 Roland S. Liblau1,2,3*
Roland S. Liblau1,2,3* Hartmut Wekerle4 and Roland M. Tisch5
Hartmut Wekerle4 and Roland M. Tisch5
- 1 INSERM, U1043, Toulouse, France
- 2 CNRS, U5282, Toulouse, France
- 3 Centre de Physiopathologie de Toulouse Purpan, Université de Toulouse, Université Paul Sabatier, Toulouse, France
- 4 Department of Neuroimmunology, Max-Planck-Institute of Neurobiology, Martinsried, Germany
- 5 Department of Microbiology and Immunology, University of North Carolina at Chapel Hill, Chapel Hill, NC, USA
T cell receptor (TCR) recognition is intrinsically polyspecific. In the field of autoimmunity, recognition of both self- and microbial peptides by a single TCR has led to the concept of molecular mimicry. However, findings made by our group and others clearly demonstrate that a given TCR can also recognize multiple distinct self-peptides. Based on experimental data we argue that recognition of several self-peptides increases the pathogenicity of an autoreactive T cell; a property we refer to as “cumulative autoimmunity.” The mechanisms of such increased pathogenicity, and the implications of cumulative autoimmunity regarding the pathophysiology of T cell-mediated autoimmune diseases will be discussed.
Polyspecificity of Autoreactive T Cells is not Limited to Recognition of Exogenous Antigens
Both theoretical considerations (Mason, 1998) and experimental evidence (Wucherpfennig and Strominger, 1995) strongly support the idea that polyspecificity is an intrinsic property of T cell receptor (TCR) recognition. A single TCR has been shown to recognize different peptides in the context of either the same, or different MHC molecules (Sospedra et al., 2006). Moreover, single amino acid substitutions involving TCR-contact residues of an antigenic peptide (so-called altered peptide ligands) can lead to markedly different TCR signaling and T cell functions (Sloan-Lancaster and Allen, 1996). The molecular basis for TCR polyspecificity is currently ill-defined but efforts in resolving the structures of MHC:peptide (pMHC) complexes have provided insight into possible mechanisms (Yin and Mariuzza, 2009). In the field of autoimmunity TCR polyspecificity has mostly been viewed in the context of molecular mimicry. The latter poses that autoimmune diseases can be triggered by microbes following lymphocyte cross-recognition of microbial- and self-derived peptides (Wucherpfennig and Strominger, 1995; Münz et al., 2009). Reports have shown in animal models of multiple sclerosis (MS) and type 1 diabetes (T1D) for instance, that self-tolerance is broken by activation of autoreactive T cells following TCR recognition of microbial antigens (Gautam et al., 1998; Harkiolaki et al., 2009; Münz et al., 2009). This ability of the TCR to recognize multiple peptides likely provides advantage to the individual despite its inherent risk of autoimmunity. For instance, TCR polyspecificity would be expected to limit the likelihood of “holes in the repertoire” and, thereby, enhance host immunity to pathogens.
A number of recent studies indicate that single TCR also bind to distinct self-peptides from different autoantigens or non-overlapping peptides derived from the same autoantigen. For instance, a study demonstrated that CD4+ T cells cloned from peripheral blood of healthy subjects and expanded in an autologous-mixed lymphocyte response recognize multiple autoantigenic epitopes (Cai and Hafler, 2007). The epitopes recognized by these CD4+ T cell clones include a panel of peptides derived from autoantigens associated with T1D and MS such as glutamic acid decarboxylase (GAD), insulin, proinsulin, and myelin basic protein (MBP). Interestingly, the majority of CD4+ T cell clones specific for viral-derived epitopes lacked significant polyspecificity, suggesting that the recognition of multiple epitopes is more common among autoreactive T cell clonotypes (Cai and Hafler, 2007). It is, however, worth noting that T cells specific for foreign antigens in order to be positively selected in the thymus interact with endogenous self-peptides:MHC complexes, at least with very low affinity (Juang et al., 2010). Londei and colleagues (Quaratino et al., 1995) reported that an HLA-DQ6-restricted self-reactive CD4+ T cell clone established from the thyroid infiltrate of a patient with Graves’ disease responded in vitro to two overlapping peptides from thyroid peroxidase (peptides 536–545 and 540–548). Molecular modeling led to the hypothesis that the two DQ6:self-peptide complexes generated similar antigenic surfaces in terms of conformation and charge for this polyspecific TCR. A study evaluated the breadth of peptide ligands recognized by a DR2 (DRB1*1501)-restricted MBP86–96 specific CD4+ T cell clone established from the blood of an MS patient using combinatorial peptide libraries (Hemmer et al., 1997). Based on this analysis, a number of stimulatory peptides derived from self-proteins could be predicted that were then confirmed experimentally. Interestingly, some of these self-peptides induced an enhanced T cell response compared to the “cognate” MBP peptide and therefore exhibited heteroclitic behavior. A similar analysis of CD4+ T cell clones derived from the cerebro-spinal fluid (CSF) of an MS patient during disease exacerbation identified stimulatory peptides from both common human viruses and several self-proteins expressed in the central nervous system (CNS; Sospedra et al., 2005). Polyspecificity has also been demonstrated within the CD8+ T cell compartment. CD8+ T clones specific for Melan-A, a melanocyte differentiation and tumor-associated antigen, originating from different HLA-A2 individuals were found to recognize multiple self-peptides. Furthermore, public patterns of self-polyspecificity, as well as private ones, were identified among these clones (Dutoit et al., 2002).
Finally, work by our group demonstrated polyspecificity among β cell-specific CD4+ T cell clones derived from non-obese diabetic (NOD) mice, a spontaneous model of T1D (Li et al., 2008). A substantial number of CD4+ T cell clones established with GAD65 protein recognized dual epitopes that were non-overlapping and had distinct amino acid sequences. Importantly, these T cell clones expressed a single TCR, and an engineered soluble polyspecific TCR was shown to interact with complexes of soluble I-Ag7 and the respective GAD65-derived peptides. Notably, some CD4+ T cell clones also exhibited inter-molecular polyspecificity by responding to GAD65 and to other β cell-derived autoantigens, further demonstrating that various forms of TCR polyspecificity exist. Collectively these findings demonstrate that several examples of polyspecificity toward self-antigens exist among autoreactive T cells in both humans and mice, arguing that this property may in fact be important for driving T cell-mediated autoimmunity.
What is the Molecular Basis for Polyspecificity?
The extent of TCR polyspecificity is very likely to be underestimated and therefore the epitopes that have been defined reflect only a portion of the cross-reactive self-peptide pool. Recognition of a peptide by a polyspecific TCR is nevertheless specific. For example, recognition by polyspecific T cell clones is sensitive to subtle structural changes in the peptide (Hagerty and Allen, 1995; Maynard et al., 2005), similar to that typically observed for monospecific T cell clones. Given the necessity of polyspecificity for thymic positive selection and its presumed importance for immune defense, the structural basis for this inherent property of the TCR has been investigated at the atomic level. Germline-encoded TCR CDR1 and CDR2 loops contact mainly the MHC helices, whereas the CDR3 loops interact with the side chains of the peptide. Cross-reactivity has been shown to rely on several alternative structural solutions (Wucherpfennig et al., 2007; Yin and Mariuzza, 2009). Structural mimicry has been well illustrated by the interactions defined between the MBP-specific Ob.1A12 TCR and a naturally processed E. coli peptide presented by the same HLA-DR2b molecule. In both cases, the binding footprint of the TCR is similar involving shared P2 and P3 TCR-contact residues (Harkiolaki et al., 2009). Analogous docking topologies have also been observed for a TCR interacting with a viral and a self-antigen bound to HLA-A2 (Borbulevych et al., 2011), and for a TCR binding self and allogenic MHC molecules (Yin and Mariuzza, 2009). A given TCR can also adopt large conformational changes of one CDR3 loop to productively interact with different peptides, with little or no sequence sharing, presented by the same MHC molecule (Reiser et al., 2003). Conformational flexibility of the peptide:MHC complex upon ligation with the TCR can also contribute to cross-reactivity. Studies have demonstrated that there can be a significant amount of flexibility in MHC-bound peptide side chains and backbone (Dessen et al., 1997; Sharma et al., 2001; Fremont et al., 2002). Accordingly, peptides exhibiting enhanced flexibility may readily adopt a conformation that enhances polyspecific recognition by a T cell clone. More drastic differences in binding strategy may also underlie recognition of two distinct peptide:MHC complexes by a given TCR. Indeed, the 2C TCR adopts very distinct orientation to interact with the allo H-2Ld:QL9 complex or with the H-2Kb:self-peptide complex (Colf et al., 2007). Weak interactions between the autoreactive 3A6 TCR and cognate HLA-DR2a:MBP84–102 ligand, which lack hydrogen bonds or salt bridges linking the TCR and the MBP peptide, are thought to result in a higher degree of tolerance for substituted peptides and, therefore, to promote cross-reactivity (Yin and Mariuzza, 2009).
MHC polymorphism has also been shown to influence peptide flexibility (Pöhlmann et al., 2004). With this in mind, one intriguing possibility is that MHC alleles associated with increased autoimmune susceptibility permit increased mobility of bound peptides, thereby promoting T cell polyspecificity. On the other hand, TCR polyspecificity can include recognition of multiple MHC molecules as noted above for alloreactive TCR. More important for autoimmunity is the possibility of an autoreactive TCR to productively engage different MHC molecules expressed by the individual. For instance, the Hy.2E11 TCR originating from an MS patient recognizes both MBP85–99 bound to HLA-DR2b and an Epstein–Barr virus peptide bound to HLA-DR2a; both DR2 molecules being encoded by the same haplotype. Crystal structure determination of these two MHC:peptide complexes revealed marked structural similarity for TCR recognition (Lang et al., 2002). A given TCR may even bind to both MHC class I: and class II:peptide complexes. Structural analysis of such a case led to the surprising observation that the TCR adopts different conformations to interact with the two ligands. A “standard” TCR binding mode occurs with the MHC class II:peptide complex, whereas the CDR1 and CDR2 loops of the Vα chain are displaced from the Vα CDR3 loop and Vβ loops when the TCR is bound to the MHC class I:peptide complex (Yin et al., 2011). The ability of a given TCR to exhibit multiple conformations is likely to further enhance polyspecificity.
The properties of TCR specific for multiple self-peptides remain ill-defined. Clues into the latter have come from crystal structures of autoreactive TCR. For instance, the study of a murine MBP-specific TCR indicates structural plasticity in TCR binding of the pMHC complex (Maynard et al., 2005). This plasticity facilitates alternative structural solutions for engagement by the autoreactive TCR. Therefore polyspecificity may reflect an enhanced capacity of a given TCR to accommodate alternative binding solutions coupled with increased conformational adaptability of a peptide bound by MHC. Alternatively, reduced surface of interaction between the TCR and the self-peptide may offer greater opportunity for cross-reactivity (Sethi et al., 2011). Interestingly, GAD65-polyspecific CD4+ T cell clones display an increased relative TCR affinity compared to the corresponding monospecific CD4+ T cell clones (Li et al., 2008). For example, relative TCR affinity for a given epitope can be increased by approximately fivefold between GAD65-polyspecific (KD = 15.1 ± 2.8 nmol/l) versus -monospecific (KD = 76.7 ± 16.9 nmol/l) CD4+ T cell clones (Li et al., 2008). Increased TCR affinity has been reported to correlate with enhanced cross-reactivity, due to flexibility in the CDR loops that bind the pMHC complex (Garcia et al., 1998; Holler et al., 2003; Reiser et al., 2003). Whether increased avidity/affinity is a common property of polyspecific TCR is an important issue that needs to be resolved.
Thymic Development of Polyspecific Autoreactive T Cells
How do polyspecific autoreactive T precursors develop in the thymus? Recognition of multiple self-peptides presented by thymic APC would a priori be expected to enhance deletion of the corresponding thymocytes due to increased pMHC complexes to drive negative selection. One explanation is that polyspecific autoreactive T precursors express TCR with reduced affinity therefore limiting the efficacy of negative selection, despite sufficient numbers of pMHC complexes. However, as noted above at least some polyspecific TCR exhibit increased avidity/affinity relative to monospecific TCR. Low thymic expression of tissue specific-antigens, and aberrant processing and/or presentation of self-peptides may be key factors favoring the development of polyspecific autoreactive thymocytes. How polyspecific autoreactive T cells escape central tolerance is an important area for future study.
Other factors associated with the polygenic nature of autoimmune diseases are likely to contribute as well to the development of polyspecific autoreactive T cells. Several gene polymorphisms have been associated with autoimmunity affecting molecules involved in TCR signaling. This could impact intra-thymic differentiation and selection of polyspecific (as well as monospecific) self-reactive T precursors. For instance, the gain-of-function variant (R620W) of the PTPN22 phosphatase, which has been linked to a number of autoimmune diseases, limits the magnitude of TCR signaling needed to promote negative selection of autoreactive (e.g., polyspecific) thymocytes (Vang et al., 2005). Similarly polymorphisms in the gene encoding Vav1, an enzyme that catalyzes the exchange of guanine nucleotides on Rho/Rac proteins in TCR signaling, are associated with susceptibility to experimental autoimmune encephalomyelitis (EAE) and MS, and may affect thymic differentiation of autoreactive T precursors by either impairing negative selection and/or commitment to the Foxp3-expressing CD4+ T cell lineage (Jagodic et al., 2009). It is conceivable that Foxp3-expressing regulatory CD4+ T cells (Tregs) also express polyspecific TCR due to enriched self-reactive specificities (Wing and Sakaguchi, 2010). The extent of polyspecificity among Tregs is currently unknown. Polyspecificity for self-antigens may enhance the likelihood of Treg activation by effectively increasing the density of TCR ligands at a given site and/or provide more sites for productive TCR:MHC:self-peptide interactions. Since Treg-mediated suppression is independent of the specificity of effector T cells, polyspecificity would be expected to expand the functional potency of Tregs. Further research should clarify whether Tregs with polyspecificity for self-antigens are more protective against autoimmunity as compared to monospecific Tregs.
Polyspecificity of Autoreactive T Cells Confers Enhanced Pathogenicity: The Concept of Cumulative Autoimmunity
The pathogenicity of T cells recognizing multiple self-epitopes has recently been examined in NOD mice and a spontaneous transgenic model of EAE. The latter model relies on 2D2-TCR mice, which are transgenic for a TCR specific for the myelin-oligodendrocyte glycoprotein (MOG) 35–55 peptide presented by I-Ab on the C57BL/6 genetic background (Bettelli et al., 2003). Unexpectedly, 2D2-TCR mice crossed with MOG-deficient (MOG−/−) animals spontaneously develop EAE despite the lack of the target myelin autoantigen [30]. The incidence, kinetics, clinical, and histological manifestations of spontaneous EAE are indistinguishable from that observed in MOG-expressing 2D2-TCR transgenic mice. To explain this paradoxical phenotype, we hypothesized that the clonotypic 2D2-TCR recognizes a neural antigen other than MOG, leading to CNS inflammation. Using a biochemical approach, neurofilament-medium (NF-M), an axonal protein, was identified as an additional self-antigen recognized by the 2D2-TCR (Krishnamoorthy et al., 2009; Figure 1). Importantly, MOG35–55-reactive CD4+ T cells from non-transgenic mice also exhibit in vitro reactivity to the NF-M peptide, both at the population and clonal levels. In addition, the MOG35–55-specific 2D2 CD4+ T cells are activated by CNS extracts from MOG−/− but not MOG−/− × NF-M−/− KO animals, indicating that the NF-M peptide is processed and presented by APC (Krishnamoorthy et al., 2009).
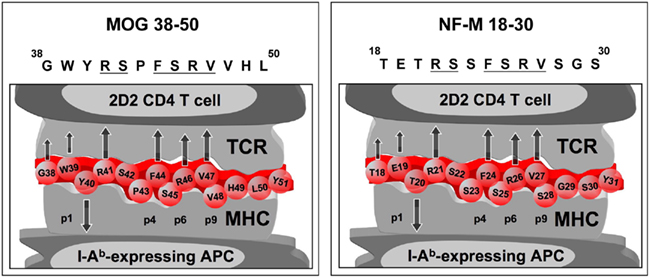
Figure 1. Schematic representation of the possible structural bases underlying the dual specificity of 2D2 T cells for MOG38–50 and NF-M18–30. The core region of the MOG38–50 peptide ranges from Y40 to V47 and contains the amino acids at position 41, 44, 46, and 47, which are known to be the contact amino acids for the 2D2-TCR and other MOG-specific T cell lines (Petersen et al., 2004; Ben-Nun et al., 2006). These amino acids are preserved at identical positions in NF-M18–30. The NF-M18–30 (TETRSSFSRVSGS) and the MOG38–50 (GWYRSPFSRVVHL) epitopes therefore share key TCR-contact residues (marked by arrows pointing upward) when presented in the context of I-Ab. The residues of the respective peptides interacting with the P1 P4, P6, and P9 pockets of the I-Ab are also indicated. Interestingly, although both peptides induce activation of CD4+ T cells from 2D2-TCR RAG−/− mice, the agonistic potential of NF-M18–30 peptide exceeds that of the MOG38–50 peptide, as revealed by proliferation and cytokine release assays.
Since 2D2 CD4+ T cells recognize two distinct self-antigens it was possible to assess the influence of TCR polyspecificity on the pathogenic potential of these T cells in vivo. CD4+ Th1 cells from 2D2-TCR RAG−/− donor mice were injected into wild-type, as well as NF-M−/−, MOG−/−, or MOG−/− × NF-M−/− KO mice, which exhibit no clinical or histological abnormalities at a young age (Jacomy et al., 1999; Delarasse et al., 2003). As expected, recipient mice expressing the two autoantigens (wild-type mice) rapidly developed lethal EAE. Conversely, mice lacking the two autoantigens (MOG−/− × NF-M−/− KO mice) remained free of CNS inflammation. Notably, MOG−/− KO mice presented an intermediate phenotype, suggesting that recognition of MOG and NF-M by the 2D2 CD4+ T cells contribute to disease phenotype and severity (Krishnamoorthy et al., 2009). We refer to this novel observation as “cumulative autoimmunity” to designate autoreactive T cell clones that target more than one cognate self-epitope and consequently exhibit additive (or synergistic) pathogenicity. Cumulative autoimmunity is also seen with GAD65-polyspecific versus monospecific CD4+ T cell clones, which exhibit increased pathogenicity upon adoptive transfer into NOD. scid recipients (Li et al., 2008).
How Does TCR Polyspecificity Influence the Pathogenicity of Autoreactive T Cells and Cumulative Autoimmunity?
Polyspecificity would be predicted to influence the pathogenicity of autoreactive T cells in a number of ways. This would include affecting the extent of: (i) T cell survival, activation, expansion, and differentiation, and (ii) T cell-mediated tissue destruction. Polyspecificity for self-peptides may promote enhanced peripheral survival and therefore persistence of the pool of autoreactive mature T cells (Wucherpfennig and Gagnon, 2009). Similarly, T cell clones exhibiting inter- or intra-molecular polyspecificity for a given tissue or autoantigen could have an advantage for clonal expansion. For instance, simultaneous recognition of multiple epitopes presented by APC may increase the local concentration of stimulatory peptides and induce efficient T cell activation, subsequent expansion and tissue retention (Cabbage et al., 2007; Lennon et al., 2009). An increased frequency of GAD65-polyspecific versus -monospecific CD4+ T cell clones residing in the pancreatic lymph nodes and islets of NOD female mice (Li et al., 2008) is consistent with such a scenario. As noted above, GAD65-polyspecific versus -monospecific CD4+ T cell clones have an increased relative TCR affinity suggesting that polyspecificity may prove to be a key factor contributing to the avidity/affinity maturation of autoreactive T effectors as autoimmunity progresses. Noteworthy is that increased avidity/affinity is a property of highly pathogenic β cell-specific T cells in NOD mice (Amrani et al., 2000). Whether these effector T cells exhibit β cell polyspecificity needs to be explored.
It is also conceivable that activated autoreactive T cells specific for one self-peptide are prone to react to a different self-peptide, due to a reduced activation threshold (Kimachi et al., 2003). T cells specific for multiple self-antigens could therefore be stimulated by the simultaneous or successive recognition of several self-peptides by local APC with a decreased dependence on co-stimulatory signals. The latter would be reflected by “epitope spread” which has been reported for the progression of various T cell-mediated autoimmune diseases (Lehmann et al., 1993). One interesting scenario is that distinct pMHC complexes presented by the same APC (or APC in the same micro-environment) adopt different structures, thereby eliciting different T cell signaling events. Since the TCR strength of signal influences commitment into the various T cell subsets, polyspecificity could have a direct bearing on the functional properties of autoreactive T cells and thereby on their propensity to initiate tissue damage.
T cell receptor polyspecificity broadens the repertoire of self-epitopes that may lead to diversification of cellular and anatomical targets, and consequently increase the severity of autoimmunity. For example, MOG is specifically expressed in CNS myelin, whereas NF-M is part of the axonal/neuronal cytoskeleton in both the CNS and peripheral nervous system (PNS). Accordingly, 2D2 CD4+ T cells typically induce inflammation in structures of both the PNS and CNS resulting in a more severe neurological phenotype. Alternatively, when several peptides from a given self-antigen or from antigens from the same anatomical structure are recognized, the increased pMHC ligand density in the targeted tissue is a likely basis by which the cross-reactive self-specific T cells exert enhanced pathogenic property (Li et al., 2008). This is potentially important since within target tissues the MHC molecules are not saturated by a given self-peptide and increased self-antigen availability can enhance local T cell activation and exacerbate disease (Odoardi et al., 2007).
Concluding Remarks
It is clear that murine and human autoreactive T cells exhibit specificity for multiple self-derived epitopes. Polyspecificity for self-antigens appears one of the parameters influencing the pathogenic potential of autoreactive T cells (Table 1). Based on our findings we argue that polyspecificity enhances the pathogenicity of autoreactive T cells through a number of possible scenarios leading to cumulative autoimmunity. The activation and expansion of polyspecific T cells may also partly explain critical events thought to drive T cell-mediated autoimmunity such as epitope spread and avidity/affinity maturation of autoreactive T cells. Nevertheless, significant work is still needed to determine the contribution of polyspecific autoreactive T cells and cumulative autoimmunity in the initiation and progression of self-tissue destruction. For instance, the frequency of autoreactive T cells displaying polyspecificity, and the properties of the peptides and corresponding TCR that promote polyspecificity are key issues that need further study. Finally, targeting polyspecific autoreactive T cells may prove to be an effective strategy for immunotherapy. For instance, the target pool of autoreactive T precursors and/or effectors would be increased with administration of a self-peptide that is recognized by polyspecific T cells. Indeed, work by our group showed that the most potent GAD65-specific peptides at inducing immunoregulatory T effectors to suppress β cell autoimmunity in NOD mice were epitopes recognized by polyspecific CD4+ T cells (Tisch et al., 1999).
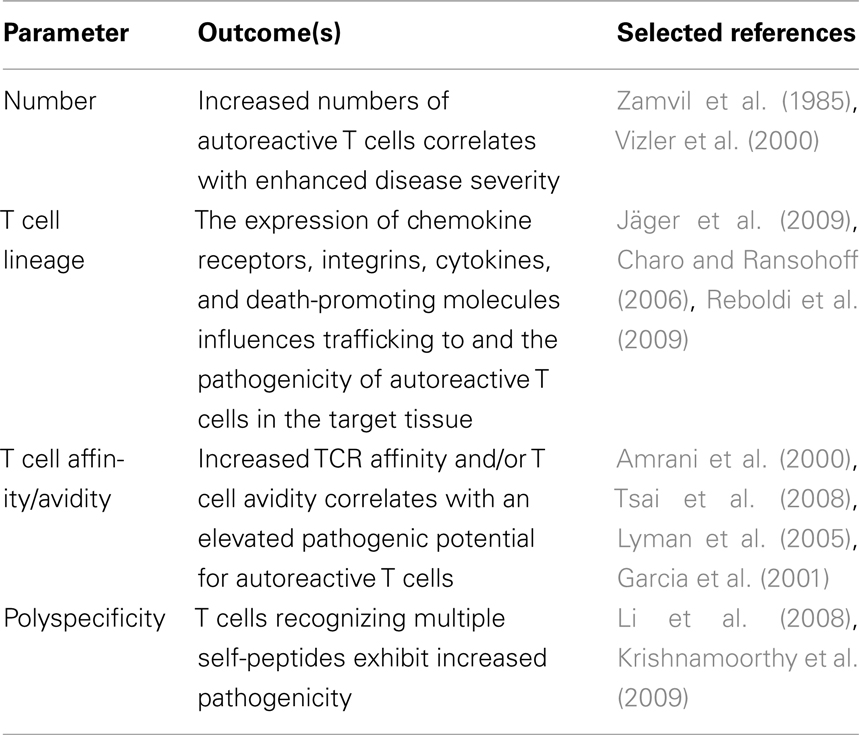
Table 1. Parameters governing the pathogenic potential of autoreactive T cells.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank their colleagues who participated to the primary research papers at the basis of this review article. The grant support of the National Institutes of Health, the Juvenile Diabetes Research Foundation, the American Diabetes Association, the European Union, the Deutsche Forschungsgemeinschaft, the Max Planck Society, the Medical Research Foundation (FRM), and the French MS society (ARSEP) is warmly acknowledged.
References
Amrani, A., Verdaguer, J., Serra, P., Tafuro, S., Tan, R., and Santamaria, P. (2000). Progression of autoimmune diabetes driven by avidity maturation of a T-cell population. Nature 406, 739–742.
Ben-Nun, A., Kerlero de Rosbo, N., Kaushansky, N., Eisenstein, M., Cohen, L., Kaye, J. F., and Mendel, I. (2006). Anatomy of T cell autoimmunity to myelin oligodendrocyte glycoprotein (MOG): prime role of MOG44F in selection and control of MOG-reactive T cells in H-2b mice. Eur. J. Immunol. 36, 478–493.
Bettelli, E., Pagany, M., Weiner, H. L., Linington, C., Sobel, R. A., and Kuchroo, V. K. (2003). Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J. Exp. Med. 197, 1073–1081.
Borbulevych, O. Y., Piepenbrink, K. H., and Baker, B. M. (2011). Conformational melding permits a conserved binding geometry in TCR recognition of foreign and self molecular mimics. J. Immunol. 186, 2950–2958.
Cabbage, S. E., Huseby, E. S., Sather, B. D., Brabb, T., Liggitt, D., and Goverman, J. (2007). Regulatory T cells maintain long-term tolerance to myelin basic protein by inducing a novel, dynamic state of T cell tolerance. J. Immunol. 178, 887–896.
Cai, G., and Hafler, D. A. (2007). Multispecific responses by T cells expanded by endogenous self-peptide/MHC complexes. Eur. J. Immunol. 37, 602–612.
Charo, I. F., and Ransohoff, R. M. (2006). The many roles of chemokines and chemokine receptors in inflammation. N. Engl. J. Med. 354, 610–621.
Colf, L. A., Bankovich, A. J., Hanick, N. A., Bowerman, N. A., Jones, L. L., Kranz, D. M., and Garcia, K. C. (2007). How a single T cell receptor recognizes both self and foreign MHC. Cell 129, 135–146.
Delarasse, C., Daubas, P., Mars, L. T., Vizler, C., Litzenburger, T., Iglesias, A., Bauer, J., Della Gaspera, B., Schubart, A., Decker, L., Dimitri, D., Roussel, G., Dierich, A., Amor, S., Dautigny, A., Liblau, R., and Pham-Dinh, D. (2003). Myelin/oligodendrocyte glycoprotein-deficient (MOG-deficient) mice reveal lack of immune tolerance to MOG in wild-type mice. J. Clin. Invest. 112, 544–553.
Dessen, A., Lawrence, C. M., Cupo, S., Zaller, D. M., and Wiley, D. C. (1997). X-ray crystal structure of HLA-DR4 (DRA*0101, DRB1*0401) complexed with a peptide from human collagen II. Immunity 7, 473–481.
Dutoit, V., Rubio-Godoy, V., Pittet, M. J., Zippelius, A., Dietrich, P. Y., Legal, F. A., Guillaume, P., Romero, P., Cerottini, J. C., Houghten, R. A., Pinilla, C., and Valmori, D. (2002). Degeneracy of antigen recognition as the molecular basis for the high frequency of naive A2/Melan-a peptide multimer(+) CD8(+) T cells in humans. J. Exp. Med. 196, 207–216.
Fremont, D. H., Dai, S., Chiang, H., Crawford, F., Marrack, P., and Kappler, J. (2002). Structural basis of cytochrome c presentation by IE(k). J. Exp. Med. 195, 1043–1052.
Garcia, K. C., Degano, M., Pease, L. R., Huang, M., Peterson, P. A., Teyton, L., and Wilson, I. A. (1998). Structural basis of plasticity in T cell receptor recognition of a self peptide-MHC antigen. Science 279, 1166–1172.
Garcia, K. C., Radu, C. G., Ho, J., Ober, R. J., and Ward, E. S. (2001). Kinetics and thermodynamics of T cell receptor-autoantigen interactions in murine experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. U.S.A. 98, 6818–6823.
Gautam, A. M., Liblau, R., Chelvanayagam, G., Steinman, L., and Boston, T. (1998). A viral peptide with limited homology to a self peptide can induce clinical signs of experimental autoimmune encephalomyelitis. J. Immunol. 161, 60–64.
Hagerty, D. T., and Allen, P. M. (1995). Intramolecular mimicry. Identification and analysis of two cross-reactive T cell epitopes within a single protein. J. Immunol. 155, 2993–3001.
Harkiolaki, M., Holmes, S. L., Svendsen, P., Gregersen, J. W., Jensen, L. T., McMahon, R., Friese, M. A., van Boxel, G., Etzensperger, R., Tzartos, J. S., Kranc, K., Sainsbury, S., Harlos, K., Mellins, E. D., Palace, J., Esiri, M. M., van der Merwe, P. A., Jones, E. Y., and Fugger, L. (2009). T cell-mediated autoimmune disease due to low-affinity crossreactivity to common microbial peptides. Immunity 30, 348–357.
Hemmer, B., Fleckenstein, B. T., Vergelli, M., Jung, G., McFarland, H., Martin, R., and Wiesmüller, K. H. (1997). Identification of high potency microbial and self ligands for a human autoreactive class II-restricted T cell clone. J. Exp. Med. 185, 1651–1659.
Holler, P. D., Chlewicki, L. K., and Kranz, D. M. (2003). TCRs with high affinity for foreign pMHC show self-reactivity. Nat. Immunol. 4, 55–62.
Jacomy, H., Zhu, Q., Couillard-Després, S., Beaulieu, J. M., and Julien, J. P. (1999). Disruption of type IV intermediate filament network in mice lacking the neurofilament medium and heavy subunits. J. Neurochem. 73, 972–984.
Jäger, A., Dardalhon, V., Sobel, R. A., Bettelli, E., and Kuchroo, V. K. (2009). Th1, Th17, and Th9 effector cells induce experimental autoimmune encephalomyelitis with different pathological phenotypes. J. Immunol. 183, 7169–7177.
Jagodic, M., Colacios, C., Nohra, R., Dejean, A. S., Beyeen, A. D., Khademi, M., Casemayou, A., Lamouroux, L., Duthoit, C., Papapietro, O., Sjöholm, L., Bernard, I., Lagrange, D., Dahlman, I., Lundmark, F., Oturai, A. B., Soendergaard, H. B., Kemppinen, A., Saarela, J., Tienari, P. J., Harbo, H. F., Spurkland, A., Ramagopalan, S. V., Sadovnick, D. A., Ebers, G. C., Seddighzadeh, M., Klareskog, L., Alfredsson, L., Padyukov, L., Hillert, J., Clames, M., Edan, G., Fontaine, B., Fournié, G. J., Kockum, I., Saoudi, A., and Olsson, T. (2009). A role for VAV1 in experimental autoimmune encephalomyelitis and multiple sclerosis. Sci. Transl. Med. 1, 10ra21.
Juang, J., Ebert, P. J., Feng, D., Garcia, K. C., Krogsgaard, M., and Davis, M. M. (2010). Peptide-MHC heterodimers show that thymic positive selection requires a more restricted set of self-peptides than negative selection. J. Exp. Med. 207, 1223–1234.
Kimachi, K., Sugie, K., and Grey, H. M. (2003). Effector T cells have a lower ligand affinity threshold for activation than naive T cells. Int. Immunol. 15, 885–892.
Krishnamoorthy, G., Saxena, A., Mars, L. T., Domingues, H. S., Mentele, R., Ben-Nun, A., Lassmann, H., Dornmair, K., Kurschus, F. C., Liblau, R. S., and Wekerle, H. (2009). Myelin-specific T cells also recognize neuronal autoantigen in a transgenic mouse model of multiple sclerosis. Nat. Med. 15, 626–632.
Lang, H. L., Jacobsen, H., Ikemizu, S., Andersson, C., Harlos, K., Madsen, L., Hjorth, P., Sondergaard, L., Svejgaard, A., Wucherpfennig, K., Stuart, D. I., Bell, J. I., Jones, E. Y., and Fugger, L. (2002). A functional and structural basis for TCR cross-reactivity in multiple sclerosis. Nat. Immunol. 3, 940–943.
Lehmann, P. V., Sercarz, E. E., Forsthuber, T., Dayan, C. M., and Gammon, G. (1993). Determinant spreading and the dynamics of the autoimmune T-cell repertoire. Immunol. Today 14, 203–208.
Lennon, G. P., Bettini, M., Burton, A. R., Vincent, E., Arnold, P. Y., Santamaria, P., and Vignali, D. A. (2009). T cell islet accumulation in type 1 diabetes is a tightly regulated, cell-autonomous event. Immunity 31, 643–653.
Li, L., Wang, B., Frelinger, J. A., and Tisch, R. (2008). T-cell promiscuity in autoimmune diabetes. Diabetes 57, 2099–2106.
Lyman, M. A., Nugent, C. T., Marquardt, K. L., Biggs, J. A., Pamer, E. G., and Sherman, L. A. (2005). The fate of low affinity tumor-specific CD8+ T cells in tumor-bearing mice. J. Immunol. 174, 2563–2572.
Mason, D. (1998). A very high level of crossreactivity is an essential feature of the T-cell receptor. Immunol. Today 19, 395–404.
Maynard, J., Petersson, K., Wilson, D. H., Adams, E. J., Blondelle, S. E., Boulanger, M. J., Wilson, D. B., and Garcia, K. C. (2005). Structure of an autoimmune T cell receptor complexed with class II peptide-MHC: insights into MHC bias and antigen specificity. Immunity 22, 81–92.
Münz, C., Lünemann, J. D., Getts, M. T., and Miller, S. D. (2009). Antiviral immune responses: triggers of or triggered by autoimmunity? Nat. Rev. Immunol. 9, 246–258.
Odoardi, F., Kawakami, N., Klinkert, W. E., Wekerle, H., and Flügel, A. (2007). Blood-borne soluble protein antigen intensifies T cell activation in autoimmune CNS lesions and exacerbates clinical disease. Proc. Natl. Acad. Sci. U.S.A. 104, 18625–18630.
Petersen, T. R., Bettelli, E., Sidney, J., Sette, A., Kuchroo, V., and Bäckström, B. T. (2004). Characterization of MHC- and TCR-binding residues of the myelin oligodendrocyte glycoprotein 38-51 peptide. Eur. J. Immunol. 34, 165–173.
Pöhlmann, T., Böckmann, R. A., Grubmüller, H., Uchanska-Ziegler, B., Ziegler, A., and Alexiev, U. (2004). Differential peptide dynamics is linked to major histocompatibility complex polymorphism. J. Biol. Chem. 279, 28197–28201.
Quaratino, S., Thorpe, C. J., Travers, P. J., and Londei, M. (1995). Similar antigenic surfaces, rather than sequence homology, dictate T-cell epitope molecular mimicry. Proc. Natl. Acad. Sci. U.S.A. 92, 10398–10402.
Reboldi, A., Coisne, C., Baumjohann, D., Benvenuto, F., Bottinelli, D., Lira, S., Uccelli, A., Lanzavecchia, A., Engelhardt, B., and Sallusto, F. (2009). C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat. Immunol. 10, 514–523.
Reiser, J. B., Darnault, C., Grégoire, C., Mosser, T., Mazza, G., Kearney, A., van der Merwe, P. A., Fontecilla-Camps, J. C., Housset, D., and Malissen, B. (2003). CDR3 loop flexibility contributes to the degeneracy of TCR recognition. Nat. Immunol. 4, 241–247.
Sethi, D. K., Schubert, D. A., Anders, A. K., Heroux, A., Bonsor, D. A., Thomas, C. P., Sundberg, E. J., Pyrdol, J., and Wucherpfennig, K. W. (2011). A highly tilted binding mode by a self-reactive T cell receptor results in altered engagement of peptide and MHC. J. Exp. Med. 208, 91–102.
Sharma, A. K., Kuhns, J. J., Yan, S., Friedline, R. H., Long, B., Tisch, R., and Collins, E. J. (2001). Class I major histocompatibility complex anchor substitutions alter the conformation of T cell receptor contacts. J. Biol. Chem. 276, 21443–21449.
Sloan-Lancaster, J., and Allen, P. M. (1996). Altered peptide ligand-induced partial T cell activation: molecular mechanisms and role in T cell biology. Annu. Rev. Immunol. 14, 1–27.
Sospedra, M., Muraro, P. A., Stefanová, I., Zhao, Y., Chung, K., Li, Y., Giulianotti, M., Simon, R., Mariuzza, R., Pinilla, C., and Martin, R. (2006). Redundancy in antigen-presenting function of the HLA-DR and -DQ molecules in the multiple sclerosis-associated HLA-DR2 haplotype. J. Immunol. 176, 1951–1961.
Sospedra, M., Zhao, Y., zur Hausen, H., Muraro, P. A., Hamashin, C., de Villiers, E. M., Pinilla, C., and Martin, R. (2005). Recognition of conserved amino acid motifs of common viruses and its role in autoimmunity. PLoS Pathog. 1, e41. doi:10.1371/journal.ppat.0010041
Tisch, R., Wang, B., and Serreze, D. V. (1999). Induction of glutamic acid decarboxylase 65-specific Th2 cells and suppression of autoimmune diabetes at late stages of disease is epitope dependent. J. Immunol. 163, 1178–1187.
Tsai, S., Shameli, A., and Santamaria, P. (2008). CD8+ T cells in type 1 diabetes. Adv. Immunol. 100, 79–124.
Vang, T., Congia, M., Macis, M. D., Musumeci, L., Orrú, V., Zavattari, P., Nika, K., Tautz, L., Taskén, K., Cucca, F., Mustelin, T., and Bottini, N. (2005). Autoimmune-associated lymphoid tyrosine phosphatase is a gain-of-function variant. Nat. Genet. 37, 1317–1319.
Vizler, C., Bercovici, N., Heurtier, A., Pardigon, N., Goude, K., Bailly, K., Combadière, C., and Liblau, R. S. (2000). Relative diabetogenic properties of islet-specific Tc1 and Tc2 cells in immunocompetent hosts. J. Immunol. 165, 6314–6321.
Wing, K., and Sakaguchi, S. (2010). Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat. Immunol. 11, 7–13.
Wucherpfennig, K. W., Allen, P. M., Celada, F., Cohen, I. R., De Boer, R., Garcia, K. C., Goldstein, B., Greenspan, R., Hafler, D., Hodgkin, P., Huseby, E. S., Krakauer, D. C., Nemazee, D., Perelson, A. S., Pinilla, C., Strong, R. K., and Sercarz, E. E. (2007). Polyspecificity of T cell and B cell receptor recognition. Semin. Immunol. 19, 216–224.
Wucherpfennig, K. W., and Gagnon, E. (2009). Positively selecting peptides: their job does not end in the thymus. Nat. Immunol. 10, 1143–1144.
Wucherpfennig, K. W., and Strominger, J. L. (1995). Molecular mimicry in T cell-mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell 80, 695–705.
Yin, L., Huseby, E., Scott-Browne, J., Rubtsova, K., Pinilla, C., Crawford, F., Marrack, P., Dai, S., and Kappler, J. W. (2011). A single t cell receptor bound to major histocompatibility complex class I and class II glycoproteins reveals switchable TCR conformers. Immunity 2011 35, 23–33.
Yin, Y., and Mariuzza, R. A. (2009). The multiple mechanisms of T cell receptor cross-reactivity. Immunity 31, 849–851.
Keywords: autoimmunity, T cell, TCR, multiple sclerosis, type 1 diabetes, MOG, GAD
Citation: Liblau RS, Wekerle H and Tisch RM (2011) Cumulative autoimmunity: T cell clones recognizing several self-epitopes exhibit enhanced pathogenicity. Front. Immun. 2:47. doi: 10.3389/fimmu.2011.00047
Received: 28 July 2011; Accepted: 10 September 2011;
Published online: 05 October 2011.
Edited by:
Luis Graca, University of Lisbon, PortugalReviewed by:
Anne L. Fletcher, Dana-Farber Cancer Institute, USADaniel Mucida, The Rockefeller University, USA
Copyright: © 2011 Liblau, Wekerle and Tisch. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Roland S. Liblau, INSERM UMR 1043, CNRS UMR 5282, Centre de Physiopathologie de Toulouse Purpan, CHU Purpan, BP 3028, 31024 Toulouse Cedex 3, France. e-mail:cm9sYW5kLmxpYmxhdUBpbnNlcm0uZnI=