 Geanncarlo Lugo-Villarino1,2
Geanncarlo Lugo-Villarino1,2 Christel Vérollet1,2
Christel Vérollet1,2 Isabelle Maridonneau-Parini1,2
Isabelle Maridonneau-Parini1,2 Olivier Neyrolles1,2*
Olivier Neyrolles1,2*- 1 CNRS, Institut de Pharmacologie et de Biologie Structurale, Toulouse, France
- 2 UPS Institut de Pharmacologie et de Biologie Structurale, Université de Toulouse, Toulouse, France
In the arms race of host–microbe co-evolution, macrophages (Mɸs) have been endowed with strategies to neutralize pathogenic challenge while preserving host integrity. During steady-states conditions, Mɸs perform multiple house-keeping functions governed by their differentiation state, tissue distribution, and signals from the microenvironment. In response to pathogenic challenge and host mediators, however, Mɸs undergo different programs of activation rendering them either pro-inflammatory and microbicidal (M1), or immunosuppressants and tissue repairers (M2). An excessive or prolonged polarization of either program may be detrimental to the host due to potential tissue injury or contribution to pathogenesis. Conversely, intracellular microbes that cause chronic diseases such as tuberculosis and acquired immunodeficiency syndrome exemplify strategies for survival in the host. Indeed, both Mycobacterium tuberculosis (Mtb) and human immunodeficiency virus (HIV-1) are successful intracellular microbes that thrive in Mɸs. Given these microbes not only co-circulate throughout the developing world but each has contributed to prevalence and mortality caused by the other, substantial insights into microbe physiology and host defenses then rest in the attempt to fully understand their influence on Mɸ polarization. This review addresses the role of Mɸ polarization in the immune response to, and pathogenesis of, Mtb and HIV.
Introduction
Pathogens have evolved ingenious strategies to circumvent the host immune response as part of the constant evolutionary process-taking place in all living organisms. Chief among these strategies is the prevention of the inflammatory response or seizure of the anti-inflammatory mechanism in place to protect tissue integrity. The manipulation of macrophage (Mɸ) polarization is one of the main targets to accomplish this, since this antigen presenting cell represents the first line of an active defense system in the host, and if successfully done, it can then undermine adaptive immunity (Benoit et al., 2008). Mɸ polarization is a dynamic process governed by mechanisms dictating their tissue distribution and functional capacities in response to endogenous and exogenous signals (Martinez et al., 2009). Polarized Mɸs are broadly classified into two groups: classical (M1) and alternative (M2) activated. On one hand, M1 program is a direct response to type-1 inflammatory conditions (e.g., IFN-Γ) and pathogen challenge, and it has been associated to resistance to intracellular pathogens and to some form of tumors. On the other hand, the M2 program is driven by type-2 inflammatory signals such as IL-4 and IL-13 (M2a); immune complexes, toll-like receptors (TLRs) agonists, or IL-1 receptors (M2b); and immunosuppressants including IL-10, transforming growth factor-β (TGF-β) or glucocorticoids (M2c; Table 1). M2 Mɸs participate in diverse activities including the suppression of inflammation, enhancement of phagocytosis, promotion of tissue remodeling and repair, elimination of parasites, and unwanted tumor angiogenesis (Sica et al., 2008; Martinez et al., 2009; Murray and Wynn, 2011). Furthermore, it is becoming clear that Mɸ polarization supports different, and in some cases, opposing biological functions, that influences tissue homeostasis, and numerous pathological situations, including infectious diseases (Benoit et al., 2008; Cairo et al., 2011). Given the pivotal role Mɸs play as sentinels of the immune system, they represent ideal cell targets for subversion by successful intracellular pathogens.
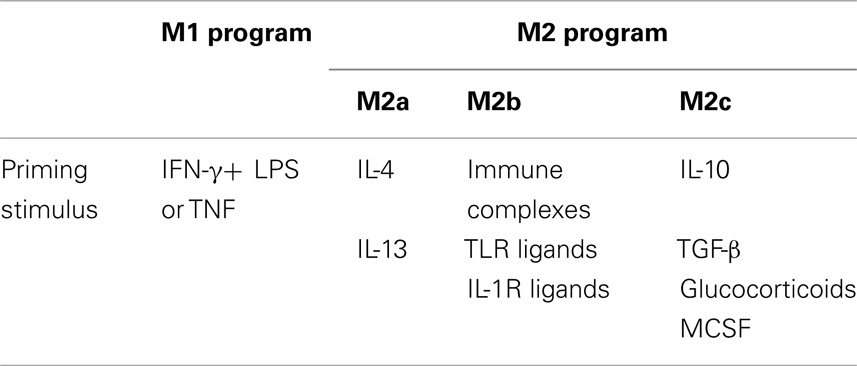
Table 1. Priming stimulus for the classical (M1) and alternative (M2a-c) activation of macrophages.
The purpose of this short review is not to provide a comprehensive summary of Mɸ polarization; others have recently reviewed this growing research area (Martinez et al., 2009; Murray and Wynn, 2011). Also, we will not address the multiple ways by which the pathogens in question circumvent the immune system, as there are excellent reviews covering this subject (Deretic et al., 2004; Carter and Ehrlich, 2008; Meena and Rajni, 2010; Hajishengallis and Lambris, 2011). Instead, we will focus exclusively on the significance of Mɸ polarization in the context of pathophysiology caused by Mycobacterium tuberculosis (Mtb) and human immunodeficiency virus (HIV).
Macrophage Polarization in Mtb Infection
The world health organization reports tuberculosis (TB) is still one of the leading causes of death due to a single infectious agent (Mtb) with 1.7 million deaths and 9.4 million new cases in 2009, and estimates that about one-third of the human population may be latently infected (WHO Global Tuberculosis Control Report 2010, 2010). Active TB may occur directly after infection or through the reactivation of latent infection that is confined in granulomas. The elaboration and maintenance of granulomas depends on a dedicated immune response, which is not fully understood. Recently, however, it was demonstrated mycobacteria exploits Mɸ activation to turn the granuloma into an effective tool for pathogenesis (Davis and Ramakrishnan, 2009; Volkman et al., 2010). Therefore, a better understanding of Mɸ polarization during Mtb infection might yield further clues about how Mtb circumvents the immune system.
As aforementioned, Mɸ polarization is mainly driven by type-1 and type-2 inflammatory signals (Table 1). Type-1 inflammatory cytokines are essential in the defense against Mtb since their expression often correlates with efficient anti-Mtb immune responses, and genetic deficiencies of these factors lead to increased TB susceptibility (Quintana-Murci et al., 2007). IFN-Γ drives the M1 program characterized by Mɸ capacity to kill most mycobacteria and restrict the replication of the remainder (Ehrt et al., 2001). The early phase of the anti-Mtb immune response is marked by M1 Mɸ polarization in multiple animal models and reminiscent of the clinical data collected from patients with active TB (Figure 1; Benoit et al., 2008). At the transcriptome level, the gene modulation induced by Mtb in Mɸs highly overlaps, and in some cases synergizes, with that induced by IFN-Γ to establish the M1 phenotype (Ehrt et al., 2001). At the granuloma level in mice, M1 Mɸ polarization is evident in mice between 7 and 30 days after Mtb infection when high levels of IFN-Γ and iNOS are also detected within this structure and around the alveolar compartment (Redente et al., 2010). All in all, polarization of M1 Mɸs is part of the “common host response” against intracellular bacteria characterized by high expression of iNOS and consequent nitric oxide (NO) production (characteristic of murine models), secretion of pro-inflammatory cytokines and chemokines, release of proteolytic enzymes and anti-microbial peptides, enhanced phagocytosis, and development of a toxic intracellular environment reflected in the fusion of microbial phagosomes with acidic and hydrolase-rich lysosomes (Ehrt et al., 2001; Deretic et al., 2004; Martinez et al., 2009; Cairo et al., 2011; Murray and Wynn, 2011). It remains to be demonstrated whether transcription factors [e.g., p65 and interferon regulatory factor (IRF5)] or regulators (e.g., SHIP1) that dictate the M1 program of macrophage polarization also play a role in TB infection (Martinez, 2011). Considering this hostile environment created by M1 Mɸs, it is not surprising Mtb has evolved strategies to interfere with M1 polarization. Indeed, Mtb inhibits IFN-Γ activation of Mɸs by secreting virulence factors such as lipoarabinomannan that halters phagosome maturation, or early secretory antigenic target-6 (ESAT-6) that prevents the activation of NF-κB and IFN-Γ regulatory factors downstream of TLR-2 (Deretic et al., 2004; Benoit et al., 2008). Indirectly, Mtb blocks M1 polarization by the transcriptional inhibition of IFN-Γ-responsive genes through a bystander effect involving IL-6 (Sibley et al., 1990; Benoit et al., 2008).
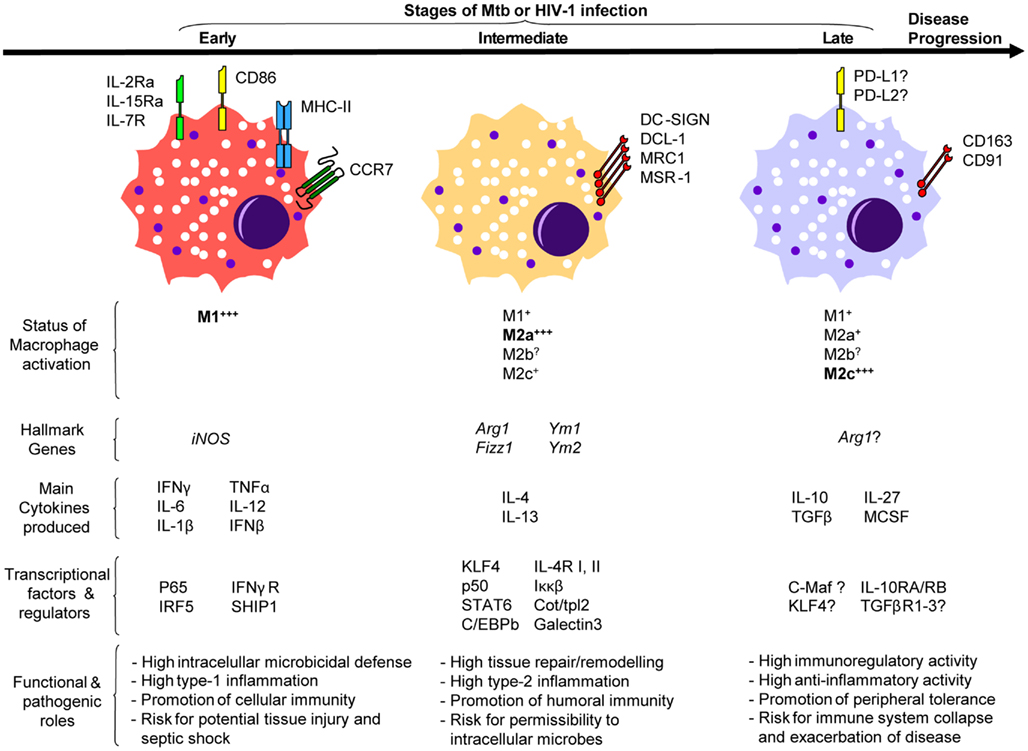
Figure 1. A model illustrating the putative involvement of macrophage polarization during Mtb or HIV-1 infection.
Perhaps the best strategy to avoid the challenges posed by M1 Mɸs is to shift their program into M2 Mɸs. TB susceptibility parallels with elevated levels of type-2 inflammatory signals (e.g., IL-4, IL-13; Kahnert et al., 2006; Raju et al., 2008; Almeida et al., 2009; Schreiber et al., 2009). Likewise, high levels of IL-10 (mostly derived from Mɸs) correlate with active TB patients (Barnes et al., 1993; Verbon et al., 1999). Interestingly, the predominant type-2 inflammatory environment shifts back to type-1 after successful treatment of pulmonary TB in infected patients (Verbon et al., 1999; Raju et al., 2008). These observations in humans parallel with those reported in Mtb-infected mice; that is, there is an early type-1 immune response characterized by IFN-Γ during the first 3 weeks after infection, followed by a type-2 immune response that contains high levels of IL-4 (Figure 1; Orme et al., 1993). A type-2 inflammatory environment drives the M2 program that renders Mɸs immunomodulatory and poorly microbicidal (Raju et al., 2008; Martinez et al., 2009). At the transcriptome level, this seems to be the case in mice since M2 Mɸs displayed a diminished inflammatory response to Mtb as reflected by a reduced NO production and increased of iron availability, suggesting these phagocytes offer a permissible intracellular environment for bacterial replication (Kahnert et al., 2006). Indeed, IFN-Γ-induced NO production is essential for host survival with respect of experimental TB, while iron-starvation is key to bacteriostasis (Ehrt et al., 2001; Forbes and Gros, 2001; Cairo et al., 2011). It remains to be seen if Mtb also influences the expression level of Kruppel-like factor 4 (KLF4) or any other transcription factor/regulator recently shown to be critical for both the establishment of the M2 program and the inhibition of M1 polarization (e.g., STAT6, Cot/tpl2; Liao et al., 2011; Martinez, 2011). At the functional level, it has been demonstrated that both IL-4 and IL-13 inhibit autophagy in M1 Mɸs resulting in enhanced survival of Mtb, an impairment that might also extend to M2a Mɸs (Harris et al., 2007). At the granuloma level in mice, iNOS continues to be expressed within this structure but a significant shift from M1 toward M2 Mɸs [iNOSneg Arginase-1 (ARG1)hi] occurs around the alveolar compartment starting at day 35 and continuing up to day 60 after Mtb infection, accompanied by high levels of type-2 inflammatory signals (Ly et al., 2007; Redente et al., 2010). Given the development of fibrosis is a key characteristic of caseous granulomas during Mtb dissemination, and that M2 Mɸs have been implicated in the inhibition of fibrosis development, the shift from M1 into M2 program might represent an attempt by the host to halter the pathophysiology caused by Mtb or a microbial strategy to shield from immune attack (Dorhoi et al., 2011).
Mycobacterium tuberculosis also reprograms M2 Mɸs through secretion of immunosuppressants such as IL-10. For instance, Mtb might influence all TLR-dependent signaling by targeting DC-SIGN to induce IL-10 and counteract the pro-inflammatory response, as shown in dendritic cells (Geijtenbeek et al., 2003; Hajishengallis and Lambris, 2011). Likewise, the mannosylated lipoarabinomannan from Mtb enhances the production of IL-10 and other immunosuppressants through recognition by the mannose receptor (MR) in immature dendritic cells (Chieppa et al., 2003). Although alveolar Mɸs express DC-SIGN and MR, their role in M2 Mɸs has yet to be demonstrated (Chroneos and Shepherd, 1995; Tailleux et al., 2005). Nevertheless, Schreiber et al. (2009) reported Mtb-induced IL-10 in Mɸs promotes the M2 polarization program displaying diminished anti-mycobacterial effector mechanisms. Indeed, Mɸ-specific overexpressing IL-10 transgenic mice were indeed susceptible to Mtb infection, displayed a specifically suppressed IL-12 in infected tissues, and were characterized by lung Mɸs with a M2 phenotype permissive to Mtb infection (Schreiber et al., 2009). These observations correlate well with another study in mice where Mtb was shown to promote its survival and ability to cause disease through a MyD88-dependent induction of ARG1. ARG1 inhibits NO production by Mɸs by competing with iNOS for arginine (the common substrate), thus rendering these cells permissive to Mtb infection (El Kasmi et al., 2008; Hajishengallis and Lambris, 2011). Taken together, these observations suggest the reprogramming toward M2 Mɸs by IL-10, and other immunosuppressants such TGF-β and glucocorticoids (Hernandez-Pando et al., 2006), might be yet another adaptation by Mtb to survive and thrive inside of Mɸs (Figure 1). However, it should be noticed that this phenomenon might also represent a control mechanism by the host to preserve the integrity of mucosal sites as uncontrolled type-1 inflammatory responses against Mtb result into lung immunopathology (Hernandez-Pando et al., 2006; Ordway et al., 2006).
Macrophage Polarization in HIV Infection
Human immunodeficiency virus-1 is another successful intracellular pathogen responsible for a worldwide pandemic. According to 2009 estimates by the United Nations, there were about 33.2 million people worldwide living with HIV-1 infection and 2.6 million individuals had been newly infected (Cohen et al., 2011). In the absence of antiviral therapy, HIV-1 infection progresses through acute and asymptomatic stages leading to the eventual failure of the host immunological functions and acquired immunodeficiency syndrome (AIDS). A reason is that HIV-1 targets cells from the mononuclear phagocyte lineage that drive an effective antiviral response and simultaneously serve as reservoirs of latent or productive infection (Goodenow et al., 2003). Among these cells, Mɸs are critical to pathogenesis because they contribute to early transmission, systemic dissemination, and persistence of HIV-1. Indeed, HIV-1 evades immune surveillance by hiding and thriving inside Mɸs despite anti-retroviral treatment, and when infected, they persist for months displaying insensitivity to viral cytopathic effects. In addition, Mɸs continuously secrete high level of viral particles over prolonged time periods by storing assembled virus in specialized endosomal compartments (Orenstein et al., 1988; Benaroch et al., 2010). Thus, they represent powerful long-term viral reservoirs (Goodenow et al., 2003; Carter and Ehrlich, 2008; Herbein and Varin, 2010; Cohen et al., 2011). In light of recent evidence suggesting that M1 and M2 Mɸs influence HIV-1 pathogenesis, there is a surging interest to study the viral effects in Mɸ polarization.
In vitro, HIV-1 infection drives Mɸs toward a M1 program. This Mɸ response includes production of type-1 pro-inflammatory cytokines (IFN-Γ, IL-2, IL-12, TNFα, IL-1β, IL-6, IL-18) and chemokines (CCL3, CCL4, MIP-α, MIP-β, RANTES), increased NO and respiratory burst, up-regulation of MHC-II molecules, and down-regulation of HIV-entry receptors (e.g., CD4, CCR5, CXCR4), and endocytic receptors (e.g., CD163, CD206; Swingler et al., 1999; Cassol et al., 2009, 2010; Herbein and Varin, 2010). Although few studies have examined thoroughly HIV-induced polarization of Mɸs in vivo, there is a predominance of Mɸs displaying a M1 phenotype during the acute stage (Figure 1; Cassol et al., 2010; Herbein and Varin, 2010; Cohen et al., 2011). Whether M1 Mɸs are beneficial to the host during HIV-1 infection remains an open question since Mɸ functions vary according to the experimental context. For instance, in vitro infection of M1 Mɸs in the presence of IFN-Γ and TNFα is associated with a suppression of HIV-1 replication, a sharp decrease in HIV-1 DNA synthesis at 48 h, and a decrease in the accumulation of HIV-1 proteins (Cassol et al., 2009). In addition, other studies demonstrate that M1 Mɸs inhibit viral entry, assembly, and budding, suggesting the M1 program can be beneficial to the host (Cassol et al., 2010; Herbein and Varin, 2010). However, it is also known that pro-inflammatory signals deriving from M1 Mɸs favor the formation of viral reservoirs with increased transcription of HIV-1 LTR (long terminal repeat), alluding M1 Mɸs might benefit HIV pathogenesis (Cassol et al., 2010; Herbein and Varin, 2010). This is supported by multiple observations that immune activation driven by Mɸs correlates with HIV-1 pathogenesis (Goodenow et al., 2003; Lamers et al., 2009; Cohen et al., 2011). Recently, Brown et al. (2008) characterized the HIV-1-induced polarization of Mɸs as “M1HIV” since it displays a pro-inflammatory state with increased production of cytokines independently of TLR-pathway. The authors argue that while HIV-1 stimulates Mɸs through a variety of signaling pathways to promote a “tailored” inflammation in its favor, the TLR recognition of viral replication is impaired and could serve as a viral evasion strategy. Given that prolonged pro-inflammatory Mɸ activation during chronic HIV-infection contributes, not only to a permissive environment for the formation of viral reservoirs with strong transcriptional activity, but also to disease progression and HIV-induced tissue damage, the proposed M1HIV polarized state may render Mɸs detrimental to the host (Goodenow et al., 2003; Brown et al., 2008; Lamers et al., 2009).
As HIV-disease progresses from the acute to asymptomatic stage, there is a switch from a type-1 toward a type-2 inflammatory environment (Figure 1; Vasilescu et al., 2003; Becker, 2004). At the transcriptional level, lymphatic tissue microarray analyses from HIV-1-infected subjects at different clinical stages revealed that each stage has a unique gene profile (Li et al., 2009). The acute phase is characterized by gene expression involved in innate and adaptive immunity. The asymptotic phase, however, down-regulates the acute phase gene profile to baseline level while it displays an increased expression of immunosuppressive genes (Li et al., 2009). Based on these immunological systemic changes, it is likely that a polarization switch occurs in Mɸs from a M1 program during the acute phase to the M2 programs through later stages. Although there is no overwhelming evidence confirming the abundance of M2 Mɸs in either the asymptotic or AIDS phase in vivo, the fact CD163 (a M2 Mɸ cell surface marker) is considered as a potential biomarker for HIV-1 disease progression may allude to the presence of M2 Mɸs in HIV-1-infected individuals (Burdo et al., 2011; Tippett et al., 2011). Similar to M1 Mɸs, it is not known whether M2 Mɸs benefits the host during HIV-1 infection. In vitro activation of M2a (IL-4-treated) Mɸs results in inhibition of virus replication (Cassol et al., 2009). Other studies have demonstrated that both IL-4 and IL-13 down-regulate viral entry receptors and HIV-1 reverse transcription in Mɸs (Cassol et al., 2010). Furthermore, activation of M2c (IL-10-treated) Mɸs strongly inhibits reverse transcription, transcription of HIV-1 LTR and viral assembly (Herbein and Varin, 2010). Based on these observations, it might be tempting to conclude that M2 Mɸs are beneficial to host immunity against HIV. However, the progression of AIDS is characterized by the loss of IL-2 and increase of IL-10 correlating with HIV viremia (Brockman et al., 2009; Sandanger et al., 2009). Moreover, the haplotypes of both IL-4 and IL-10 genes have been associated recently with AIDS progression (Vasilescu et al., 2003). Therefore, the switch toward a M2 Mɸ program might simply be part of a defensive mechanism by the host to control HIV-induced tissue damage since they participate in suppression of inflammation and promotion of tissue repair (Figure 1; Martinez et al., 2009; Murray and Wynn, 2011). Recently, a functional proteomic analysis of HIV-infected Mɸs in the presence of regulatory T cells showed that a deviation of M1 to M2 Mɸ program is associated with neuroprotection in the case of HIV-associated neurocognitive disorders, suggesting M2 Mɸs may curtail the M1HIV polarized activity resulting in tissue damage (Huang et al., 2010). Conversely, the switch toward the M2 Mɸ program might also occur as an evasion strategy by HIV to promote its own survival. A recent study demonstrated that HIV up-regulates both programmed cell death ligand 1 (PD-L1) and PD-L2 expression, members of the B7:CD28 family, and PD-1 ligands, in Mɸs (Porichis et al., 2011). Given the importance of these molecules in T cell exhaustion during HIV infection, the ability of IL-10 to both activate the M2c Mɸ program and induce PD-L1, and the fact that IL-10 production and increased expression of PD-L1 correlate in HIV-infected patients, the authors propose the manipulation of PDL expression in Mɸs as a strategy to evade immune responses (Trabattoni et al., 2003; Porichis et al., 2011). Whatever the true role of M2 Mɸs in HIV infection, it is clear they influence the establishment of HIV pathogenesis, and more studies are needed to examine thoroughly HIV-induced polarization of Mɸs in vivo.
Conclusion
Tuberculosis is the most common opportunistic infection in AIDS and often used as a clinical parameter for undiagnosed AIDS cases (Deretic et al., 2004). While the synergy between Mtb and HIV is evident at the clinical level, the mechanisms accounting for it are poorly understood. Deretic et al. (2004) proposed the interference with endosomal sorting machine as a molecular mechanism contributing to the synergy between these two pathogens. Likewise, we envision the pathogenic modulation of Mɸ polarization as a cellular mechanism that might influence this synergism. As aforementioned, it is estimated that about one-third of the human population may be latently infected with Mtb (WHO Global Tuberculosis Control Report 2010, 2010), suggesting that one in three of the 2.6 million people newly infected with HIV-1 in 2009 (Cohen et al., 2011), for example, would also be coinfected with Mtb. Latent Mtb is confined in solid granulomas composed of mainly by Mɸs and T cells that maintain their stability. The coinfection with HIV-1 results in a dramatic increase in the odds of latently infected people progressing into overt TB to a staggering annual risk of 10% (Deretic et al., 2004; Swaminathan et al., 2010). HIV-driven immune perturbation, reflected in the loss of CD4+ T cells and abnormal low levels of TNFα causes the loss of granuloma integrity and efficiency in anti-microbial containment leading to post-primary reactivation state (Paige and Bishai, 2010). These events may increase both Mɸ necrosis and release of intracellular bacilli accounting for the extrapulmonary TB manifestation diagnosed in patients with HIV-driven immunosuppression (Swaminathan et al., 2010). The awakened Mtb then might induce M1 Mɸs to drive an excessive TNFα response (together with other mechanisms such as MMP secretion) to deliberately promote parasitic granuloma formation, resulting in the recruitment of additional naïve Mɸs and the tissue pathology (Davis and Ramakrishnan, 2009; Paige and Bishai, 2010; Volkman et al., 2010). Excessive levels of TNFα, may not only contribute to the classical symptoms of cachexia in TB, but also to the augmentation of HIV-1 transcription and accelerated formation of viral reservoirs (Deretic et al., 2004). In the absence of an efficient adaptive immune response due to HIV-driven impairment, uncontrolled inflammation can result in lung immunopathology, and consequently, the host may induce tissue repair responses. The shift from M1 to M2 Mɸ program may become pronounced and prolonged in the sterile attempt to restore tissue integrity, elevating the level of IL-10 that is typical of disease progression by both pathogens, and thus contributing to the failure of all immunological functions and clinical collapse. While highly speculative, this scenario highlights the importance to understand Mɸ polarization in the context of immune activation and pathogen-driven disease, and its potential to be yet another convergence point targeted by Mtb and HIV to circumvent the host immune system.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors received no specific funding for this work. The Neyrolles laboratory is supported by the Centre National de la Recherche Scientifique, the European Union (7th Framework Programme), the Agence Nationale de la Recherche (ANR), and the Fondation pour la Recherche Médicale. Olivier Neyrolles is a fellow of a CNRS-ATIP fellowship. The Maridonneau-Parini laboratory was supported in part by Association pour la Recherche sur le Cancer #2010-120-1733, ANR 2010-01301, ANRS 2010-061, and Sidaction AI 22-1-01892. Christel Vérollet was supported by Sidaction.
References
Almeida, A. S., Lago, P. M., Boechat, N., Huard, R. C., Lazzarini, L. C., Santos, A. R., Nociari, M., Zhu, H., Perez-Sweeney, B. M., Bang, H., Ni, Q., Huang, J., Gibson, A. L., Flores, V. C., Pecanha, L. R., Kritski, A. L., Lapa e Silva, J. R., and Ho, J. L. (2009). Tuberculosis is associated with a down-modulatory lung immune response that impairs Th1-type immunity. J. Immunol. 183, 718–731.
Barnes, P. F., Lu, S., Abrams, J. S., Wang, E., Yamamura, M., and Modlin, R. L. (1993). Cytokine production at the site of disease in human tuberculosis. Infect. Immun. 61, 3482–3489.
Becker, Y. (2004). The changes in the T helper 1 (Th1) and T helper 2 (Th2) cytokine balance during HIV-1 infection are indicative of an allergic response to viral proteins that may be reversed by Th2 cytokine inhibitors and immune response modifiers – a review and hypothesis. Virus Genes 28, 5–18.
Benaroch, P., Billard, E., Gaudin, R., Schindler, M., and Jouve, M. (2010). HIV-1 assembly in macrophages. Retrovirology 7, 29.
Benoit, M., Desnues, B., and Mege, J. L. (2008). Macrophage polarization in bacterial infections. J. Immunol. 181, 3733–3739.
Brockman, M. A., Kwon, D. S., Tighe, D. P., Pavlik, D. F., Rosato, P. C., Sela, J., Porichis, F., Le Gall, S., Waring, M. T., Moss, K., Jessen, H., Pereyra, F., Kavanagh, D. G., Walker, B. D., and Kaufmann, D. E. (2009). IL-10 is up-regulated in multiple cell types during viremic HIV infection and reversibly inhibits virus-specific T cells. Blood 114, 346–356.
Brown, J. N., Kohler, J. J., Coberley, C. R., Sleasman, J. W., and Goodenow, M. M. (2008). HIV-1 activates macrophages independent of toll-like receptors. PLoS ONE 3, e3664. doi: 10.1371/journal.pone.0003664
Burdo, T. H., Lentz, M. R., Autissier, P., Krishnan, A., Halpern, E., Letendre, S., Rosenberg, E. S., Ellis, R. J., and Williams, K. C. (2011). Soluble CD163 made by monocyte/macrophages is a novel marker of hiv activity in early and chronic infection prior to and after anti-retroviral therapy. J. Infect. Dis. 204, 154–163.
Cairo, G., Recalcati, S., Mantovani, A., and Locati, M. (2011). Iron trafficking and metabolism in macrophages: contribution to the polarized phenotype. Trends Immunol. 32, 241–247.
Carter, C. A., and Ehrlich, L. S. (2008). Cell biology of HIV-1 infection of macrophages. Annu. Rev. Microbiol. 62, 425–443.
Cassol, E., Cassetta, L., Alfano, M., and Poli, G. (2010). Macrophage polarization and HIV-1 infection. J. Leukoc. Biol. 87, 599–608.
Cassol, E., Cassetta, L., Rizzi, C., Alfano, M., and Poli, G. (2009). M1 and M2a polarization of human monocyte-derived macrophages inhibits HIV-1 replication by distinct mechanisms. J. Immunol. 182, 6237–6246.
Chieppa, M., Bianchi, G., Doni, A., Del Prete, A., Sironi, M., Laskarin, G., Monti, P., Piemonti, L., Biondi, A., Mantovani, A., Introna, M., and Allavena, P. (2003). Cross-linking of the mannose receptor on monocyte-derived dendritic cells activates an anti-inflammatory immunosuppressive program. J. Immunol. 171, 4552–4560.
Chroneos, Z., and Shepherd, V. L. (1995). Differential regulation of the mannose and SP-A receptors on macrophages. Am. J. Physiol. 269, L721–726.
Cohen, M. S., Shaw, G. M., Mcmichael, A. J., and Haynes, B. F. (2011). Acute HIV-1 Infection. N. Engl. J. Med. 364, 1943–1954.
Davis, J. M., and Ramakrishnan, L. (2009). The role of the granuloma in expansion and dissemination of early tuberculous infection. Cell 136, 37–49.
Deretic, V., Vergne, I., Chua, J., Master, S., Singh, S. B., Fazio, J. A., and Kyei, G. (2004). Endosomal membrane traffic: convergence point targeted by Mycobacterium tuberculosis and HIV. Cell. Microbiol. 6, 999–1009.
Dorhoi, A., Reece, S. T., and Kaufmann, S. H. (2011). For better or for worse: the immune response against Mycobacterium tuberculosis balances pathology and protection. Immunol. Rev. 240, 235–251.
Ehrt, S., Schnappinger, D., Bekiranov, S., Drenkow, J., Shi, S., Gingeras, T. R., Gaasterland, T., Schoolnik, G., and Nathan, C. (2001). Reprogramming of the macrophage transcriptome in response to interferon-gamma and Mycobacterium tuberculosis: signaling roles of nitric oxide synthase-2 and phagocyte oxidase. J. Exp. Med. 194, 1123–1140.
El Kasmi, K. C., Qualls, J. E., Pesce, J. T., Smith, A. M., Thompson, R. W., Henao-Tamayo, M., Basaraba, R. J., Konig, T., Schleicher, U., Koo, M. S., Kaplan, G., Fitzgerald, K. A., Tuomanen, E. I., Orme, I. M., Kanneganti, T. D., Bogdan, C., Wynn, T. A., and Murray, P. J. (2008). Toll-like receptor-induced arginase 1 in macrophages thwarts effective immunity against intracellular pathogens. Nat. Immunol. 9, 1399–1406.
Forbes, J. R., and Gros, P. (2001). Divalent-metal transport by NRAMP proteins at the interface of host-pathogen interactions. Trends Microbiol. 9, 397–403.
Geijtenbeek, T. B., Van Vliet, S. J., Koppel, E. A., Sanchez-Hernandez, M., Vandenbroucke-Grauls, C. M., Appelmelk, B., and Van Kooyk, Y. (2003). Mycobacteria target DC-SIGN to suppress dendritic cell function. J. Exp. Med. 197, 7–17.
Goodenow, M. M., Rose, S. L., Tuttle, D. L., and Sleasman, J. W. (2003). HIV-1 fitness and macrophages. J. Leukoc. Biol. 74, 657–666.
Hajishengallis, G., and Lambris, J. D. (2011). Microbial manipulation of receptor crosstalk in innate immunity. Nat. Rev. Immunol. 11, 187–200.
Harris, J., De Haro, S. A., Master, S. S., Keane, J., Roberts, E. A., Delgado, M., and Deretic, V. (2007). T helper 2 cytokines inhibit autophagic control of intracellular Mycobacterium tuberculosis. Immunity 27, 505–517.
Herbein, G., and Varin, A. (2010). The macrophage in HIV-1 infection: from activation to deactivation? Retrovirology 7, 33.
Hernandez-Pando, R., Orozco-Esteves, H., Maldonado, H. A., Aguilar-Leon, D., Vilchis-Landeros, M. M., Mata-Espinosa, D. A., Mendoza, V., and Lopez-Casillas, F. (2006). A combination of a transforming growth factor-beta antagonist and an inhibitor of cyclooxygenase is an effective treatment for murine pulmonary tuberculosis. Clin. Exp. Immunol. 144, 264–272.
Huang, X., Stone, D. K., Yu, F., Zeng, Y., and Gendelman, H. E. (2010). Functional proteomic analysis for regulatory T cell surveillance of the HIV-1-infected macrophage. J. Proteome Res. 9, 6759–6773.
Kahnert, A., Seiler, P., Stein, M., Bandermann, S., Hahnke, K., Mollenkopf, H., and Kaufmann, S. H. (2006). Alternative activation deprives macrophages of a coordinated defense program to Mycobacterium tuberculosis. Eur. J. Immunol. 36, 631–647.
Lamers, S. L., Salemi, M., Galligan, D. C., De Oliveira, T., Fogel, G. B., Granier, S. C., Zhao, L., Brown, J. N., Morris, A., Masliah, E., and Mcgrath, M. S. (2009). Extensive HIV-1 intra-host recombination is common in tissues with abnormal histopathology. PLoS ONE 4, e5065. doi: 10.1371/journal.pone.0005065
Li, Q., Smith, A. J., Schacker, T. W., Carlis, J. V., Duan, L., Reilly, C. S., and Haase, A. T. (2009). Microarray analysis of lymphatic tissue reveals stage-specific, gene expression signatures in HIV-1 infection. J. Immunol. 183, 1975–1982.
Liao, X., Sharma, N., Kapadia, F., Zhou, G., Lu, Y., Hong, H., Paruchuri, K., Mahabeleshwar, G. H., Dalmas, E., Venteclef, N., Flask, C. A., Kim, J., Doreian, B. W., Lu, K. Q., Kaestner, K. H., Hamik, A., Clement, K., and Jain, M. K. (2011). Kruppel-like factor 4 regulates macrophage polarization. J. Clin. Invest. 121, 2736–2749.
Ly, L. H., Russell, M. I., and Mcmurray, D. N. (2007). Microdissection of the cytokine milieu of pulmonary granulomas from tuberculous guinea pigs. Cell. Microbiol. 9, 1127–1136.
Martinez, F. O., Helming, L., and Gordon, S. (2009). Alternative activation of macrophages: an immunologic functional perspective. Annu. Rev. Immunol. 27, 451–483.
Meena, L. S., and Rajni, (2010). Survival mechanisms of pathogenic Mycobacterium tuberculosis H37Rv. FEBS J. 277, 2416–2427.
Murray, P. J., and Wynn, T. A. (2011). Obstacles and opportunities for understanding macrophage polarization. J. Leukoc. Biol. 89, 557–563.
Ordway, D., Harton, M., Henao-Tamayo, M., Montoya, R., Orme, I. M., and Gonzalez-Juarrero, M. (2006). Enhanced macrophage activity in granulomatous lesions of immune mice challenged with Mycobacterium tuberculosis. J. Immunol. 176, 4931–4939.
Orenstein, J. M., Meltzer, M. S., Phipps, T., and Gendelman, H. E. (1988). Cytoplasmic assembly and accumulation of human immunodeficiency virus types 1 and 2 in recombinant human colony-stimulating factor-1-treated human monocytes: an ultrastructural study. J. Virol. 62, 2578–2586.
Orme, I. M., Roberts, A. D., Griffin, J. P., and Abrams, J. S. (1993). Cytokine secretion by CD4 T lymphocytes acquired in response to Mycobacterium tuberculosis infection. J. Immunol. 151, 518–525.
Paige, C., and Bishai, W. R. (2010). Penitentiary or penthouse condo: the tuberculous granuloma from the microbe’s point of view. Cell. Microbiol. 12, 301–309.
Porichis, F., Kwon, D. S., Zupkosky, J., Tighe, D. P., Mcmullen, A., Brockman, M. A., Pavlik, D. F., Rodriguez-Garcia, M., Pereyra, F., Freeman, G. J., Kavanagh, D. G., and Kaufmann, D. E. (2011). Responsiveness of HIV-specific CD4 T cells to PD-1 blockade. Blood 118, 965–974.
Quintana-Murci, L., Alcais, A., Abel, L., and Casanova, J. L. (2007). Immunology in natura: clinical, epidemiological and evolutionary genetics of infectious diseases. Nat. Immunol. 8, 1165–1171.
Raju, B., Hoshino, Y., Belitskaya-Levy, I., Dawson, R., Ress, S., Gold, J. A., Condos, R., Pine, R., Brown, S., Nolan, A., Rom, W. N., and Weiden, M. D. (2008). Gene expression profiles of bronchoalveolar cells in pulmonary TB. Tuberculosis (Edinb) 88, 39–51.
Redente, E. F., Higgins, D. M., Dwyer-Nield, L. D., Orme, I. M., Gonzalez-Juarrero, M., and Malkinson, A. M. (2010). Differential polarization of alveolar macrophages and bone marrow-derived monocytes following chemically and pathogen-induced chronic lung inflammation. J. Leukoc. Biol. 88, 159–168.
Sandanger, O., Ryan, L., Bohnhorst, J., Iversen, A. C., Husebye, H., Halaas, O., Landro, L., Aukrust, P., Froland, S. S., Elson, G., Visintin, A., Oktedalen, O., Damas, J. K., Sundan, A., Golenbock, D., and Espevik, T. (2009). IL-10 enhances MD-2 and CD14 expression in monocytes and the proteins are increased and correlated in HIV-infected patients. J. Immunol. 182, 588–595.
Schreiber, T., Ehlers, S., Heitmann, L., Rausch, A., Mages, J., Murray, P. J., Lang, R., and Holscher, C. (2009). Autocrine IL-10 induces hallmarks of alternative activation in macrophages and suppresses antituberculosis effector mechanisms without compromising T cell immunity. J. Immunol. 183, 1301–1312.
Sibley, L. D., Adams, L. B., and Krahenbuhl, J. L. (1990). Inhibition of interferon-gamma-mediated activation in mouse macrophages treated with lipoarabinomannan. Clin. Exp. Immunol. 80, 141–148.
Sica, A., Larghi, P., Mancino, A., Rubino, L., Porta, C., Totaro, M. G., Rimoldi, M., Biswas, S. K., Allavena, P., and Mantovani, A. (2008). Macrophage polarization in tumour progression. Semin. Cancer Biol. 18, 349–355.
Swaminathan, S., Padmapriyadarsini, C., and Narendran, G. (2010). HIV-associated tuberculosis: clinical update. Clin. Infect. Dis. 50, 1377–1386.
Swingler, S., Mann, A., Jacque, J., Brichacek, B., Sasseville, V. G., Williams, K., Lackner, A. A., Janoff, E. N., Wang, R., Fisher, D., and Stevenson, M. (1999). HIV-1 Nef mediates lymphocyte chemotaxis and activation by infected macrophages. Nat. Med. 5, 997–103.
Tailleux, L., Pham-Thi, N., Bergeron-Lafaurie, A., Herrmann, J. L., Charles, P., Schwartz, O., Scheinmann, P., Lagrange, P. H., De Blic, J., Tazi, A., Gicquel, B., and Neyrolles, O. (2005). DC-SIGN induction in alveolar macrophages defines privileged target host cells for mycobacteria in patients with tuberculosis. PLoS Med. 2, e381. doi: 10.1371/journal.pmed.0020381
Tippett, E., Cheng, W. J., Westhorpe, C., Cameron, P. U., Brew, B. J., Lewin, S. R., Jaworowski, A., and Crowe, S. M. (2011). Differential expression of CD163 on monocyte subsets in healthy and HIV-1 infected individuals. PLoS ONE 6, e19968. doi: 10.1371/journal.pone.0019968
Trabattoni, D., Saresella, M., Biasin, M., Boasso, A., Piacentini, L., Ferrante, P., Dong, H., Maserati, R., Shearer, G. M., Chen, L., and Clerici, M. (2003). B7-H1 is up-regulated in HIV infection and is a novel surrogate marker of disease progression. Blood 101, 2514–2520.
Vasilescu, A., Heath, S. C., Ivanova, R., Hendel, H., Do, H., Mazoyer, A., Khadivpour, E., Goutalier, F. X., Khalili, K., Rappaport, J., Lathrop, G. M., Matsuda, F., and Zagury, J. F. (2003). Genomic analysis of Th1-Th2 cytokine genes in an AIDS cohort: identification of IL4 and IL10 haplotypes associated with the disease progression. Genes Immun. 4, 441–449.
Verbon, A., Juffermans, N., Van Deventer, S. J., Speelman, P., Van Deutekom, H., and Van Der Poll, T. (1999). Serum concentrations of cytokines in patients with active tuberculosis (TB) and after treatment. Clin. Exp. Immunol. 115, 110–113.
Keywords: macrophage, Mycobacteria, tuberculosis, HIV, AIDS, polarization
Citation: Lugo-Villarino G, Vérollet C, Maridonneau-Parini I and Neyrolles O (2011) Macrophage polarization: convergence point targeted by Mycobacterium tuberculosis and HIV. Front. Immun. 2:43. doi: 10.3389/fimmu.2011.00043
Received: 29 June 2011; Paper pending published: 20 July 2011;
Accepted: 25 August 2011; Published online: 15 September 2011.
Edited by:
Amiram Ariel, University of Haifa, IsraelReviewed by:
Andreas Ludwig, RWTH Aachen University, GermanyPaola Allavena, Clinical Institute Humanitas, Italy
Copyright: © 2011 Lugo-Villarino, Vérollet, Maridonneau-Parini and Neyrolles. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Olivier Neyrolles, CNRS, Institut de Pharmacologie et de Biologie Structurale, 205 Route de Narbonne, F-31077 Toulouse, France. e-mail:b2xpdmllci5uZXlyb2xsZXNAaXBicy5mcg==