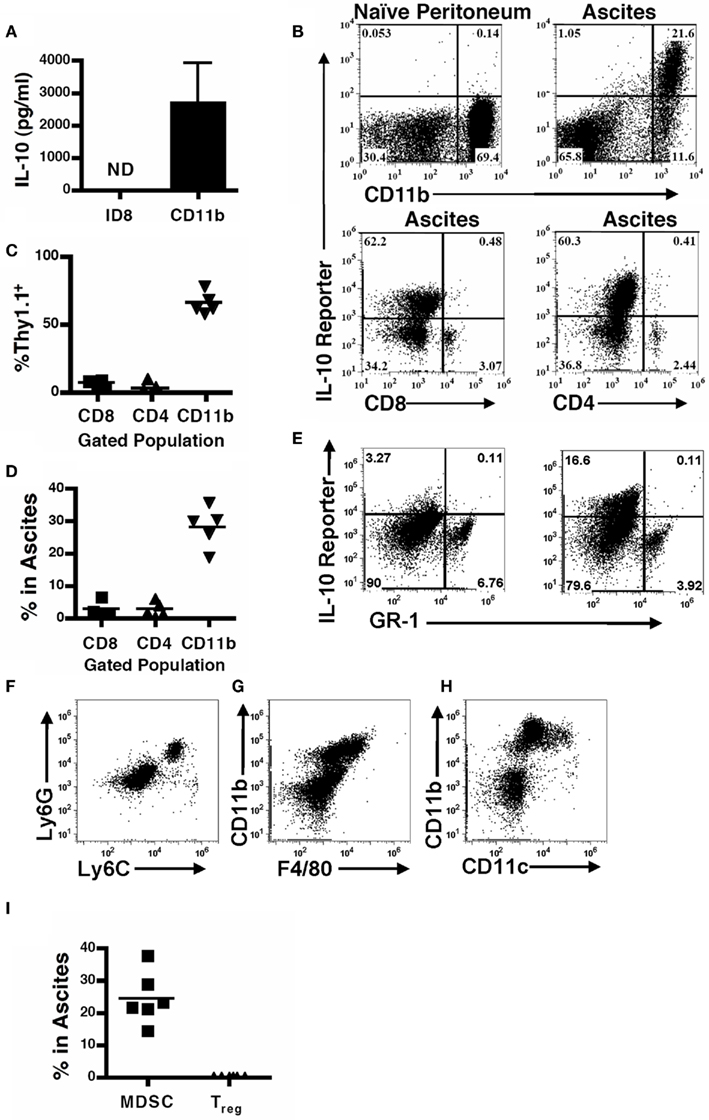
Figure 1. Leukocytes are the predominant cellular source of IL-10 in the tumor microenvironment. (A) Supernatants from 106 ID8 tumor cells, or CD11b+ MDSC cultured for 24 h were analyzed for IL-10 by ELISA. SD is shown (n = 5). (B) Naïve and tumor-bearing mice were analyzed for IL-10 reporter staining on CD11b+ myeloid cells (top panels, pre-gated on total live cell forward and side scatter), and CD4+ or CD8+ leukocytes (bottom panels, pre-gated for lymphocytes based on forward and side scatter) in the peritoneum of naïve and tumor-bearing mice was assessed. Quantification of reporter mice analyses (C) and percentages of gated populations within the ascites (D). (E) IL-10 reporter staining on GR-1+ cells in the peritoneum. (F) Phenotypic analysis of Ly6C and Ly6G expression on cells from the ascites pre-gated on CD11b. (G) Expression of F4/80 on the CD11b+ population in the ascites. (H) Expression of CD11c on the CD11b+ population in the ascites. (I) Percentage composition in the ascites of FOXP3 T regulatory cells by successive gating on CD45, CD4, and FOXP3-GFP, and MDSC gated on CD115-GFP and CD11b staining in FOXP3/MAFIA mice. ND, not detected.
To identify cellular sources of IL-10 in the tumor microenvironment we used 10BiT reporter mice (Maynard et al., 2007), which have the Thy1.1 antigen driven under control of the IL-10 promoter (Maynard et al., 2007). Compared to naïve mice, tumor-bearing reporter mice exhibited a substantial portion of cells that were positive for Thy1.1 in the peritoneum (Figure 1B). Notably, the CD11b+ compartment in the ascites, which we have previously identified as functional MDSC (Bak et al., 2008; Hart et al., 2009), not only robustly expressed the reporter but also represented the vast majority of the cells that stained positive for the reporter, and also make up the bulk of the total leukocyte infiltrate (Figures 1C,D). Both CD4- and CD8-positive cells showed limited expression of IL-10 in the tumor ascites (Figure 1B). To further characterize the IL-10-producing population, since CD11b-positive cells (Figure 1B) derive from both myeloid and granulocytic lineages, we assessed IL-10 expression versus GR-1 expression. The IL-10 reporter revealed that IL-10 expression lies in the GR-1-negative myeloid population (Figure 1E). Granulocytes express both the Ly6C and Ly6G proteins (the components of GR-1; Fleming et al., 1993), while myeloid cells do not express Ly6G though Ly6C expression has been described on a population of inflammatory monocytes (Geissmann et al., 2003). Therefore, to further clarify the myeloid populations within the ascites we independently assessed Ly6C and Ly6G expression on the CD11b population and identified that the majority of the cells express neither epitope, and those that do are primarily double positive (Figure 1F). There are a small percentage of cells that singly express Ly6C, which our previous phenotypic analysis of the ascites indicates are tumor-infiltrating monocytes (Hart et al., 2009). Additionally, we found that the CD11b-positive cells expressed low levels of F4/80 (Figure 1G). We also found a similar intermediate expression pattern of CD11c on the CD11b-positive cells, with a small percentage expressing higher levels of CD11c (Figure 1H). This data indicates that the CD11b+ IL-10-producing cells in the ascites are comprised of a myeloid population distinct from granulocytes, with phenotypic markers of monocytes, macrophages, and dendritic cells.
T regulatory cells (Treg), another suppressive population known to produce IL-10 in cancer (Liyanage et al., 2002; Marshall et al., 2004; Kawaida et al., 2005; Kryczek et al., 2006a; Mougiakakos et al., 2010), were comparatively assessed with the use of MAFIA/FOXP3-GFP mice in which both the CD115+ and FOXP3+ cells express GFP. MDSCs were identified as CD115-GFP+CD11b+ cells, and Treg were identified as CD45+CD4+FOXP3-GFP+ cells within the ascites. In contrast to the substantial MDSC population, the Treg population was present at extremely low frequencies (Figure 1I). In combination with the relative minutia of CD4 T cells staining for the IL-10 reporter, this indicates that the Treg population is not a predominant source of IL-10 in these tumors.
Leukocytes Preferentially Induce STAT3 Signaling in Response to IL-10 within the Tumor Microenvironment
Having delineated the origin of IL-10, we next identified the cellular network that responds to IL-10 within the tumor microenvironment. To directly identify IL-10 responsive cells in the microenvironment, we analyzed STAT3 phosphorylation, a requisite downstream signaling component from the IL-10 receptor complex (Donnelly et al., 1999; Moore et al., 2001; Mosser and Zhang, 2008). To do so we created chimeric mice by reconstituting lethally irradiated mice with equal amounts of bone marrow from wild-type (WT) and congenic IL-10R−/− mice (Figure 2A); reconstitution was subsequently assessed by staining with the CD45.1 congenic marker (Figure 2B). ID8 ovarian tumors were grown in these mice and the CD11b+ cells harvested from the ascites. CD11b-positive wild-type cells, but not the IL-10 receptor knockout cells derived from the same tumor-bearing mouse and exposed to identical treatment, exhibited phospho-STAT3 staining after ex vivo IL-10 stimulation (Figure 2C). As a confirmatory and complimentary method, we assessed relative IL-10-stimulated phosphorylation of STAT3 by Western analysis. Consistent with our FACS results, CD11b-positive Ly6G-depleted cells, but not the Ly6G-positive cells, exhibited IL-10-induced STAT3 phosphorylation (Figure 2D). This indicates that the same CD11b-positive myeloid cells in the tumor that make IL-10 are responding to IL-10, and that this signaling is distinct from the granulocyte population. Our analyses of IL-10-stimulated ascites cells did not detect any STAT3 phosphorylation in the CD45-negative fraction of the ascites which includes the tumor cells (Figure 2E). These results demonstrate that CD11b-positive immune cells distinct from the Ly6G population in the tumor microenvironment selectively initiate downstream signaling events in response to IL-10 stimulation, that the MDSC population in the tumor is a specific mediator of IL-10 signaling, and that IL-10 mediates its effects through immune modulation rather than by direct effect on tumor cells.
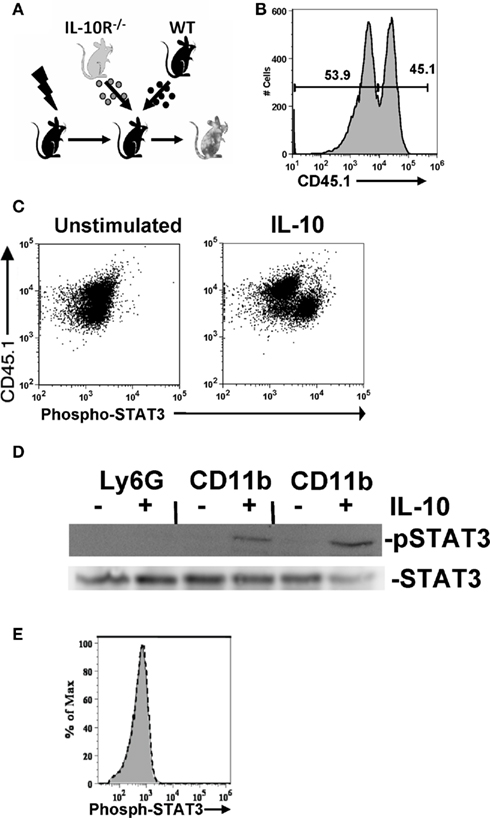
Figure 2. IL-10-elicited STAT3 signaling is preferentially induced in tumor-associated leukocytes. (A) Mixed chimeric mice were generated by reconstituting lethally irradiated wild-type mice with three million bone marrow cells from both wild-type and IL-10R−/− mice. (B) Relative chimerism of reconstituted mice shown by congenic marker staining. (C) IL-10 responsive cells in the ascites of ovarian tumor-bearing mice were identified by comparing phospho-STAT3 staining after a 10-min ex vivo stimulation with IL-10 (100 ng/ml). Comparison of phospho-STAT3 staining CD11b+ cells from congenic IL-10R deficient (CD45.1+) and wild-type (CD45.1−) cells isolated from the ascites of an IL-10R knockout and wild-type mixed bone marrow chimeric mouse. (D) Western blot for phospho-STAT3 from 106 unstimulated or IL-10 (10 ng/ml for 30 min) stimulated Ly6G bead-selected cells (pooled from the ascites of two mice), and Ly6G-depleted CD11b+ cells from the same two mice; total STAT3 is shown as a loading control. (E) CD45-negative cells are shown, with IL-10 treated (black dotted line) compared to untreated (gray) cells.
IL-10 Dictates the Immunosuppressive Phenotype of MDSCs in the Tumor Microenvironment
Since the tumor-infiltrating MDSCs are both the primary producers and responders to IL-10, we hypothesized that this may represent a novel autocrine or paracrine network by which MDSC function is enabled and enforced. However, MDSCs are known to produce a variety of immunosuppressive molecules and thus the functional and potentially redundant role of IL-10 on these cells is unknown. To test this, we returned to the chimeric mouse system. We asked whether the congenic WT and IL-10R−/− MDSCs, derived from the same tumor, only differing in their expression of the IL-10R, exhibited phenotypic and functional differences. FACS analysis showed an increased expression of MHC-II and CD86 on both the CD11chi cells (Figure 3A) and the total CD11b+ cells (Figure 3B) from the IL-10R knockout compartment compared to the congenic wild-type compartment in the ascites of these mice. This is indicative that IL-10 is an important in vivo modulator of myeloid maturation and activation in the tumor microenvironment. Importantly, we assessed the functional consequences of IL-10 signaling on the myeloid populations in the tumor microenvironment by sorting wild-type and IL-10R knockout CD11b+Ly6G− MO-MDSC populations, based on expression of the CD45.1 congenic marker, from tumor-bearing chimeric mice to test their suppressive capacity. The IL-10R−/− MDSCs were significantly impaired in their ability to suppress T cell activation, as assessed by IFN-γ production, compared to the wild-type MDSCs when titrated into a mixed splenocyte reaction (Figure 3C). To determine if the decrease in suppressive capacity from the IL-10R deficient MDSC was dependent on IL-10 production by the MDSC, we first compared IL-10 production by sorted MDSC from each genotype by ELISA and did not detect a significant difference (Figure 4A). Furthermore, IL-10 production by the MDSC does not appear to be a direct means of suppression in our in vitro suppression assay since blockade of IL-10R signaling by antibody (Figure 4B) as well as the use of IL-10R knockout responder splenocytes did not alleviate relative suppression (Figure 4C). Therefore the IL-10 dependent differences in suppression from the sorted chimeric MDSC are not reliant on differential IL-10 production. From this we propose that signaling through the IL-10 receptor is a required component to establish the phenotypic and functional characteristics of MDSC in tumor-bearing hosts, and IL-10 is non-redundant in aspects of programming myeloid populations within the tumor.
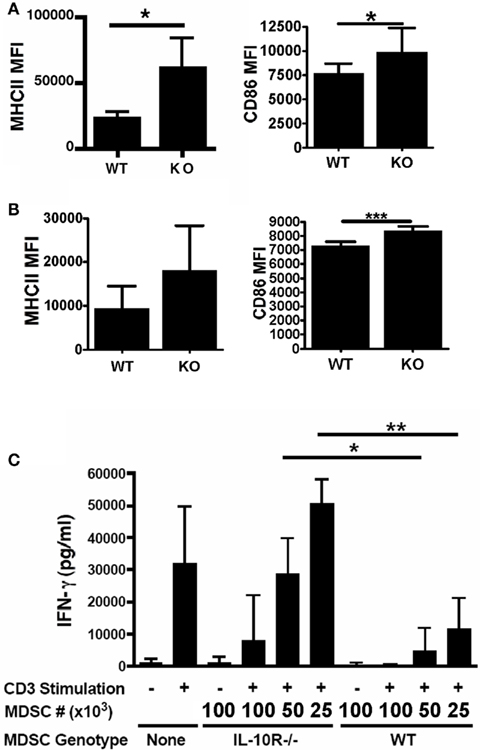
Figure 3. Acquisition of tumor phenotype in myeloid cells requires the IL-10 receptor. Chimeric mice were injected with tumors and analyzed for relative expression of MHC Class II and CD86 on tumor-associated (A) CD11b+CD11c+ cells and (B) total CD11b+ cells, from either wild-type or IL-10R−/− (identified by congenic CD45.1) background. (C) To assess the suppressive capacity of the MDSC populations from each background, CD11b+Ly6G− MDSCs were sorted and titrated into mixed splenocyte reactions stimulated with anti-CD3 (1 μg/ml). Interferon-γ secretion into the supernatants was analyzed by ELISA. n = 3 for all experiments, SD is shown, statistical significance (*p < 0.05, **p < 0.01) was determined by Student’s t Test.
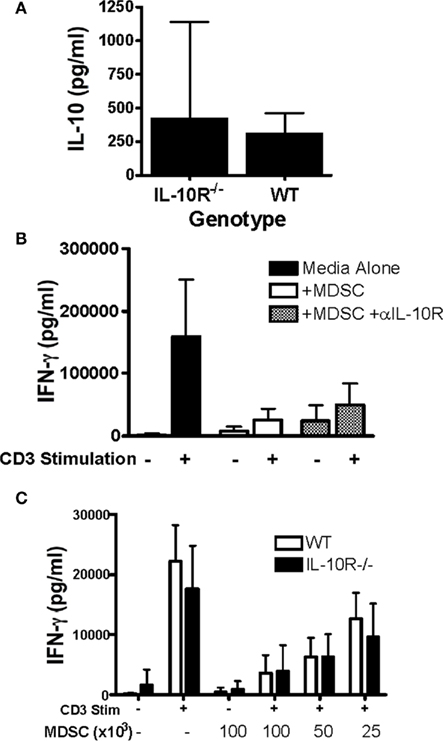
Figure 4. In vitro suppression does not require IL-10 production by MDSC. (A) IL-10 production from sorted WT and IL-10R−/− CD11b+Ly6G− MDSC derived from chimeric mice was assessed by ELISA from 48-h culture supernatants. To assess the requirement for IL-10 in the suppressive capacity of MDSC, CD11b+Ly6G− MDSCs were (B) selected and added at 1:10 ratio with or without anti-IL-10R (100 μg/ml), or (C) titrated as indicated into mixed splenocyte reactions with either WT or IL-10R−/− splenocytes stimulated with anti-CD3 (1 μg/ml). Interferon-γ secretion into the supernatants was analyzed by ELISA. n ≥ 3 for (A,B) and n ≥ 2 for (C), SD is shown.
Therapeutic Blockade of the IL-10 Receptor Inhibits Ovarian Tumor Progression
The critical role for IL-10 signaling to the MDSC tumor-promoting phenotype and function led us to hypothesize that it might be an efficacious therapeutic target to inhibit tumor progression. We have previously shown that elimination of MDSCs in the ovarian tumor microenvironment reduces tumor progression and restores immune function (Bak et al., 2007), therefore we hypothesized that IL-10 receptor blockade may provide an alternative method to induce the therapeutic efficacy observed with MDSC depletion. To determine if IL-10 signaling blockade inhibited ID8 progression, we measured cellularity in the tumor-associated ascites and, most importantly, survival. Mice were injected with ID8 tumors and subsequently received five intraperitoneal injections of anti-IL-10R antibody, spaced every third day, starting 14 days post-tumor inoculation (Figure 5A). At 1-week post-treatment, control mice had developed discernable ascites. However, compared to mice that received PBS or IgG injections, treated mice had significantly less total cellularity (Figure 5B) and significantly fewer MDSCs in the ascites (Figure 5C). Additionally, cytokine analyses of the peritoneal lavages indicate that IL-10 levels were reduced in the treated mice (Figures 5D,F), which is in accord with the reduction in IL-10-producing MDSCs within the peritoneum. Importantly, treated mice had significantly increased IL-12p70 levels, indicating an important pro-inflammatory alteration in the peritoneum of the treated mice (Figures 5E,G).
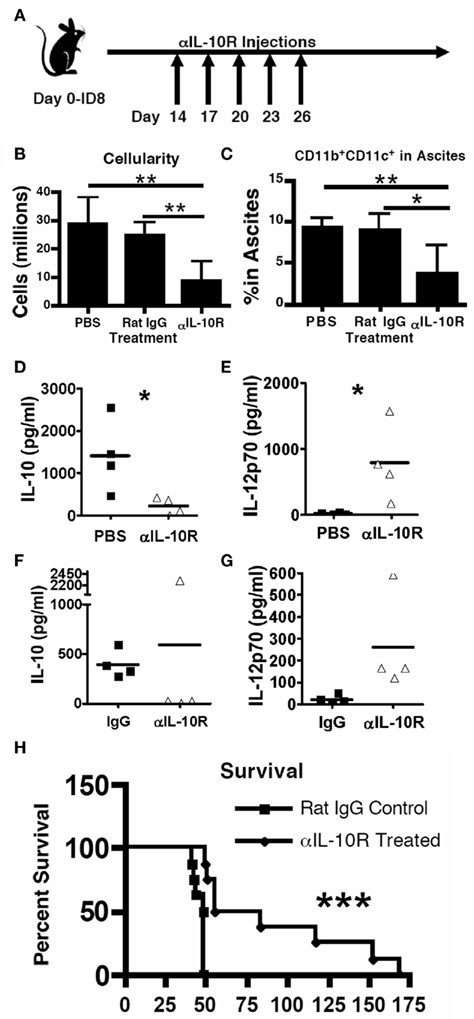
Figure 5. Therapeutic blockade of the IL-10 receptor inhibits ovarian tumor progression. (A) ID8 tumor-bearing mice received PBS, 100 μg rat IgG, or 100 μg anti-IL-10 receptor antibody every third day for 2 weeks starting 2 weeks post-ID8 injection. Mice were analyzed 2 weeks later for total cellularity (B), presence of the CD11b+CD11c+ MDSC in the peritoneum (C), and for the presence of IL-10 (D,F), and IL-12p70 (E,G) in the ascites (Luminex analysis); similar results were observed regardless of whether the control injection was PBS (D,E) or non-specific IgG (F,G). (H) Mice receiving 2 weeks of either rat IgG or anti-IL-10 receptor antibody starting 1 week after ID8 injection. Statistical significance (*p < 0.05, **p < 0.01, ***p < 0.0001) was determined by Student’s t Test for (B–D), and by Kaplan–Meier analysis for survival (n ≥ 8).
To determine if IL-10 receptor blockade provides a therapeutic survival advantage in tumor-bearing mice, we used the same injection strategy, instead starting 1-week post-tumor inoculation (similar strategy as in Figure 5A). Mice that received the IL-10R blocking therapy showed significantly and substantially enhanced survival over mice receiving injections of rat IgG control antibody (Figure 5H). These data support a critical role for MDSC in tumor progression and identify a crucial role for the MDSC-produced IL-10 in the tumor microenvironment. Importantly, this is the first demonstration that IL-10 receptor blockade has therapeutic efficacy against an aggressive model of ovarian cancer.
We hypothesized that the MDSC reduction (Figure 5C) was due either to antibody-mediated depletion of this cell population, or a reduction in the recruitment of this population. Confirming previous reports that the anti-IL-10R antibody is blocking but non-depleting, naïve mice that received an i.p. injection of 100 μg anti-IL-10R antibody had no measurable cellular depletion (Figure 6); as a positive control, anti-CD8 depleted mice exhibited a complete eradication of the CD8+ T cell population.
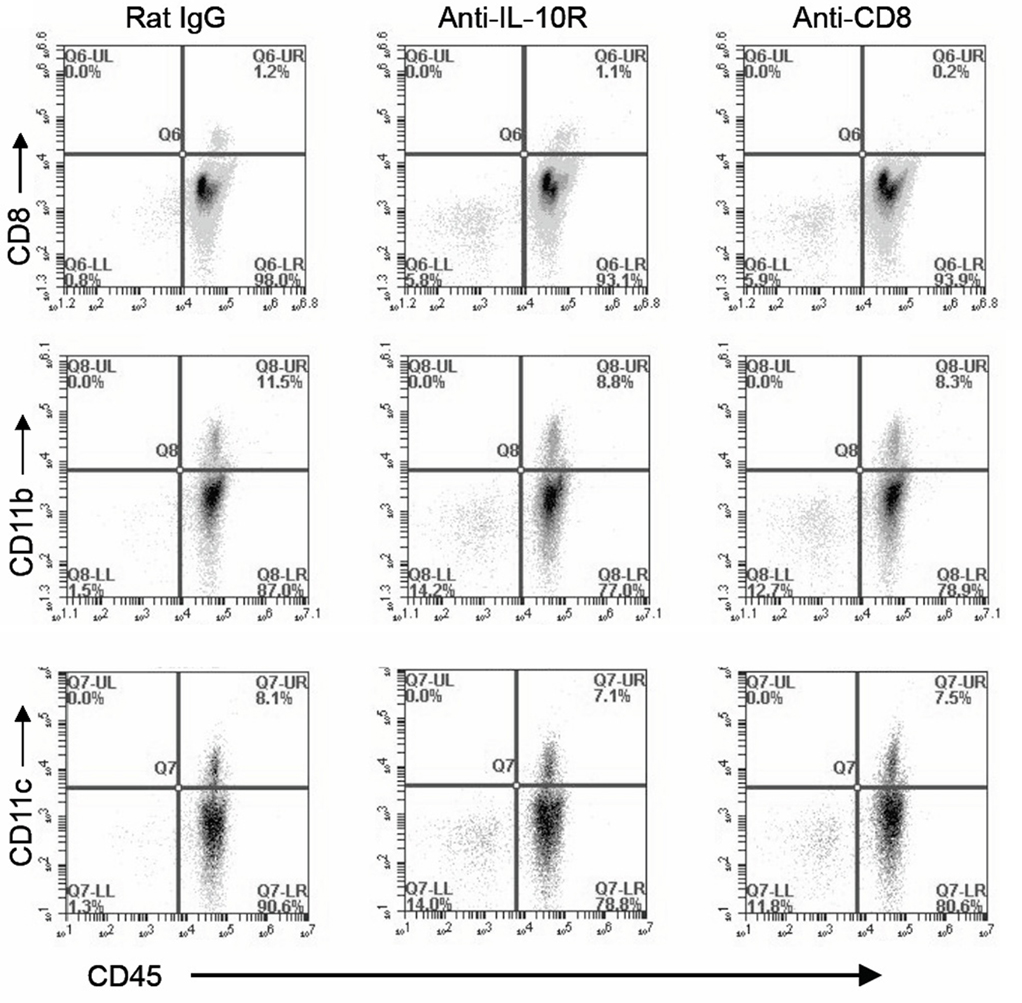
Figure 6. The anti-IL-10R antibody does not induce cellular depletion. Naïve mice were injected i.p. with 100 ng of either depleting anti-CD8 antibody, anti-IL-10R antibody, or rat IgG control antibody. Mice were subsequently sacrificed and peritoneal lavages were collected, stained for CD45, CD8, CD11b, or CD11c as indicated, and analyzed by FACS.
IL-10 Signaling Modulates T Cell Phenotypes in the Tumor Microenvironment
While it is known that the MDSC population in the tumor can produce several immunosuppressive molecules that potentially alter T cell activity, the direct effect(s) from the high levels of IL-10 present in the tumor microenvironment on T cells is unknown and cannot be ascertained from the previous anti-IL-10R experiments. Two alternate, and not mutually exclusive possibilities include that any differential in T cell anti-tumor function could be due to the direct action of IL-10 on the T cells, or indirect effects on T cells mediated through altered MDSC function.
We next determined if IL-10 in the tumor microenvironment has a direct effect on infiltrating T cells or only affects them by modulating MDSC function. We returned to the IL-10R chimeric mice to evaluate differences between IL-10R knockout and wild-type T cells derived from the same tumor-bearing mouse. Analogous to the methods used in Figure 3 to identify differential, IL-10R-dependent, MDSC activity, we analyzed PD-1 and CD62L levels on T cells from the ascites of chimeric tumor-bearing mice as markers of activation status. In normal tumor-bearing mice, T cells in the ascites are skewed CD62Llo (Figure 7A). While the population of CD62Llo T cells in the wild-type mice might suggest further differentiation and a more activated status, these cells are clearly ineffective at responding to the tumor. In the ascites of chimeric mice, the percent of CD62LloCD8+ T cells from the IL-10R−/− cells, identified by expression of the CD45.1 congenic marker, was significantly less than from the wild-type cells (Figures 7B,C left). In contrast, the peritoneal CD8+ T cells from naïve chimeric mice from both the wild-type and IL-10R knockout background were dominantly CD62Lhi, indicating that this phenotype is specific to the tumor environment and is not inherent in the IL-10R knockout T cells (Figure 7D). Similar trends were observed in the CD4+ T cell pools of chimeras although the differences in CD4 T cells expressing low levels of CD62L did not reach significance (Figures 7A,C,D right panels). Additionally, we did not detect differences in PD-1 expression regardless of IL-10R expression on CD8+ T cells in the ascites of tumor-bearing chimeric mice (Figure 7E). To determine if IL-10 is sufficient to cause the down-regulation of CD62L in a purified system, we selected CD8 T cells from the spleens of naïve IL-10R knockout and wild-type mice, and found that 72-h IL-10 stimulation in vitro was able to induce a modest down-regulation of CD62L surface expression on the CD62L high cells from wild-type, but not IL-10R knockout spleens (Figures 7F–H); the in vitro changes were more modest than those in vivo (Figures 7A,B), which suggests additional in vivo factors also influence this effect. Matsuzaki et al. (2010) recently described a population of antigen specific tumor-associated CD8+ T cells in human epithelial ovarian cancer, with impaired effector functions, and showed that IL-10 was able to induce the expression of the inhibitory marker LAG-3 on healthy peripheral blood lymphocytes (PBL). We further characterized the T cells in chimeric mice by analyzing LAG-3 and CD44 expression, and found the CD44+CD8+ population from the IL-10R−/− background expressed significantly less of the inhibitory marker LAG-3 than wild-type cells in the same tumor (Figures 8A,B). Furthermore, upon gating on the CD44+CD62L populations of each genotype (Figure 8C, Gate 1 and 2), we find that LAG-3 expression on the CD62Llo cells (Figure 8C, right panels) was consistent with the CD62Llo population we observed at an increased frequency in the wild-type population in Figure 7C. Furthermore, T cells from the IL-10R−/− background that expressed LAG-3 expressed it at lower levels by MFI (Figure 8C, gate 1). This indicates that IL-10 signaling is required for normal generation of a CD62LloLAG-3+CD8+CD44+ population in vivo at the tumor site, and demonstrates that IL-10 signaling can directly influence T cells by driving LAG-3 expression in the murine tumor. To determine if LAG-3 expression correlates with functional differences from T cell populations, we comparatively assessed LAG-3-expressing and -absent T cells from tumor-bearing chimeric mice for IFN-γ production. We found that directly ex vivo, there are no significant differences in the frequency of IFN-γ producing CD8 T cells from the LAG-3 positive and negative populations from either IL-10R knockout cells or WT cells (Figure 8D left side), which may represent suppression enforced by other means in the tumor. Following a 5-h anti-CD3 stimulation we did detect significantly more IFN-γ producing cells in the LAG-3 negative T cells compared to the LAG-3 positive cells (Figure 8D right side). While similar proportions of the wild-type and IL-10R knockout CD8 T cell populations were capable of producing IFN-γ, the increased ability of the LAG-3 negative cells, which are more prevalent in the IL-10R knockout compartment, indicates that IL-10 drives the development of a LAG-3 expressing population of T cells whose production of functional IFN-γ is impeded compared to the LAG-3 negative cells.
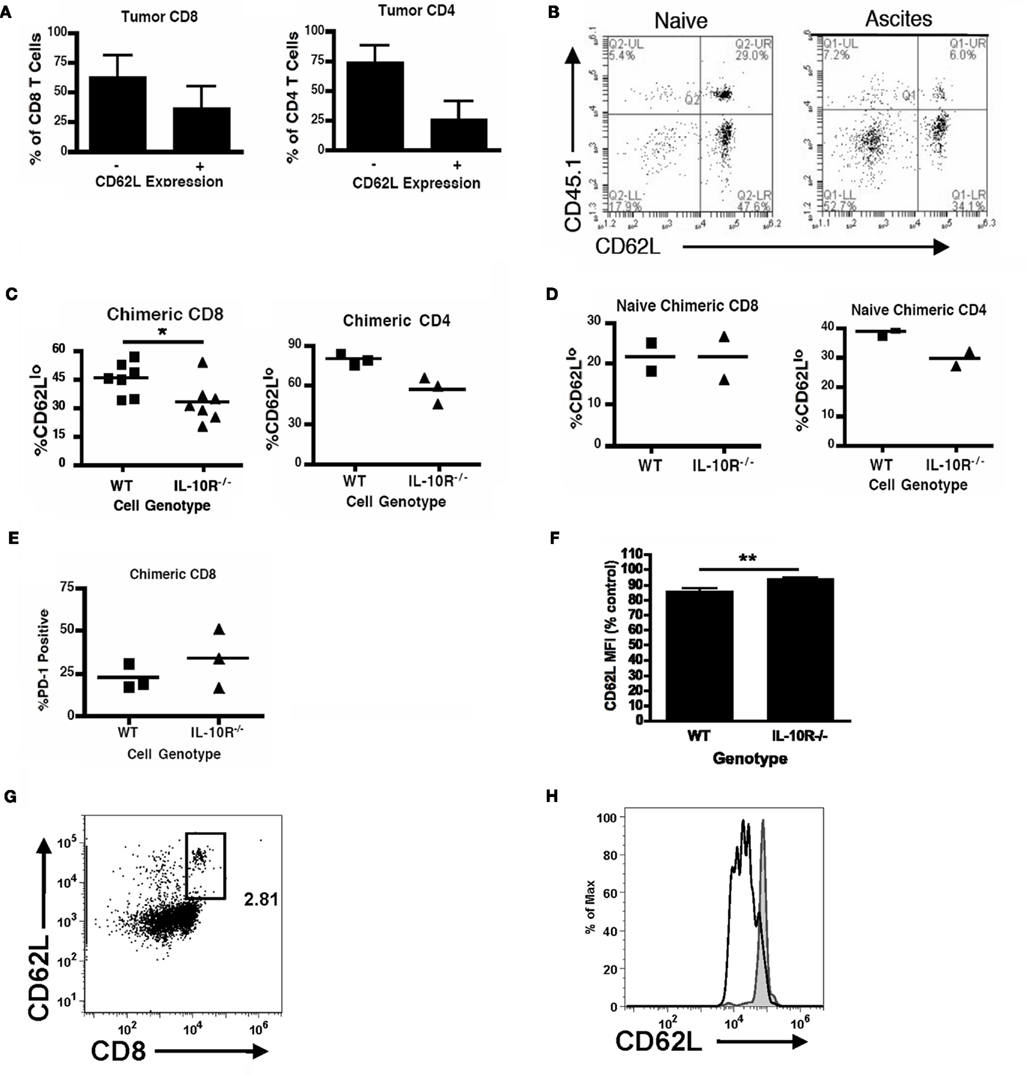
Figure 7. IL-10 signaling modulates T cell phenotypes in the ovarian tumor microenvironment. (A) The CD62L status of peritoneal CD8+ and CD4+ T cells from wild-type mice bearing ID8 tumors was analyzed (n = 6). Naïve [(B), left] and tumor-bearing [(B), right] mixed (IL-10R−/− and wild-type) chimeric mice were analyzed for CD62L expression on IL-10R-deficient (CD45.1+) or -expressing (CD45.2+) congenically marked CD8 T cells from the peritoneum. (C) The percentages of CD8 (left) and CD4 (right) T cells expressing low levels of CD62L from the wild-type or IL-10R−/− background were quantified. (D) CD62Llo percentages of CD8+ (left) and CD4+ (right) T cells from naïve chimeric mice from the wild-type and IL-10R knockout compartments were assessed. (E) Additionally, CD8 T cells from tumor-bearing mixed chimeric mice were analyzed for the phenotypic marker PD-1. (F) Relative CD62L status of WT and IL-10R−/− CD8 T cells stimulated with 10 ng/ml IL-10 for 72-h, assessed by FACS analyses (n ≥ 3). (G) Representative CD8 CD62L staining with gate for CD62Lhi population shown in (H). (H) CD62L on IL-10-stimulated (black empty histogram) and -unstimulated (gray filled histogram) CD8 T cells. Statistical significance (*p < 0.05, **p < 0.01, ***p < 0.0001) was determined by Student’s t Test.
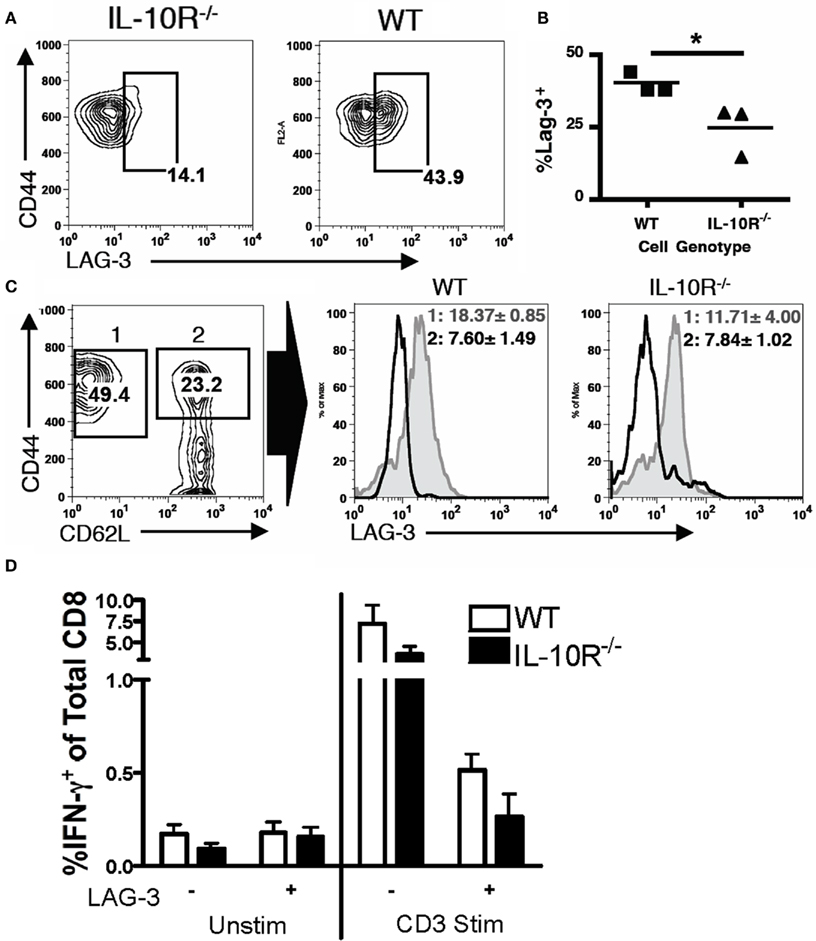
Figure 8. CD8 T cells require IL-10 signaling to obtain tumor-associated LAG-3 expression levels. (A,B) The percentage of LAG-3+ cells from the CD8+CD44+ tumor-associated T cells of each background was analyzed. (C) CD8+ tumor-associated T cells in a chimeric mouse were analyzed for CD44 and CD62L (left, wild-type T cells shown). CD44 high cells were gated as shown, and the LAG-3 expression on the CD62L hi (black line) or lo (gray filled line) populations was determined for each genotype (right panels). The LAG-3 MFI (geom. mean) with SD for the respective CD62L hi and lo populations is shown. (D) Percentage of IFN-γ producing cells in the CD8 population from WT or IL-10R−/− backgrounds, gated on LAG-3 expression, unstimulated (left) or stimulated for 5 h with 10 μg/ml anti-CD3 (right; n = 3). Statistical significance (*p < 0.05, **p < 0.01, ***p < 0.0001) was determined by Student’s t Test.
Therapeutic Efficacy of IL-10R Blockade Requires T Cells
Our previous studies demonstrated systemic MDSC-mediated suppression of T cells in sites distal to the tumor, including the spleen (Peter et al., 2009). To ascertain if the IL-10R therapy (Figure 5), which reduced the levels of MDSCs accumulating within the tumor microenvironment, impacted the ability of T cells to respond in these mice, we analyzed splenic CD8+ T cells from mice treated with IL-10R antibody or PBS starting 2 weeks post-tumor injection by stimulating ex vivo and analyzing for intracellular IFN-γ production. Significantly more CD8+ T cells from the treated mice produced IFN-γ than the same cells in control IgG treated mice (Figure 9A). Therefore, even in mice treated after 2 weeks of tumor progression, therapy was able to restore systemic T cell function.
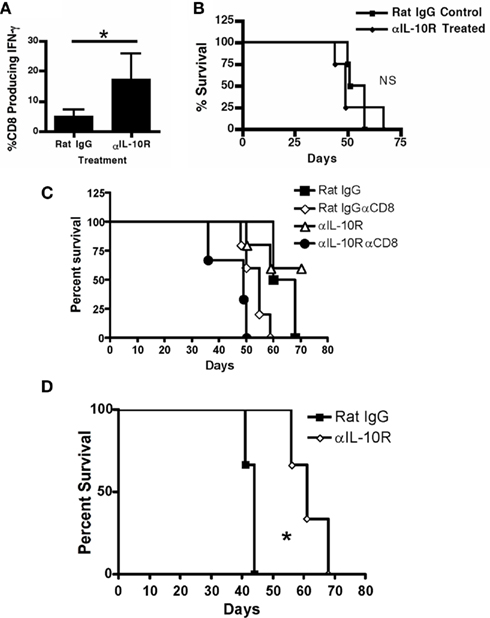
Figure 9. Therapeutic efficacy of IL-10R blockade requires T cells. (A) Splenic CD8+ T cells from treated mice (treatment scheme from Figure 4) were analyzed by intracellular staining for IFN-γ production after anti-CD3 stimulation. (B) Tumor-bearing TCRα knockout mice were treated with anti-IL-10R or, rat IgG 1 week after tumor injection every other day for 2 weeks and then survival was assessed. (C) Tumor-bearing WT mice, or WT mice receiving weekly injections of CD8 depleting antibody, were treated with anti-IL-10R or rat IgG 1 week after tumor injection every other day for 2 weeks and then survival was assessed. (D) Mixed bone marrow chimeric (IL-10R knockout: TCRα knockout, 20:80 respectively) mice were treated with anti-IL-10R or rat IgG 1 week after tumor injection every other day for 2 weeks and then survival was assessed. Statistical significance (*p < 0.05) was determined by Student’s t Test and by Kaplan–Meier analysis for survival. NS, not significant.
Since IL-10R blockade is able to reduce systemic suppression in tumor-bearing mice, and the phenotypic changes on T cells seen in the chimeric mice show direct IL-10 influence on T cells, we tested the therapy on TCRα knockout mice, devoid of both CD4+ and CD8+ T cells. The knockout mice received the same anti-IL-10R treatment regimen as in Figure 5D starting 1 week after tumor injection. The lack of T cells resulted in no significant difference in tumor growth or survival in the TCRα knockout mice regardless of whether they received the IL-10R therapy (Figure 9B). To further clarify the requirement for T cells in the efficacy of the IL-10R therapy, we repeated the therapy in mice receiving concurrent injections of CD8 depleting antibody (Figure 9C). Therapeutic efficacy is lost in mice without CD8 T cells, in demonstration of a direct requirement for CD8 T cells in the efficacy of the IL-10 receptor blocking therapy. To further dissect the independent effects of IL-10 modulation of MDSC accumulation and function from IL-10 alterations detected on T cells, we generated chimeric mice reconstituted with an 20:80 ratio of bone marrow from IL-10R knockout mice and TCRα knockout mice, respectively, which yields mice that have both wild-type and IL-10R knockout MDSC, but all T cells are IL-10R deficient. Survival studies of these tumor-bearing mice indicate that loss of IL-10R on T cells does not affect tumor growth or mouse survival since the tumors in these mice progressed with expected kinetics (Figure 9D). The ability of IL-10-ignorant T cells in these mice to confer survival indicates that the functionality of the T cells in these mice is likely being inhibited through additional mechanisms. Furthermore, the anti-IL-10R therapy was still sufficient to extend survival in these mice (Figure 9D), indicating that the observed IL-10 effects on T cells are secondary to the role of IL-10 in MDSC function. This demonstrates that IL-10 can influence T cell phenotype and function through both IL-10-driven MDSC inhibition and direct IL-10 receptor ligation resulting in an ineffective T cell pool incapable of responding to the tumor.
Discussion
Myeloid-derived suppressor cells and the cytokine IL-10 are emerging as key immunosuppressive mediators and enforcers present in a myriad of cancers and in other chronic diseases. A preponderance of data from a variety of systems now support that both of these modulators are potently immunosuppressive in vitro, and that their presence corresponds with tumor progression and severity in vivo. Specifically, in both murine models of ovarian cancer and in the clinical disease the tumor recruits massive numbers of immunosuppressive leukocytes into the peritoneal ascites, and IL-10 levels correlate with clinical disease severity (Loercher et al., 1999; Santin et al., 2001; Kryczek et al., 2006b; Mustea et al., 2009; Nowak et al., 2010). Despite this, the precise contributions and roles of MDSCs and IL-10 within the in vivo tumor microenvironment remain indistinct, in regard to IL-10 it is not clear if there is functional redundancy with other immunosuppressive molecules and, perhaps most importantly, the interrelationship between the MDSC and IL-10 immunosuppressive networks and mechanisms are unknown. Here we demonstrate that IL-10 production by MDSCs, and MDSC responsiveness to IL-10, plays a critical role in the failure of the immune system to control ovarian tumor growth, resulting from a breakdown in multiple integral components required for effective anti-tumor immunity.
It is well known that MDSC are involved in a plethora of diseases and disease models including infection (Sander et al., 2010), cancer (Marigo et al., 2008; Ostrand-Rosenberg and Sinha, 2009), transplantation (Adeegbe et al., 2010; Highfill et al., 2010), and autoimmunity (Yin et al., 2010), and are thought to provide specific inhibition of immune responses by rendering T cells impotent through production of a number of inhibitory factors such as iNOS, arginase, and peroxynitrites (Ochoa et al., 2007; Marigo et al., 2008; Gabrilovich and Nagaraj, 2009). Less understood are the specific signals required for the generation of MDSC, however some of the key factors in this process are now beginning to be identified. IL-6, G-CSF, and GM-CSF have been used in the in vitro generation of MDSC (Dolcetti et al., 2010; Lechner et al., 2010) and additional factors that appear to be sufficient to regulate aspects of MDSC expansion and function include COX-2, PGE, and VEGF (Marigo et al., 2008; Ribechini et al., 2010). While some of these signals that are sufficient to induce MDSC phenotype and function have been identified, the signals that are necessary for the in vivo generation of MDSC and, correspondingly, those that could be used to inhibit MDSC generation or function, are not at all clear. Previous in vitro experiments support that IL-10 is capable of modulating myeloid activation states, and it has been hypothesized that IL-10 may play a regulatory role in MDSC biology (Sinha et al., 2007; Ribechini et al., 2010), however the specific requirements for IL-10 for in vivo MDSC programming and function have not previously been explored. Here we demonstrate that IL-10 is a requisite and direct functional contributor that establishes the suppressive phenotype and function of MDSCs. We have previously reported the presence of a massive MDSC population in the ID8 ovarian cancer model capable of suppressing in vitro T cell responses and that are required in vivo for normal tumor progression (Bak et al., 2007, 2008). With the use of chimeric mice that contain both IL-10-responsive and -unresponsive MDSC, we now provide an in vivo demonstration that loss of IL-10 signaling can singularly shape the function of these MDSC and thereby functionally tilt the balance of the host environment in favor of tumor growth. This experimental model represents the first in vivo demonstration that concurrent MDSC production of, and responsiveness to, IL-10 is necessary for enforcement of immunosuppression by these cells and elucidates a novel paracrine MDSC IL-10 signaling network in ovarian cancer. We propose that the IL-10 pathway represents a potentially efficacious target in the diseases listed above where MDSC related disease biology could be altered by modulation of MDSC function.
To determine the therapeutic efficacy of blockade of the tumor IL-10 network, we used anti-IL-10 receptor antibody to block IL-10 signaling during tumor progression. This was deemed preferable to using IL-10 or IL-10 receptor knockout mice since antibody blockade is therapeutically relevant; and these knockout mice have well-documented inherent inflammation (Kuhn et al., 1993; Spencer et al., 1998). Mice treated i.p. with anti-IL-10R antibody show reduced tumor burden and peritoneal cellularity and, most importantly, extended survival (Figure 5). Since our STAT3 phosphorylation analyses only detected CD45+ cells in the ascites responding to IL-10 stimulation, we conclude that therapeutic efficacy lies in alleviation of suppression of the immune system rather than a direct effect on tumor cell growth or survival. The improved IFN-γ responsiveness from CD8+ splenic T cells in treated mice led us to ask if IL-10 was mediating these effects solely through a myeloid intermediary or if IL-10 could also induce changes in T cells. The substantially altered CD62L and LAG-3 expression from IL-10R knockout T cells in the ascites of the IL-10R chimeric mice provide formal evidence of IL-10-mediated phenotypic changes on T cells in the ascites of tumor-bearing mice. Recent reports suggest that CD62L-expressing T cells might represent less differentiated early effectors with greater proliferation capacity and increased survival capabilities (Klebanoff et al., 2005; Gattinoni et al., 2006; Diaz-Montero et al., 2008). Additionally, reports indicate that skewing of CD62Llo expression corresponds with MDSC levels, and is proposed to limit T cell migration into sites of tumor growth, thereby reducing T cell responsiveness (Hanson et al., 2009). Based on the CD62L skewing on IL-10R−/− T cells in chimeric mice, MDSC-produced IL-10 may represent a potential mechanism for control of CD62L expression on T cells and therefore the ability of T cells to mount an effective immune response against the tumor. While data presented here indicates that IL-10 signaling networks as a whole are crucial for the systematic inhibition of T cell responsiveness, and blockade of these is sufficient to restore T cell functionality, the individual contribution of IL-10 effects on myeloid activation versus direct effects on T cells remains to be elucidated.
The highly immunosuppressive microenvironment established by progressive ovarian tumors is an important and often unaccounted-for obstacle to the success of many immunotherapeutic strategies. Previous studies have shown that elimination of suppressive, tumor-supporting leukocytes in the tumor can be efficacious (Bak et al., 2007; Huarte et al., 2008; Vincent et al., 2010), however the identification of selective pathways to neutralize their suppressive capacity may provide quicker and more specific therapies. Of direct relevance, studies taking advantage of the phenotypic plasticity of myeloid cells by re-activating them through TLR and CD40 (Scarlett et al., 2009), or block suppressive mechanisms through phospho-diesterase-5 inhibition (Serafini et al., 2006) or triterpenoid (Nagaraj et al., 2010) have had considerable success. Thus, the identification of IL-10 as a master regulator of the suppressive phenotype of the tumor is of critical importance. Notably, therapeutic IL-10 pathway inhibitors have been developed and established as safe in clinical trials for autoimmunity (Llorente et al., 2000). Data herein identify the IL-10 pathway as a non-redundant network critical to ovarian tumor-associated immunosuppression and, concomitantly, as a therapeutic target that may enhance the efficacy of current and emergent immunotherapies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank the Dartmouth Englert Cell Analysis Laboratory for assistance with FACS, the Dartmouth IML for help with Luminex analyses, and Drs. Mary Jo Turk and Fekl S. Leib for helpful reagents and discussions. This research was supported by NIH grant R01 AI067405 and American Cancer Society grant RSG-10-229-01-LIB (Brent Berwin) and T32 GM08704 (Kevin M. Hart).
References
Adeegbe, D., Serafini, P., Bronte, V., Zoso, A., Ricordi, C., and Inverardi, L. (2010). In vivo induction of myeloid suppressor cells and CD4Foxp3 T regulatory cells prolongs skin allograft survival in mice. Cell Transplant. (in press).
Bak, S. P., Alonso, A., Turk, M. J., and Berwin, B. (2008). Murine ovarian cancer vascular leukocytes require arginase-1 activity for T cell suppression. Mol. Immunol. 46, 258–268.
Bak, S. P., Walters, J. J., Takeya, M., Conejo-Garcia, J. R., and Berwin, B. L. (2007). Scavenger receptor-A-targeted leukocyte depletion inhibits peritoneal ovarian tumor progression. Cancer Res. 67, 4783–4789.
Berger, S., Siegert, A., Denkert, C., Kobel, M., and Hauptmann, S. (2001). Interleukin-10 in serous ovarian carcinoma cell lines. Cancer Immunol. Immunother. 50, 328–333.
Burnett, S. H., Kershen, E. J., Zhang, J., Zeng, L., Straley, S. C., Kaplan, A. M., and Cohen, D. A. (2004). Conditional macrophage ablation in transgenic mice expressing a Fas-based suicide gene. J. Leukoc. Biol. 75, 612–623.
Carr, T. M., Adair, S. J., Fink, M. J., and Hogan, K. T. (2008). Immunological profiling of a panel of human ovarian cancer cell lines. Cancer Immunol. Immunother. 57, 31–42.
Conejo-Garcia, J. R., Benencia, F., Courreges, M. C., Kang, E., Mohamed-Hadley, A., Buckanovich, R. J., Holtz, D. O., Jenkins, A., Na, H., Zhang, L., Wagner, D. S., Katsaros, D., Caroll, R., and Coukos, G. (2004). Tumor-infiltrating dendritic cell precursors recruited by a beta-defensin contribute to vasculogenesis under the influence of Vegf-A. Nat. Med. 10, 950–958.
Diaz-Montero, C. M., El Naggar, S., Al Khami, A., El Naggar, R., Montero, A. J., Cole, D. J., and Salem, M. L. (2008). Priming of naive CD8+ T cells in the presence of IL-12 selectively enhances the survival of CD8+ CD62Lhi cells and results in superior anti-tumor activity in a tolerogenic murine model. Cancer Immunol. Immunother. 57, 563–572.
Dolcetti, L., Peranzoni, E., Ugel, S., Marigo, I., Fernandez Gomez, A., Mesa, C., Geilich, M., Winkels, G., Traggiai, E., Casati, A., Grassi, F., and Bronte, V. (2010). Hierarchy of immunosuppressive strength among myeloid-derived suppressor cell subsets is determined by GM-CSF. Eur. J. Immunol. 40, 22–35.
Donnelly, R. P., Dickensheets, H., and Finbloom, D. S. (1999). The interleukin-10 signal transduction pathway and regulation of gene expression in mononuclear phagocytes. J. Interferon Cytokine Res. 19, 563–573.
Fleming, T. J., Fleming, M. L., and Malek, T. R. (1993). Selective expression of Ly-6G on myeloid lineage cells in mouse bone marrow. RB6-8C5 mAb to granulocyte-differentiation antigen (Gr-1) detects members of the Ly-6 family. J. Immunol. 151, 2399–2408.
Fontenot, J. D., Gavin, M. A., and Rudensky, A. Y. (2003). Foxp3 programs the development and function of CD4+ CD25+ regulatory T cells. Nat. Immunol. 4, 330–336.
Friedline, R. H., Brown, D. S., Nguyen, H., Kornfeld, H., Lee, J., Zhang, Y., Appleby, M., Der, S. D., Kang, J., and Chambers, C. A. (2009). CD4+ regulatory T cells require CTLA-4 for the maintenance of systemic tolerance. J. Exp. Med. 206, 421–434.
Gabrilovich, D. I., and Nagaraj, S. (2009). Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 9, 162–174.
Gattinoni, L., Powell, D. J. Jr., Rosenberg, S. A., and Restifo, N. P. (2006). Adoptive immunotherapy for cancer: building on success. Nat. Rev. Immunol. 6, 383–393.
Geissmann, F., Jung, S., and Littman, D. R. (2003). Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 19, 71–82.
Giuntoli, R. L. II, Webb, T. J., Zoso, A., Rogers, O., Diaz-Montes, T. P., Bristow, R. E., and Oelke, M. (2009). Ovarian cancer-associated ascites demonstrates altered immune environment: implications for antitumor immunity. Anticancer Res. 29, 2875–2884.
Gotlieb, W. H., Abrams, J. S., Watson, J. M., Velu, T. J., Berek, J. S., and Martinez-Maza, O. (1992). Presence of interleukin 10 (IL-10) in the ascites of patients with ovarian and other intra-abdominal cancers. Cytokine 4, 385–390.
Hanson, E. M., Clements, V. K., Sinha, P., Ilkovitch, D., and Ostrand-Rosenberg, S. (2009). Myeloid-derived suppressor cells down-regulate L-selectin expression on CD4+ and CD8+ T cells. J. Immunol. 183, 937–944.
Hart, K. M., Bak, S. P., Alonso, A., and Berwin, B. (2009). Phenotypic and functional delineation of murine CX(3)CR1 monocyte-derived cells in ovarian cancer. Neoplasia 11, 564–573, 561 following 573.
Highfill, S. L., Rodriguez, P. C., Zhou, Q., Goetz, C. A., Koehn, B. H., Veenstra, R., Taylor, P. A., Panoskaltsis-Mortari, A., Serody, J. S., Munn, D. H., Tolar, J., Ochoa, A. C., and Blazar, B. R. (2010). Bone marrow myeloid-derived suppressor cells (MDSCs) inhibit graft-versus-host disease (GVHD) via an arginase-1-dependent mechanism that is up-regulated by interleukin-13. Blood 116, 5738–5747.
Huarte, E., Cubillos-Ruiz, J. R., Nesbeth, Y. C., Scarlett, U. K., Martinez, D. G., Buckanovich, R. J., Benencia, F., Stan, R. V., Keler, T., Sarobe, P., Sentman, C. L., and Conejo-Garcia, J. R. (2008). Depletion of dendritic cells delays ovarian cancer progression by boosting antitumor immunity. Cancer Res. 68, 7684–7691.
Kawaida, H., Kono, K., Takahashi, A., Sugai, H., Mimura, K., Miyagawa, N., Omata, H., Ooi, A., and Fujii, H. (2005). Distribution of CD4+ CD25high regulatory T-cells in tumor-draining lymph nodes in patients with gastric cancer. J. Surg. Res. 124, 151–157.
Klebanoff, C. A., Gattinoni, L., Torabi-Parizi, P., Kerstann, K., Cardones, A. R., Finkelstein, S. E., Palmer, D. C., Antony, P. A., Hwang, S. T., Rosenberg, S. A., Waldmann, T. A., and Restifo, N. P. (2005). Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc. Natl. Acad. Sci. U.S.A. 102, 9571–9576.
Kryczek, I., Wei, S., Zou, L., Zhu, G., Mottram, P., Xu, H., Chen, L., and Zou, W. (2006a). Cutting edge: induction of B7-H4 on APCs through IL-10: novel suppressive mode for regulatory T cells. J. Immunol. 177, 40–44.
Kryczek, I., Zou, L., Rodriguez, P., Zhu, G., Wei, S., Mottram, P., Brumlik, M., Cheng, P., Curiel, T., Myers, L., Lackner, A., Alvarez, X., Ochoa, A., Chen, L., and Zou, W. (2006b). B7-H4 expression identifies a novel suppressive macrophage population in human ovarian carcinoma. J. Exp. Med. 203, 871–881.
Kuhn, R., Lohler, J., Rennick, D., Rajewsky, K., and Muller, W. (1993). Interleukin-10-deficient mice develop chronic enterocolitis. Cell 75, 263–274.
Lech-Maranda, E., Bienvenu, J., Michallet, A. S., Houot, R., Robak, T., Coiffier, B., and Salles, G. (2006). Elevated IL-10 plasma levels correlate with poor prognosis in diffuse large B-cell lymphoma. Eur. Cytokine Netw. 17, 60–66.
Lechner, M. G., Liebertz, D. J., and Epstein, A. L. (2010). Characterization of cytokine-induced myeloid-derived suppressor cells from normal human peripheral blood mononuclear cells. J. Immunol. 185, 2273–2284.
Liyanage, U. K., Moore, T. T., Joo, H. G., Tanaka, Y., Herrmann, V., Doherty, G., Drebin, J. A., Strasberg, S. M., Eberlein, T. J., Goedegebuure, P. S., and Linehan, D. C. (2002). Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J. Immunol. 169, 2756–2761.
Llorente, L., Richaud-Patin, Y., Garcia-Padilla, C., Claret, E., Jakez-Ocampo, J., Cardiel, M. H., Alcocer-Varela, J., Grangeot-Keros, L., Alarcon-Segovia, D., Wijdenes, J., Galanaud, P., and Emilie, D. (2000). Clinical and biologic effects of anti-interleukin-10 monoclonal antibody administration in systemic lupus erythematosus. Arthritis Rheum. 43, 1790–1800.
Loercher, A. E., Nash, M. A., Kavanagh, J. J., Platsoucas, C. D., and Freedman, R. S. (1999). Identification of an IL-10-producing HLA-DR-negative monocyte subset in the malignant ascites of patients with ovarian carcinoma that inhibits cytokine protein expression and proliferation of autologous T cells. J. Immunol. 163, 6251–6260.
Marigo, I., Dolcetti, L., Serafini, P., Zanovello, P., and Bronte, V. (2008). Tumor-induced tolerance and immune suppression by myeloid derived suppressor cells. Immunol. Rev. 222, 162–179.
Marshall, N. A., Christie, L. E., Munro, L. R., Culligan, D. J., Johnston, P. W., Barker, R. N., and Vickers, M. A. (2004). Immunosuppressive regulatory T cells are abundant in the reactive lymphocytes of Hodgkin lymphoma. Blood 103, 1755–1762.
Matsuzaki, J., Gnjatic, S., Mhawech-Fauceglia, P., Beck, A., Miller, A., Tsuji, T., Eppolito, C., Qian, F., Lele, S., Shrikant, P., Old, L. J., and Odunsi, K. (2010). Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc. Natl. Acad. Sci. U.S.A. 107, 7875–7880.
Maynard, C. L., Harrington, L. E., Janowski, K. M., Oliver, J. R., Zindl, C. L., Rudensky, A. Y., and Weaver, C. T. (2007). Regulatory T cells expressing interleukin 10 develop from Foxp3+ and Foxp3− precursor cells in the absence of interleukin 10. Nat. Immunol. 8, 931–941.
Moore, K. W., De Waal Malefyt, R., Coffman, R. L., and O’garra, A. (2001). Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol. 19, 683–765.
Mosser, D. M., and Zhang, X. (2008). Interleukin-10: new perspectives on an old cytokine. Immunol. Rev. 226, 205–218.
Mougiakakos, D., Choudhury, A., Lladser, A., Kiessling, R., and Johansson, C. C. (2010). Regulatory T cells in cancer. Adv. Cancer Res. 107, 57–117.
Murray, P. J. (2006). Understanding and exploiting the endogenous interleukin-10/STAT3-mediated anti-inflammatory response. Curr. Opin. Pharmacol. 6, 379–386.
Mustea, A., Braicu, E. I., Koensgen, D., Yuan, S., Sun, P. M., Stamatian, F., Lichtenegger, W., Chen, F. C., Chekerov, R., and Sehouli, J. (2009). Monitoring of IL-10 in the serum of patients with advanced ovarian cancer: results from a prospective pilot-study. Cytokine 45, 8–11.
Mustea, A., Konsgen, D., Braicu, E. I., Pirvulescu, C., Sun, P., Sofroni, D., Lichtenegger, W., and Sehouli, J. (2006). Expression of IL-10 in patients with ovarian carcinoma. Anticancer Res. 26, 1715–1718.
Nagaraj, S., Youn, J. I., Weber, H., Iclozan, C., Lu, L., Cotter, M. J., Meyer, C., Becerra, C. R., Fishman, M., Antonia, S., Sporn, M. B., Liby, K. T., Rawal, B., Lee, J. H., and Gabrilovich, D. I. (2010). Anti-inflammatory triterpenoid blocks immune suppressive function of MDSCs and improves immune response in cancer. Clin. Cancer Res. 16, 1812–1823.
Nash, M. A., Lenzi, R., Edwards, C. L., Kavanagh, J. J., Kudelka, A. P., Verschraegen, C. F., Platsoucas, C. D., and Freedman, R. S. (1998). Differential expression of cytokine transcripts in human epithelial ovarian carcinoma by solid tumour specimens, peritoneal exudate cells containing tumour, tumour-infiltrating lymphocyte (TIL)-derived T cell lines and established tumour cell lines. Clin. Exp. Immunol. 112, 172–180.
Nowak, M., Glowacka, E., Szpakowski, M., Szyllo, K., Malinowski, A., Kulig, A., Tchorzewski, H., and Wilczynski, J. (2010). Proinflammatory and immunosuppressive serum, ascites and cyst fluid cytokines in patients with early and advanced ovarian cancer and benign ovarian tumors. Neuro Endocrinol. Lett. 31, 375–383.
Ochoa, A. C., Zea, A. H., Hernandez, C., and Rodriguez, P. C. (2007). Arginase, prostaglandins, and myeloid-derived suppressor cells in renal cell carcinoma. Clin. Cancer Res. 13, 721s–726s.
Ostrand-Rosenberg, S., and Sinha, P. (2009). Myeloid-derived suppressor cells: linking inflammation and cancer. J. Immunol. 182, 4499–4506.
Peter, S., Bak, G., Hart, K., and Berwin, B. (2009). Ovarian tumor-induced T cell suppression is alleviated by vascular leukocyte depletion. Transl. Oncol. 2, 291–299.
Ribechini, E., Greifenberg, V., Sandwick, S., and Lutz, M. B. (2010). Subsets, expansion and activation of myeloid-derived suppressor cells. Med. Microbiol. Immunol. 199, 273–281.
Roby, K. F., Taylor, C. C., Sweetwood, J. P., Cheng, Y., Pace, J. L., Tawfik, O., Persons, D. L., Smith, P. G., and Terranova, P. F. (2000). Development of a syngeneic mouse model for events related to ovarian cancer. Carcinogenesis 21, 585–591.
Sander, L. E., Sackett, S. D., Dierssen, U., Beraza, N., Linke, R. P., Muller, M., Blander, J. M., Tacke, F., and Trautwein, C. (2010). Hepatic acute-phase proteins control innate immune responses during infection by promoting myeloid-derived suppressor cell function. J. Exp. Med. 207, 1453–1464.
Santin, A. D., Bellone, S., Ravaggi, A., Roman, J., Smith, C. V., Pecorelli, S., Cannon, M. J., and Parham, G. P. (2001). Increased levels of interleukin-10 and transforming growth factor-beta in the plasma and ascitic fluid of patients with advanced ovarian cancer. BJOG 108, 804–808.
Scarlett, U. K., Cubillos-Ruiz, J. R., Nesbeth, Y. C., Martinez, D. G., Engle, X., Gewirtz, A. T., Ahonen, C. L., and Conejo-Garcia, J. R. (2009). In situ stimulation of CD40 and toll-like receptor 3 transforms ovarian cancer-infiltrating dendritic cells from immunosuppressive to immunostimulatory cells. Cancer Res. 69, 7329–7337.
Serafini, P., Meckel, K., Kelso, M., Noonan, K., Califano, J., Koch, W., Dolcetti, L., Bronte, V., and Borrello, I. (2006). Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J. Exp. Med. 203, 2691–2702.
Sinha, P., Clements, V. K., Bunt, S. K., Albelda, S. M., and Ostrand-Rosenberg, S. (2007). Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J. Immunol. 179, 977–983.
Spencer, S. D., Di Marco, F., Hooley, J., Pitts-Meek, S., Bauer, M., Ryan, A. M., Sordat, B., Gibbs, V. C., and Aguet, M. (1998). The orphan receptor CRF2-4 is an essential subunit of the interleukin 10 receptor. J. Exp. Med. 187, 571–578.
Vincent, J., Mignot, G., Chalmin, F., Ladoire, S., Bruchard, M., Chevriaux, A., Martin, F., Apetoh, L., Rebe, C., and Ghiringhelli, F. (2010). 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 70, 3052–3061.
Yin, B., Ma, G., Yen, C. Y., Zhou, Z., Wang, G. X., Divino, C. M., Casares, S., Chen, S. H., Yang, W. C., and Pan, P. Y. (2010). Myeloid-derived suppressor cells prevent type 1 diabetes in murine models. J. Immunol. 185, 5828–5834.
Keywords: MDSC, ovarian cancer, IL-10, T cell
Citation: Hart KM, Byrne KT, Molloy MJ, Usherwood EM and Berwin B (2011) IL-10 immunomodulation of myeloid cells regulates a murine model of ovarian cancer. Front. Immun. 2:29. doi: 10.3389/fimmu.2011.00029
Received: 30 March 2011;
Accepted: 07 July 2011;
Published online: 21 July 2011.
Edited by:
Shigeo Koyasu, Keio University, JapanReviewed by:
Toshinori Nakayama, Graduate School of Medicine, JapanYasuharu Nishimura, Kumamoto University, Japan
Copyright: © 2011 Hart, Byrne, Molloy, Usherwood and Berwin. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Brent Berwin, Department of Microbiology and Immunology, Dartmouth Medical Center, HB7556, 1 Medical Center Drive, Lebanon, NH 03756, USA. e-mail:YmVyd2luQGRhcnRtb3V0aC5lZHU=