 Anne L. Fletcher1
Anne L. Fletcher1 Deepali Malhotra1,2 Sophie E. Acton1
Deepali Malhotra1,2 Sophie E. Acton1 Veronika Lukacs-Kornek1 Angelique Bellemare-Pelletier1 Mark Curry3 Myriam Armant4
Veronika Lukacs-Kornek1 Angelique Bellemare-Pelletier1 Mark Curry3 Myriam Armant4 Shannon J. Turley1,5*
Shannon J. Turley1,5*- 1 Department of Cancer Immunology and AIDS, Dana–Farber Cancer Institute, Boston, MA, USA
- 2 Division of Medical Sciences, Harvard Medical School, Boston, MA, USA
- 3 Flow Cytometry Core Facility, Dana–Farber Cancer Institute, Boston, MA, USA
- 4 Center for Human Cell Therapy, Immune Disease Institute, Boston, MA, USA
- 5 Department of Microbiology and Immunobiology, Harvard Medical School, Boston, MA, USA
Within lymph nodes, non-hematopoietic stromal cells organize and interact with leukocytes in an immunologically important manner. In addition to organizing T and B cell segregation and expressing lymphocyte survival factors, several recent studies have shown that lymph node stromal cells shape the naïve T cell repertoire, expressing self-antigens which delete self-reactive T cells in a unique and non-redundant fashion. A fundamental role in peripheral tolerance, in addition to an otherwise extensive functional portfolio, necessitates closer study of lymph node stromal cell subsets using modern immunological techniques; however this has not routinely been possible in the field, due to difficulties reproducibly isolating these rare subsets. Techniques were therefore developed for successful ex vivo and in vitro manipulation and characterization of lymph node stroma. Here we discuss and validate these techniques in mice and humans, and apply them to address several unanswered questions regarding lymph node composition. We explored the steady-state stromal composition of lymph nodes isolated from mice and humans, and found that marginal reticular cells and lymphatic endothelial cells required lymphocytes for their normal maturation in mice. We also report alterations in the proportion and number of fibroblastic reticular cells (FRCs) between skin-draining and mesenteric lymph nodes. Similarly, transcriptional profiling of FRCs revealed changes in cytokine production from these sites. Together, these methods permit highly reproducible stromal cell isolation, sorting, and culture.
Introduction
Lymph nodes are important secondary lymphoid organs located at lymphatic intersections in the body, allowing efficient interaction between antigen-presenting cells and naïve T and B cells and promoting effective initiation of immune responses (Warnock et al., 1998; Mebius, 2003; Katakai et al., 2004a,b; Sixt et al., 2005; Bajenoff et al., 2006; Junt et al., 2008). Lymph node stromal cells also play a crucial and arguably underappreciated role in promoting the maintenance of naïve T cells (Link et al., 2007) and the deletion of autoreactive T cells (Lee et al., 2007; Nichols et al., 2007; Gardner et al., 2008; Magnusson et al., 2008; Cohen et al., 2010; Fletcher et al., 2010).
The structure of the lymph node consists of afferent lymphatics emptying their contents into an “antigen sampling” subcapsular zone (Junt et al., 2008), which meets paracortical B cell follicles surrounding a central cortical T cell zone, in turn leading to a largely macrophage-filled medulla at the lymph node’s efferent face (Mebius, 2003; Willard-Mack, 2006). A conduit network of microchannels also expedites delivery of small proteins and antigens from subcapsule to deep cortex (Sixt et al., 2005). Distinct stromal subtypes impose this structure and maintain each microniche (Bajenoff et al., 2006).
T cell zone fibroblastic reticular cells (FRCs) express chemokines CCL19 and CCL21 to attract T cells and dendritic cells (DCs), and lay down an intricate meshwork of collagens and extracellular matrix on which these cells crawl (Ansel et al., 2000; Luther et al., 2000; Katakai et al., 2004b; Bajenoff et al., 2006; Link et al., 2007; Woolf et al., 2007). Analogous to FRCs but specialized for B cell attraction, follicular dendritic cells (FDCs) are a rare stromal subset specialized for B cell attraction and antigen presentation. FDCs are only found within B cell follicles, which they create through CXCL13 secretion (Cyster et al., 2000). The lymph node capsule is essentially an enlarged lymphatic vessel constructed from lymphatic endothelial cells (LECs), which carry interstitial fluid to the node and empty it into the parenchyma. It filters through the paracortex and cortex, eventually reaching efferent lymphatics in the medulla, which convey lymph further upstream (Mebius, 2003; Willard-Mack, 2006). Blood endothelial cells (BECs) construct cortical blood vessels and capillaries, including high endothelial venules specialized to attract naïve T cells from the bloodstream (Mebius, 2003; Willard-Mack, 2006).
Several other stromal subsets have been reported in the literature but are poorly studied, with their lineages and major functions undefined (reviewed by Fletcher et al., 2011). Marginal reticular cells (MRCs) are a subset of CXCL13-producing stroma growing at the cortical face of the subcapsular sinus (Katakai et al., 2008). Their origin is unknown though the cells show similarities to FRCs, including podoplanin expression (Katakai et al., 2008). In the spleen, an analogous stromal subset is important for migration of immature B cells into follicles (Ansel et al., 2000). It is hypothesized that MRCs represent the postnatal equivalent of the lymphoid tissue organizer (LTo) subset (Katakai et al., 2008) which is responsible for lymph node organogenesis (Coles et al., 2010). Indeed, it has been convincingly shown that postnatal podoplanin+ splenic stroma are capable of regenerating through interaction with hematopoietic lymphoid tissue inducer cells (Scandella et al., 2008).
Very little is known about medullary lymph node stroma (Eikelenboom et al., 1978; Willard-Mack, 2006). Similarly, the recently described “double negative” (DN) stromal subset has been identified using flow cytometry, but its function and location within the lymph node are unknown (Cohen et al., 2010; Fletcher et al., 2010).
Recent advances have shown that lymph node stroma play an important role in self-tolerance induction (Lee et al., 2007; Nichols et al., 2007; Gardner et al., 2008; Magnusson et al., 2008; Cohen et al., 2010; Fletcher et al., 2010). FRCs (Fletcher et al., 2010) and LECs (Cohen et al., 2010), in particular, express an array of self-antigens from diverse tissues and organs and are capable of deleting naïve CD8 T cells with high efficiency if they are autoreactive to these proteins. Any role in tolerizing CD4 T cells is unreported, and the major driving mechanisms are unknown.
Traditionally, studies of lymph node stromal cells have relied heavily on tissue sections and microscopy. However, our understanding of the roles that lymph node stroma play in maintaining immunity has dramatically expanded in recent years, and the field has begun to require purification of lymph node stromal subsets for more sophisticated explorations of their biology. Isolation of stromal cells provides greater challenges than isolation of leukocyte populations, requiring different techniques and reagents. Well-established methods such as cell sorting remain challenging to most laboratories when applied to these rare stromal populations, for reasons including cell size, sorting pressure, and purity. The stroma must be isolated using a combination of enzymatic digestion and careful agitation to disrupt extracellular matrix and cell–cell contacts. The cells are sensitive to both over-exposure to enzymes and over-agitation; speed and technique are each critical requirements for success. For these reasons, stromal cell isolation skills have thus far been limited to very few laboratories. However, once successfully isolated, we have found FRCs and LECs in particular to be very pliable across a range of immunological assays.
We have refined and applied a comprehensive suite of techniques for the study of lymph node stroma. Surprisingly, we found that skin-draining and mesenteric lymph nodes vary significantly in stromal composition, and that, unlike thymic stroma, most lymph node stromal cell subsets do not require lymphocytic cross-talk to develop and expand or to survive in culture. Moreover, human lymph node stroma were markedly similar to murine stroma both in composition and amenability to cell culture.
Materials and Methods
Mice
Male C57Bl/6 mice aged 4–6 week were obtained from the Jackson Laboratory. Mice used for ImmGen sorting were precisely age-matched to 6 weeks. Male C57BL/6 Rag−/− mice were obtained from Taconic. All mice were rested for 5 days post-transport, and were specific pathogen free and cared for in accordance with institutional and National Institutes of Health guidelines. Experimental procedures were conduced with the approval of the Research Animal Care subcommittee at the Dana–Farber Cancer Institute.
Human Lymph Nodes
Human lymph nodes were procured from cadaveric donors through the National Disease Research Interchange (NDRI) resource center (Philadelphia, USA). Intact lymph nodes were transported in DMEM on ice, and processed for flow cytometry or cell culture within 24 h.
Antibodies
For flow cytometry, cell sorting and cryosection staining of mouse lymph node stroma, the following antibodies were used: anti-CD45 (clone 40-F11, BD Biosciences), anti-podoplanin (clone 8.1.1, Developmental Studies Hybridoma Bank), anti-CD31 (clone MEC13.3, Biolegend), anti-Lyve-1 (clone 10.1.1, a kind gift from Dr. Andrew Farr), and anti-MadCAM (clone MECA-367, eBioscience). Propidium iodide and clone TER119 (Biolegend) were used where appropriate to exclude dead cells and erythrocytes. For staining human cells, antibodies used were: anti-CD45 (clone HI30, Biolegend), anti-CD31 (clone WM59, BD Biosciences), and anti-podoplanin (clone NZ-1, AngioBio Co), detected with highly cross-adsorbed anti-rat IgG (H + L) Alexa-488 (Invitrogen).
Enzymatic Digestion of Lymph Nodes from Individual Mice
For flow cytometric analysis or cell culture, lymph nodes from individual mice were dissected, pierced once with fine forceps, and placed in 5 ml of RPMI-1640 on ice. Where use of skin-draining lymph nodes was specified, we dissected axillary, brachial and inguinal lymph nodes. After all lymph nodes were dissected, RPMI-1640 was removed and replaced with 2 ml of freshly made enzyme mix comprised of RPMI-1640 containing 0.8 mg/ml Dispase and 0.2 mg/ml Collagenase P (both from Roche), and 0.1 mg/ml DNase I (Invitrogen). Tubes were incubated at 37°C in a waterbath and gently inverted at 5 min intervals to ensure the contents were well-mixed. After 20 min, lymph nodes were very gently aspirated and expirated using a 1-ml pipette, which disrupted the capsule and released most leukocytes. The mixture was replaced in the waterbath and large fragments were allowed to settle for 30 s, after which the enzyme mix was removed and added to 10 ml of ice-cold FACS buffer (2% FCS, 5 mM EDTA in PBS) and centrifuged (300 g, 4 min, 4°C). Two milliliter of fresh enzyme mix was added to the digestion tube, the contents gently mixed using a 1-ml pipette, and incubated, with regular gentle mixing using a 1-ml pipette. After 10 min, the cells were mixed vigorously for 30 s using a 1-ml pipette. Fragments were again allowed to settle, the supernatant was removed and added to the previously spun cell pellet, and 2 ml of fresh enzyme mix was added to the digestion tube. From here, the digestion mix was vigorously mixed using a 1-ml pipette every 5 min until, when held up to light, it was clear that all remaining lymph node fragments were completely digested. This procedure, from the time of first incubation to complete digestion, usually took 50 min, and never longer than 60 min. Supernatants were centrifuged after each removal (300 g, 4 min, 4°C) until finally, each collection tube contained the entire cellular contents of an individual mouse’s harvested lymph nodes. Cells were filtered through 80 μm nylon mesh, counted using a hemocytometer, and assessed for viability using trypan blue. Viability exceeded 95%. The 5 × 106 cells per stain were then incubated with 50 μl diluted antibodies for 20 min at 4°C in ice-cold FACS buffer (2% FCS, 5 mM EDTA in PBS) before acquisition on a FACSCalibur or FACSAria IIu (BD Biosciences).
Enzymatic Digestion and Stromal Enrichment of Lymph Nodes from Pooled Mice for Cell Sorting and Large-Scale Isolation
Digestion of multiple pooled mice generally proceeded as described in Section “Enzymatic Digestion of Lymph Nodes From Individual Mice,” with changes as follows (depicted graphically in Figure 1): lymph nodes from 4 to 10 mice were pooled in a single tube and digested using 5 ml of enzyme mix per digestion step. After lymph nodes were fully digested, and the cells centrifuged and counted, the single cell preparation was enriched for non-hematopoietic stromal cells using CD45 microbeads and the autoMACS system (both from Miltenyi). We used 7 μl beads per 107 cells, incubated in FACS buffer for 20 min on a rotating wheel at 4°C. The labeled fraction was then depleted using the Depletes program, according to the manufacturer’s instructions. Enriched stroma were then counted and stained using a high volume of antibody (100 μl of diluted antibody cocktail per 106 cells) for cell sorting.
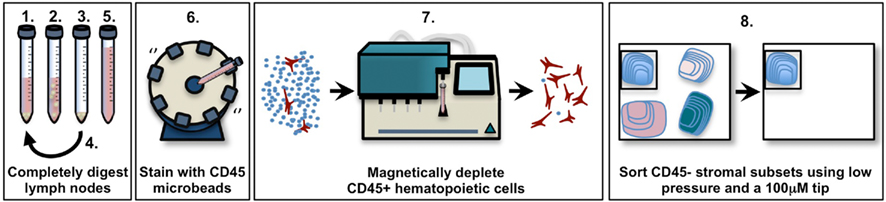
Figure 1. Schematic diagram of pooled murine lymph node stromal cell digestion and preparation for cell sorting. 1. Add enzyme mix (RPMI-1640 with 0.8 mg/ml Dispase, 0.2 mg/ml Collagenase P, 0.1 mg/ml DNase I) to tissue, incubate in 37°C waterbath. 2. After 10–20 min (see Materials and Methods), agitate lymph node fragments using 1 ml pipette, then incubate until the fragments settle. 3. Remove the supernatant containing released cells, centrifuge it and store it on ice. 4. Repeat steps 1–3 until all fragments are fully digested (no more than 60 min). 5. Pool all supernatant fractions, centrifuge, filter, and count. 6. Add anti-CD45 microbeads, incubate at 4°C on rotating wheel. 7. Use MACS or AutoMACS to deplete CD45+ cells, count. 8. Stain enriched CD45− stroma with antibodies; sort to high purity using 100 μm tip at 20 psi.
Enzymatic Digestion of Human Lymph Nodes
Human lymph nodes were carefully cleaned of fat and connective tissue, then cut into small pieces (<0.5 cm) using fine scissors. Digestion proceeded according to Section “Enzymatic Digestion of Lymph Nodes From Individual Mice,” however enzyme concentration was increased to 2.4 mg/ml Dispase and 0.6 mg/ml Collagenase P (both from Roche), and 0.3 mg/ml DNase I (Invitrogen) in RPMI-1640. In some cases we did not digest the entire organ, and instead took a random selection of approx. 20 mixed fragments, representing all areas of the node. After 60 min, lymph node fragments were completely digested, and were then filtered, counted and where required stained, as detailed in Section “Enzymatic Digestion of Lymph Nodes From Individual Mice.”
Low-Pressure Flow Cytometric Stromal Cell Sorting
Flow cytometric sorting of stromal cells is improved when a specialized low pressure, large aperture setup is used; however there are technical challenges inherent in resetting a sorter to a non-standard specification and then successfully sorting pure populations. We used FACSAria or upgraded FACSAria IIu machines utilizing FACSDiva software (all from BD Bioscience) set to 20 psi and fitted with a 100 μm tip. While each machine will differ, we have found that the following approximate specifications have applied to at least four different FACSAria machines set up for stromal sorting. These differ from the suggested setups specified by the manufacturer.
Decreased stream pressure increases droplet size, such that only 5–6 droplets are visible through the stream visualization window at 20 psi, instead of 10–12 droplets at 70 psi. While the droplet breakoff point directly affects sort success and should always be visible, it is possible for the satellite resolution point to occur below view while sorting at low pressure, even after increasing the droplet frequency. This does not preclude a successful sort; however, it is important to be confident that satellite resolution occurs no more than one to two droplets below the screen. This can be reasonably established in two ways: first, by temporarily adjusting the droplet breakoff point higher and out of view, so that the satellite resolution point becomes visible, and counting the number of droplets between breakoff and resolution before readjusting downwards; and second, by looking at the decreasing distance between satellite and droplet, and estimating the number of droplets until resolution occurs. The satellites, if not resolved on-screen, should be very close to resolution.
Critically, deciding whether to apply the attenuation control (which dampens droplet amplitude) alters subsequent specifications considerably. Sample setup sort reports both with and without attenuation are provided for reference (Table 1). BD Biosciences manual recommends attenuation not be applied when sorting at 20 psi; however, we found it necessary at times with the FACSAria (but not FACSAria IIu) to generate a good stream. While a manual adjustment of the 100 μm tip was required to generate a good stream using the FACSAria, we have not had to move the tip or utilize attenuation for the FACSAria IIu system, where the o-ring is permanently fitted.
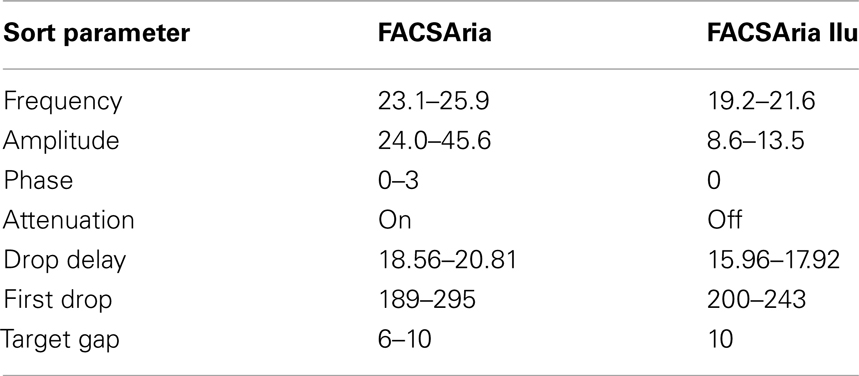
Table 1. A sample range of low-pressure stromal sorting parameters for 100 μm tip in the FACSAria and FACSAria IIu (BD Biosciences). n = 4–8 independent experiments.
Lastly, we have noted that the 633-nm laser delay can suddenly change when low pressure is engaged. The reasons for this are unclear, but we recommend using CST beads to set delays, or testing laser function using SPHERO Rainbow particles (BD Bioscience; or any similar calibration check). Note that a lack of signal from the 633-nm laser may require re-calibration of the laser delay.
Compensation for a stromal cell sort should be strictly performed using enriched stromal cells. Stroma are large cells with a degree of accompanying autofluorescence. A machine compensated using leukocytes yields suboptimal resolution of populations when stroma are sorted. Enriched stroma should be used to compensate, because in unenriched populations, stroma are usually rare, and we find that the autocompensation software cannot differentiate between normal levels of autofluorescence in a digested single cell suspension and true stromal staining.
When the machines are optimally calibrated, sorting proceeds with excellent resolution of sorting streams and good efficiency. Sort streams should resolve well as bright, discrete dots in the sort window; “spraying” of the sorting streams should not be evident.
AutoMACS-enriched stroma were routinely sorted to high purity (>95%) into TRIzol (Invitrogen). After gating on CD45− propidium iodide− stroma, subsets were sorted as follows: FRCs, podoplanin+CD31−; LECs, podoplanin+CD31−; BECs, podoplanin−CD31+; double negative stromal cells (DNs), podoplanin−CD31−.
Microarray Assays and Analysis
RNA from 10,000 to 15,000 sorted cells was isolated as previously described (Yamagata et al., 2004), then amplified and hybridized to Affymetrix GeneChip Mouse Gene 1.0 ST Arrays (Santa Clara, CA, USA) using the GeneChip Whole Transcript Sense Target Labeling Assay according to the manufacturer’s instructions. This yielded whole-transcript information using probes designed to bind in multiple locations across each gene. Raw data was normalized using the robust multi-array average (RMA) preprocessing algorithm (performing background adjustment, quantile normalization, and summarization; Irizarry et al., 2003), in the Expression File Creator module of GenePattern. Probe lists were analyzed for expression in at least one subset (excluded if mean expression value <120 across all subsets), and for low variance among replicates (excluded if coefficient of variation >0.5 in either population). We used the Multiplot module of GenePattern to calculate differences between skin lymph node FRCs and mesenteric lymph node FRCs. Differences were considered for further analysis if the fold-change in expression was >2 and P < 0.05 (Student’s T-test). Output data lists were ranked using Microsoft Excel, and gene lists were analyzed for enrichment across KEGG pathways using the DAVID program’s Functional Annotation tool (NIAID, david.abcc.ncifcrf.gov; Dennis et al., 2003), and with Affymetrix exon background set to the MoGene-1_0-st-v-1 chip. KEGG pathway analyses with T-test P-values < 0.05 after multiple hypothesis (Benjamini) correction were considered significant. The NCBI GEO accession number for the data series is GSE15907.
Lymph Node Stromal Cell Culture Techniques
Lymph nodes were digested as detailed above, then counted, and plated at a concentration of 5 × 105 cells/cm2. Cell culture media was αMEM supplemented with 10% batch-tested, low Ig FCS, and 1% Penicillin/Streptomycin. Plates were washed after 24 h to remove non-adherent cells. After 5 days, mouse cell cultures primarily contained LECs and FRCs. Human cell cultures grew more slowly and were harvested after 7 days. Human lymph node stromal cell cultures primarily contained FRCs and DNs. A non-standard harvesting protocol was routinely utilized, consisting of a short incubation in low-concentration Trypsin with EDTA to minimize trypsin-based shearing of important cell surface markers (Table 2). Cells were washed with PBS to remove residual protein, and harvest buffer (0.2% Trypsin with 5 mM EDTA in PBS) was added to culture plates. Cells were incubated in harvest buffer at 37°C for 2 min. At this point, their morphology became rounder, and an equal volume of complete media was immediately added to wells. Cells were washed from the plate with gentle agitation using a 1-ml pipette, and supernatant was added to another equal volume of complete media for centrifuging (300 g, 3 min, 4°C). FRCs and LECs were replated, sorted, or MACS-purified from this preparation as required.
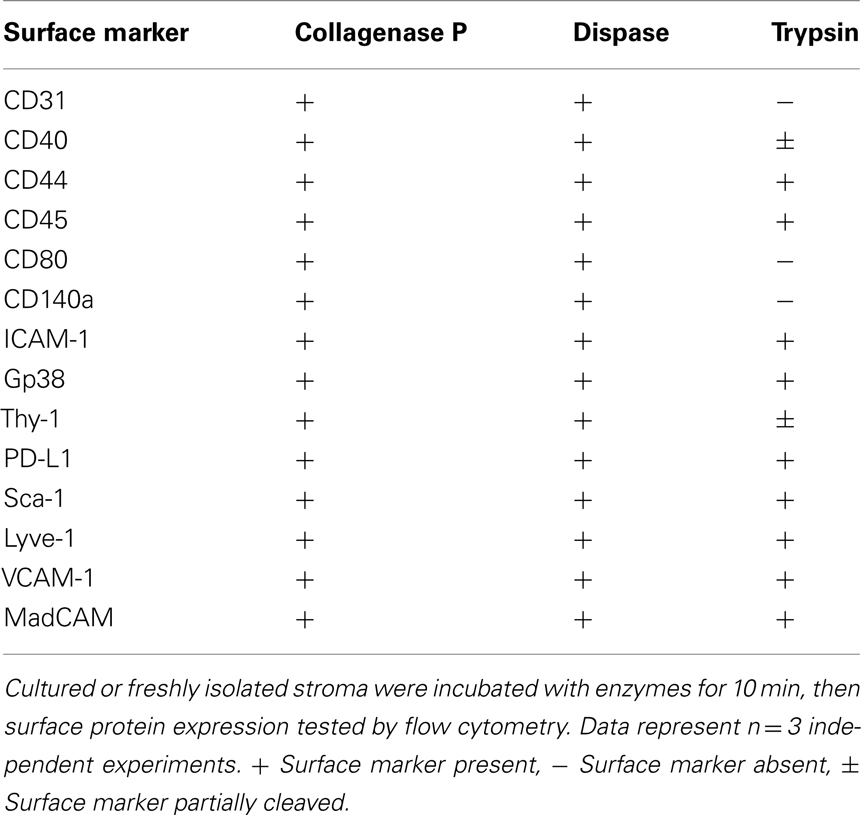
Table 2. Enzyme sensitivity of surface markers used to characterize non-hematopoietic lymph node stromal cells.
For three-dimensional (3D) cultures and network analyses, cells were cultured in deformable matrices constructed from 3.2 mg/ml of high concentration rat tail Collagen I (BD Biosciences), 1.8 mg/ml Matrigel basement membrane matrix (BD Bioscience), 8.3% (v/v) of 5× αMEM stock made in-house from αMEM powder (Invitrogen), and 30% (v/v) culture media containing cells to be plated. Gel components were mixed on ice and 200 μl plated onto plastic, or glass-bottomed culture dishes for imaging (MatTek). Gels were left to set at 37°C for 10 min before being covered with culture media. For contraction assays, gels were created in flat-bottomed 96 well culture plates and photographed after 15 h. Contraction was measured using the following equation: area contracted gel (cm2)/area well (cm2) *100 = % contraction. For time-lapse live cell imaging, bone-marrow derived DCs or B cells were mixed at a 5:1 ratio with purified cultured FRCs. During imaging, gels were incubated at 37°C with 10% CO2 in αMEM containing 10% FCS. Movies depict time-lapse imaging with 1 frame acquired every 90 s.
Immunohistology and Confocal Microscopy
Lymph nodes were freshly dissected and fixed in 10% paraformaldehyde for 4 h, then left in 10% sucrose solution overnight. Blocks were then embedded in OCT compound (VWR) and snap-frozen, then stored at −80°C. Lymph nodes were cryosectioned (7 μm) using a Leica cryostat, and acetone fixed, then blocked for 30 min using 2% BSA. After washing in PBS, sections were stained with primary antibody for 60 min, then washed and stained with secondary antibody for a further 60 min. Slides were mounted with a coverslip using fluorescent mounting medium (DAKO) and stored at 4°C. Images were acquired using a Leica Sp5 confocal microscope.
Statistical Analysis
GenePattern’s Multiplot program was used to calculate T-test P-values on normalized, array data where the coefficient of variance between replicates for any sample was <0.5. A P-value of <0.05 was considered significant if the fold-change observed was also <2. Genes were tested for KEGG pathway association using the DAVID program (david.abcc.ncifcrf.gov), which compared data using a modified Fisher’s Exact Test, and a T-test with a Benjamini multiple hypothesis correction, to show enrichment of two or more genes in a particular list. The non-parametric Mann–Whitney U-test was used to compare flow cytometric data (Prism).
Results
Enzymatic Isolation of Mouse and Human Lymph Node Stromal Cells
The study of lymph node stromal cells has gathered recent momentum, with several high-impact papers dissecting their newly discovered roles in maintaining (Link et al., 2007) and deleting (Lee et al., 2007; Nichols et al., 2007; Gardner et al., 2008; Magnusson et al., 2008; Yip et al., 2009; Cohen et al., 2010; Fletcher et al., 2010) naïve T cells. Methodological capabilities, however, have been a limiting factor.
We therefore aimed to develop a low-mortality enzymatic digestion protocol which would enable highly reproducible isolation of lymph node stromal cells with low variability. Using a combination of enzymatic and physical dissociation (Figure 1), we routinely isolated stromal cells with >95% viability (Figure 2A) using trypan blue on whole lymph node suspensions (1.5% stroma) immediately after isolation. Six hours after isolation and stromal enrichment, the average viability of stroma using propidium iodide was 87.8 ± 0.8% (mean ± SD, n = 3 experiments). This high viability also allowed us to characterize and compare the number of each stromal subset present in individual mice, and was a key factor in successfully sorting and culturing these cells. We used CD45, podoplanin, CD31, and MadCAM (Figure 2B) to study five major subsets of lymph node stroma. The MadCAM+ MRC subset, in particular, has not previously been isolated for flow cytometric analysis.
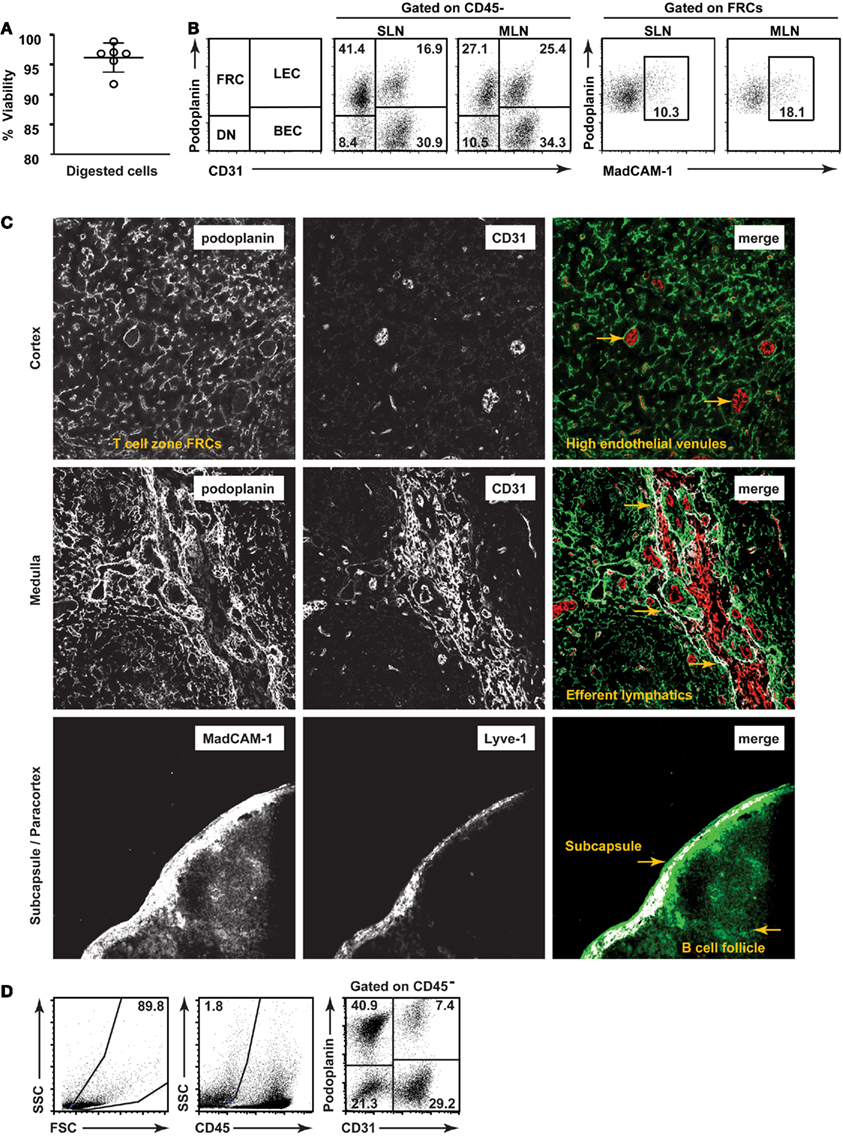
Figure 2. Validation of a low-mortality method for isolation of lymph node stromal subsets. (A) Viability of single cell suspensions prepared using a low-mortality enzymatic digestion method. n = 6, mean + SD. (B) Typical flow cytometric profiles of lymph node stromal subsets freshly isolated from skin-draining or mesenteric lymph nodes of individual mice. (C) Lymph node stromal subsets identified in situ on cryosections. Top panel: T cell zone FRCs (podoplanin+ CD31−) and high endothelial venules (podoplanin−CD31+ with cuboidal morphology). Two examples of HEVs are designated using arrows. Middle panel: medullary LECs (podoplanin+CD31+) and large medullary blood vessels (podoplanin−CD31+). Medullary lymphatic vessel/s are shown using arrows. Bottom panel: Capsular afferent LECs (Lyve-1+ MadCAM+), MRCs (subcapsular, Lyve-1− MadCAM+), and FDCs (follicular, Lyve-1−, MadCAM+). Colocalization overlay shown in white on merged images. Original magnification: 200×.(D) Human lymph nodes (non-mesenteric origin) were digested and stained for flow cytometric analysis. Dotplots are representative of n = 4 independent experiments.
We first directly compared our flow cytometric strategies to previously defined in situ histological analyses of stromal subsets (Figure 2C). Our aim was to establish methods that would enable changes in stromal architecture to be monitored in parallel with changes to number or proportion of cell types, mRNA profile, and other techniques requiring cell isolation.
Accordingly, we found that FRCs, as defined by flow cytometry, created a reticular network throughout the T cell zone as expected (top panel). BECs expressed CD31 but not podoplanin and were primarily present in the cortex (top panel), with high endothelial venules distinguishable by their small size and cuboidal morphology. LECs, defined by coexpression of podoplanin and CD31, lined large lymphatic vessels in the hilar region of the medulla (middle panel) and subcapsule (data not shown). This hilar region also contained large blood vessels (CD31+ podoplanin−). MadCAM+ reticular cells lined the subcapsule (bottom panel) and MadCAM staining was also present in the B cell zone, as previously reported. We found that subcapsular LECs (shown here expressing Lyve-1) also expressed MadCAM (bottom panel), making the subcapsule a MadCAM-rich region of the lymph node. Phenotypically, MRCs formed a subset within the podoplanin+ CD31− FRC gate by flow cytometry (Figure 2B).
We hypothesized that our isolation methods could be applied to isolate human lymph node stroma, allowing the creation of useful ex vivo experimental systems. Human lymph node stromal architecture is well described by histology (Link et al., 2011) but subsets have not been isolated for flow cytometry or other immunological studies. It is unknown whether human lymph node stromal cells maintain naive T cells or tolerize autoreactive T cells as murine lymph node stromal cells do.
We acquired human lymph nodes of non-mesenteric origin from cadaveric donors. While increased enzyme concentration was required to digest the lymph nodes (see Materials and Methods), once digested, their stromal composition was markedly similar to mice (compare Figures 2B,D). These results suggest that murine lymph node stromal cell techniques may be equally applicable to human studies.
Mesenteric Lymph Nodes Contain Fewer FRCs than Skin-Draining Lymph Nodes
To further validate our digestion, we tested its reproducibility by assessing how much the proportional makeup of lymph node stromal cell subsets varied between age-matched mice. We also compared the stromal composition of skin-draining and mesenteric lymph nodes. While these lymph nodes have well-established structural similarities and are well-studied by histology, their ontogenies occur at different times and are dependent on different signaling pathways (Mebius, 2003). Additionally, mesenteric lymph nodes are continually exposed to gut and gastromucosally derived antigens, while skin-draining lymph nodes are not.
Surprisingly, we found that while similar numbers of cells were isolated from each skin-draining or mesenteric lymph node (Figure 3A), the stromal composition significantly differed (Figure 3B). FRCs, which grow throughout the T cell zone, were present at greater frequency (Figures 2B and 3B), and were more numerous (Figure 3C) in skin-draining lymph nodes than mesenteric lymph nodes. LECs, BECs, DNs, and MRCs were present at similar numbers in both sites (Figure 3C), making the difference FRC-specific.
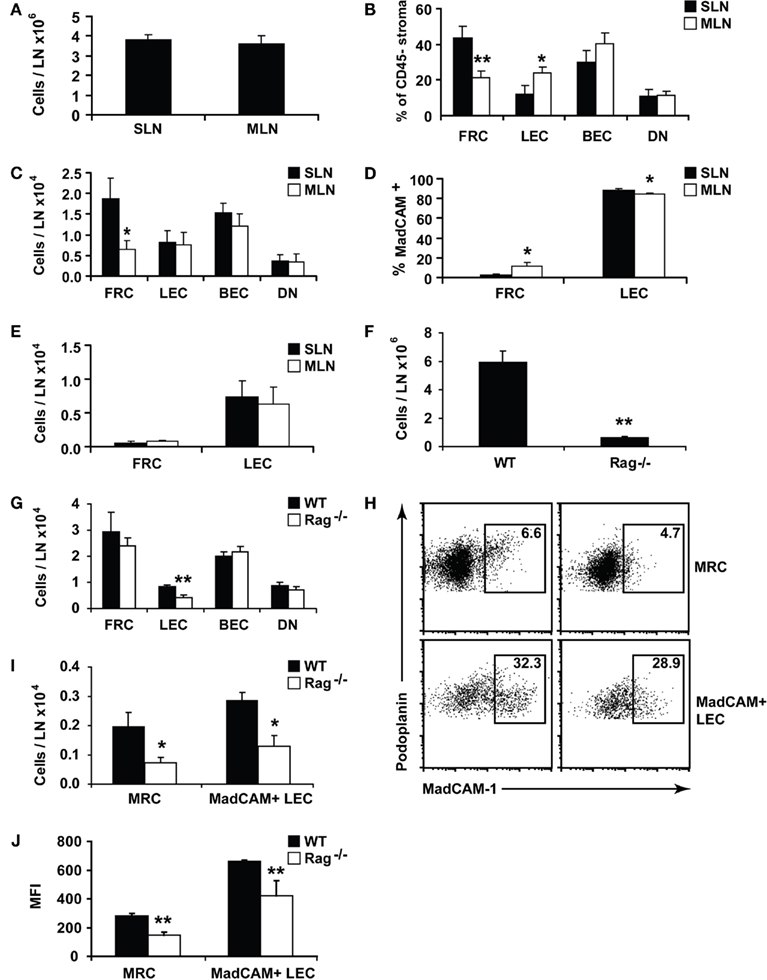
Figure 3. Lymph nodes show site-specific alterations in stromal composition. Lymph nodes from individual C57Bl/6 mice were digested and stained for flow cytometry. (A) Lymph node cellularity. (B) Proportion of CD45 negative lymph node stromal subsets. (C) Number of lymph node stromal subsets. (D) MadCAM+ cells shown as a proportion of total FRCs or LECs. (E) Total number of MadCAM+ FRCs or LECs. (F) Cellularity of skin-draining lymph nodes isolated from age-matched Rag−/− or WT (B6) mice. (G) Number of lymph node stroma from age-matched Rag−/− or WT mice. (H) The proportion, or (I) number of MadCAM+ FRCs (MRCs) or LECs in SLN of age-matched Rag−/− or WT mice. Plots are representative of n = 5 mice. (J) Mean fluorescence intensity (MFI) of MadCAM staining in MRCs or MadCAM+ LECs. WT, wildtype. *P < 0.05; **P < 0.01 Mann–Whitney U-test. n = 5–10 mice from two to three independent experiments.
We asked whether this difference was due to skewing in the proportions of the MRC subset (Katakai et al., 2008) which, we found, falls within the FRC gate (Figure 2B). About 10–20% of cells within the FRC gate expressed MadCAM in 5 week old male mice, with a higher proportion in mesenteric lymph nodes than skin-draining lymph nodes (Figures 2B and 3D). However, while MRC proportions were significantly altered between sites, similar to LECs, their overall numbers were not (Figure 3E). This suggests that MRC numbers and FRC numbers are differently regulated, since MRCs, like the other stromal subsets, are numerically similar between sites, while FRCs are reduced in mesenteric lymph nodes.
In some lymphoid organs, such as the thymus, the numbers of predominant stromal cell subsets expand or shrink in parallel with developing T cells. We tested whether this applied to the FRC population using Rag−/− mice, which lack T and B cells. FRCs develop in Rag−/− lymph nodes, but it is not known whether they expand and contract in response to lymphocyte numbers. This question is particularly relevant to infection, since lymph nodes must expand dramatically in a short time to contain proliferating lymphocytes.
While Rag−/− mice had smaller lymph nodes (Figure 3F), they possessed normal numbers of FRCs compared to age-matched wildtype controls (Figure 3G). The number of LECs, however, was significantly reduced, suggesting that their numerical homeostasis is linked to the presence of mature lymphocytes. Rag−/− FRCs did not differ from WT in rates of cell turnover (data not shown), and after injecting LPS into mice, the number of FRCs did not expand alongside T and B cells (data not shown). FRC numbers were therefore maintained completely independent of mature lymphocytes under lymphopenic, steady-state, and inflammatory conditions.
Surprisingly, we found that the MRC subset was underdeveloped in Rag−/− mice (Figures 3H–J). It is reported that MadCAM+ MRCs exist by histology in Rag−/− mice (Katakai et al., 2008), but in our hands, both the numbers of MadCAM+ MRCs and the amount of MadCAM protein per MRC were significantly reduced (Figures 3H–J). This also applied to MadCAM+ LECs (Figures 3H–J), suggesting that upregulation of MadCAM expression in both cell types denotes a developmental stage requiring the presence of lymphocytes, though expression of the protein itself can occur in their absence. While the proportion of MadCAM+ LECs was similar between WT and Rag−/− (Figure 3H), the significant reduction in total LECs (Figure 3G) translated to reduced numbers of MadCAM+ LECs (Figure 3I).
Enriching and Sorting Lymph Node Stromal Cell Subsets
Following the successful digestion of lymph nodes, we next adapted the means to successfully sort and culture the stromal cells. Sorting stromal cells can be technically difficult (see Materials and Methods) and we used MACS-based depletion of hematopoietic cells to decrease sorting times and increase the resulting purity. This additional step yielded minimal loss of stroma (Figures 4A,B) and did not appreciably alter the stromal composition; we saw no selective loss of stromal subsets post-MACS (Figure 4C) and, after sorting, achieved excellent purity (Figure 4D).
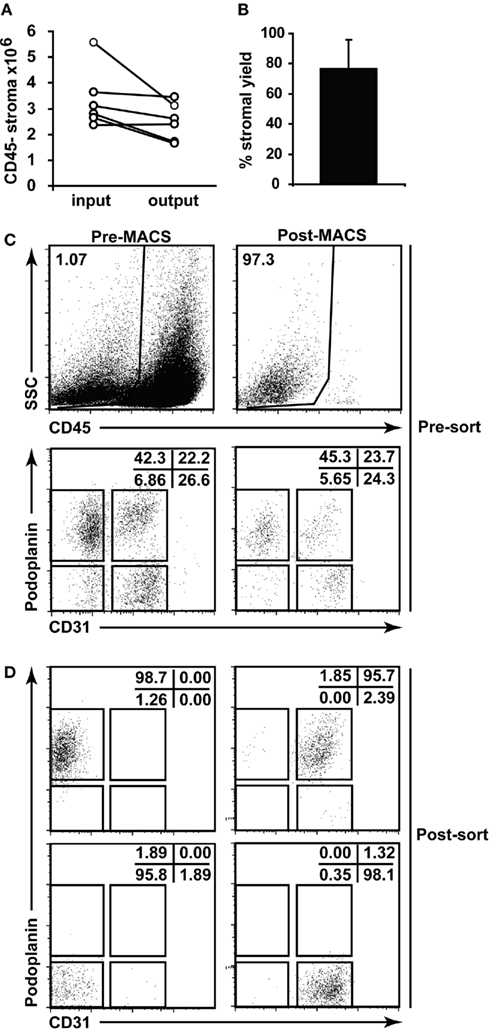
Figure 4. Lymph node stromal cell enrichment and sorting. (A) Skin-draining lymph nodes from 6 to 10 C57Bl/6 mice were enzymatically digested and enriched for CD45− stromal cells using autoMACS. The number of CD45− stroma added to the column (input) and retrieved from the column (output) were charted. Data depict n = 5 independent experiments. (B) Percent stromal cell yield for autoMACS enrichment of stroma was calculated. (C) Flow cytometric profiles of stromal cells pre- and post-autoMACS enrichment. (D) Sorting strategy and post-sort purity for major lymph node stromal cell subsets. Dotplots gated on CD45− propidium iodide− stroma.
Using this method, we sorted FRCs from skin-draining and mesenteric lymph nodes as part of the multicenter ImmGen Consortium (www.immgen.org). This involved transcriptomic analysis of cells sorted under highly controlled, minimally variable conditions, with the overall aim of generating a public database to provide a comprehensive, cross-referenced map of the immunological genome.
Fibroblastic reticular cells from skin-draining and mesenteric lymph nodes were analyzed for expression of 25,194 probes to yield a list of 11,162 probes expressed above an arbitrary threshold (mean expression value >120) in either skin-draining or mesenteric lymph nodes. These probes were also filtered for a low (<0.5) coefficient of variance between replicates.
We first confirmed that FRCs from these different locations shared expression of previously published FRC-specific markers such as CD140a, CCL19, and IL7 (data not shown), then analyzed the data for differences. Comparing skin-draining versus mesenteric lymph node FRCs, a volcano plot depicting fold-change and P-value showed that 130 probes were significantly upregulated in FRCs from skin-draining compared to mesenteric lymph nodes, while expression of 97 probes was significantly higher in mesenteric lymph node FRCs compared to skin-draining (>2-fold-change, P < 0.05; Figure 5A). After mapping the probes to unique genes, these data corresponded to upregulation of 126 genes in skin-draining lymph node FRCs (Table S1 in Supplementary Material) and 54 genes in mesenteric lymph node FRCs (Table S2 in Supplementary Material).
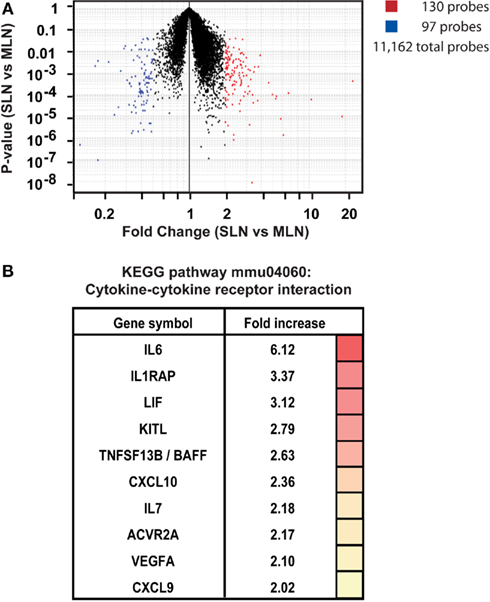
Figure 5. Site-specific transcriptional upregulation of cytokines in FRCs from skin-draining lymph nodes. (A) Microarray analysis comparing FRCs sorted from skin-draining (SLN) or mesenteric lymph nodes (MLN). Dotplot depicts P-value versus fold-change for SLN versus MLN for 15,486 selected probes. Probes upregulated in SLN are shown in red, while probes upregulated in MLN are blue; (P < 0.05 and fold-change > 2; T-test). n = 4–5 independent replicates. (B) Genes enriched in SLNs encoding cytokines and cytokine receptors (KEGG pathway mmu04060; P < 0.015; modified Fisher’s exact test with Benjamini correction). The fold-increase in mean expression in SLNs compared to MLNs is shown.
We were interested in whether these lists contained evidence for activation of particular gene pathways, or gene networks uniquely operating in either cell type, as a clue to any site-specific specialization. Using the free online program DAVID (version 6.7; National Institute of Allergy and Infectious Diseases), we used a network analysis tool (KEGG) to comb the lists for known biological connections between these genes, then used statistical analysis tools to assess whether the connected genes appeared together at a frequency greater than would be expected by chance. The KEGG pathway mmu04060, which covers cytokine–cytokine receptor interaction, was significantly enriched in FRCs from skin-draining lymph nodes compared to mesenteric (genes appeared at a frequency 4.8-fold higher than expected by chance, Benjamini corrected P-value = 0.0055). This list included 10 genes of immunological relevance (Figure 5B) upregulated in skin-draining lymph node FRCs by at least twofold. Genes upregulated in mesenteric lymph nodes were not significantly enriched in an identifiable KEGG pathway, but notably included genes involved in regulating the growth of fibroblasts, genes relevant to the gut microenvironment, such as those involved in retinoic acid metabolism or MadCAM expression, endothelial cell cross-talk, extracellular matrix secretion, and outgrowth of peripheral neurons (Table 3).
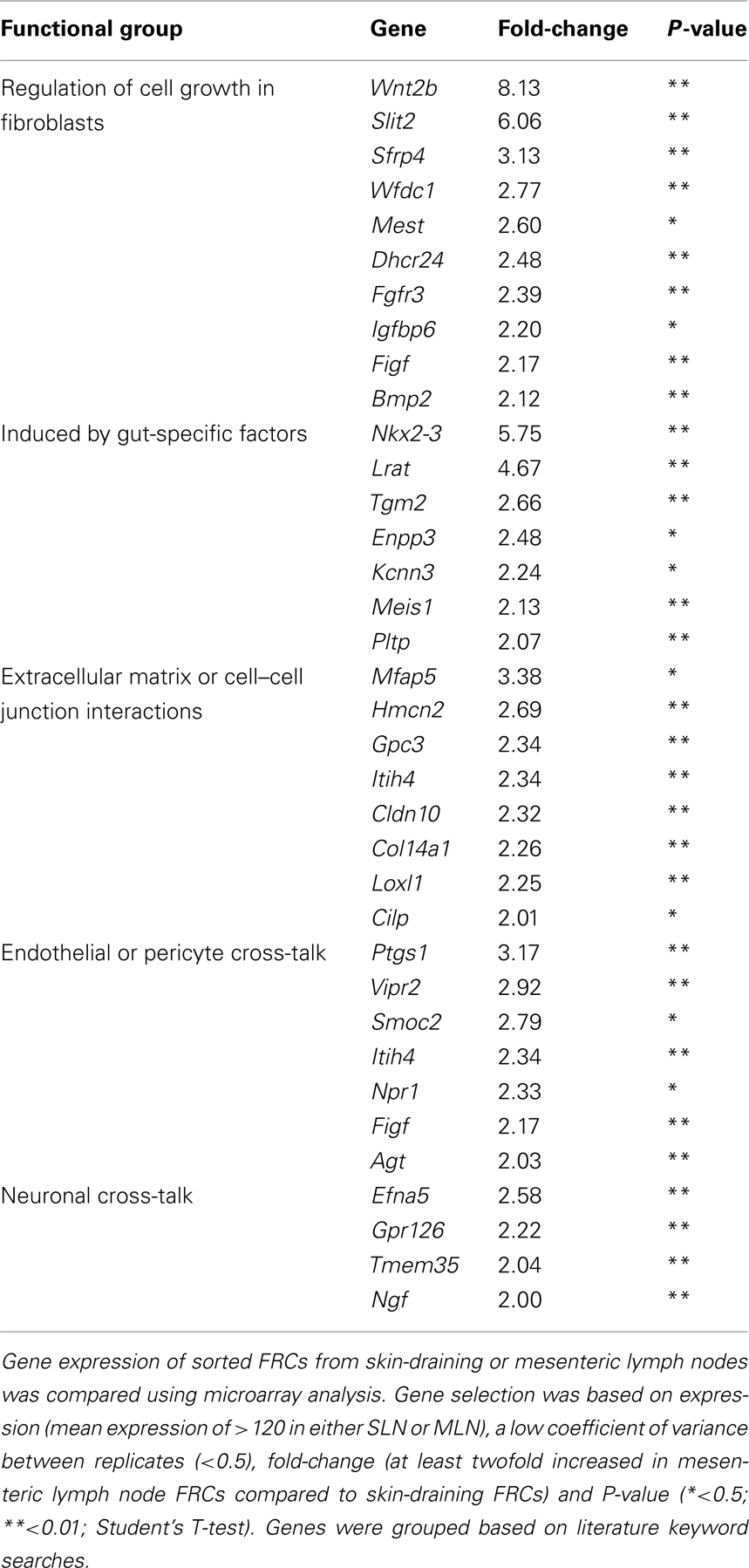
Table 3. Genes upregulated in FRCs from mesenteric lymph nodes compared to FRCs from skin-draining lymph nodes, grouped by functional similarity.
Cultured FRCs Mimic in vivo Function and Support Leukocyte Migration
We next tested the ability of lymph node stroma to grow in culture. We found that stroma from a successfully digested lymph node cell suspension will readily adhere to tissue culture plates within 2 h of plating and begin to expand at the expense of CD45+ leukocytes, which form less than 10% of the culture after 5 days (Figure 6A). FRCs and LECs readily grew in culture, while BECs and DN cells did not (Figure 6A). FDCs did not grow in culture (Figure 6B). Importantly, our low trypsin, high EDTA harvesting protocol allowed retention of important surface markers such as CD31 (Figure 6C) and CD140a (not shown; see Table 2). We found that 5 × 105 cells/cm2, containing approx. 5 × 103 stromal cells, routinely yielded approx. 1.5–2 × 104 stroma/cm2 after 5 days of culture. This culture system enabled easy expansion and manipulation of FRCs, LECs, and MRCs.
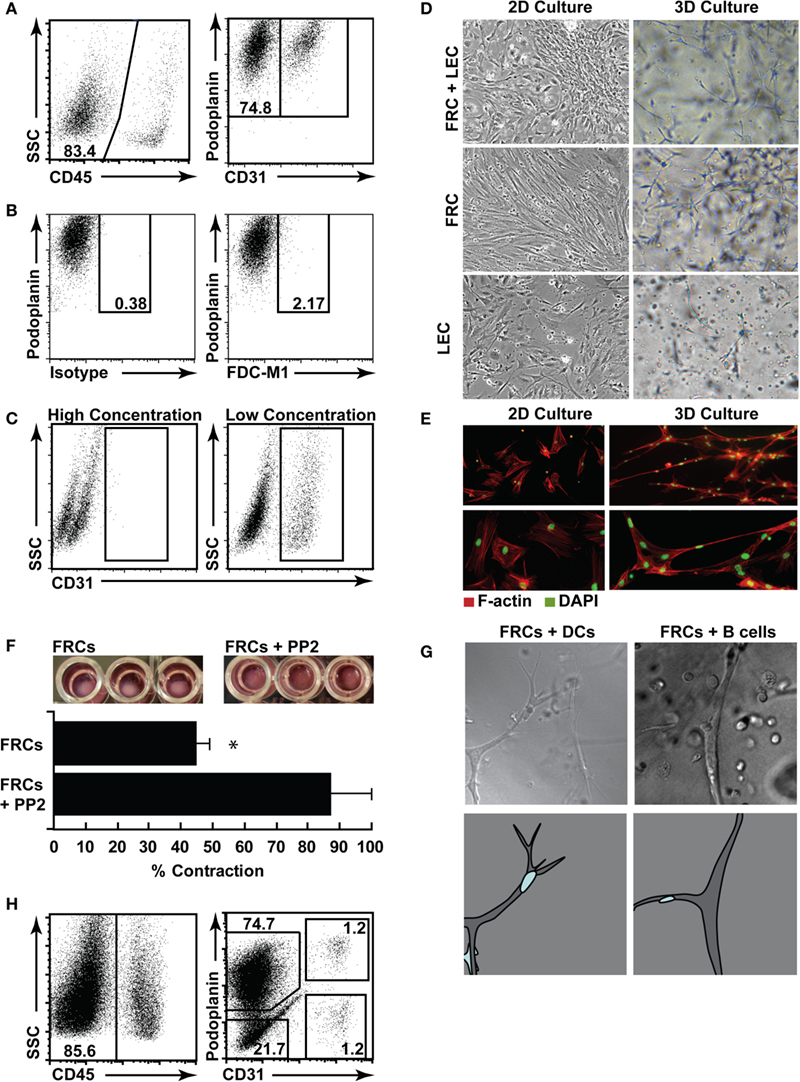
Figure 6. Three-dimensional (3D) culture of FRCs mimics in vivo function. (A) Lymph nodes were digested, and single cell suspensions put into culture for 5 days, then trypsinized and stained for flow cytometric profiling. Stromal cells were identified using CD45 (left panel), then stained with CD31 and podoplanin (right panel, gated on CD45− stroma). (B) Cultured stromal cells stained for FDC-M1 or a relevant isotype control. (C) CD31 staining in cultured stromal cells harvested using a high trypsin or low trypsin protocol. Dotplots represent n = 3 independent experiments. (D) Cultured stromal cells were harvested, then purified using autoMACS. FRCs, LECs, or mixed FRCs and LECs were plated in 2D (plastic tissue culture plates), or 3D (a matrigel and collagen gel) and cultured for a further 3 days prior to imaging. Original magnification 10×. Images represent n = 2 independent experiments. (E) F-actin (red) and DAPI (green) were used to stain purified FRCs in 2D or 3D to highlight networks. Images represent n = 3 independent experiments. (F) Purified FRCs were placed in 3D culture in a 96 well plate with or without PP2. After 24–36 h, gel contraction was quantified. Images are representative of n = 3 independent experiments. Bar graph depicts mean + SD, P < 0.05, T-test. (G) Purified FRCs were placed in 3D cultured with purified dendritic cells (DCs) or B cells, then images were sequentially acquired every 90 s. Stills from Supplemental Movies 1 (DCs) and 2 (B cells) are shown (upper panel), original magnification: 20×. Lower panel shows a cartoon of the imaged stromal cell (gray) and the relevant migrating leukocyte (blue). (H) Human lymph nodes were digested, and single cell suspensions put into culture for 7 days, then trypsinized and stained for flow cytometric profiling.
Fibroblastic reticular cells in particular became highly polarized in two-dimensional (2D) culture (Figure 6D) showing strong F-actin stress fibers (Figure 6E). This did not mimic their morphology in vivo; so, using an optimized mix of Collagen I, matrigel, and enriched αMEM, we also explored 3D culture of FRCs and LECs, as a tool to examine their function when permitted to expand and connect in 3Ds as they do in vivo. We found that FRCs stretched and formed extensive networks when grown in 3D (Figures 6D,E), while LECs preferred a flat surface and grew more successfully in 2D culture (Figure 6D). Interestingly, neither cell type grew to overconfluence under 3D conditions (data not shown), suggesting an innate regulation of cell density that did not occur in 2D culture. To validate the biological relevance of this in vitro FRC network, we performed two experiments. First, we placed a high number of FRCs into a small matrigel plug, and monitored their ability to contract the gel (Figure 6F). FRCs express high levels of alpha smooth muscle actin and are highly contractile (Link et al., 2007). We found that FRCs contracted the gel with high efficiency, and that this contraction was prevented when the Src tyrosine kinase inhibitor PP2 was added to the gel. Second, we added bone-marrow derived DCs or spleen-derived lymphocytes to a gel containing FRCs, and used a live-imaging approach to find that, as reported in vivo, DCs and lymphocytes crawled efficiently and preferentially along the FRC network (stills are shown in Figure 6G with full time-lapse movies shown in Movies S1 and S2 in Supplementary Material).
Using the same methods, we successfully cultured human lymph node stromal cell subsets. Unlike murine cells, BECs and DN cells grew in vitro alongside LECs and FRCs (Figure 6H).
Together, these results showed that murine lymph node stroma were readily isolated for flow cytometry and sorting, and could be cultured in the absence of leukocytes, yet created extensive 3D networks on which leukocytes would readily crawl, as reported in vivo. Our data suggested that human lymph node stroma were similarly amenable to study using murine techniques.
Together, these results describe the validation of a suite of techniques applicable to the study of lymph node stromal cells in mice and humans, under a variety of ex vivo and in vitro conditions.
Discussion
Techniques routinely employed in immunology and cell biology, such as generating viable single cell suspensions of reproducible composition from primary tissues, have created difficulties when applied to lymph node stroma. As such, histology and whole-organ PCR, or in vivo systems involving lymphocytic readouts have been the field’s standard. These have lead to several important advances regarding the role of lymph node stromal cell subsets in steady-state tolerance and immunity; however, there are many unexplored questions of direct biological and medical relevance. We believe that modern immunological and cell biological techniques, refined and adapted for use with these rare and delicate cell types, will be required to answer these.
Highlighting this unmet need, DN stroma in particular remain almost completely undefined. Several groups have now reported the existence of this non-hematopoietic stromal cell subset (which may or may not be homogeneous; Link et al., 2007; Cohen et al., 2010; Fletcher et al., 2010) but despite comprising >10% of the lymph node stromal niche and harboring an Aire-expressing cell type (Cohen et al., 2010; Fletcher et al., 2010), it has remained almost entirely uncharacterized. Such fundamental characteristics as morphology, lineage, surface phenotype, and function are unreported. These deficits speak directly to the difficulties inherent in studying lymph node stromal cells.
One such difficulty in studying DN cells is that dying or poorly stained lymphocytes appear CD45-low or negative, appearing in the stromal cell gate with the same phenotype (CD31−podoplanin−) as DN stroma. This greatly skews the proportions of true stromal subsets, making it difficult to tie down even the most basic proportional composition of the lymph node stromal niche. Indeed, since stroma comprise only 1–2% of lymph node cells, non-viable lymphocytes routinely outnumber stroma even in a highly viable (>95% live) single cell suspension. In our experience, inclusion of a dead cell stain such as propidium iodide, does not adequately remove cells that appear CD45-low post-digestion.
For this reason, we felt that the key to reproducible isolation of stroma lay in developing a low-mortality enzymatic digestion protocol. Using a combination of dispase, DNase I, and collagenase P, we routinely isolated a high number of stromal cells with >95% viability. This also allowed us to compare each stromal subset across individual mice and humans, and to successfully sort rare subsets, including DN cells and MRCs, which will be the subject of further study. It has not yet been possible to identify DN stroma by histology, since no unique or differentially expressed surface markers are reported. However, the sorting strategy described herein could be used to perform the unbiased array-based approaches required to unmask this rare subset.
Similarly, these methods allowed us to isolate the four major populations of human lymph node stromal cells directly from primary tissue, opening the door for sophisticated clinical and pre-clinical studies. It is now well established that mouse lymph node stromal cells express many clinically relevant self-antigens and directly present these to T cells to tolerize them (Lee et al., 2007; Nichols et al., 2007; Gardner et al., 2008; Magnusson et al., 2008; Cohen et al., 2010; Fletcher et al., 2010). It is possible that the same mechanisms may be at play in humans. Studies have shown that FRCs and LECs retain the ability to present PTAs to naïve T cells while in culture, mimicking interactions seen in vivo (Cohen et al., 2010; Fletcher et al., 2010), though it is unclear whether this is due to continual transcription of the PTAs in question, or retention of peptide-MHC. These techniques could be used to address this and many other outstanding questions pertaining to human lymph node biology.
Further, the development of 3D culture systems for lymph nodes will allow the creation of highly controlled experimental systems mimicking conditions and interactions in vivo. We found that DCs and lymphocytes readily and preferentially migrated on FRC networks in vitro, as has been observed in vivo. Such techniques are particularly applicable to studies with human cells.
We found strikingly low variation in stromal cell subsets between young male age-matched C57Bl/6 mice. In testing whether cross-talk between lymphocytes and stroma was required for the expansion of particular stromal subsets, we found that FRCs, LECs, BECs, and DN stroma were present at normal proportions in Rag−/− mice, showing no requirement for T or B cells in their development or expansion. The MRC (MadCAM-1+) stromal subset has not previously been isolated for flow cytometric analysis, and we found that MRCs formed a subset within the podoplanin+ CD31− FRC gate. While MRCs show similarities to FRCs, expressing VCAM-1 and ICAM-1, and secreting ECM components such as laminin, they additionally produce high amounts of CXCL13 and can be identified through expression of MadCAM-1 (Katakai et al., 2008)
Strikingly, the number of MRCs was significantly reduced in Rag−/− mice. The level of MadCAM protein per cell was also lower. By histology, MadCAM+ MRCs are visible in skin-draining Rag−/− lymph nodes (Katakai et al., 2008), however numerical and quantitative flow cytometric analysis of MadCAM expression and MRC numbers has not previously been possible. These data suggest that lymphocytes are indeed required for normal development of MRCs. Interestingly, LECs from Rag−/− mice also developed into MadCAM+ cells at reduced proportions, and showed less protein per cell. It is as yet unclear whether MRCs and MadCAM+ LECs fail to expand normally in Rag−/− mice, or just fail to upregulate MadCAM. The role for MadCAM+ LECs is also undescribed.
Since MRCs show the same phenotype as LTo cells (Katakai et al., 2008), which construct the lymph node downstream of lymphotoxin signaling and an NF-κB2 transcription program (Mebius, 2003; Coles et al., 2010), it has been postulated that MRCs may play a role in postnatal maintenance or regeneration of the lymph node stroma (Katakai et al., 2008). Our results show that normal expansion and/or maturation of MadCAM+ MRCs is not required for normal expansion of FRCs, BECs, and DN stroma. However, further functional studies would be required to determine if MRC function (expression of chemokines, for example) and MadCAM expression were inexorably linked.
An unexpected finding was that the FRCs of skin-draining and mesenteric lymph nodes significantly differed in proportion, number, and transcriptional profile. We found significantly fewer FRCs in mesenteric lymph nodes, containing reduced expression of several important cytokines and chemokines: IL6, IL7, BAFF, CXCL9, CXCL10, IL1 receptor accessory protein (IL1RAP), Activin receptor IIA, VEGFa, LIF, and cKIT-ligand. Statistically, the pathway analysis showed that these genes were highly unlikely to have clustered by chance. It will be interesting to determine whether these changes occur as the result of a congenital developmental difference, or following exposure to different inflammatory cues, such as differences in antigen exposure frequency or dose. Skin-draining and mesenteric lymph nodes are exposed to markedly different microenvironmental stimuli. Unless skin barriers are breached, FRCs residing in skin-draining lymph nodes from this study could be expected to encounter large doses of microbe-sourced antigen relatively rarely. Mesenteric FRCs, however, are constantly exposed to a barrage of pathogen-associated molecular patterns through normal functioning of the gut, with important steady-state roles in Treg induction and oral tolerance (Hadis et al., 2011). These day-to-day factors are reasonably be expected to alter expression profile, to unknown effect.
Developmental differences in skin-draining lymph nodes and mesenteric lymph nodes have been reported: a full complement of mesenteric lymph nodes are present in Cxcl13−/− and Cxcr5−/− mice, while many skin-draining lymph nodes are missing, or develop variably in particular locations (Ansel et al., 2000). Similarly, Ltb−/− mice develop mesenteric lymph nodes (albeit abnormally) but lack many skin-draining lymph nodes (Koni et al., 1997). Since lymph node development is initiated during embryogenesis, these results suggest that cytokines capable of compensating for CXCL13 or LTβ are more highly available in prenatal mesenteric lymph nodes than skin-draining lymph nodes. Similarly, a lower proportion of presumptive lymph node organizer cells are present in the skin-draining lymph nodes of neonates compared with mesenteric lymph nodes (Cupedo et al., 2004). Finally, after transplanting skin-draining lymph nodes to the mesentery, it was found that MadCAM− BECs do not adopt the mesenteric MadCAM+ phenotype, suggesting the difference is developmental and not a result of exposure to gut-derived antigens (Ahrendt et al., 2008). A recent study also interestingly suggests that stroma from transplanted peripheral and mesenteric lymph nodes may differ in their ability to induce oral tolerance, assuming that carryover hematopoietic cells, progressively replaced by donor cells after transplant, are not influential (Buettner et al., 2011).
In the skin-draining lymph nodes of 6 week old mice, we found higher expression of several cytokines with known roles in lymph nodes. IL6, upregulated sixfold in FRCs from skin-draining lymph nodes, is a cytokine subject to complex regulation. When produced early in the immune response after exposure to proinflammatory IL1, IL6 is potently immunostimulatory, but can also exert anti-inflammatory functions through dampening of TNFα and IL1 signaling, and upregulation of IL10. IL10 was not expressed by stroma (data not shown). Instead, we found >3-fold upregulation of the IL1 receptor accessory protein (IL1Rap) in skin-draining lymph nodes, suggesting that skin-draining lymph node FRCs possess the capacity to increase IL6 production early in infection. Accordingly, during steady-state, FRCs from skin-draining lymph nodes also expressed high levels of IL1R1, the high-affinity binding partner of IL1Rap, and did not express the decoy analog IL1-RN (Dinarello, 1996; data not shown). Supporting these findings, we have previously reported that FRCs respond to TLR3 signaling within hours (Fletcher et al., 2010), suggesting that FRCs respond swiftly to infection. It is also worth noting that IL6 is produced in an anti-inflammatory capacity by smooth muscle cells following stretch (Pedersen, 2006), as a means of phosphorylating PDGF-receptors and stimulating cell contraction (Hu et al., 1998; Zampetaki et al., 2005). Similarly, in dermal myofibroblasts, IL6 modulates alpha smooth muscle actin to enhance contraction and wound closure (Gallucci et al., 2006). Like myofibroblasts, FRCs are highly contractile cells expressing large amounts of alpha smooth muscle actin (Link et al., 2007 and data not shown). IL6 shares a receptor with LIF, which was also upregulated in FRCs from skin-draining lymph nodes. Like IL6, LIF is upregulated in contracting myocytes as a means of inducing autocrine or paracrine muscle cell proliferation. IL6, IL1Rap, and LIF represented the three genes most upregulated by FRCs from skin-draining lymph nodes in the identified network.
With direct relevance for lymphocyte function, we found that IL7 and BAFF were upregulated by more than twofold in skin-draining FRCs. IL7 is a known product of FRCs, and functions to maintain naïve T cells (Link et al., 2007). BAFF is a potent survival factor and costimulatory activator for B cells. In secondary lymphoid organs, it is known to be produced by non-hematopoietic FDCs, but has not previously been reported in FRCs. However, the expression of BAFF by stromal cells outside lymph nodes is well-reported, though this seems to require chronic inflammation and has not previously been linked to steady-state stroma (Mackay and Schneider, 2009). Our cell sorts did not contain FDC contamination, since during transcriptional analysis, we found no evidence for expression of Fcγ receptors, and <1% of lymph node stromal cells stain positive for FDC-M1 or CD35 (data not shown). FDCs are confined to the B cell follicle, while FRCs surround, and occasionally extend into the B cell follicle. However, both cell types are thought to develop from a common precursor (Mebius, 2003). This result supports and extends the list of noted similarities between these cell types.
Strikingly, our results indicated that CXCL9 and CXCL10 are expressed by FRCs in skin-draining and mesenteric lymph nodes. These chemokines both signal through CXCR3, expressed by activated T cells, B cells, and NK cells, and their transcription is upregulated by IFNγ, IFNα, and other proinflammatory stimuli (Muller et al., 2010). While a role for CXCL10 in lymph nodes during steady-state has not been described, during some infections CXCL10 has an important role retaining Th1 lymphocytes in lymph nodes, a mechanism which encourages newly activated T cells to also become Th1, creating a polarized, microbe-appropriate response (Yoneyama et al., 2002). However, to date, CXCL10 production has only been identified in mature DCs within lymph nodes (Yoneyama et al., 2002). These data show that CXCL10 mRNA is also expressed by steady-state FRCs, with unknown function.
CXCL9 is poorly studied, but has been observed in DCs from cancer-associated lymphoid tissues and draining lymph nodes (Ohtani et al., 2009), any role in steady-state regulation of cell migration in lymph nodes is unreported.
Interestingly, FRCs from both mesenteric and skin-draining lymph nodes expressed genes involved in signaling to endothelial cells. It is known that FRCs are the primary producers of VEGF in skin-draining lymph nodes under steady-state conditions (Chyou et al., 2008). Surprisingly we found that FRCs from skin-draining lymph nodes expressed higher levels of VEGFa than those from mesenteric lymph nodes. Indeed, mesenteric lymph nodes showed upregulation of a relatively short list of genes compared to skin-draining lymph nodes, and the results did not map to a functional pathway when our robust statistical correction for multiple hypotheses was performed. However, we observed upregulation of known gut-related genes such as Nkx2-3, required for MadCAM expression and lymphocyte segregation in mesenteric lymph nodes (Pabst et al., 2000); as well as upregulation of Lrat, Tgm2, Meis1, and Pltp. These genes are either involved in the retinoic acid metabolism cycle, or upregulated by retinoic acid, (Matsuura and Ross, 1993; Cao et al., 2002; Galdones et al., 2006; Rebe et al., 2009) which is processed in mesenteric lymph nodes to impose a gut-specific homing phenotype on T cells (Iwata, 2009). We also found expression of several vasodilators (Vipr2, Itih4, Npr1) and a vasoconstrictor (Agt) suggesting that FRCs actively communicate with lymph node blood vessels. Similarly, upregulation of genes that induce neurite outgrowth (Efna5, gpr126, Tmem35, and Ngf) suggests a potential role in maintaining the normal sympathetic innervation of the lymph node (Panuncio et al., 1999). Further studies would be required to validate these results.
Taken together, altered expression of cytokines and chemokines in skin-draining lymph nodes suggests a novel picture of microenvironmental regulation by FRCs. Expression of BAFF and CXCR3 ligands imply previously unreported mechanisms of cross-talk with the hematopoietic system. Similarly, the upregulation of IL6, IL1Rap, and LIF suggests that skin-draining lymph node FRCs may be more contractile, and/or actively dividing faster than their mesenteric lymph node counterparts, similarly influencing endothelial cells to keep pace by transcribing increased VEGFα.
These transcriptional events may not be occurring equally across all FRCs, however. Lymph nodes contain a number of poorly understood FRC microniches. The best studied of these is the cortical T cell zone FRC, (sometimes called a TRC), which produces IL7, CCL19, CCL21, maintains the conduit system, and is used as a scaffold for the migration of DCs, T cells, and B cells. However, fibroblastic gp38+CD31− cells exist throughout the lymph node: in the subcapsular region abutting B cell follicles; as pericytes; and throughout the medulla. It is not yet possible to differentiate between these different FRC types, and the differences between skin-draining lymph nodes and mesenteric lymph nodes may involve a change in their proportion, if they differentially express the cytokines and chemokines identified.
Together, these data represent a comprehensive catalog of techniques adapted for lymph node stromal cells.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the National Institutes of Health (DK074500, AI045757, to Shannon J. Turley, and R24 AI072073 to the Immunological Genome Project), and NHMRC Australia (Overseas Postdoctoral Training Fellowship 546259, to Anne L. Fletcher). The authors wish to thank members of the ImmGen consortium for technical help and discussions.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/Immunological_Tolerance/10.3389/fimmu.2011.00035/abstract
References
Ahrendt, M., Hammerschmidt, S. I., Pabst, O., Pabst, R., and Bode, U. (2008). Stromal cells confer lymph node-specific properties by shaping a unique microenvironment influencing local immune responses. J. Immunol. 181, 1898–1907.
Ansel, K. M., Ngo, V. N., Hyman, P. L., Luther, S. A., Forster, R., Sedgwick, J. D., Browning, J. L., Lipp, M., and Cyster, J. G. (2000). A chemokine-driven positive feedback loop organizes lymphoid follicles. Nature 406, 309–314.
Bajenoff, M., Egen, J. G., Koo, L. Y., Laugier, J. P., Brau, F., Glaichenhaus, N., and Germain, R. N. (2006). Stromal cell networks regulate lymphocyte entry, migration and territoriality in lymph nodes. Immunity 25, 989–1001.
Buettner, M., Pabst, R., and Bode, U. (2011). Lymph node stromal cells strongly influence immune response suppression. Eur. J. Immunol. 41, 624–633.
Cao, G., Beyer, T. P., Yang, X. P., Schmidt, R. J., Zhang, Y., Bensch, W. R., Kauffman, R. F., Gao, H., Ryan, T. P., Liang, Y., Eacho, P. I., and Jiang, X. C. (2002). Phospholipid transfer protein is regulated by liver x receptors in vivo. J. Biol. Chem. 277, 39561–39565.
Chyou, S., Ekland, E. H., Carpenter, A. C., Tzeng, T. C., Tian, S., Michaud, M., Madri, J. A., and Lu, T. T. (2008). Fibroblast-type reticular stromal cells regulate the lymph node vasculature. J. Immunol. 181, 3887–3896.
Cohen, J. N., Guidi, C. J., Tewalt, E. F., Qiao, H., Rouhani, S. J., Ruddell, A., Farr, A. G., Tung, K. S., and Engelhard, V. H. (2010). Lymph node-resident lymphatic endothelial cells mediate peripheral tolerance via aire-independent direct antigen presentation. J. Exp. Med. 207, 681–688.
Coles, M., Kioussis, D., and Veiga-Fernandes, H. (2010). Cellular and molecular requirements in lymph node and peyer’s patch development. Prog. Mol. Biol. Transl. Sci. 92, 177–205.
Cupedo, T., Vondenhoff, M. F., Heeregrave, E. J., De Weerd, A. E., Jansen, W., Jackson, D. G., Kraal, G., and Mebius, R. E. (2004). Presumptive lymph node organizers are differentially represented in developing mesenteric and peripheral nodes. J. Immunol. 173, 2968–2975.
Cyster, J. G., Ansel, K. M., Reif, K., Ekland, E. H., Hyman, P. L., Tang, H. L., Luther, S. A., and Ngo, V. N. (2000). Follicular stromal cells and lymphocyte homing to follicles. Immunol. Rev. 176, 181–193.
Dennis, G. Jr., Sherman, B. T., Hosack, D. A., Yang, J., Gao, W., Lane, H. C., and Lempicki, R. A. (2003). David: database for annotation, visualization, and integrated discovery. Genome Biol. 4, P3.
Eikelenboom, P., Nassy, J. J., Post, J., Versteeg, J. C., and Langevoort, H. L. (1978). The histogenesis of lymph nodes in rat and rabbit. Anat. Rec. 190, 201–215.
Fletcher, A. L., Lukacs-Kornek, V., Reynoso, E. D., Pinner, S. E., Bellemare-Pelletier, A., Curry, M. S., Collier, A. R., Boyd, R. L., and Turley, S. J. (2010). Lymph node fibroblastic reticular cells directly present peripheral tissue antigen under steady-state and inflammatory conditions. J. Exp. Med. 207, 689–697.
Fletcher, A. L., Malhotra, D., and Turley, S. J. (2011). Lymph node stroma broaden the peripheral tolerance paradigm. Trends Immunol. 32, 12–18.
Galdones, E., Lohnes, D., and Hales, B. F. (2006). Role of retinoic acid receptors alpha1 and gamma in the response of murine limbs to retinol in vitro. Birth Defects Res. Part A Clin. Mol. Teratol. 76, 39–45.
Gallucci, R. M., Lee, E. G., and Tomasek, J. J. (2006). IL-6 modulates alpha-smooth muscle actin expression in dermal fibroblasts from IL-6-deficient mice. J. Invest. Dermatol. 126, 561–568.
Gardner, J. M., Devoss, J. J., Friedman, R. S., Wong, D. J., Tan, Y. X., Zhou, X., Johannes, K. P., Su, M. A., Chang, H. Y., Krummel, M. F., and Anderson, M. S. (2008). Deletional tolerance mediated by extrathymic aire-expressing cells. Science 321, 843–847.
Hadis, U., Wahl, B., Schulz, O., Hardtke-Wolenski, M., Schippers, A., Wagner, N., Muller, W., Sparwasser, T., Forster, R., and Pabst, O. (2011). Intestinal tolerance requires gut homing and expansion of foxp3+ regulatory t cells in the lamina propria. Immunity 34, 237–246.
Hu, Y., Bock, G., Wick, G., and Xu, Q. (1998). Activation of pdgf receptor alpha in vascular smooth muscle cells by mechanical stress. FASEB J. 12, 1135–1142.
Irizarry, R. A., Hobbs, B., Collin, F., Beazer-Barclay, Y. D., Antonellis, K. J., Scherf, U., and Speed, T. P. (2003). Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4, 249–264.
Iwata, M. (2009). Retinoic acid production by intestinal dendritic cells and its role in t-cell trafficking. Semin. Immunol. 21, 8–13.
Junt, T., Scandella, E., and Ludewig, B. (2008). Form follows function: lymphoid tissue microarchitecture in antimicrobial immune defence. Nat. Rev. Immunol. 8, 764–775.
Katakai, T., Hara, T., Lee, J. H., Gonda, H., Sugai, M., and Shimizu, A. (2004a). A novel reticular stromal structure in lymph node cortex: an immuno-platform for interactions among dendritic cells, T cells and B cells. Int. Immunol. 16, 1133–1142.
Katakai, T., Hara, T., Sugai, M., Gonda, H., and Shimizu, A. (2004b). Lymph node fibroblastic reticular cells construct the stromal reticulum via contact with lymphocytes. J. Exp. Med. 200, 783–795.
Katakai, T., Suto, H., Sugai, M., Gonda, H., Togawa, A., Suematsu, S., Ebisuno, Y., Katagiri, K., Kinashi, T., and Shimizu, A. (2008). Organizer-like reticular stromal cell layer common to adult secondary lymphoid organs. J. Immunol. 181, 6189–6200.
Koni, P. A., Sacca, R., Lawton, P., Browning, J. L., Ruddle, N. H., and Flavell, R. A. (1997). Distinct roles in lymphoid organogenesis for lymphotoxins alpha and beta revealed in lymphotoxin beta-deficient mice. Immunity 6, 491–500.
Lee, J. W., Epardaud, M., Sun, J., Becker, J. E., Cheng, A. C., Yonekura, A. R., Heath, J. K., and Turley, S. J. (2007). Peripheral antigen display by lymph node stroma promotes T cell tolerance to intestinal self. Nat. Immunol. 8, 181–190.
Link, A., Hardie, D. L., Favre, S., Britschgi, M. R., Adams, D. H., Sixt, M., Cyster, J. G., Buckley, C. D., and Luther, S. A. (2011). Association of T-zone reticular networks and conduits with ectopic lymphoid tissues in mice and humans. Am. J. Pathol. 178, 1662–1675.
Link, A., Vogt, T. K., Favre, S., Britschgi, M. R., Acha-Orbea, H., Hinz, B., Cyster, J. G., and Luther, S. A. (2007). Fibroblastic reticular cells in lymph nodes regulate the homeostasis of naive t cells. Nat. Immunol. 8, 1255–1265.
Luther, S. A., Tang, H. L., Hyman, P. L., Farr, A. G., and Cyster, J. G. (2000). Coexpression of the chemokines ELC and SLC by T zone stromal cells and deletion of the ELC gene in the plt/plt mouse. Proc. Natl. Acad. Sci. U.S.A. 97, 12694–12699.
Magnusson, F. C., Liblau, R. S., von Boehmer, H., Pittet, M. J., Lee, J. W., Turley, S. J., and Khazaie, K. (2008). Direct presentation of antigen by lymph node stromal cells protects against CD8 T-cell-mediated intestinal autoimmunity. Gastroenterology 134, 1028–1037.
Matsuura, T., and Ross, A. C. (1993). Regulation of hepatic lecithin: retinol acyltransferase activity by retinoic acid. Arch. Biochem. Biophys. 301, 221–227.
Muller, M., Carter, S., Hofer, M. J., and Campbell, I. L. (2010). Review: the chemokine receptor cxcr3 and its ligands cxcl9, cxcl10 and cxcl11 in neuroimmunity – a tale of conflict and conundrum. Neuropathol. Appl. Neurobiol. 36, 368–387.
Nichols, L. A., Chen, Y., Colella, T. A., Bennett, C. L., Clausen, B. E., and Engelhard, V. H. (2007). Deletional self-tolerance to a melanocyte/melanoma antigen derived from tyrosinase is mediated by a radio-resistant cell in peripheral and mesenteric lymph nodes. J. Immunol. 179, 993–1003.
Ohtani, H., Jin, Z., Takegawa, S., Nakayama, T., and Yoshie, O. (2009). Abundant expression of CXCL9 (MIG) by stromal cells that include dendritic cells and accumulation of CXCR3+ T cells in lymphocyte-rich gastric carcinoma. J. Pathol. 217, 21–31.
Pabst, O., Forster, R., Lipp, M., Engel, H., and Arnold, H. H. (2000). NKX2.3 is required for madcam-1 expression and homing of lymphocytes in spleen and mucosa-associated lymphoid tissue. EMBO J. 19, 2015–2023.
Panuncio, A. L., De La Pena, S., Gualco, G., and Reissenweber, N. (1999). Adrenergic innervation in reactive human lymph nodes. J. Anat. 194(Pt 1), 143–146.
Pedersen, B. K. (2006). The anti-inflammatory effect of exercise: its role in diabetes and cardiovascular disease control. Essays Biochem. 42, 105–117.
Rebe, C., Raveneau, M., Chevriaux, A., Lakomy, D., Sberna, A. L., Costa, A., Bessede, G., Athias, A., Steinmetz, E., Lobaccaro, J. M., Alves, G., Menicacci, A., Vachenc, S., Solary, E., Gambert, P., and Masson, D. (2009). Induction of transglutaminase 2 by a liver X receptor/retinoic acid receptor alpha pathway increases the clearance of apoptotic cells by human macrophages. Circ. Res. 105, 393–401.
Scandella, E., Bolinger, B., Lattmann, E., Miller, S., Favre, S., Littman, D. R., Finke, D., Luther, S. A., Junt, T., and Ludewig, B. (2008). Restoration of lymphoid organ integrity through the interaction of lymphoid tissue-inducer cells with stroma of the T cell zone. Nat. Immunol. 9, 667–675.
Sixt, M., Kanazawa, N., Selg, M., Samson, T., Roos, G., Reinhardt, D. P., Pabst, R., Lutz, M. B., and Sorokin, L. (2005). The conduit system transports soluble antigens from the afferent lymph to resident dendritic cells in the T cell area of the lymph node. Immunity 22, 19–29.
Warnock, R. A., Askari, S., Butcher, E. C., and von Andrian, U. H. (1998). Molecular mechanisms of lymphocyte homing to peripheral lymph nodes. J. Exp. Med. 187, 205–216.
Willard-Mack, C. L. (2006). Normal structure, function, and histology of lymph nodes. Toxicol. Pathol. 34, 409–424.
Woolf, E., Grigorova, I., Sagiv, A., Grabovsky, V., Feigelson, S. W., Shulman, Z., Hartmann, T., Sixt, M., Cyster, J. G., and Alon, R. (2007). Lymph node chemokines promote sustained T lymphocyte motility without triggering stable integrin adhesiveness in the absence of shear forces. Nat. Immunol. 8, 1076–1086.
Yamagata, T., Mathis, D., and Benoist, C. (2004). Self-reactivity in thymic double-positive cells commits cells to a CD8 alpha alpha lineage with characteristics of innate immune cells. Nat. Immunol. 5, 597–605.
Yip, L., Su, L., Sheng, D., Chang, P., Atkinson, M., Czesak, M., Albert, P. R., Collier, A. R., Turley, S. J., Fathman, C. G., and Creusot, R. J. (2009). Deaf1 isoforms control the expression of genes encoding peripheral tissue antigens in the pancreatic lymph nodes during type 1 diabetes. Nat. Immunol. 10, 1026–1033.
Yoneyama, H., Narumi, S., Zhang, Y., Murai, M., Baggiolini, M., Lanzavecchia, A., Ichida, T., Asakura, H., and Matsushima, K. (2002). Pivotal role of dendritic cell-derived CXCL10 in the retention of T helper cell 1 lymphocytes in secondary lymph nodes. J. Exp. Med. 195, 1257–1266.
Keywords: stromal cells, lymph node stromal cells, fibroblastic reticular cells, endothelial cells, tolerance, human lymph nodes, mouse lymph nodes
Citation: Fletcher AL, Malhotra D, Acton SE, Lukacs-Kornek V, Bellemare-Pelletier A, Curry M, Armant M and Turley SJ (2011) Reproducible isolation of lymph node stromal cells reveals site-dependent differences in fibroblastic reticular cells. Front. Immun. 2:35. doi: 10.3389/fimmu.2011.00035
Received: 12 July 2011; Paper pending published: 25 July 2011;
Accepted: 10 August 2011; Published online: 12 September 2011.
Edited by:
Daniel Hawiger, Saint Louis University, USAReviewed by:
C. Garrison (Garry) Fathman, Stanford University Medical School, USAKang Liu, Columbia University, USA
Copyright: © 2011 Fletcher, Malhotra, Acton, Lukacs-Kornek, Bellemare-Pelletier, Curry, Armant and Turley. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Shannon J. Turley, Department of Cancer Immunology and AIDS, Dana–Farber Cancer Institute, 450 Brookline Avenue, Boston, MA 02115, USA. e-mail:c2hhbm5vbl90dXJsZXlAZGZjaS5oYXJ2YXJk. edu