
- 1 Department of Immunohematology and Blood Transfusion, Leiden University Medical Center, Leiden, Netherlands
- 2 Department of Pathology, VU University Medical Center, Amsterdam, Netherlands
Major histocompatibility complex (MHC)-I and MHC-II molecules play an essential role in the immune response to pathogens by virtue of their ability to present peptides to CD8+ and CD4+ T cells, respectively. Given this critical role, MHC-I and MHC-II genes are regulated in a tight fashion at the transcriptional level by a variety of transcription factors that interact with conserved cis-acting regulatory promoter elements. In addition to the activities of these regulatory factors, modification of chromatin also plays an essential role in the efficient transcription of these genes to meet with local requirement for an effective immune response. The focus of this review is on the transcription factors that interact with conserved cis-acting promoter elements and the epigenetic mechanisms that modulate induced and constitutive expression of these MHC genes.
Introduction
The products of the MHC class I (MHC-I) and MHC class II (MHC-II) genes encode cell-surface glycoproteins involved in the binding and presentation of peptides to the T cell receptors (TCRs) of T lymphocytes. Major histocompatibility complex (MHC)-I proteins present peptides from endogenous sources, such as those derived from viruses, to CD8+ T cells, whereas MHC-II molecules mainly present peptides from exogenous sources, such as those derived from extracellular pathogens, to CD4+ T cells. These tri-molecular interactions of MHC, peptide, and TCR are central to the generation of antigen-specific immune responses.
The MHC-I gene cluster encodes the highly polymorphic classical MHC-I molecules (human leukocyte antigen, HLA-A, -B, and -C), which play essential roles in the detection and elimination of virus-infected cells, tumor cells and transplanted allogeneic cells and the less polymorphic non-classical MHC-Ib molecules (HLA-E, -F, and -G). These latter MHC-Ib molecules have specialized immune regulatory functions (Van den Elsen et al., 2004). All cell-surface expressed MHC-I and MHC-Ib molecules are associated with the non-polymorphic β2-microglobulin.
The MHC-II genes encode the polymorphic HLA-DR, -DQ, and -DP molecules, which are expressed as α- and β-chain heterodimers on the cell surface. MHC-II molecules are central in the initiation of cellular and humoral immune responses, but they have also been implicated as contributing factors for a variety of autoimmune disorders. In contrast to the classical MHC-I molecules, which are expressed in a constitutive fashion on almost all nucleated cells, the constitutive expression of MHC-II molecules is tissue-specific and is restricted to professional antigen presenting cells (APCs, i.e., dendritic cells, macrophages, and B cells) and in thymic epithelial cells (Van den Elsen et al., 2004). All other cell types lack constitutive expression of MHC-II molecules, but their expression can be induced by exposure to cytokines of which interferon γ (IFNγ) is the most potent, or upon activation, such as in human T cells (Holling et al., 2002; Wong et al., 2002). Because of their crucial role in the immune response, the genes encoding MHC-I and MHC-II molecules are tightly regulated at the transcriptional level both by genetic and epigenetic mechanisms.
Transcriptional Regulation of MHC Genes
Activation of MHC-I genes, with the exception of HLA-G, is mediated by several conserved cis-acting regulatory promoter elements: i.e., the enhancer A, interferon-stimulated response element (ISRE) and the SXY-module (comprising the S/W, X1, X2, and Y-boxes; Figure 1). These conserved regulatory elements play an important role in the inducible and constitutive expression of MHC-I genes (Van den Elsen et al., 2004). Interestingly, these regulatory elements are also involved in the transcriptional activation of the β2-microglobulin promoter but not of the promoters of the genes encoding the transporter associated with antigen processing (TAP) and the large multifunctional protease (LMP), which are essential components in the MHC-I antigen processing and presentation pathway (Gobin et al., 1997, 2001; Van den Elsen et al., 2004). In MHC-I promoters, the enhancer A is bound by nuclear factor (NF)-κB, while the ISRE is bound by interferon regulatory factor (IRF) family members (Figure 1; Gobin et al., 1998, 1999). The transcription factors NF-κB and IRF-1 are mediators of the TNFα and IFNγ (Janus-family kinase/signal transducer and activator of transcription, Jak/STAT) routes of gene activation, respectively, which account for the induced MHC-I transcription. Additionally, binding sites for upstream-stimulatory factor (USF)-1, -2 and for the transcription factor Sp-1 can also be found within these upstream regulatory promoter elements in a locus and allele-specific fashion (Figure 1; Gobin et al., 1998, 1999). As a result of nucleotide sequence variation in the enhancer A and the ISRE in the different MHC-I promoters the level of promoter activation induced by these pathways differs among the various MHC-I loci (Gobin et al., 1998, 1999; Girdlestone, 2000; Johnson, 2003).
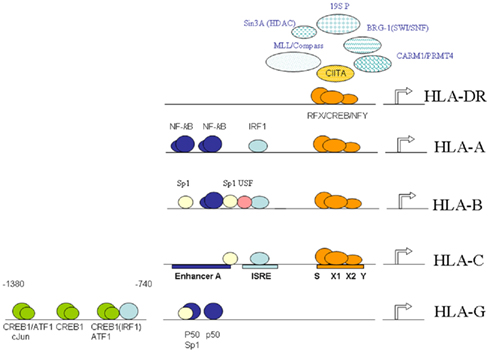
Figure 1. Schematic overview of the elements, interacting factors and epigenetic events governing MHC-I and MHC-II transcription. Shown is the shared proximal SXY-module, and the upstream enhancer A and ISRE typical for MHC-I promoters with the exception of HLA-G. The proximal SXY-module is bound by the MHC-enhanceosome.
Expression of HLA-G is under normal circumstances confined primarily to fetal trophoblast cells and thymic epithelium. This suggest an alternative transcriptional control of HLA-G than that of the classical MHC-I genes. Indeed, while encoding similar regulatory elements in its 5′ DNA, HLA-G is not regulated by the upstream ISRE and κB sites or the SXY regulatory module (Gobin and Van den Elsen, 1999). Instead, the HLA-G promoter can be transactivated by the cyclic-AMP response element binding protein (CREB)-1 (Gobin et al., 2002). This transactivation is mediated by further upstream positioned CRE sites, which can bind CREB-1, activating transcription factor (ATF)-1 and c-Jun in vitro (Figure 1; Gobin et al., 2002). Since these factors are ubiquitously expressed, the lack of HLA-G expression in cell types other than trophoblast cells must be regulated in a different manner such as by alternative regulatory factors that suppress expression of HLA-G or by epigenetic mechanisms. In this respect it was recently shown that Ras-responsive binding protein-1 (RREB-1) is a transcriptional repressor of HLA-G, which exerts its function through chromatin remodeling of the HLA-G locus by virtue of its interaction with subunits of the CtBP complex in cells that lack expression of HLA-G (Flajollet et al., 2009). Additionally, it has also been demonstrated that treatment of various types of cells lacking HLA-G expression with the DNA methyltransferase inhibitor 5′-AZA-deoxycytidine results in restoration of HLA-G transcription (Moreau et al., 2003). Besides DNA methylation, histone acetylation modifications have also been implicated in the transcriptional control of HLA-G in trophoblast cell lines (Holling et al., 2009).
The SXY-module is also present is the promoters of MHC-II and its accessory genes (invariant chain, HLA-DM and HLA-DO). However, MHC-II promoters differ from MHC-I promoters in that they lack the typical enhancer A and ISRE (Figure 1). The sequence and stereo-specific alignment of the various boxes in the SXY-module is highly conserved and critical for its functioning in constitutive and inducible-transcriptional activation of MHC-I and MHC-II genes (Gobin et al., 2001; Ting and Trowsdale, 2002). The SXY-module is cooperatively bound by a multi-protein complex containing regulatory factor X (RFX; consisting of RFX5, RFXB/ANK, and RFXAP; Steimle et al., 1995; Durand et al., 1997; Masternak et al., 1998; Nagarajan et al., 1999), CREB/ATF (Moreno et al., 1999; Gobin et al., 2001), and nuclear factor-Y (NF-Y; Louis-Plence et al., 1997; Jabrane-Ferrat et al., 2002). This complex acts as an enhanceosome driving transactivation of these genes (Masternak et al., 2000; Gobin et al., 2001; Choi et al., 2011). The presence of the RFX components is crucial for the assembly of this enhanceosome, a notion that has been derived from studies with cell lines established from MHC-II deficiency patients (Reith and Mach, 2001). In addition to these factors that assemble directly to the X1, X2, and Y-box sequences the class II transactivator (CIITA), which acts as a co-activator, is also required. CIITA is essential for MHC-II transcription (Steimle et al., 1993), while it contributes to the activation of MHC-I promoters (Gobin et al., 1997; Martin et al., 1997). CIITA belongs to the large NLR (nucleotide binding domain, leucine-rich repeat containing) family of proteins that play multiple functions in innate immune responses (Harton et al., 2002; Ting et al., 2008). Despite the fact that CIITA activates MHC-I promoters in vitro, its in vivo contribution to MHC-I gene transcription remained enigmatic. It is therefore of note that more recently the NLR family member NLRC5 was identified to associate with and activate the promoters of MHC-I genes and not MHC-II genes in vivo (Meissner et al., 2010). Like CIITA, NLRC5 also induced the expression of the gene encoding β2-microglobulin. However, in contrast to CIITA, NLRC5 was also found to control the expression of the genes encoding TAP and LMP. These observations reveal that NLRC5 appears to be a transcriptional regulator, which orchestrates the concerted expression of critical components in the MHC-I antigen presentation pathway (Meissner et al., 2010).
Given the essential role of CIITA in MHC-II transcription, constitutive expression of CIITA coincides with constitutive MHC-II molecule expression in APCs. In non-immune cells expression of CIITA can be induced by IFNγ resulting in inducible MHC-II expression at the cell surface. CIITA therefore can be regarded as a molecular switch for MHC-II expression. Transcriptional activation of MHC-II genes also involves modulation of covalent histones modifications and chromatin remodeling (Choi et al., 2011). As an example, IFNγ-induced MHC-II expression results in an increase in active histone marks, i.e., acetylation of histone H3 and H4, and 3meK4-H3 at the MHC-II promoter, while at the same time a decrease in the repressive 3meK9-H3 histone mark is noted (Chou and Tomasi, 2008).
CIITA Transactivation and Epigenetic Activities
Class II transactivator exerts its transactivating function through protein–protein interactions with the components of the MHC-enhanceosome bound to the proximal SXY regulatory module in MHC promoters (Figure 1; Masternak et al., 2000; Zhu et al., 2000; Jabrane-Ferrat et al., 2003). This interaction of CIITA with the MHC-enhanceosome allows for the subsequent recruitment of the lysine acetyltransferases (KATs) p300 (KAT3b)/CREB binding protein (CBP or KAT3a) and p300/CBP-associated factor (PCAF or KAT2b), which promote transcription of MHC-I and MHC-II genes by providing a more open chromatin structure (Kretsovali et al., 1998; Fontes et al., 1999; Spilianakis et al., 2000; Gobin et al., 2001). Furthermore, CIITA also recruits the co-activator-associated arginine methyltransferase-1/protein arginine N-methyltransferase 4 (CARM1/PRMT4; Zika and Ting, 2005; Zika et al., 2005; Figure 1). Besides acting as a platform for recruitment of KAT activities, CIITA itself contains intrinsic KAT activity. The CIITA-mediated transactivation of MHC promoters was found to rely on this intrinsic KAT activity, which maps to a region in its N-terminus (Raval et al., 2001). This KAT activity of CIITA is regulated by its C-terminal GTP-binding domain and is stimulated by GTP (Raval et al., 2001). Interestingly, the CIITA KAT activity was found to bypass TATA box binding protein (TBP)-associated factor 250 kDa (TAFII250) in MHC-I promoter activation (Raval et al., 2001). Moreover, acetylation of CIITA itself by CBP and/or PCAF at specific lysine residues within the bipartite nuclear localization signal in the amino-terminal region of CIITA governs its nuclear accumulation (Spilianakis et al., 2000).
Class II transactivator also interacts with histone deacetylases (HDACs), which were found to interfere with CIITA function. These activities that acetylate/deacetylate lysine residues act as molecular switches for CIITA-mediated transcriptional activation/silencing of MHC genes. In this respect, it was found that HDAC1 and HDAC2 interfere in the transcriptional transactivation function of CIITA following IFNγ induction (Zika et al., 2003; Kong et al., 2009). In mice the HDAC1/HDAC2-associated repressor SIN3 homolog A (mSin3A) amplifies this inhibition in CIITA function (Zika et al., 2003). HDAC2 has the potential to deacetylate CIITA through its interaction with CIITA (Kong et al., 2009). This results in targeting of CIITA to the proteasomal degradation machinery and decreased interaction of CIITA with the RFX component RFX5 (Kong et al., 2009). Together, these observations reveal that these HDAC activities affect CIITA function on the one hand by disrupting assembly of the MHC-enhanceosome, while on the other hand they interfere in CIITA interactions with the MHC-enhanceosome. The switch/sucrose non-fermentable (SWI/SNF) ATPase Brahma-related gene-1 (BRG-1) also associates with CIITA and is required for the CIITA-mediated induction of MHC-II genes (Mudhasani and Fontes, 2002). The association of CIITA and BRG-1 suggest that the ATP-dependent chromatin remodeling SWI/SNF complex is recruited by CIITA to MHC-II promoters to control transcription of MHC-II genes. The ATPase Sug 1, a component of the 19S proteasome (19S P) complex, was found to be involved in increased levels of acetylation at MHC-II promoters and appeared to be essential for CIITA stability and MHC-II expression (Bhat et al., 2008, 2010a; Koues et al., 2008). More recently, another subunit of the 19S proteosome complex, the ATPase S6a (S6′/Tat-binding protein 1), was found to be crucial for regulating cytokine-inducible transcription of CIITA thereby indirectly modulation MHC-II transcription (Truax et al., 2010). Other activities involved monoubiquitination of CIITA, which was shown to stabilize CIITA at MHC-II promoters (Bhat et al., 2010b). In addition MLL/COMPASS subunits, which are involved in establishing active histone H3-K4 methylation marks, were found also to be recruited to MHC-II promoters following IFNγ treatment of cells (Koues et al., 2010; Figure 1).
Long-Range Promoter Interactions
The appropriate temporal and spatial expression of MHC-II genes in vivo also requires the involvement of additional, long-range regulatory elements. The more distal X–Y or X-box like sequences in the MHC-II region play an important role in these processes (Gomez et al., 2005). It has been found that interactions between the proximal elements and more distal X–Y or X-box like sequences (2.3 kb upstream of the HLA-DRA promoter) result in epigenetic changes at the MHC-II promoter (Masternak et al., 2003; Wright and Ting, 2006; Choi et al., 2011). In one model, RFX and CIITA can interact with the proximal SXY-module and with distal X–Y or X-box like sequences to form a chromatin loop (Masternak et al., 2003). This chromatin loop results in enhanced histone acetylation (Krawczyk et al., 2004). Likewise, the transcriptional insulator factor CCCTC binding factor insulator (CTCF) was found to control MHC-II gene expression through long-distance chromatin interactions (Majumder et al., 2008). The intergenic DNA of the HLA-DRB1 and HLA-DQA1 genes hosts a region that was bound by CTCF and acts as a potent enhancer-blocking element (Majumder et al., 2006). This element and its bound factors were found to interact with HLA-DRB1 and HLA-DQA1 (Majumder et al., 2008). Subsequently it was demonstrated that CTCF associates with CIITA and RFX5 suggesting that the CTCF bound region and the flanking HLA-DRB1 and HLA-DQA1 proximal promoters may interact (Majumder et al., 2008). More recently it was shown by RNAi depletion of CTCF that all CIITA-regulated genes within the MHC-II locus required CTCF for maximal expression (Majumder and Boss, 2010).
Epigenetic Regulation of MHC2TA Transcription
Transcriptional regulation of MHC2TA, the gene encoding CIITA, is mediated through the activity of four independent promoter units (CIITA-PI through CIITA-PIV; Muhlethaler-Mottet et al., 1997; Figure 2). These promoter units are employed in a cell type- and activation-specific manner. CIITA-PI and CIITA-PIII are used for the constitutive expression in dendritic cells and in B cells, respectively (Muhlethaler-Mottet et al., 1997). CIITA-PIV has been shown to be the promoter predominantly involved in IFNγ-inducible expression (Muhlethaler-Mottet et al., 1998; Piskurich et al., 1998, 1999). In addition, in human non-B cells, CIITA-PIII can also be activated by IFNγ through an element located 2 kb upstream of the core CIITA-PIII promoter (Piskurich et al., 1999; Van der Stoep et al., 2002a, 2007). CIITA-PIII has also been shown to be employed by human T cells upon activation (Holling et al., 2002; Wong et al., 2002). The promoter function of CIITA-PII is still ill-defined. The various MHC2TA promoters each transcribe a unique first exon and are located within a region of approximately 14 kb (Muhlethaler-Mottet et al., 1997).
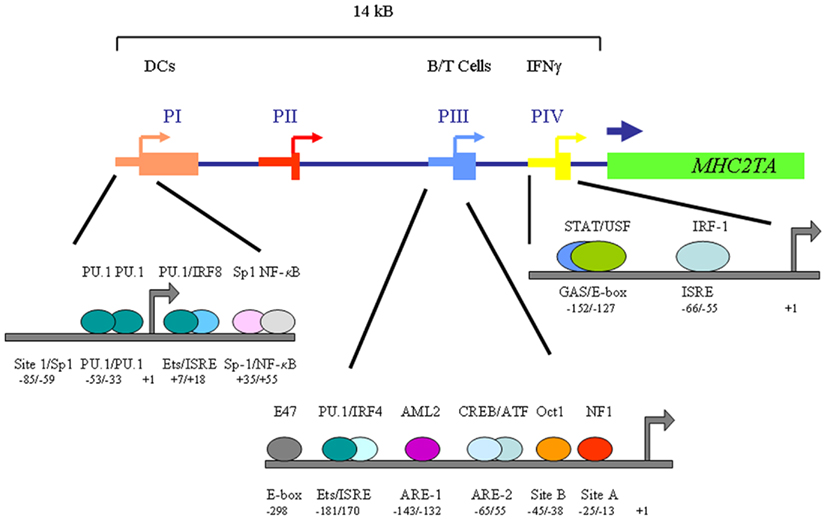
Figure 2. Schematic overview of the MHC2TA multi-promoter region. Shown are the factors and elements governing transactivation of CIITA-PI in immature dendritic cells, CIITA-PIII in B cells and CIITA-PIV following IFNγ stimulation. The localization of the various protein/DNA-binding elements is indicated relative to the transcription start site.
Over the past years, several regulatory elements in MHC2TA promoters and interacting factors that are important for transcriptional activation have been identified (Figure 2). IFNγ-mediated activation of CIITA-PIV requires occupation of the gamma-activated sequence (GAS)-box and the ISRE in CIITA-PIV by STAT-1 and the STAT-1 target gene IRF-1 (Muhlethaler-Mottet et al., 1998). Importantly, STAT-1 needs to interact with USF-1 bound to the E-box adjacent to the GAS-box for stable interaction (Muhlethaler-Mottet et al., 1998; Figure 2). The IFNγ-mediated activation of CIITA-PIV also results in increased histone H3 and H4 acetylation at CIITA-PIV (Morris et al., 2002). Interestingly, this increase in histone acetylation in CIITA-PIV chromatin is already noted prior to recruitment of IRF-1 to the CIITA-PIV promoter (Morris et al., 2002). In addition to the factors interacting with these CIIT-PIV promoter elements, the SWI/SNF ATPase BRG-1 was also found to be an important factor in the IFNγ-mediated transcriptional activation of CIITA-PIV because cells that lack expression of BRG-1 failed to induce CIITA expression following exposure to IFNγ (Pattenden et al., 2002).
Class II transactivator-PIII is employed in B cells and activated T cells in humans (Ghosh et al., 1999; Holling et al., 2002; Van der Stoep et al., 2002a; Wong et al., 2002). Activation of CIITA-PIII requires interaction of the transcription factor CREB-1 with CRE-binding sites in the activation response element (ARE)-2 and, depending on the cellular context, also with CRE-binding sites in the 5′-UTR of CIITA-PIII (Holling et al., 2002; Van der Stoep et al., 2002a; Wong et al., 2002). The CREB-1 mediated activation of CIITA-PIII in B cells was amplified by the KAT CBP (Van der Stoep et al., 2002a). CIITA-PIII also contains a composite Ets/ISRE-consensus element (Site C) and 2 E-box motifs, which were found to play a crucial role in its B cell-specific transcriptional regulation (Van der Stoep et al., 2004). In B cells the Ets/ISRE-consensus element is bound by PU.1 and IRF-4, whereas the basic helix-loop-helix factor E47 interacts with the E-box motifs. When bound respectively to the Ets/ISRE and E-boxes PU.1 and IRF-4, and E47 synergize to direct B cell-specific activation of CIITA-PIII (Figure 2; Van der Stoep et al., 2004). The involvement of these factors in B cell-specific activation of CIITA-PIII is of interest because PU.1, IRF-4, and E47 also play an important role in B cell differentiation and activation.
In vivo footprint analysis of CIITA-PI in immature dendritic cells revealed multiple protein/DNA interactions that were lost upon maturation of dendritic cells. CIITA-PI was found to contain binding sites for PU.1, Sp-1 NF-κB, and a composite Ets/ISRE (Figure 2; Smith et al., 2011). Transcription mediated by CIITA-PI in immature dendritic cells requires binding of PU.1, IRF-8, NF-κB, and Sp-1 to the promoter (Smith et al., 2011). Interestingly, for binding of PU.1 to the composite Ets/ISRE heterodimerization with IRF-8 is a prerequisite. Mutational analysis of the PU.1, IRF-8, and NF-κB sites showed that these sites were critical for transcriptional activity of CIITA-PI. Of note is also that mice lacking IRF-8 displayed an unoccupied CIITA-PI, which was restored by reconstitution with IRF-8 in vitro (Smith et al., 2011). Together, these observations reveal an important role for PU.1/IRF-8 in the activation of CIITA-PI in immature dendritic cells.
Plasma B cells lack expression of CIITA (Silacci et al., 1994). This extinction of CIITA and resulting MHC-II molecule expression during differentiation of B cells into plasma B cells is mediated by the transcriptional repressor B lymphocyte-induced maturation protein-1 (Blimp-1, also known as positive regulatory domain I-binding factor 1, PRDI-BF1; Piskurich et al., 2000; Ghosh et al., 2001). The silencing of CIITA expression in plasma cells is most likely resulting from binding of Blimp-1 to the Ets/ISRE-consensus element in CIITA-PIII thereby disrupting the interaction of PU.1/IRF-4 to this element (Piskurich et al., 2000; Ghosh et al., 2001; Wright and Ting, 2006). Interestingly, besides its repressive activity on CIITA-PIII transactivation, there is more recent evidence that PRDI-BF1 mediates also repression of CIITA-PIV (Chen et al., 2007). Transcriptional repression of MHC2TA mediated by Blimp-1/PRDI-BF1 is also associated with histone deacetylase and lysine methyltransferase (KMT) activities (Yu et al., 2000; Gyory et al., 2004). In particular the activities of the histone deacetylases HDAC1 and HDAC2, and the lysine methyltransferase KMT1C (also known as G9a), are required in these epigenetic silencing processes. Indeed it was demonstrated by chromatin immunoprecipitation (ChIP) that differences between B cells and plasma cells exist in the levels of activating and repressive histone marks in CIITA-PIII chromatin (Green et al., 2006). In plasma cells lacking CIITA expression, histone marks associated with gene transcription such as acetylated histone H3 and H4, and 2meK4-H3 and 3meK4-H3 are lost at CIITA-PIII, while the repressive 2meK9-H3 mark is increased (Green et al., 2006). Interestingly these histone marks were found also to exist at CIITA-PI, CIITA-PII and CIITA-PIV, revealing the involvement of the entire MHC2TA multi-promoter region. As a consequence of the repressive histone marks and resulting chromatin inaccessibility, the binding of CIITA-PIII interacting transcription factors (i.e., Sp-1, CREB-1, E47, PU.1, IRF-4) was lost in plasma cells (Green et al., 2006).
In dendritic cell maturation chromatin remodeling also plays an important role in the control of MHC2TA transcription (Landmann et al., 2001; Choi et al., 2009). During differentiation of monocytes into immature dendritic cells the CIITA-PI isoform is induced. In immature dendritic cells, MHC-II molecules are largely retained in intracellular compartments. Upon maturation of dendritic cells, the peptide/MHC-II complexes are assembled and transported to the cell surface. The increase of transported MHC-II molecules at the cell surface is accompanied by rapid transcriptional silencing of MHC2TA in matured dendritic cells (Landmann et al., 2001). Like in plasma B cells, the transcriptional inactivation of CIITA-PI is also accompanied by global histone deacetylation involving not only CIITA-PI but also CIITA-PIII and CIITA-PIV (Landmann et al., 2001). In a mouse model activation of CIITA-PI, during differentiation of monocytes into dendritic cells by mGM-CSF, is accompanied by an increase in the levels of acetylated histone H3 and H4 (Choi et al., 2009). IL-10 could block the increase in histone H3 and H4 acetylation during differentiation resulting in inhibition of MHC2TA transcription (Choi et al., 2009). Recently the involvement of the transcriptional repressor PRD1-BF1/Blimp-1 in these CIITA-PI silencing processes was revealed (Smith et al., 2011). It was shown that during dendritic cell maturation binding to CIITA-PI of the transcriptional activators PU.1, IRF-8, NF-κB, and Sp-1 is lost while at the same time the transcriptional repressor PRD1-BF1/Blimp-1 and its associated co-repressors KMT1C and HDAC2 are recruited to CIITA-PI (Smith et al., 2011). This results in a loss of histone acetylation and acquisition of histone K9-H3 dimethylation and heterochromatin protein 1γ (HP1γ) at CIITA-PI (Smith et al., 2011). Together, the transcriptional repressor PRD1-BF1/Blimp-1 appears to play a central role in the extinction of the various CIITA promoters and therefore can be regarded as a central regulator of MHC-II antigen presentation (Smith et al., 2011).
Distal elements and chromatin remodeling also play an essential role in the transcriptional regulation of MHC2TA (Ni et al., 2008). Transcriptional activation of CIITA-PIV by IFNγ relies on the interaction with distal elements at −50 and −8 kb (Ni et al., 2008). Contact was also detected between elements at −50 and −16 kb. In these long-range interactions, BRG-1, the ATPase driving the chromatin remodeling complex SWI-SNF (also called BAF), was constitutively bound to sites at −50, −16, −8, and +59 kb, and also CIITA-IV (Ni et al., 2008). Thus BRG-1 not only is an important factor in the CIITA-mediated activation of MHC-II genes, but also controls the transcriptional activation of MHC2TA itself through long-range chromatin and promoter interactions.
Extinction of MHC-II Expression in Cancer
As discussed in the previous sections lack of MHC-II molecule expression coincides with lack of detectable levels of CIITA. In cancer the transcriptional silencing of MHC2TA and resulting extinction of MHC-II gene expression is frequently noted (Murphy and Tomasi, 1998; Yazawa et al., 1999; Meissner et al., 2008; Berghuis et al., 2009). The silencing of CIITA is mostly mediated by chromatin modifications involving methylation of DNA and modifications of histones. The lack of CIITA expression in several cancer types is associated with CpG dinucleotide methylation of CIITA-PIV and of CIITA-PIII DNA (Morris et al., 2000; Van den Elsen et al., 2000, 2003; Van der Stoep et al., 2002b; Holling et al., 2004, 2006; Morimoto et al., 2004; Satoh et al., 2004; De Lerma Barbaro et al., 2008; Meissner et al., 2008). Besides CpG dinucleotide methylation, the lack of IFNγ-induced transcription of MHC2TA is also associated with histone deacetylase activities (Magner et al., 2000; Murphy et al., 2002; Holtz et al., 2003; Kanaseki et al., 2003; Chou et al., 2005).
In a uveal melanoma tumor cell line it was demonstrated for the first time that histone methylation played an important role in MHC2TA transcriptional silencing (Holling et al., 2007). The strongly reduced expression levels of CIITA after IFNγ-induction were found to correlate with high levels of the repressive 3meK27-H3 histone modification in CIITA-PIV chromatin in the absence of CpG dinucleotide methylation of CIITA-PIV DNA (Holling et al., 2007). Moreover, the KMTase Enhancer of Zeste Homolog 2 (EZH2, or KMT6) was recruited to relative high levels into CIITA-PIV chromatin (Holling et al., 2007). KMT6 is the catalytic subunit of the polycomb repressive complex 2 (PRC2), which plays an important role in transcriptional silencing and maintenance of cellular identity. Consistent with the transcriptional silent state of MHC2TA was the lack of RNA polymerase II recruitment into CIITA-PIV chromatin after IFNγ-induction (Holling et al., 2007). RNA interference-mediated silencing of expression of KMT6 resulted in an increment in CIITA mRNA expression levels after IFNγ induction. These observations suggest that KMT6 is involved in the transcriptional downregulation of IFNγ-induced expression of CIITA in uveal melanoma (Holling et al., 2007). Notably, the transcriptional silencing of MHC2TA by histone methylation in the absence of CpG dinucleotide methylation is in line with the observation that the 3meK27-H3 modification pre-marks genes for de novo methylation in cancer (Schlesinger et al., 2007). It could therefore be argued that the epigenetic make-up of the CIITA-PIV region in uveal melanoma reflects pre-marking for de novo methylation of DNA, and that this reflects an intermediate epigenetic state of MHC2TA in the complete shut down of MHC-II mediated antigen presentation functions. More recently, lack of CIITA expression was also found associated with high levels of the 3meK27-H3 repressive histone mark in the relative absence of activating histone acetylation and methylation marks in CIITA-PIII chromatin in T leukemia cell lines lacking MHC-II molecule expression (Van Eggermond et al., 2011). In contrast, CIITA and MHC-II molecule expressing T lymphoma cells displayed the opposite phenotype. As detailed above, in T leukemia cells these repressive histone marks were also found to be associated with the other CIITA promoters in the MHC2TA multi-promoter region, while the opposite was noted for T lymphoma cells (Van Eggermond et al., 2011). The high levels of the repressive histone mark 3meK27-H3 were accompanied by relative high levels of KMT6 at the various CIITA promoters in T leukemia cells, while CIITA expressing T lymphoma cells displayed a general lack of KMT6 (Van Eggermond et al., 2011). Additionally, in HeLa cervical carcinoma cells a role for KMT6 in transcriptional silencing of IFNγ-inducible expression of CIITA was also revealed (Mehta et al., 2011). Together, these observations provide a clear link with expression of Polycomb Group proteins and transcriptional silencing of MHC2TA and resulting lack of expression of MHC-II molecules in cancer.
Conclusion
Constitutive and induced transcription of MHC-I and MHC-II genes is mediated by a set of conserved regulatory elements in their promoters and interacting transcription factors of which the SXY-module is shared by both sets of genes. CIITA plays an essential role in the control of constitutive and induced MHC-II gene transcription through its interaction with the MHC-enhanceosome bound to the conserved SXY-module in MHC-II promoters. When bound to the MHC-enhanceosome, CIITA acts as a platform recruiting various activities involved in histone acetylation/deacetylation and methylation. CIITA is also central to recruitment of more general chromatin remodeling activities and long-range chromatin interactions of MHC-II promoters with distal elements. These activities mediated by CIITA provide strict control of transcription of these important immune genes. An additional level of transcriptional control for MHC-II gene expression is at the level of MHC2TA, the gene encoding CIITA, which is also tightly regulated by both genetic and epigenetic mechanisms. Because of the involvement of epigenetic mechanisms in the transcriptional control of MHC genes, deviations in these epigenetic mechanisms as observed under pathological conditions such as in cancer and autoimmune disease might provide an opportunity for pharmacological interference targeting the enzymes that modify DNA and histones.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Support of the research of PJvdE was obtained from the Dutch Cancer Society and the Dutch MS Research Foundation. I greatly acknowledge the contributions of all the past and present members of the laboratory, and national and international collaborators.
References
Berghuis, D., De Hooge, A. S., Santos, S. J., Horst, D., Wiertz, E. J., Van Eggermond, M. C., Van den Elsen, P. J., Taminiau, A. H., Ottaviano, L., Schaefer, K. L., Dirksen, U., Hooijberg, E., Mulder, A., Melief, C. J., Egeler, R. M., Schilham, M. W., Jordanova, E. S., Hogendoorn, P. C., and Lankester, A. C. (2009). Reduced human leukocyte antigen expression in advanced-stage Ewing sarcoma: implications for immune recognition. J. Pathol. 218, 222–231.
Bhat, K. P., Truax, A. D., Brooks, J. K., and Greer, S. F. (2010a). Association of the 19S proteasomal ATPases with the ATPase-binding domain of CIITA is essential for CIITA stability and MHC class II expression. Immunol. Cell Biol. 88, 807–816.
Bhat, K. P., Truax, A. D., and Greer, S. F. (2010b). Phosphorylation and ubiquitination of degron proximal residues are essential for class II transactivator (CIITA) transactivation and major histocompatibility class II expression. J. Biol. Chem. 285, 25893–25903.
Bhat, K. P., Turner, J. D., Myers, S. E., Cape, A. D., Ting, J. P.-Y., and Greer, S. F. (2008). The 19S proteasome ATPase Sug1 plays a critical role in regulating MHC class II transcription. Mol. Immunol. 45, 2214–2224.
Chen, H., Gilbert, C. A., Hudson, J. A., Bolick, S. C., Wright, K. L., and Piskurich, J. F. (2007). Positive regulatory domain I-binding factor 1 mediates repression of the MHC class II transactivator (CIITA) type IV promoter. Mol. Immunol. 44, 1461–1470.
Choi, N. M., Majumder, P., and Boss, J. M. (2011). Regulation of major histocompatibility complex class II genes. Curr. Opin. Immunol. 23, 81–87.
Choi, Y. E., Yu, H. N., Yoon, C. H., and Bae, Y. S. (2009). Tumor-mediated down-regulation of MHC class II in DC development is attributable to the epigenetic control of the CIITA type I promoter. Eur. J. Immunol. 39, 858–868.
Chou, S. D., Khan, A. N., Magner, W. J., and Tomasi, T. B. (2005). Histone acetylation regulates the cell type specific CIITA promoters, MHC class II expression and antigen presentation in tumor cells. Int. Immunol. 17, 1483–1494.
Chou, S. D., and Tomasi, T. B. (2008). Spatial distribution of histone methylation during MHC class II expression. Mol. Immunol. 45, 971–980.
De Lerma Barbaro, A., De Ambrosis, A., Banelli, B., Li Pira, G., Aresu, O., Romani, M., Ferrini, S., and Accolla, R. S. (2008). Methylation of CIITA promoter IV causes loss of HLA-II inducibility by IFN-gamma in promyelocytic cells. Int. Immunol. 20, 1457–1466.
Durand, B., Sperisen, P., Emery, P., Barras, E., Zufferey, M., Mach, B., and Reith, W. (1997). RFXAP, a novel subunit of the RFX DNA binding complex is mutated in MHC class II deficiency. EMBO J. 16, 1045–1055.
Flajollet, S., Poras, I., Carosella, E. D., and Moreau, P. (2009). RREB-1 is a transcriptional repressor of HLA-G. J. Immunol. 183, 6948–6959.
Fontes, J. D., Kanazawa, S., Jean, D., and Peterlin, B. M. (1999). Interactions between the class II transactivator and CREB binding protein increase transcription of major histocompatibility complex class II genes. Mol. Cell. Biol. 19, 941–947.
Ghosh, N., Gyory, I., Wright, G., Wood, J., and Wright, K. L. (2001). Positive regulatory domain I binding factor 1 silences class II transactivator expression in multiple myeloma cells. J. Biol. Chem. 276, 15264–15268.
Ghosh, N., Piskurich, J. F., Wright, G., Hassani, K., Ting, J. P.-Y., and Wright, K. L. (1999). A novel element and a TEF-2-like element activate the major histocompatibility complex class II transactivator in B-lymphocytes. J. Biol. Chem. 274, 32342–32350.
Girdlestone, J. (2000). Synergistic induction of HLA class I expression by RelA and CIITA. Blood 95, 3804–3808.
Gobin, S. J., Peijnenburg, A., Keijsers, V., and Van den Elsen, P. J. (1997). Site alpha is crucial for two routes of IFN gamma-induced MHC class I transactivation: the ISRE-mediated route and a novel pathway involving CIITA. Immunity 6, 601–611.
Gobin, S. J. P., Biesta, P., De Steenwinkel, J. E. M., Datema, G., and Van den Elsen, P. J. (2002). CREB provides for an alternative pathway of HLA-G transactivation. J. Biol. Chem. 277, 39525–39531.
Gobin, S. J. P., Keijsers, V., Van Zutphen, M., and Van den Elsen, P. J. (1998). The role of enhancer A in the locus-specific transactivation of classical and non-classical MHC class I genes by NF-κB. J. Immunol. 161, 2276–2283.
Gobin, S. J. P., and Van den Elsen, P. J. (1999). The regulation of HLA class I expression: is HLA-G the odd one out? Semin. Cancer Biol. 9, 55–59.
Gobin, S. J. P., Van Zutphen, M., Westerheide, S. D., Boss, J. M., and Van den Elsen, P. J. (2001). The MHC-specific enhanceosome and its role in MHC class I and beta(2)-microglobulin gene transactivation. J. Immunol. 167, 5175–5184.
Gobin, S. J. P., Van Zutphen, M., Woltman, A. M., and Van den Elsen, P. J. (1999). Transactivation of classical and non-classical HLA class I genes through the interferon-stimulated response element. J. Immunol. 163, 1428–1434.
Gomez, J. A., Majumder, P., Nagarajan, U. M., and Boss, J. M. (2005). X box-like sequences in the MHC class II region maintain regulatory function. J. Immunol. 175, 1030–1040.
Green, M. R., Yoon, H., and Boss, J. M. (2006). Epigenetic regulation during B cell differentiation controls CIITA promoter accessibility. J. Immunol. 177, 3865–3873.
Gyory, I., Wu, J., Fejér, G., Seto, E., and Wright, K. L. (2004). PRDI-BF1 recruits the histone H3 methyltransferase G9a in transcriptional silencing. Nat. Immunol. 5, 299–308.
Harton, J. A., Linhoff, M. W., Zhang, J., and Ting, J. P. (2002). Cutting edge: CATERPILLER: a large family of mammalian genes containing CARD, pyrin, nucleotide-binding, and leucine-rich repeat domains. J. Immunol. 169, 4088–4093.
Holling, T. M., Bergevoet, M. W., Wierda, R. J., Van Eggermond, M. C., and Van den Elsen, P. J. (2009). Genetic and epigenetic control of the major histocompatibility complex class Ib gene HLA-G in trophoblast cell lines. Ann. N. Y. Acad. Sci. 1173, 538–544.
Holling, T. M., Bergevoet, M. W., Wilson, L., Van Eggermond, M. C., Schooten, E., Steenbergen, R. D., Snijders, P. J., Jager, M. J., and Van den Elsen, P. J. (2007). A role for EZH2 in silencing of IFN-gamma inducible MHC2TA transcription in uveal melanoma. J. Immunol. 179, 5317–5325.
Holling, T. M., Schooten, E., Langerak, A. W., and Van den Elsen, P. J. (2004). Regulation of MHC class II expression in human T-cell malignancies. Blood 103, 1438–1444.
Holling, T. M., Van der Stoep, N., Quinten, E., and Van den Elsen, P. J. (2002). Activated human T cells accomplish MHC class II expression through T cell-specific occupation of class II transactivator promoter III. J. Immunol. 168, 763–770.
Holling, T. M., Van Eggermond, M. C., Jager, M. J., and Van den Elsen, P. J. (2006). Epigenetic silencing of MHC2TA transcription in cancer. Biochem. Pharmacol. 72, 1570–1576.
Holtz, R., Choi, J. C., Petroff, M. G., Piskurich, J. F., and Murphy, S. P. (2003). Class II transactivator (CIITA) promoter methylation does not correlate with silencing of CIITA transcription in trophoblasts. Biol. Reprod. 69, 915–924.
Jabrane-Ferrat, N., Nekrep, N., Tosi, G., Esserman, L., and Peterlin, B. M. (2003). MHC class II enhanceosome: how is the class II transactivator recruited to DNA-bound activators? Int. Immunol. 15, 467–475.
Jabrane-Ferrat, N., Nekrep, N., Tosi, G., Esserman, L. J., and Peterlin, B. M. (2002). Major histocompatibility complex class II transcriptional platform: assembly of nuclear factor Y and regulatory factor X (RFX) on DNA requires RFX5 dimers. Mol. Cell. Biol. 22, 5616–5625.
Johnson, D. R. (2003). Locus-specific constitutive and cytokine induced HLA class I gene expression. J. Immunol. 170, 1894–1902.
Kanaseki, T., Ikeda, H., Takamura, Y., Toyota, M., Hirohashi, Y., Tokino, T., Himi, T., and Sato, N. (2003). Histone deacetylation, but not hypermethylation, modifies class II transactivator and MHC class II gene expression in squamous cell carcinomas. J. Immunol. 170, 4980–4985.
Kong, X., Fang, M., Li, P., Fang, F., and Xu, Y. (2009). HDAC2 deacetylates class II transactivator and suppresses its activity in macrophages and smooth muscle cells. J. Mol. Cell. Cardiol. 46, 292–299.
Koues, O. I., Dudley, R. K., Truax, A. D., Gerhardt, D., Bhat, K. P., McNeal, S., and Greer, S. F. (2008). Regulation of acetylation at the major histocompatibility complex class II proximal promoter by the 19S proteasomal ATPase Sug1. Mol. Cell. Biol. 28, 5837–5850.
Koues, O. I., Mehta, N. T., Truax, A. D., Dudley, R. K., Brooks, J. K., and Greer, S. F. (2010). Roles for common MLL/COMPASS subunits and the 19S proteasome in regulating CIITA pIV and MHC class II gene expression and promoter methylation. Epigenetics Chromatin 3, 5.
Krawczyk, M., Peyraud, N., Rybtsova, N., Masternak, K., Bucher, P., Barras, E., and Reith, W. (2004). Long distance control of MHC class II expression by multiple distal enhancers regulated by regulatory factor X complex and CIITA. J. Immunol. 173, 6200–6210.
Kretsovali, A., Agalioti, T., Spilianakis, C., Tzortzakaki, E., Merika, M., and Papamatheakis, J. (1998). Involvement of CREB binding protein in expression of major histocompatibility complex class II genes via interaction with the class II transactivator. Mol. Cell. Biol. 18, 6777–6783.
Landmann, S., Muhlethaler-Mottet, A., Bernasconi, L., Suter, T., Waldburger, J. M., Masternak, K., Arrighi, J. F., Hauser, C., Fontana, A., and Reith, W. (2001). Maturation of dendritic cells is accompanied by rapid transcriptional silencing of class II transactivator (CIITA) expression. J. Exp. Med. 194, 379–391.
Louis-Plence, P., Moreno, C. S., and Boss, J. M. (1997). Formation of a regulatory factor X/X2 box-binding protein/nuclear factor-Y multiprotein complex on the conserved regulatory regions of HLA class II genes. J. Immunol. 159, 3899–3909.
Magner, W. J., Kazim, A. L., Stewart, C., Romano, M. A., Catalano, G., Grande, C., Keiser, N., Santaniello, F., and Tomasi, T. B. (2000). Activation of MHC class I, II, and CD40 gene expression by histone deacetylase inhibitors. J. Immunol. 165, 7017–7024.
Majumder, P., and Boss, J. M. (2010). CTCF controls expression and chromatin architecture of the human major histocompatibility complex class II locus. Mol. Cell. Biol. 30, 4211–4223.
Majumder, P., Gomez, J. A., and Boss, J. M. (2006). The human major histocompatibility complex class II HLA-DRB1 and HLA-DQA1 genes are separated by a CTCF-binding enhancer-blocking element. J. Biol. Chem. 281, 18435–18443.
Majumder, P., Gomez, J. A., Chadwick, B. P., and Boss, J. M. (2008). The insulator factor CTCF controls MHC class II gene expression and is required for the formation of long-distance chromatin interactions. J. Exp. Med. 205, 785–798.
Martin, B. K., Chin, K. C., Olsen, J. C., Skinner, C. A., Dey, A., Ozato, K., and Ting, J. P. (1997). Induction of MHC class I expression by the MHC class II transactivator CIITA. Immunity 6, 591–600.
Masternak, K., Barras, E., Zufferey, M., Conrad, B., Corthals, G., Aebersold, R., Sanchez, J. C., Hochstrasser, D. F., Mach, B., and Reith, W. (1998). A gene encoding a novel RFX-associated transactivator is mutated in the majority of MHC class II deficiency patients. Nat. Genet. 20, 273–277.
Masternak, K., Muhlethaler-Mottet, A., Villard, J., Zufferey, M., Steimle, V., and Reith, W. (2000). CIITA is a transcriptional coactivator that is recruited to MHC class II promoters by multiple synergistic interactions with an enhanceosome complex. Genes Dev. 14, 1156–1166.
Masternak, K., Peyraud, N., Krawczyk, M., Barras, E., and Reith, W. (2003). Chromatin remodeling and extragenic transcription at the MHC class II locus control region. Nat. Immunol. 4, 132–137.
Mehta, N. T., Truax, A. D., Boyd, N. H., and Greer, S. F. (2011). Early epigenetic events regulate the adaptive immune response gene CIITA. Epigenetics 6, 516–525.
Meissner, M., Whiteside, T. L., Van Kuik-Romein, P., Valesky, E. M., Van den Elsen, P. J., Kaufmann, R., and Seliger, B. (2008). Loss of interferon-gamma inducibility of the MHC class II antigen processing pathway in head and neck cancer: evidence for post-transcriptional as well as epigenetic regulation. Br. J. Dermatol. 158, 930–940.
Meissner, T. B., Li, A., Biswas, A., Lee, K. H., Liu, Y. J., Bayir, E., Iliopoulos, D., Van den Elsen, P. J., and Kobayashi, K. S. (2010). NLR family member NLRC5 is a transcriptional regulator of MHC class I genes. Proc. Natl. Acad. Sci. U.S.A. 107, 13794–13799.
Moreau, P., Mouillot, G., Rousseau, P., Marcou, C., Dausset, J., and Carosella, E. D. (2003). HLA-G gene repression is reversed by demethylation. Proc. Natl. Acad. Sci. U.S.A. 100, 1191–1196.
Moreno, C. S., Beresford, G. W., Louis-Plence, P., Morris, A. C., and Boss, J. M. (1999). CREB regulates MHC class II expression in a CIITA-dependent manner. Immunity 10, 143–151.
Morimoto, Y., Toyota, M., Satoh, A., Murai, M., Mita, H., Suzuki, H., Takamura, Y., Ikeda, H., Ishida, T., Sato, N., Tokino, T., and Imai, K. (2004). Inactivation of class II transactivator by DNA methylation and histone deacetylation associated with absence of HLA-DR induction by interferon-gamma in haematopoietic tumour cells. Br. J. Cancer 90, 844–852.
Morris, A. C., Beresford, G. W., Mooney, M. R., and Boss, J. M. (2002). Kinetics of a gamma interferon response: expression and assembly of CIITA promoter IV and inhibition by methylation. Mol. Cell. Biol. 22, 4781–4791.
Morris, A. C., Spangler, W. E., and Boss, J. M. (2000). Methylation of class II trans-activator promoter IV: a novel mechanism of MHC class II gene control. J. Immunol. 164, 4143–4149.
Mudhasani, R., and Fontes, J. D. (2002). The class II transactivator requires brahma-related gene 1 to activate transcription of major histocompatibility complex class II genes. Mol. Cell. Biol. 22, 5019–5026.
Muhlethaler-Mottet, A., Di Berardino, W., Otten, L. A., and Mach, B. (1998). Activation of the MHC class II transactivator CIITA by interferon-gamma requires cooperative interaction between Stat1 and USF-1. Immunity 8, 157–166.
Muhlethaler-Mottet, A., Otten, L. A., Steimle, V., and Mach, B. (1997). Expression of MHC class II molecules in different cellular and functional compartments is controlled by differential usage of multiple promoters of the transactivator CIITA. EMBO J. 16, 2851–2860.
Murphy, S. P., Holtz, R., Lewandowski, N., Tomasi, T. B., and Fuji, H. (2002). DNA alkylating agents alleviate silencing of class II transactivator gene expression in L1210 lymphoma cells. J. Immunol. 169, 3085–3093.
Murphy, S. P., and Tomasi, T. B. (1998). Absence of MHC class II antigen expression in trophoblast cells results from a lack of class II transactivator (CIITA) gene expression. Mol. Reprod. Dev. 51, 1–12.
Nagarajan, U. M., Louis-Plence, P., DeSandro, A., Nilsen, R., Bushey, A., and Boss, J. M. (1999). RFX-B is the gene responsible for the most common cause of the bare lymphocyte syndrome, an MHC class II immunodeficiency. Immunity 10, 153–162.
Ni, Z., Abou El Hassan, M., Xu, Z., Yu, T., and Bremner, R. (2008). The chromatin-remodeling enzyme BRG1 coordinates CIITA induction through many interdependent distal enhancers. Nat. Immunol. 9, 785–793.
Pattenden, S. G., Klose, R., Karaskov, E., and Bremner, R. (2002). Interferon-gamma-induced chromatin remodeling at the CIITA locus is BRG1 dependent. EMBO J. 21, 1978–1986.
Piskurich, J. F., Lin, K. I., Lin, Y., Wang, Y., Ting, J. P.-Y., and Calame, K. (2000). BLIMP-I mediates extinction of major histocompatibility class II transactivator expression in plasma cells. Nat. Immunol. 1, 526–532.
Piskurich, J. F., Linhoff, M. W., Wang, Y., and Ting, J. P.-Y. (1999). Two distinct gamma interferon-inducible promoters of the major histocompatibility complex class II transactivator gene are differentially regulated by STAT1, interferon regulatory factor 1, and transforming growth factor beta. Mol. Cell. Biol. 19, 431–440.
Piskurich, J. F., Wang, Y., Linhoff, M. W., White, L. C., and Ting, J. P.-Y. (1998). Identification of distinct regions of 5′ flanking DNA that mediate constitutive, IFN-gamma, STAT1, and TGF-beta-regulated expression of the class II transactivator gene. J. Immunol. 160, 233–240.
Raval, A., Howcroft, T. K., Weissman, J. D., Kirshner, S., Zhu, X. S., Yokoyama, K., Ting, J., and Singer, D. S. (2001). Transcriptional coactivator, CIITA, is an acetyltransferase that bypasses a promoter requirement for TAF(II)250. Mol. Cell 7, 105–115.
Reith, W., and Mach, B. (2001). The bare lymphocyte syndrome and the regulation of MHC expression. Annu. Rev. Immunol. 19, 331–373.
Satoh, A., Toyota, M., Ikeda, H., Morimoto, Y., Akino, K., Mita, H., Suzuki, H., Sasaki, Y., Kanaseki, T., Takamura, Y., Soejima, H., Urano, T., Yanagihara, K., Endo, T., Hinoda, Y., Fujita, M., Hosokawa, M., Sato, N., Tokino, T., and Imai, K. (2004). Epigenetic inactivation of class II transactivator (CIITA) is associated with the absence of interferon-gamma-induced HLA-DR expression in colorectal and gastric cancer cells. Oncogene 23, 8876–8886.
Schlesinger, Y., Straussman, R., Keshet, I., Farkash, S., Hecht, M., Zimmerman, J., Eden, E., Yakhini, Z., Ben-Shushan, E., Reubinoff, B. E., Bergman, Y., Simon, I., and Cedar, H. (2007). Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat. Genet. 39, 232–236.
Silacci, P., Mottet, A., Steimle, V., Reith, W., and Mach, B. (1994). Developmental extinction of major histocompatibility complex class II gene expression in plasmocytes is mediated by silencing of the transactivator gene CIITA. J. Exp. Med. 180, 1329–1336.
Smith, M. A., Wright, G., Wu, J., Tailor, P., Ozato, K., Chen, X., Wei, S., Piskurich, J. F., Ting, J. P.-Y., and Wright, K. L. (2011). Positive regulatory domain I (PRDM1) and IRF8/PU.1 counter-regulate MHC class II transactivator (CIITA) expression during dendritic cell maturation. J. Biol. Chem. 286, 7893–7904.
Spilianakis, C., Papamatheakis, J., and Kretsovali, A. (2000). Acetylation by PCAF enhances CIITA nuclear accumulation and transactivation of major histocompatibility complex class II genes. Mol. Cell. Biol. 20, 8489–8498.
Steimle, V., Durand, B., Barras, E., Zufferey, M., Hadam, M. R., Mach, B., and Reith, W. (1995). A novel DNA-binding regulatory factor is mutated in primary MHC class II deficiency (bare lymphocyte syndrome). Genes Dev. 9, 1021–1032.
Steimle, V., Otten, L. A., Zufferey, M., and Mach, B. (1993). Complementation cloning of an MHC class II transactivator mutated in hereditary MHC class II deficiency (or bare lymphocyte syndrome). Cell 75, 135–146.
Ting, J. P.-Y., and Trowsdale, J. (2002). Genetic control of MHC class II expression. Cell 109, S21–S33.
Ting, J. P.-Y., Willingham, S. B., and Bergstralh, D. T. (2008). NLRs at the intersection of cell death and immunity. Nat. Rev. Immunol. 8, 372–379.
Truax, A. D., Koues, O. I., Mentel, M. K., and Greer, S. F. (2010). The 19S ATPase S6a (S6′/TBP1) regulates the transcription initiation of class II transactivator. J. Mol. Biol. 395, 254–269.
Van den Elsen, P. J., Holling, T. M., Kuipers, H. F., and Van der Stoep, N. (2004). Transcriptional regulation of antigen presentation. Curr. Opin. Immunol. 16, 67–75.
Van den Elsen, P. J., Holling, T. M., Van der Stoep, N., and Boss, J. M. (2003). DNA methylation and expression of major histocompatibility complex class I and class II transactivator genes in human developmental tumor cells and in T cell malignancies. Clin. Immunol. 109, 46–52.
Van den Elsen, P. J., Van der Stoep, N., Viëtor, H. E., Wilson, L., Van Zutphen, M., and Gobin, S. J. P. (2000). Lack of CIITA expression is central to the absence of antigen presentation functions of trophoblast cells and is caused by methylation of the IFN-gamma inducible promoter (PIV) of CIITA. Hum. Immunol. 61, 850–862.
Van der Stoep, N., Quinten, E., Alblas, G., Plancke, A., Van Eggermond, M. C., Holling, T. M., and Van den Elsen, P. J. (2007). Constitutive and IFNgamma-induced activation of MHC2TA promoter type III in human melanoma cell lines is governed by separate regulatory elements within the PIII upstream regulatory region. Mol. Immunol. 44, 2036–2046.
Van der Stoep, N., Quinten, E., Marcondes Rezende, M., and Van den Elsen, P. J. (2004). E47, IRF-4, and PU.1 synergize to induce B-cell-specific activation of the class II transactivator promoter III (CIITA-PIII). Blood 104, 2849–2857.
Van der Stoep, N., Quinten, E., and Van den Elsen, P. J. (2002a). Transcriptional regulation of the MHC class II trans-activator (CIITA) promoter III: identification of a novel regulatory region in the 5′-untranslated region and an important role for cAMP-responsive element binding protein 1 and activating transcription factor-1 in CIITA-promoter III transcriptional activation in B lymphocytes. J. Immunol. 169, 5061–5071.
Van der Stoep, N., Biesta, P., Quinten, E., and Van den Elsen, P. J. (2002b). Lack of IFN-gamma-mediated induction of the class II transactivator (CIITA) through promoter methylation is predominantly found in developmental tumor cell lines. Int. J. Cancer 97, 501–507.
Van Eggermond, M. C., Boom, D. R., Klous, P., Schooten, E., Marquez, V. E., Wierda, R. J., Holling, T. M., and Van den Elsen, P. J. (2011). Epigenetic regulation of CIITA expression in human T-cells. Biochem. Pharmacol. PMID: 21664896. [Epub ahead of print] .
Wong, A. W., Ghosh, N., McKinnon, K. P., Reed, W., Piskurich, J. F., Wright, K. L., and Ting, J. P.-Y. (2002). Regulation and specificity of MHC2TA promoter usage in human primary T lymphocytes and cell line. J. Immunol. 169, 3112–3119.
Wright, K. L., and Ting, J. P.-Y. (2006). Epigenetic regulation of MHC-II and CIITA genes. Trends Immunol. 27, 405–412.
Yazawa, T., Kamma, H., Fujiwara, M., Matsui, M., Horiguchi, H., Satoh, H., Fujimoto, M., Yokoyama, K., and Ogata, T. (1999). Lack of class II transactivator causes severe deficiency of HLA-DR expression in small cell lung cancer. J. Pathol. 187, 191–199.
Yu, J., Angelin-Duclos, C., Greenwood, J., Liao, J., and Calame, K. (2000). Transcriptional repression by blimp-1 (PRDI-BF1) involves recruitment of histone deacetylase. Mol. Cell. Biol. 20, 2592–2603.
Zhu, X. S., Linhoff, M. W., Li, G., Chin, K. C., Maity, S. N., and Ting, J. P.-Y. (2000). Transcriptional scaffold: CIITA interacts with NF-Y, RFX, and CREB to cause stereospecific regulation of the class II major histocompatibility complex promoter. Mol. Cell. Biol. 20, 6051–6061.
Zika, E., Fauquier, L., Vandel, L., and Ting, J. P.-Y. (2005). Interplay among coactivator-associated arginine methyltransferase 1, CBP, and CIITA in IFN-gamma-inducible MHC-II gene expression. Proc. Natl. Acad. Sci. U.S.A. 102, 16321–16326.
Zika, E., Greer, S. F., Zhu, X. S., and Ting, J. P.-Y. (2003). Histone deacetylase 1/mSin3A disrupts gamma interferon-induced CIITA function and major histocompatibility complex class II enhanceosome formation. Mol. Cell. Biol. 23, 3091–3102.
Keywords: MHC-I, MHC-II, CIITA, transcription, epigenetics
Citation: van den Elsen PJ (2011) Expression regulation of major histocompatibility complex class I and class II encoding genes. Front. Immun. 2:48. doi: 10.3389/fimmu.2011.00048
Received: 31 August 2011;
Paper pending published: 13 September 2011;
Accepted: 14 September 2011;
Published online: 04 October 2011.
Edited by:
Johan van der Vlag, Radboud University Nijmegen Medical Centre, NetherlandsReviewed by:
Johan van der Vlag, Radboud University Nijmegen Medical Centre, NetherlandsJurgen Dieker, Nijmegen Center for Molecular Life Sciences, Netherlands
Copyright: © 2011 van den Elsen. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Peter J. van den Elsen, Department of Immunohematology and Blood Transfusion, Leiden University Medical Center, Building 1, E3-Q, Albinusdreef 2, 2333 ZA Leiden, Netherlands. e-mail:cGp2ZGVsc2VuQGx1bWMubmw=