

 Karin V. Greco
Karin V. Greco
- William Harvey Research Institute, Barts and The London School of Medicine, Queen Mary University of London, London, UK
Macrophages have crucial functions in initiating the inflammatory reaction in a strict temporal and spatial manner to provide a “clear-up” response required for resolution. Hormonal peptides such as melanocortins modulate macrophage reactivity and attenuate inflammation ranging from skin inflammation to joint disease and reperfusion injury. The melanocortins (e.g., adrenocorticotrophin, ACTH and αMSH) elicit regulatory properties through activation of a family of GPCRs, the melanocortin (MC) receptors; MC1–MC5. Several studies have focused on MC1 and MC3 as anti-inflammatory receptors expressed on cells of the macrophage lineage. We review here elements of the melanocortin pathway with particular attention to macrophage function in anti-inflammatory and pro-resolving inflammatory settings. Evidence shows that ACTH, αMSH, and other MC agonists can activate MC1 and MC3 on macrophage through cAMP and/or NFκB-dependent mechanisms to abrogate pro-inflammatory cytokines, chemokines, and NO and enhance anti-inflammatory mediators such as IL-10 and HO-1. Melanocortins and their receptors regulate inflammation by inhibiting leukocyte recruitment to and interaction with inflamed tissue. An intensely exciting addition to this field of research has been the ability of an αMSH analog; AP214 to activate MC3 expressed on macrophage to enhance their clearance of both zymosan particles and apoptotic neutrophils thus putting melanocortins in line with other pro-resolving mediators. The use of mouse colonies mutated or nullified for MC1 or MC3, respectively as well as availability of selective MC receptor agonist/antagonists have been key to deciphering mechanisms by which elements of the melanocortin system play a role in these phenomena. We review here melanocortin pathway components with attention to the macrophage, reiterating receptor targets required for pro-resolving properties. The overall outcome will be identification of selective MC agonists as a strategy for innovative anti-inflammatory therapeutics.
Introduction
An inflammatory reaction is characterized by cellular recruitment within a tissue that firstly involves an acute phase whereby neutrophils act as invaders to release toxins to kill and eliminate foreign encounters. Macrophages play a crucial role in the latter phase of this inflammatory reaction. At the site of injury their ultimate goal is counterbalance the acute phase to cease inflammation and clear-up detrimental artifacts including pathogens and debris as well as apoptotic cells from the environment. To disperse inflammation macrophage must complete complex reactions, including chemotaxis to move to the site of inflammation, phagocytose particles or apoptotic cells (process of efferocytosis) and secrete cytokines/chemokines, lipid mediators, reactive oxygen species, and other factors to underpin an immune response facilitating healing/repair and return to normal homeostatic physiology. Pathological conditions can arise as a consequence of disruption to the sequence of events leading to resolution of inflammation. There has been a recent spate in research to identify and understand these biochemical pathways and mediators enhancing the resolution of inflammation by macrophage in the hope to discover new therapeutics that “switch on” this protective arm of inflammation. Within the umbrella of anti-inflammatory and pro-resolution mediators being investigated are the omega-3 fatty acid derived resolvins, aspirin-related lipoxins, and the glucocorticoid induced protein Annexin A1. One other such pathway gaining attention within this field includes the melanocortin peptides and their counterpart melanocortin receptors.
The Melanocortin System
Melanocortins are derived from proteolytic cleavage of pro-opiomelanocortin (POMC), a hormone first thought to be expressed only within the pituitary. Evidence now shows that POMC is also expressed within peripheral cells and tissues (Blalock, 1985). The enzymes proprotein convertase 1 (PC1) and 2 (PC2) belong to a conserved family of serine proteinases of the subtilisin/kexin-type that also include furin, PC4, PACE4, PC5/6, and PC7/LPC/PC8. Their action upon single and/or pairs of dibasic residues within the POMC sequence generate the melanocortin peptides; adrenocorticotrophin (ACTH), α-, β-, Γ-melanocyte stimulating hormone. PC1 leads to generation of full-length pro-ACTH1–39 and β-lipotropin. PC1 then further cleaves β-lipotropin to generate Γ-lipotropin and β-endorphin, and pro-ACTH to generate N-pro-opiocortin (POC), joining peptide (JP), and ACTH. The down-stream actions of PC2 result in production of ACTH1–17 and corticotrophin-like intermediate lobe peptide (from ACTH), ΓMSH (from N-POC), and β-MSH (from β-lipotropin). Alpha-MSH (αMSH) is generated by the combined actions of carboxypeptidase (CPE), peptidylglycine alpha-amidating mono-oxygenase (PAM), and N-acetyltransferase (N-AT) on ACTH1–17 (Mountjoy, 2010; Figure 1).
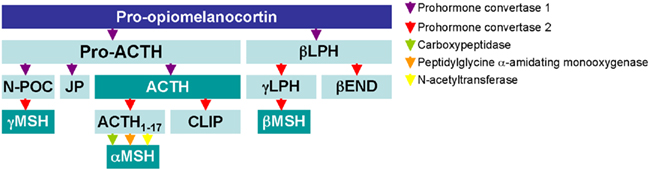
Figure 1. Biochemical processing of the POMC gene product as detailed in the pituitary. The processing of POMC and products by posttranslational modification enzymes is depicted by arrows. Agonists of the melanocortin receptors MC1–MC5 are highlighted.
The biological activity of the ACTH and MSH species occurs through activation of melanocortin receptors (MC) of which five have been cloned so far. All are seven transmembrane coupled to s-type G-proteins thus positively coupled to adenylate cyclase; their activation leading to increases in intracellular cAMP and, possibly partly independently from it, down-regulation of nuclear factor-kappa beta activation (Wikberg et al., 2000; Gantz and Fong, 2003). MC1 is the receptor expressed on melanocytes, responsible for MSH control over skin pigmentation, whereas MC2 is the canonical ACTH receptor, expressed on adrenal cells and responsible for glucocorticoid synthesis and release. The other MCs have less clear-cut biological functions. Within the central nervous system, MC4 is actively studied for its role in feed control, pain, and sexual health; MC5 might modulate exocrine gland activity (Gantz and Fong, 2003). MC3 might exert a control over energy metabolism (Butler et al., 2000), but our own work has indicated an important modulatory role for this receptor in the control of the host inflammatory response (Getting et al., 2002). Of interest here, there is now ample evidence that MC, especially MC1, MC3, and MC5 are distributed on peripheral cells (Catania et al., 2010).
Production of αMSH and Expression of MC Receptors by Macrophage
Melanocortins are expressed and functionally active on the macrophage. Star et al. (1995) reported increases in cAMP accumulation in RAW 264.7 macrophage (shown to express MC1 and MC3 but not MC5) upon αMSH (pan agonist) incubation, suggestive of the presence of functionally active MCs on the cell surface (Star et al., 1995; Li and Taylor, 2008). Production of αMSH by these cells was also noted with further augmentation of the response upon TNFα stimulation. These two lines of evidence along with the anti-inflammatory action of αMSH to reduce nitric oxide release by RAW 264.7 macrophage suggest that αMSH may act in an autocrine/paracrine manner by counteracting pro-inflammatory effect of cytokines (Star et al., 1995). A similar autocrine circuit emerged in a latter study with human monocyte/macrophage THP-1 cells. Here, incubation of resting macrophage with an antibody recognizing MC1 on its own increased TNFα release, with the addition of αMSH reducing this response. Furthermore the presence of the anti-MC1 attenuated this inhibitory signal caused by αMSH. Collectively, these data indicate that MC receptor agonists limit pro-inflammatory cytokine production from macrophages, possibly being pivotal also in peripheral autocrine circuits (Taherzadeh et al., 1999).
Expression of melanocortin receptors is not only restricted to macrophage cell lines, as MC receptor mRNA has been detected in primary rodent macrophage of alveolar (MC1 and MC3), knee joint (MC3), mesentery (MC1 and MC3), and peritoneal (MC1, MC3, and MC5) origin and human macrophage blood derived macrophage (MC1, MC3, and MC5; Getting et al., 2002, 2003, 2008; Andersen et al., 2005; Leoni et al., 2008; Montero-Melendez et al., 2011).
Melanocortins Receptor Activation on Macrophage Augment Anti-Inflammatory Responses
In inflammatory/tissue injury settings, activation of MCs can influence a number of fundamental macrophage functions including:
(1) antagonizing the release of pro-inflammatory cytokines, chemokines, nitric oxide
(2) inhibiting leukocyte chemoattraction
(3) inducing release of anti-inflammatory cytokines and expression of heme oxygenase-1
(4) augmenting phagocytosis and efferocytosis.
It should be noted that MC receptor activation can also affect macrophage responsiveness indirectly. For instance, the existence of a circuit involving MC3 and MC4 within the CNS that activates descending cholinergic fibers to activate anti-inflammatory nicotinic receptors have been described (Catania, 2007). Here we will restrict our overview on the direct effects of MC receptor on this cell type and discuss now some of the properties listed above in more detail.
Melanocortins Inhibit Cytokine Production
The ability of melanocortin peptides to reduce macrophage release of pro-inflammatory mediators and increase anti-inflammatory mediators is a well-understood phenomenon with a number of groups contributing to this field. For example Capsoni et al. (2009) stimulated peripheral blood derived monocytes in vitro with monosodium urate crystals in the presence of αMSH (pan agonist) or (CKPV)2 (a biologically potent Lys-Pro-Val peptide linked by Cys–Cys residues) and found significant reductions in IL-1β, IL-8, and TNFα release into supernatants (Capsoni et al., 2009). We treated the RAW264.7 mouse macrophage cell line with ACTH1–39 (pan agonist) and MTII, amore stable αMSH derivative with higher affinity for MC3 and MC4 promoted an increase in cAMP accumulation and release of anti-inflammatory cytokine IL-10, the later effect being abrogated when cells were treated with H-89 (protein kinase A [PKA] inhibitor) suggesting signaling down-stream of MC3 and not MC4 (as RAW264.7 did not express MC4) was through the cAMP–PKA pathway (Lam et al., 2006). In similar experiments both ACTH and MTII elicited in a cAMP–PKA dependent manner cytoprotective and anti-inflammatory heme oxygenase-1 (HO-1) but not heme oxygenase-2, heat shock protein 70 and 90 in RAW267.4 cells (Lam et al., 2005). Mandrika et al. (2001) showed dual signaling mechanism by which αMSH inhibited nitric oxide production by RAW267.4 macrophage. One pathway dependent of MC1/cAMP activation and the other causing inhibition of NFκB translocation in a cAMP-independent manner. Other specialized cells of the macrophage lineage such as microglia treated with αMSH or ACTH substantially reduced TNFα, IL-6, and nitric oxide increases caused by LPS + IFNΓ activation, suggesting that MC peptides exert their anti-inflammatory actions on peripheral as well as central cells of the phagocytic lineage. There is evidence of active MC receptors because the same study reported that αMSH could induce cAMP accumulation in both resting and activated microglia (Delgado et al., 1998).
Rat macrophage isolated from gouty knee joints also accumulated intracellular cAMP upon treatment with melanocortin peptide; ACTH (Getting et al., 2002). More recently functionality of MC receptors by cAMP readout on murine alveolar macrophage was confirmed after incubation with αMSH, [D-Trp8]ΓMSH (MC3/MC5 agonist), and MSO5 (MC1 agonist; Getting et al., 2008; Joseph et al., 2010). Together these studies suggest that the cAMP pathway represents a common underlying mechanism for melanocortin to deliver anti-inflammatory effects. Manna and Aggarwal stimulated human monocytes with various inflammatory agents including TNFα, LPS, ceramide, and okadaic acid to obtain NFκB activation, which was diminished in cells treated with αMSH (Manna and Aggarwal, 1998). The agonistic effect appeared to be cAMP-dependent such that inhibitors of adenylate cyclase and PKA reversed the inhibitory effect of αMSH. Furthermore αMSH inhibition of degradation of the NFκB inhibitory subunit IκBα and nuclear translocation of p65 subunit were also noted (Manna and Aggarwal, 1998).
Collectively these data appear to show that the anti-inflammatory actions of αMSH are dependent on MC1, MC3, and/or MC5 receptor activation and triggers inhibition of NFκB in a cAMP-dependent and independent manner through molecular links which are yet to be deciphered.
Melanocortins Inhibit Chemotaxis of Leukocytes
As discussed so far, melanocortins are able to dampen macrophage release of pro-inflammatory mediators in inflammatory settings. These phenomena were exploited further by Capsoni et al. (2009) who showed that melanocortins could influence the monocytes ability to recruit and prime neutrophils. Chemotaxis of neutrophils treated with αMSH or (CKPV)2 was examined using a boyden chamber assay and supernatants collected from monosodium urate crystal-stimulated monocytes as the source of chemoattractants. Chemotaxis was significantly reduced in neutrophils treated with agonist compared to controls. Furthermore reduced chemiluminescence (as a marker of reactive oxygen intermediates) production by pre-treated primed neutrophils was observed, in line with other studies showing melanocortin peptides can inhibit chemotaxis and generation of reactive oxygen species (Catania et al., 1996; Getting et al., 1999a; Capsoni et al., 2009).
To decipher what role MC3 has on leukocyte interaction with the inflamed mesentery, experiments were performed using pharmacological agents and mice nullified for MC3 where the superior mesenteric artery was occluded for 35 min followed by reopening to allow for blood reperfusion. Treatment with [D-Trp8]ΓMSH, a MC3/MC5 agonist did not alter cell rolling but decreased cell adhesion and emigration compared to vehicle control an effect not observed in Mc3r−/− mice. These data were mirrored by augmented KC and MCP-1 responses in mesenteric tissues of Mc3r−/− mice suggesting MC3 is able to regulate leukocyte interaction with postcapillary venules and regulate levels of pro-inflammatory molecules in ischemia reperfusion injured tissues (Leoni et al., 2008). Moreover, whilst agonizing more than one MC receptor, the vasculoprotective properties of [D-Trp8]ΓMSH are solely transduced by MC3.
Of interest, the modulation of chemokines occurred in the tissue and not with respect to plasma levels, moreover, levels of the cytokines IL-1β and TNFα were not affected by pharmacological treatment with [D-Trp8]ΓMSH or absence of the MC3 gene. The “selective” effect on tissue chemokine levels may be secondary to inhibition of activation of resident cells, such as tissue macrophages and connective tissue type mast cells (Kubes and Granger, 1996; Tailor et al., 1999).
In similar experimental settings, the effects of a small molecule MC1 agonist, compound BMS-470539 (Kang et al., 2006) inhibited cell adhesion, and emigration, an effect that was lacking in yellowe/e mice. Interestingly, unlike Mc3r−/− mice, the yellowe/e mice (expressing non-functional MC1) displayed a comparable leukocyte adhesion and emigration response to wild types. Collectively these two studies identify a complex scenario whereby although both MC1 and MC3 are expressed in the inflamed mesentery, MC1 may not be endogenously activated in ischemia reperfusion injury whereas MC3 is both exogenously and endogenously activated in this setting (Leoni et al., 2010). Future studies addressing the hypothetical generation of selective agonists post-ischemia reperfusion might help deciphering this apparent discrepancy.
Activation of MC3 is a Key Mediator of Anti-Inflammatory Effects in Macrophages
Until a decade ago, the mainstream school of thought was that the anti-inflammatory actions of melanocortin agonists were solely through MC1 activation (Lipton and Catania, 1997; Luger et al., 2003). We used two mouse colonies, one bearing a non-functional MC1 (yellowe/e mice) and the other knocked out for MC3 (Mc3r−/− mice), to conclude that MC3 can be engaged by agonists with anti-inflammatory properties. Cultured primary peritoneal macrophage from yellowe/e mice were treated with [D-Trp8]ΓMSH displaying a dose-dependent increase in cAMP accumulation, an effect reversed in the presence of an antibody to MC3. The release of the chemokine KC was abrogated in the presence of [D-Trp8]ΓMSH with the antagonist agouti related protein (AGRP) abolishing the inhibitory effect.
In another study mice pre-treated with ACTH were injected with MSU crystals to induce peritonitis. A reduced accumulation of PMN was observed in the peritoneal cavity of mice treated with ACTH compared to vehicle control, data that was mirrored by decreased levels of KC also within the cavity. Interestingly co-administration of SHU9119 (an MC3/MC4 antagonist) with ACTH inhibited the agonists effects suggesting ACTH was acting through MC3 and further confirmed by the detection of MC3 but not MC4 at the mRNA transcript level on peritoneal macrophage (Getting et al., 1999b). At a latter date, Getting et al. (2003) in the same peritonitis model using more selective MC3 agonists and yellowe/e mice confirmed that indeed agonism at MC3 inhibited pro-inflammatory cytokines (IL-1) and chemokines (KC) and accumulation of neutrophils in the inflamed peritoneal cavity (Getting et al., 2003). Collectively these data suggest MC3 more than MC1 (at least in mouse) to be pivotal in bringing about the anti-inflammatory effects observed following treatment with these agonists (Getting et al., 1999b, 2003, 2006).
MC3 Regulates Macrophage Differentiation to Osteoclasts during Inflammatory Arthritis
More recently MC3 has been implicated in the regulation of macrophage precursor differentiation to osteoclasts. Inflammatory arthritis was induced by injection of an arthritogenic serum into wild type and Mc3r−/−, observing a higher prevalence and severity of disease observed in the latter genotype (Patel et al., 2010a). In a series of real time PCR analyses of extracts from the mouse ankle joint, a discrete set of inflammatory genes (13 out of 96) were upregulated in Mc3r−/− including IL-1β, IL-6, NOS2, CCR4, CXCR3, CCL2 as compared to wild types. It is worthy to note here that major macrophage secretory products include IL-1β and IL-6 (Gordon, 2003).
Upon microscopic analyses of the ankle joints, a significantly higher number of joints were affected by bone erosion, as indicated by histological scores and number of TRAP-positive osteoclasts within the Mc3r−/− joint. This observation was corroborated by the higher levels of RANKL (a key driver of osteoclast formation) mRNA in Mc3r−/− ankle joints (Lacey et al., 1998). Although the exact mechanism was not characterized, some conclusion could be reached by the study of osteoclast formation in vitro. Bone marrow-derived macrophage from wild type and Mc3r−/− mice were differentiated to osteoclasts in the presence of M-CSF and RANKL. A defect was noted such that a higher number of osteoclasts were generated from macrophage absent of the MC3 gene compared to MC3 sufficient cells. The Mc3r−/− osteoclasts displayed an increased “eating” ability such that when cultured on calcium phosphate coated wells significantly more resorption pits could be observed and quantified. Interestingly Mc3r−/− cells had increased and sustained RANKL-mediated NFκB signaling compared to wild types; this finding could provide mechanistic support to the increased CCL2 synthesis observed by these cells (Patel et al., 2010a). Together with the study by Cornish et al. (2003), our work implicates a role for melanocortins and their melanocortin receptors in the regulation of macrophage differentiation to specialized cells, example being here the osteoclast. It is plausible that MCs may modulate macrophage differentiation in other specialized cells such as the microglia or Kupffer cells, with further implications for their therapeutic potential.
MC3 Activation Enhances Resolution of Inflammation
Resolution of inflammation is an important process required to reset tissue/cells to a state of normalization after insult/injury. During this process a number of endogenous pathways are activated in order to regain homeostatic balance after inflammation. An exciting enhancement to the field of melanocortin biology in inflammation has been the recent finding that AP214, a modified αMSH analog, possesses prophagocytic and pro-resolving effects (Montero-Melendez et al., 2011), in line with the profile reported for resolvins and lipoxins (Schwab et al., 2007).
AP214 inhibits neutrophil recruitment in the zymosan peritonitis model, an effect that could be due, at least in part, to a modulation of macrophage phagocytic abilities of the particles. In vitro, AP214-treated biogel-elicited macrophages were incubated with zymosan particles to determine the percentage of phagocytic cells and number of ingested particles, observing an increment in both parameters. Such an effect of AP214 was also observed with respect to efferocytosis since this MSH analog augmented phagocytosis of human apoptotic neutrophils by mouse macrophages, an effect that was absent when Mc3r−/− macrophages were used. In in vivo settings, injection of apoptotic neutrophils into murine peritoneal cavities pre-treated with AP214 led to an increase in macrophage ingestion of neutrophils compared to vehicle control. This recent study has uncovered a new angle in which melanocortins and their receptors can affect the inflammatory reaction providing strong evidence for genuine pro-resolving activities centered on tight regulation of macrophage functions.
Conclusion
In this review we have highlighted the importance of melanocortins and their receptors in modulating the macrophage function in inflammation and tissue injury (see Figure 2). We have seen that MC activation does not merely reduce production of pro-inflammatory mediators, but can regulate cell differentiation as well as chemotaxis of leukocytes. Excitingly, melanocortins can now be added to the plethora of pro-resolution mediators by way of their ability to augment phagocytosis and efferocytosis; we predict these portfolio of properties will define other functions/biological properties of melanocortins in other aspects of resolution of inflammation in the years to come.
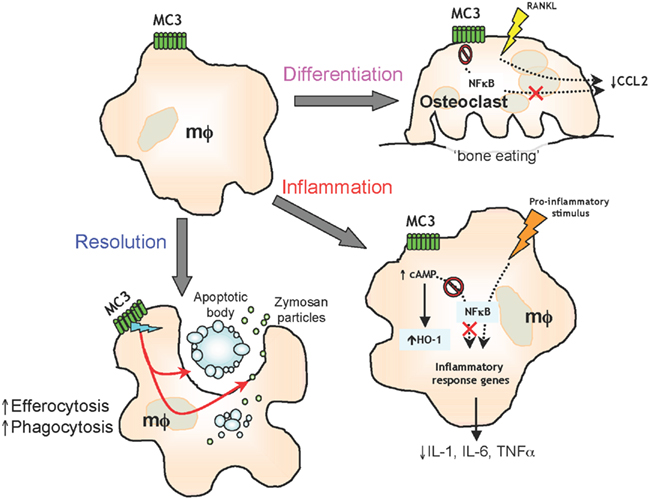
Figure 2. Melanocortin receptor type 3 activation on macrophage (mφ) functions Activation of MC3 by endogenous or selective synthetic agonists leads to regulation of osteoclast generation, control of pro-inflammatory and anti-inflammatory mediators and resolution of inflammation by efferocytosis and phagocytosis.
What does this mean for the development of melanocortin-based therapies? Noteworthy, ACTH has been used as early as the 1940s for the treatment of gouty and rheumatoid arthritis, yet as discussed above never were the mechanisms of actions deciphered until the recent decade (Hench et al., 1949; Gutman and Yu, 1950). What does remain in its infancy is the pattern in which MCs may be expressed in specific disease, nevertheless this area of research and drug development has become particularly eye-catching as seen with the development of AP214 for the treatment of post-surgical kidney injury following cardiac heart surgery in phase II trials (www.clinicaltrials.gov). Other melanocortin-based therapies are in the development pipeline for biotechnology companies with emphasis on obesity and sexual dysfunction as disease targets (Patel et al., 2010b).
We envisage over the next decade new drugs based on the melanocortin peptides – specifically targeting MC3 – to be developed and designed for the treatment of inflammatory conditions. These drugs would be better tolerated with a lower burden of side-effects as they would mimic the body’s way of naturally abating inflammation to promote pro-resolving and anti-inflammatory mediators.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Work on melanocortin peptides and their receptors produced in the laboratory of the Authors has been funded, over the years, by several grants from the Arthritis Research UK (active project number 18049) and by one collaborative project between the William Harvey Research Foundation and Action Pharma A/S.
References
Andersen, G. N., Hagglund, M., Nagaeva, O., Frangsmyr, L., Petrovska, R., Mincheva-Nilsson, L., and Wikberg, J. E. (2005). Quantitative measurement of the levels of melanocortin receptor subtype 1, 2, 3 and 5 and pro-opio-melanocortin peptide gene expression in subsets of human peripheral blood leucocytes. Scand. J. Immunol. 61, 279–284.
Blalock, J. E. (1985). Proopiomelanocortin-derived peptides in the immune system. Clin. Endocrinol. (Oxf.) 22, 823–827.
Butler, A. A., Kesterson, R. A., Khong, K., Cullen, M. J., Pelleymounter, M. A., Dekoning, J., Baetscher, M., and Cone, R. D. (2000). A unique metabolic syndrome causes obesity in the melanocortin-3 receptor-deficient mouse. Endocrinology 141, 3518–3521.
Capsoni, F., Ongari, A. M., Reali, E., and Catania, A. (2009). Melanocortin peptides inhibit urate crystal-induced activation of phagocytic cells. Arthritis Res. Ther. 11, R151.
Catania, A., Lonati, C., Sordi, A., Carlin, A., Leonardi, P., and Gatti, S. (2010). The melanocortin system in control of inflammation. ScientificWorldJournal 10, 1840–1853.
Catania, A., Rajora, N., Capsoni, F., Minonzio, F., Star, R. A., and Lipton, J. M. (1996). The neuropeptide alpha-MSH has specific receptors on neutrophils and reduces chemotaxis in vitro. Peptides 17, 675–679.
Cornish, J., Callon, K. E., Mountjoy, K. G., Bava, U., Lin, J. M., Myers, D. E., Naot, D., and Reid, I. R. (2003). alpha -Melanocyte-stimulating hormone is a novel regulator of bone. Am. J. Physiol. Endocrinol. Metab. 284, E1181–E1190.
Delgado, R., Carlin, A., Airaghi, L., Demitri, M. T., Meda, L., Galimberti, D., Baron, P., Lipton, J. M., and Catania, A. (1998). Melanocortin peptides inhibit production of proinflammatory cytokines and nitric oxide by activated microglia. J. Leukoc. Biol. 63, 740–745.
Gantz, I., and Fong, T. M. (2003). The melanocortin system. Am. J. Physiol. Endocrinol. Metab. 284, E468–E474.
Getting, S. J., Christian, H. C., Flower, R. J., and Perretti, M. (2002). Activation of melanocortin type 3 receptor as a molecular mechanism for adrenocorticotropic hormone efficacy in gouty arthritis. Arthritis Rheum. 46, 2765–2775.
Getting, S. J., Christian, H. C., Lam, C. W., Gavins, F. N., Flower, R. J., Schioth, H. B., and Perretti, M. (2003). Redundancy of a functional melanocortin 1 receptor in the anti-inflammatory actions of melanocortin peptides: studies in the recessive yellow (e/e) mouse suggest an important role for melanocortin 3 receptor. J. Immunol. 170, 3323–3330.
Getting, S. J., Flower, R. J., and Perretti, M. (1999a). Agonism at melanocortin receptor type 3 on macrophages inhibits neutrophil influx. Inflamm. Res. 48(Suppl. 2), S140–S141.
Getting, S. J., Gibbs, L., Clark, A. J., Flower, R. J., and Perretti, M. (1999b). POMC gene-derived peptides activate melanocortin type 3 receptor on murine macrophages, suppress cytokine release, and inhibit neutrophil migration in acute experimental inflammation. J. Immunol. 162, 7446–7453.
Getting, S. J., Lam, C. W., Leoni, G., Gavins, F. N., Grieco, P., and Perretti, M. (2006). [D-Trp8]-gamma-melanocyte-stimulating hormone exhibits anti-inflammatory efficacy in mice bearing a nonfunctional MC1R (recessive yellow e/e mouse). Mol. Pharmacol. 70, 1850–1855.
Getting, S. J., Riffo-Vasquez, Y., Pitchford, S., Kaneva, M., Grieco, P., Page, C. P., Perretti, M., and Spina, D. (2008). A role for MC3R in modulating lung inflammation. Pulm. Pharmacol. Ther. 21, 866–873.
Gutman, A. B., and Yu, T. F. (1950). Effects of adrenocorticotropic hormone (ACTH) in gout. Am. J. Med. 9, 24–30.
Hench, P. S., Kendall, E. C., Slocumb, C. H., and Polley, H. F. (1949). The effect of a hormone of the adrenal cortex (17-hydroxy-11-dehydrocorticosterone; compound E) and of pituitary adrenocorticotropic hormone on rheumatoid arthritis. Mayo Clin. Proc. 24, 181–197.
Joseph, C. G., Yao, H., Scott, J. W., Sorensen, N. B., Marnane, R. N., Mountjoy, K. G., and Haskell-Luevano, C. (2010). gamma-Melanocyte stimulation hormone (gamma-MSH) truncation studies results in the cautionary note that gamma-MSH is not selective for the mouse MC3R over the mouse MC5R. Peptides 31, 2304–2313.
Kang, L., McIntyre, K. W., Gillooly, K. M., Yang, Y., Haycock, J., Roberts, S., Khanna, A., Herpin, T. F., Yu, G., Wu, X., Morton, G. C., Tuerdi, H., Koplowitz, B., Walker, S. G., Wardwell-Swanson, J., Macor, J. E., Lawrence, R. M., and Carlson, K. E. (2006). A selective small molecule agonist of the melanocortin-1 receptor inhibits lipopolysaccharide-induced cytokine accumulation and leukocyte infiltration in mice. J. Leukoc. Biol. 80, 897–904.
Kubes, P., and Granger, D. N. (1996). Leukocyte-endothelial cell interactions evoked by mast cells. Cardiovasc. Res. 32, 699–708.
Lacey, D. L., Timms, E., Tan, H. L., Kelley, M. J., Dunstan, C. R., Burgess, T., Elliott, R., Colombero, A., Elliott, G., Scully, S., Hsu, H., Sullivan, J., Hawkins, N., Davy, E., Capparelli, C., Eli, A., Qian, Y. X., Kaufman, S., Sarosi, I., Shalhoub, V., Senaldi, G., Guo, J., Delaney, J., and Boyle, W. J. (1998). Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 93, 165–176.
Lam, C. W., Getting, S. J., and Perretti, M. (2005). In vitro and in vivo induction of heme oxygenase 1 in mouse macrophages following melanocortin receptor activation. J. Immunol. 174, 2297–304.
Lam, C. W., Perretti, M., and Getting, S. J. (2006). Melanocortin receptor signaling in RAW264.7 macrophage cell line. Peptides 27, 404–412.
Leoni, G., Patel, H. B., Sampaio, A. L., Gavins, F. N., Murray, J. F., Grieco, P., Getting, S. J., and Perretti, M. (2008). Inflamed phenotype of the mesenteric microcirculation of melanocortin type 3 receptor-null mice after ischemia-reperfusion. FASEB J. 22, 4228–4238.
Leoni, G., Voisin, M. B., Carlson, K., Getting, S., Nourshargh, S., and Perretti, M. (2010). The melanocortin MC(1) receptor agonist BMS-470539 inhibits leucocyte trafficking in the inflamed vasculature. Br. J. Pharmacol. 160, 171–180.
Li, D., and Taylor, A. W. (2008). Diminishment of alpha-MSH anti-inflammatory activity in MC1r siRNA-transfected RAW264.7 macrophages. J. Leukoc. Biol. 84, 191–198.
Lipton, J. M., and Catania, A. (1997). Anti-inflammatory actions of the neuroimmunomodulator alpha-MSH. Immunol. Today 18, 140–145.
Luger, T. A., Scholzen, T. E., Brzoska, T., and Bohm, M. (2003). New insights into the functions of alpha-MSH and related peptides in the immune system. Ann. N. Y. Acad. Sci. 994, 133–140.
Mandrika, I., Muceniece, R., and Wikberg, J. E. (2001). Effects of melanocortin peptides in lipopolysaccharide/interferon-gamma-induced NF-kappaB DNA binding and nitric oxide production in macrophage-like RAW 264.7 cells: evidence for dual mechanisms of action. Biochem. Pharmacol. 61, 613–621.
Manna, S. K., and Aggarwal, B. B. (1998). Alpha-melanocyte-stimulating hormone inhibits the nuclear transcription factor NF-kappa B activation induced by various inflammatory agents. J. Immunol. 161, 2873–2880.
Montero-Melendez, T., Patel, H. B., Seed, M., Nielsen, S., Jonassen, T. E. N., and Perretti, M. (2011). The melanocortin agonist AP214 exerts anti-inflammatory and pro-resolving properties: evidence for a major involvement of the melanocortin receptor type 3. Am. J. Pathol. 179, 259–269.
Mountjoy, K. G. (2010). Functions for pro-opiomelanocortin-derived peptides in obesity and diabetes. Biochem. J. 428, 305–324.
Patel, H. B., Bombardieri, M., Sampaio, A. L., D’Acquisto, F., Gray, M., Grieco, P., Getting, S. J., Pitzalis, C., and Perretti, M. (2010a). Anti-inflammatory and antiosteoclastogenesis properties of endogenous melanocortin receptor type 3 in experimental arthritis. FASEB J. 24, 4835–4843.
Patel, H. B., Leoni, G., Melendez, T. M., Sampaio, A. L., and Perretti, M. (2010b). Melanocortin control of cell trafficking in vascular inflammation. Adv. Exp. Med. Biol. 681, 88–106.
Schwab, J. M., Chiang, N., Arita, M., and Serhan, C. N. (2007). Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature 447, 869–874.
Star, R. A., Rajora, N., Huang, J., Stock, R. C., Catania, A., and Lipton, J. M. (1995). Evidence of autocrine modulation of macrophage nitric oxide synthase by alpha-melanocyte-stimulating hormone. Proc. Natl. Acad. Sci. U.S.A. 92, 8016–8020.
Taherzadeh, S., Sharma, S., Chhajlani, V., Gantz, I., Rajora, N., Demitri, M. T., Kelly, L., Zhao, H., Ichiyama, T., Catania, A., and Lipton, J. M. (1999). alpha-MSH and its receptors in regulation of tumor necrosis factor-alpha production by human monocyte/macrophages. Am. J. Physiol. 276(Pt 2), R1289–R1294.
Tailor, A., Tomlinson, A., Salas, A., Panes, J., Granger, D. N., Flower, R. J., and Perretti, M. (1999). Dexamethasone inhibition of leucocyte adhesion to rat mesenteric postcapillary venules: role of intercellular adhesion molecule 1 and KC. Gut 45, 705–712.
Keywords: inflammation, resolution, macrophage, melanocortins, melanocortin receptor, melanocyte stimulating hormone, G-protein coupled receptor, anti-inflammatory therapeutics
Citation: Patel HB, Montero-Melendez T, Greco KV and Perretti M (2011) Melanocortin receptors as novel effectors of macrophage responses in inflammation. Front. Immun. 2:41. doi: 10.3389/fimmu.2011.00041
Received: 30 June 2011; Accepted: 22 August 2011;
Published online: 13 September 2011.
Edited by:
Amiram Ariel, University of Haifa, IsraelReviewed by:
Paola Allavena, Clinical Institute Humanitas, ItalyAnna Catania, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico di Milano, Italy
Copyright: © 2011 Patel, Montero-Melendez, Greco and Perretti. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Hetal B. Patel, William Harvey Research Institute, Barts and The London School of Medicine, Queen Mary University of London, Charterhouse Square, London EC1M 6BQ, UK. e-mail:aC5iLnBhdGVsQHFtdWwuYWMudWs=