 Arnaud M. Labrousse1,2
Etienne Meunier1,2†
Julien Record1,2†
Anna Labernadie1,2,3,4
Amèlie Beduer3,4
Christophe Vieu3,4
Thouraya Ben Safta1,2
Arnaud M. Labrousse1,2
Etienne Meunier1,2†
Julien Record1,2†
Anna Labernadie1,2,3,4
Amèlie Beduer3,4
Christophe Vieu3,4
Thouraya Ben Safta1,2 Isabelle Maridonneau-Parini1,2*
Isabelle Maridonneau-Parini1,2*- 1 UMR5089, CNRS, Institut de Pharmacologie et de Biologie Structurale, Toulouse, France
- 2 Université de Toulouse, Université Paul Sabatier, Toulouse, France
- 3 CNRS; LAAS; 7 avenue du colonel Roche, F-31077 Toulouse, France
- 4 Université de Toulouse; UPS, INSA, INP, ISAE; UT1, UTM, LAAS; F-31077 Toulouse, France
Lysosome mobilization is a key cellular process in phagocytes for bactericidal activities and trans-matrix migration. The molecular mechanisms that regulate lysosome mobilization are still poorly known. Lysosomes are hard to track as they move toward phagosomes throughout the cell volume. In order to anticipate cell regions where lysosomes are recruited to, human and RAW264.7 macrophages were seeded on surfaces that were micro-patterned with immune complexes (ICs) as 4 μm-side squares. Distances between IC patterns were adapted to optimize cell spreading in order to constrain lysosome movements mostly in two dimensions. FcΓ receptors triggered local frustrated phagocytosis, frustrated phagosomes appeared as rings of F-actin dots around the IC patterns as early as 5 min after cells made contact with the substratum. Frustrated phagosomes recruited actin-associated proteins (vinculin, paxillin, and gelsolin). The fusion of lysosomes with frustrated phagosomes was shown by the release of beta-hexosaminidase and the recruitment of Lamp1 to frustrated phagosomes. Lysosomes of RAW264.7 macrophages were labeled with cathepsin-D-mCherry to visualize their movements toward frustrated phagosomes. Lysosomes saltatory movements were markedly slowed down compared to cells layered on non-opsonized patterns. In addition, the linearity of the trajectories and the frequency and duration of contacts of lysosomes with frustrated phagosomes were measured. Our experimental set-up is the first step toward deciphering molecular mechanisms which are involved in lysosome movements in the cytoplasm (speed, directionality, and interaction with phagosomes), and opens the door to approaches such as RNA interference, pharmacological inhibition, or mutant expression.
Introduction
Secretion of lysosomes and related granules is a cellular process that is essential for the bactericidal functions of specialized immune cells such as phagocytes (see Luzio et al., 2007 for review). In macrophages, these secretory lysosomes constitute a population of vesicles which is different from the classical ubiquitous lysosomes dedicated to the digestion of nutriments (Rabinowitz et al., 1992; Claus et al., 1998; Astarie-Dequeker et al., 2002). Lysosomes are also at least in part associated with the biogenesis of podosomes that are F-actin rich structures responsible for adhering to and degrading the extracellular environment of phagocytes. Lysosome mobilization is therefore a crucial process for phagocyte migration and bactericidal function, but the molecular mechanisms that control this process remain unclear. It was shown that lysosomes and lysosome-related organelles travel over long distances along microtubules within the cell cytoplasm. This movement mainly entails kinesin motors, with a switch to actin rails when lysosomes reach the cell periphery (Barral and Seabra, 2004). However, detailed characterization of lysosome movements during phagocytosis has not yet been carried out, probably because of the technical challenge of tracking vesicles in three dimensions within the cytoplasm, where phagosomes themselves move around. Here, in order to (1) anticipate where lysosomes are recruited to, and (2) spread cells out as much as possible to neglect the third dimension during vesicle tracking, we set-up frustrated phagocytosis on micro-patterned immune complex surfaces. Frustrated phagocytosis has already been used in the past in immobilized phagocytosing macrophages (Wright and Silverstein, 1984; Takemura et al., 1986; Bainton et al., 1989). More recently, Eng et al. (2007) were able to demonstrate and measure the reorientation of the Golgi apparatus during frustrated phagocytosis in live RAW264.7 cells. In a more sophisticated experimental set-up, patterned antigen arrays were used to study exocytosis in mast cells in response to local activation of the IgE receptor (Wu et al., 2007). In that study, the authors used total internal reflection fluorescence microscopy (TIRF) to differentiate the location of exocytosis of early endosomes and lysosomes in response to local activation of IgE receptors. TIRF and frustrated phagocytosis have been combined (Touret et al., 2005) to visualize whether the endoplasmic reticulum fuses with the plasma membrane in response to the activation of phagocytic receptors. None of the above studies, however, explored the regulation of the mobilization of lysosomes during their upstream movement through the cell cytoplasm, toward their fusion with the target membrane. Here, for the first time, we have been able to follow and characterize lysosome movements during phagocytosis in live macrophages.
Materials and Methods
Products and Antibodies
Ovalbumin, Nocodazole, Cytochalasin-D1, and Latrunculin-A, antibodies against gelsolin (1/100), paxillin (1/100) and vinculin (1/100) were purchased from Sigma-Aldrich (France). Lysotracker Red, phalloidin-Alexa488 and -Alexa350 were from Molecular Probes (Fisher Scientific, France) and used at 50 nM and a 1/500 dilution, respectively. Anti-CD16/CD32 monoclonal antibodies were from Immunostep (clone 2.4G2, Caltag medsystems, UK). Rabbit anti-ovalbumin serum was home-made by classical intradermal immunization and used at a 1/10 dilution to generate immune complexes (ICs). Bovine serum albumin (BSA, fraction V) was from Euromedex (France).
Cell Culture
RAW264.7 stably expressing actin-GFP (RAW–GFP) were kindly provided by S. Grinstein (Canada). They were cultured as described (Verollet et al., 2010). RAW–GFP cells were transfected with 2 μg of the cathepsin-D-mCherry construct kindly provided by F. Darchen (Paris) and with the Amaxa electroporator apparatus according to the manufacturer’s indications. Human monocyte-derived macrophages (hMDM) were prepared from healthy donors and cultured as described (Van Goethem et al., 2010). Bone-marrow derived macrophages (BMDM) were prepared from wild-type C57/bl6 mice and cultured as described (Cougoule et al., 2005).
Micro-contact Printing and Opsonization of the Substrata
PDMS stamps patterned with squares of 4 μm edges spaced by 5 μm were prepared as described (Labernadie et al., 2010). Stamps were incubated for 20 min at room temperature (RT) in a phosphate-buffer saline (PBS) solution containing 50 μg/mL of either unlabeled ovalbumin or a 8:2 mixture of unlabeled ovalbumin and ovalbumin-TRITC (OVA-TRITC), or human IgG, as indicated in the text. Stamps were then dried under a nitrogen flow and brought into contact with glass cover slips for 1 min. These cover slips had been pre-cleaned in HCl/methanol solution 1/2 vol/vol for 2 h, then rinsed for 1 h in distilled water, for 30 min in ethanol (75% vol), and dried under airflow and sterilized. For studies of fixed samples, patterned cover slips of 12-mm diameter were placed at the bottom of 24-wells plates (VWR, France) and incubated with cell culture medium for 30 min at 37°C and 5% CO2 before adding the cells. For live cell imaging, we patterned 24-mm cover slips that had been glued to the bottom of pre-drilled plastic Petri dishes (3-cm diameter). Alternatively, two-chamber Labtek-II (Nunc, Fisher Scientific, France) were micro-patterned. Ovalbumin-patterned cover slips were then opsonized with home-made anti-ovalbumin sera from rabbit at a 1/10 dilution for 30 min at RT, leading to patterned ICs. IgG-patterned cover slips were used directly. Cells were trypsinized at 37°C, washed in PBS and directly added to the prepared cover slips, either for later fixation and immunofluorescence studies or for live imaging.
Frustrated Phagocytosis
RAW cells or hMDM were trypsinized, washed in PBS and layered on micro-patterned cover slips at a density of 5 × 104 cells/mL or 104 cells/mL, respectively. For blocking experiments, RAW cells were treated with the anti-CD16/CD32 antibodies at a 1/50 dilution for 1 h at RT, before being washed in PBS and layered on the micro-patterned surfaces.
Immunofluorescence
Cells were fixed for 45 min at RT with 3.7% paraformaldehyde in PBS containing 15 mM sucrose. Unreacted aldehyde functions were quenched with 50 mM NH4Cl in PBS for 2 min at RT. Cells were then permeabilized with PBS containing 0.3% TX100 for 10 min at RT before being saturated with PBS containing 5% BSA for 1 h at RT. Cells were then stained with primary antibodies at indicated dilutions (see above) in the presence of 5% BSA (PBS/BSA) for 1 h at RT, washed three times in PBS/BSA and incubated with a mixture of the corresponding secondary antibodies and fluorescent phalloidin for 30 min at RT in the dark. Samples were then mounted on slides with the fluorescent mounting medium from DAKO (France). Immunofluorescent samples were observed with a DM-RB up-right microscope (Leica Microsystems, Paris, France) equipped with a CoolSnap HQ camera (Roper Scientific, France) and the appropriate FITC or TRITC filter cubes.
Atomic Force Microscopy
Height measurements and profiles of micro-patterned ovalbumin alone or ICs were obtained by atomic force microscopy (AFM) in contact mode in liquid at RT. See (Labernadie et al., 2010) for technical details.
Live Cell Imaging and Image Processing
Cells performing frustrated phagocytosis on micro-patterned surfaces were imaged with a DM-IRB inverted microscope equipped with a 63× objective (n.a. = 1.3), a heated stage and a chamber to maintain temperature and CO2 at constant values. Images of CathD-mCherry- or LysoTracker Red-positive lysosomes were acquired every 250 ms and images of actin-GFP were acquired every 10 s. Actin images (green) were used to delineate frustrated phagosome areas on the lysosomes images (red). Distances were calibrated using a graduated slide (1 pixel corresponds to 0.102 μm) and lysosome movements were tracked with the manual tracking plug in imbedded in the Image J software version 1.44F. Instant velocities were calculated between two consecutive frames. Linearity of trajectories was measured as the correlation coefficient (r2) of the linear regression of sets of three consecutive positions, sliding along individual tracks.
Statistics
A t test or one-way ANOVA analysis with Tukey’s multiple comparisons posttest was performed using GraphPad Prism. Statistical significance is indicated as follows: ***P < 0.001;**P < 0.01; *P < 0.05.
Results
Preparation of the Substrata
Glass cover slips were micro-texturized with ovalbumin (OVA) or OVA-TRITC by micro-contact printing as described recently, and outlined briefly in the Section “Materials and Methods” (Labernadie et al., 2010). Patterns were then opsonized with anti-ovalbumin rabbit serum. We used AFM to characterize the OVA-TRITC patterns, and found out that they have an average height of 7 nm, whereas height was about 14 nm when anti-OVA IgGs were added (Figures 1A,C). Their shape was very regular with very sharp and vertical edges (Figure 1B). As anticipated from the AFM results, fluorescence microscopy confirmed that anti-ovalbumin IgGs did not bind outside the OVA patterns (Figure 1D).
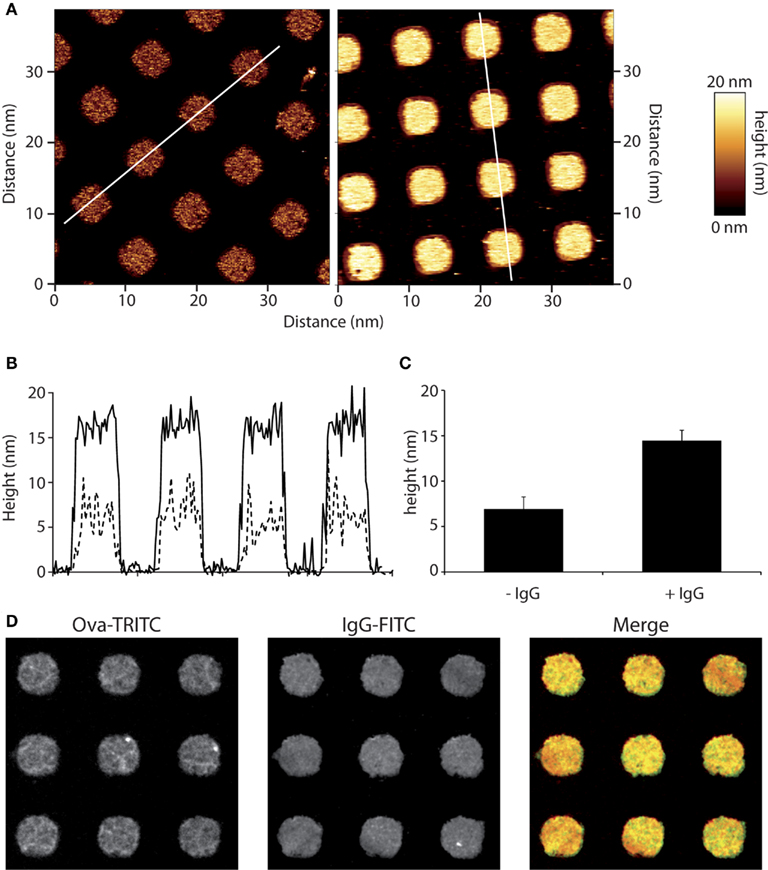
Figure 1. Micro-contact printing of immune complexes. Micro-patterns of ovalbumin (OVA) were applied to glass cover slips and those were then incubated with anti-OVA antibodies. (A) Micro-patterned surfaces were scanned by AFM. Squares of 4 μm-sides are separated by 5 μm. Heights were measured as detailed in the Section “Materials and Methods” for OVA alone (left) and OVA-IgG (right) patterns.” (B) Profiles of OVA patterns (dotted line) are compared to that of immune complexes patterns (solid line). (C) Heights are measured for more than 50 patterns and mean ± SD are plotted. (D) Immune-detection of micro-patterned ovalbumin-TRITC using an anti-OVA primary serum and Alexa488-coupled secondary antibodies revealed a homogenous repartition of immune complexes on micro-patterned surfaces.
Frustrated Phagocytosis on Opsonized Ovalbumin Patterns
When RAW264.7 cells were layered on IgG-ovalbumin patterns, actin rearranged rapidly as dots mainly present around the patterns (Figure 2A). As early as 5 min after cell suspension was added to the micro-texturized cover slips, 50% of cells presented such actin rearrangements, and this number rose up to 83% after 30 min of adhesion. Formation of frustrated phagosomes was dependent on the FcΓ receptors since it was not observed in the absence of opsonization (Figure 2B) or when macrophages were pre-incubated withanti-CD16/CD32 blocking antibodies (Figure A1 in Appendix). In our experimental conditions, about 85% of the patterns that were covered by a cell triggered a frustrated phagosome (not shown). Frustrated phagocytosis was also obtained with human macrophages derived from monocytes (Figure 2C) and mouse BMDM (see Figure 5). These larger cells could form up to 30 frustrated phagosomes with the actin rings sometimes surrounding several patterns (Figure 2C). As expected, biogenesis of frustrated phagosomes was dependent on actin dynamics since it was completely abolished when cells where pre-treated for 15 min with 0.5 μM Latrunculin A or 10 μM Cytochalasin-D1 (data not shown). Their stability was also dependent on actin polymerization since frustrated phagosomes disassembled after a 10-min treatment with either drug (not shown).
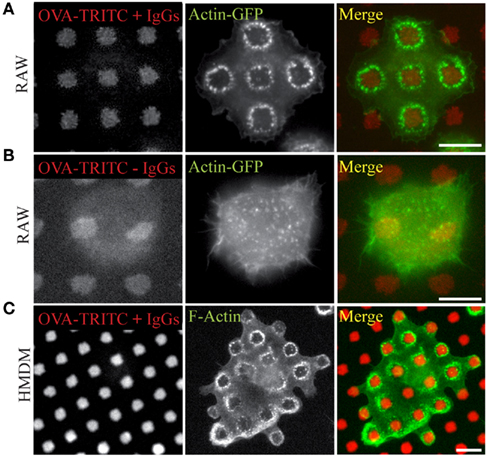
Figure 2. Frustrated phagocytosis of macrophages on micro-texturized surfaces. Micro-patterns of OVA-TRITC were laid on glass cover slips as in Figure 1, and then incubated (A,C) or not (B) with anti-OVA antibodies. RAW–GFP (A,B) or hMDMs (C) were layered on the substrata for 15 min, then fixed and stained (C) with phalloidin-FITC and imaged. OVA-TRITC and actin-GFP (A,B). Bars = 5 μm.
In order to observe frustrated phagocytosis in live cells, we used RAW264.7 cells that constitutively express actin-GFP, hereafter designated as RAW–GFP. The process could be divided into three phases. First, actin concentrated as dots at the point of contact of the cells with the substratum; second, actin dots moved rapidly to the cells’ periphery like a growing belt as the cells spread on the cover-slip, and third, dots rearranged into rings around patterns when the cells were completely spread out (see Movie S1 in Supplementary Material). These actin rings could last as long as 10 min, but could also disassemble as cells moved to another position. Sometimes, one could observe a dynamic exchange of actin between two rings (see Movie S2 in Supplementary Material). Whatever the behavior of the ring was, each actin dot proved to be very dynamic by itself, with cycles of appearance/disappearance of about 2 min 40 s (not shown).
Characterization of the Frustrated Phagosomes
We further went on to characterize the rings of actin dots by staining cells undergoing frustrated phagocytosis for several actin-linked proteins that are classically found at phagosomes. Figure 3 shows that vinculin, paxillin, and gelsolin were present at the actin rings. Paxillin described a clear outline of the rings, whereas vinculin was found around each dot of actin and gelsolin colocalized with actin.
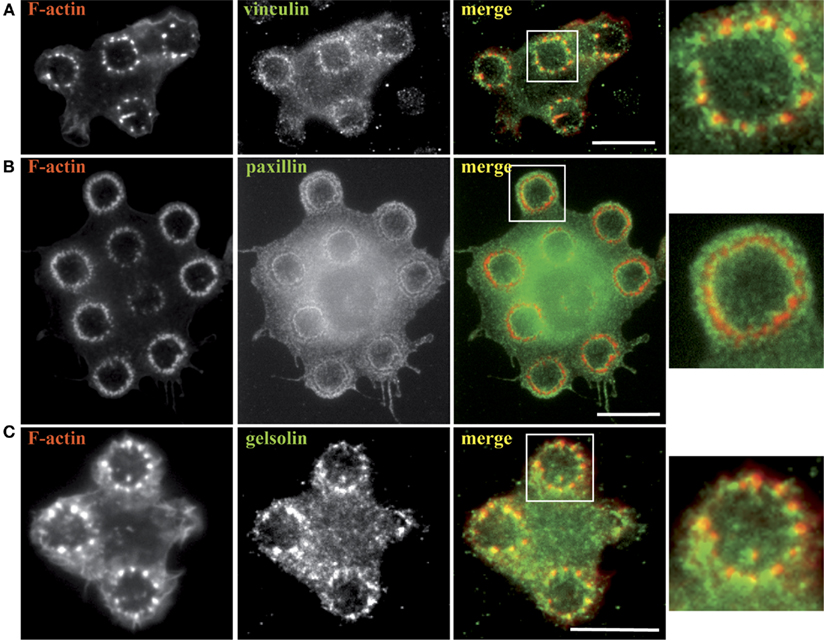
Figure 3. Actin-associated proteins are recruited to the frustrated phagosomes. RAW–GFP (A,C) or hMDM (B) were plated on micro-patterned immune complexes for 15 min, fixed and stained for F-actin (B), vinculin (A), paxillin (B), and gelsolin (C). Actin in RAW cells was visualized thanks to GFP fluorescence (A,C). Zoomed images correspond to the areas indicated by white boxes in the corresponding merged images. Bars = 5 μm.
Since we wanted to use this model to measure lysosome mobilization in live cells, we first looked for evidence that lysosomes fused with the frustrated phagosomes. As a first indirect approach, we measured the release of the lysosomal enzyme beta-hexosaminidase in the extracellular environment. We observed that RAW264.7 macrophages increased their secretion activity by 25.8 ± 4.4% (mean ± SD) when they were layered on IgG-opsonized patterns for 1 h, as compared to non-opsonized patterns (not shown, two independent experiments with measurements in triplicates). However, this protocol did not tell us whether secretion was occurring at the frustrated phagosomes or not. If lysosomes fuse with the frustrated phagosome, then we should detect the accumulation of lamp1 at the level of the patterns. Figure 4 shows that Lamp1 was indeed recruited to the membrane of the frustrated phagosome as early as 10 min after cells spread on the micro-patterned ICs.
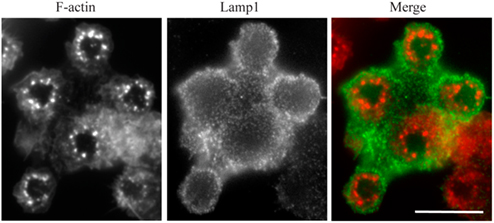
Figure 4. Lamp1 is recruited to the frustrated phagosomes. RAW–GFP cells were added to micro-patterned immune complexes for 10 min before being fixed and stained for Lamp1 (red) without cell permeabilization. Actin was visualized thanks to GFP fluorescence (green). Bar = 5 μm.
It has been described that the integrity of the microtubule network was dispensable for the biogenesis of FcΓR-triggered phagosomes (Newman et al., 1991) and we wanted to check whether this holds true for frustrated phagosomes. Mouse bone-marrow derived macrophages were treated with nocodazole either pre- or post-adhesion on patterned ICs, and we observed that neither of these treatments had an effect on the formation or the stability of frustrated phagosomes (Figure 5).
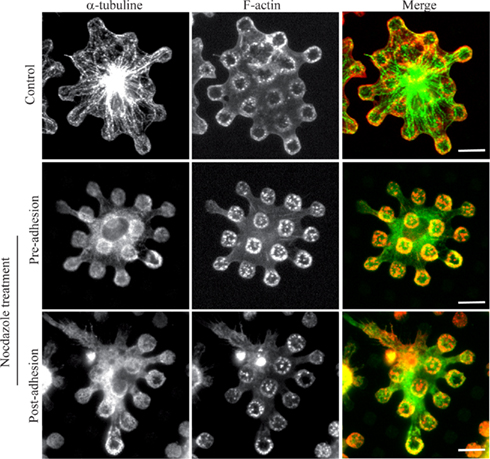
Figure 5. Microtubules are dispensable for biogenesis of frustrated phagosomes and their stability. Bone-marrow derived macrophages from wild-type mice were treated or not (Control) with 10 μM nocodazole either before (pre-adhesion) or after (post-adhesion) being layered on micro-patterned immune complexes. Cells were then fixed, permeabilized, and stained for F-actin and α-tubulin. Bars = 5 μm.
Tracking Lysosomes in Live Macrophages
The advantages of this experimental set-up are (1) to have all the phagocytosis-related signaling pathways initiated at the ventral side of the cell; (2) to anticipate where lysosomes get recruited to fuse with the phagosomal membrane; (3) to have the cells as spread out as possible in order to minimize the third dimension during characterization of the vesicle movements. RAW cells spread reasonably well on 5 μm-spaced patterns. However, they would form frustrated phagosomes that were too close to each other to unambiguously evaluate the direction of moving lysosomes. Thus, we looked for the appropriate distance between patterns to obtain cells that would be sufficiently spread out to form frustrated phagosomes which are sufficiently distant from each other. We compared our initial 5 μm-spaced patterns to patterns with spaces of 7, 9, 15, or 20 μm (not shown). We found out that a distance of 7 μm between two frustrated phagosomes was not enough to track lysosome efficiently, and RAW cells layered on 15 μm-spaced patterns did not form more than one frustrated phagosome (not shown). When the distance between patterns was 9 μm, about 25% of the cells formed two to four frustrated phagosomes and lysosome tracking was feasible (not shown). Thus, 9 μm was a good compromise and we carried out all lysosome tracking with such a pattern.
For the following experiments, we used RAW–GFP macrophages either stably expressing the lysosomal protease Cathepsin-D (CathD)-mCherry or stained with LysoTracker Red®. Since the same results were obtained with both methods, only the results with CathD-mCherry are shown. Lysosomes are numerous in macrophages and even in cells that are extensively spread out on the substratum, we found that we could not use automated tracking software (see Movie S3 in Supplementary Material). Thus we decided to track vesicles manually, using the “manual tracking” PlugIn of the ImageJ software, after calibration of the images to have the pixel-to-μm correspondence. The actin/lysosome double staining allowed us to visualize frustrated phagosomes and lysosomes at the same time, hence to sort lysosomes moving “toward” a frustrated phagosome from those moving “outward” or “on” a frustrated phagosome, i.e., inside a ring of actin dots. Lysosomes with no clear destination over the time span of the experiment were called “Off” (Figure 6A and see also Movie S3 in Supplementary Material). Only lysosomes that could be individually tracked over a minimum of four frames, which corresponds to 1 s, were considered. Most of the time, tracking was stopped because lysosomes encountered an area of the cell over-crowded with lysosomes and could not be distinguished as an individual vesicle anymore. Very rarely, we had to stop tracking because the vesicle went out of focus. This observation comforted us into our decision to neglect the Z dimension in our measurements. As a control, we tracked lysosomes of resting macrophages that were layered on glass, in the absence of IgGs. We specifically chose “control” cells that were as spread as possible to be able to track lysosomes.
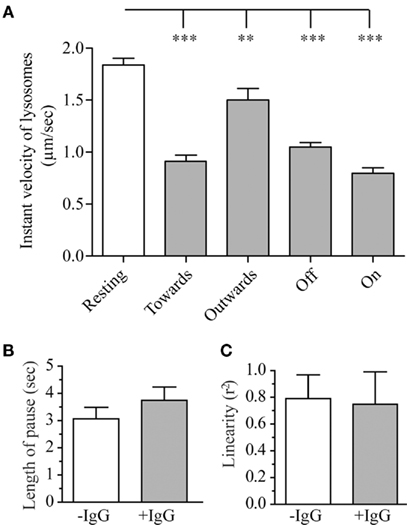
Figure 6. Parameters of the lysosomal movement during frustrated phagocytosis and roles of Src kinases. RAW–GFP expressing cathepsin-D-mCherry were layered on glass cover slips that were micro-patterned with OVA alone (−IgG, “Resting”) or OVA + anti-OVA antibodies (+IgG). Tracking of lysosomes was carried out as described in the Section “Materials and Methods.” (A) Instant velocity of lysosomes as a function of their direction relative to the position of frustrated phagosomes (see text for details). (B) Length of the pauses of lysosomes during their saltatory movements. (C) Linearity of the trajectories for lysosomes moving toward a frustrated phagosome. For all graphs, 15–30 lysosomes were measured on three to six different cells per conditions; mean ± SEM.
Activation of Phagocytosis Receptors Decreases Lysosome Mobility
As shown on Figure 6A, lysosomes in resting macrophages had an instant speed of about 1.8 μm/s. Surprisingly when macrophages were performing frustrated phagocytosis, lysosomes velocity was significantly lower than in resting macrophages (Figure 6A). This slowing down was observed for all lysosomes, but, lysosomes moving away from a frustrated phagosome were found to be less affected than lysosomes moving toward, on or off a frustrated phagosome. The duration of pauses during lysosomes saltatory movements (see Movie S3 in Supplementary Material) was slightly longer during frustrated phagocytosis but this difference remained below statistical significance (Figure 6B), and no difference was detected in the linearity of the trajectories (Figure 6C). This parameter is shown for lysosomes moving toward a frustrated phagosome but no difference was observed for lysosomes moving outward either (not shown). We showed that the integrity of the microtubule network was required for lysosome movement since no movement at all could be tracked when cells were treated with 10 μM nocodazole after they had formed frustrated phagosomes (not shown). We were also able to measure the frequency of lysosome arrival in the vicinity of a frustrated phagosome, and observed that, under our experimental conditions, 3.7 ± 0.5 lysosomes per minute (mean ± SEM) come into contact with the same frustrated phagosome. We did not look at the fusion process itself, but we could measure the length of time that a lysosome stays in contact with a frustrated phagosome. This parameter was called “interaction” and was measured to be 7.9 ± 0.8 s (mean ± SEM).
Discussion
In this study, we describe a new experimental set-up to measure several parameters of lysosome movements in macrophages. When layered on micro-patterned ICs, macrophages spread, and formed frustrated phagosomes. As a consequence, we were able to anticipate where lysosomes were recruited to, to image them in live cells and measure their speed, saltatory movements, directionality, and interaction with frustrated phagosomes.
A frustrated phagosome was defined as a ring of actin dots surrounding an immune complex pattern. Previous experiments involving frustrated phagocytosis had been designed on glass supports uniformly coated with ICs. Under these conditions, macrophages adhere very strongly to the surface, forming a tight sealing zone that separates the area underneath the cell from the rest of the medium, where secreted material is confined (Wright and Silverstein, 1984; Heiple et al., 1990), much like the sealing zone described for the macrophage-derived osteoclasts (Saltel et al., 2008). In our present study, we stained F-actin and noticed that frustrated phagosomes were delineated by F-actin dots triggered by activation of FcΓ receptors. The actin cores positioned just outside the IgG patterns and the space that was often visible between the dots and the pattern underneath may represent the scaffold of actin-linked proteins that are necessary to maintain the structure. Vinculin and paxillin did not co-localize with F-actin cores but displayed the same organization as in podosomes (Linder and Aepfelbacher, 2003; Van Goethem et al., 2011). The reason why actin rearranged as individual dots around the IgG patterns remains to be clarified. Under conditions of frustrated phagocytosis, actin polymerization is unable to form pseudopodia which normally appear in three dimensions around IgG-coated particles. Thus the actin dots could constitute “stable vestiges” of the protrusive machinery of frustrated pseudopodia.
We chose to work with the macrophage cell line RAW264.7 for its amenability to molecular manipulations. Our experimental procedure can also be applied to hMDMs and BMDMs. However, when we tried to label lysosomes of hMDMs with LysoTracker, most of the labeled compartments were tubular and interconnected, as previously described (Knapp and Swanson, 1990; not shown), and tracking lysosomes in such cells was not as easy as in RAW264.7 cells.
The optimization of the micro-pattern geometry for RAW264.7 cells led us to use squares that were spaced by 9 μm for optimal imaging of lysosomes. With this set-up, we could characterize for the first time the movement of lysosomes in phagocytic macrophages by anticipating where lysosomes are recruited to and by tracking lysosomes almost exclusively in two dimensions as cells spread strongly on patterned ICs and became very flat. The instant speed of lysosomes in macrophages adhering on glass was about 2 μm/s. We do not know of any previously published data documenting lysosome speed in macrophages. However, it appeared to be about five times greater than Hck-positive lysosomes in NIH-3T3 fibroblasts (Vincent et al., 2007) and twice as fast as the most rapid Lamp1-positive lysosomes in immortalized skin fibroblasts (Falcon-Perez et al., 2005). This, together with the observation that lysosomes are slowing down in macrophages that formed frustrated phagosomes thus suggests that lysosomes may be moving through some active, microtubule-dependent mechanism in the cytoplasm of non-phagocytic macrophages, and that these movements could then become hindered by the lysosomes becoming tethered to the cytoskeleton upon activation of phagocytosis. The decrease in the velocity of lysosomes that we observed during the frustrated phagocytosis process may be a feature of polarized secretion. We will modify our set-up to micro-texturize cover slips with proteins that induce the formation of podosomes (Labernadie et al., 2010), we will see whether lysosomes addressed to podosomes move also slowlier, when compared to lysosomes that are not targeted to podosomes.
Lysosome movements in macrophages were found to be dependent on intact microtubules, in agreement with previous studies (Astarie-Dequeker et al., 2002; Harrison et al., 2003; Huynh et al., 2007). The saltatory property of lysosome movement is clearly the same in resting and phagocytic macrophages. Pauses during movement have been described in vitro as time that vesicles spend at across-road of two microtubules or a microtubule and a microfilament before “choosing” to go on the same track or to switch track (Ross et al., 2008; Schroeder et al., 2010). However, such mechanisms have not been shown to exist in live cells, as yet.
Frustrated phagosomes being easily identified by the ring of actin dots, two additional parameters were measured: the frequency and the duration of lysosome/phagosome interactions. We called “interaction” the presence of a motionless lysosome in the vicinity of a frustrated phagosome, i.e., detected either inside the ring of actin dots or apparently touching the ring. Interaction of lysosomes with the frustrated phagosome lasted about 8 s, which is very close to the duration of the interaction of MT1-MMP-positive vesicles with podosomes actin rich structures where MT1-MMP is supposed to be delivered (Wiesner et al., 2010). Most of the time, interactions between lysosomes and phagosomes were transient and the same incoming lysosome was observed moving away from the phagosome. Although we could detect lysosomal enzymes in the extracellular medium, whether lysosomes delivered part of their content during the docking time in a kiss-and-run process (Desjardins, 1995) is not clear yet. Sometimes, cathepsin-D-positive lysosomes disappeared when the vesicles interacted with frustrated phagosomes, as if fusion occurred. In order to visualize the kiss-and-run and the fusion processes properly, we plan to use TIRF microscopy in future experiments, which should bring significant improvement to our current experimental set-up. This should also provide an answer to the questions raised by others as to whether lysosomes need to interact with actin structures before fusion with the phagosomes (Kjeken et al., 2004; Liebl and Griffiths, 2009).
The molecular mechanisms which control the movements and fusion of lysosomes in response to IgG receptor activation is of particular interest in the context of infections with pathogens which target these mechanisms to survive in the host (see Kumar and Valdivia, 2009 for review). Moreover, lysosomes are also secreted in a spatially controlled manner in different contexts than IgG-mediated phagocytosis. They fuse to the plasma membrane to repair injuries induced by migration on glass supports (Reddy et al., 2001); they fuse at podosomes to release their lytic content and degrade the extracellular matrix (Cougoule et al., 2005, 2010; Tu et al., 2008; Linder et al., 2010); and polarized exocytosis of secretory lysosomes takes place at the immunological synapse in cytotoxic lymphocytes and Natural Killers (see for Holt et al., 2006 review). Our set-up will be used to examine which parameter of lysosome movements actors of the lysosome trafficking such as Lyst [lysosomal trafficking regulator; (Stinchcombe et al., 2000)] or Hck (Cougoule et al., 2005; Vincent et al., 2007; Guiet et al., 2008) are involved in, using, for instance, sh- or siRNA-mediated knock-down approaches.
In conclusion, the experimental approach described here will facilitate studies dedicated to the identification of the actors which play a role in lysosome movement, directionality, speed, docking, and also fusion once TIRF microscopy is coupled to the present set-up.
Taking into account that lysosome secretion is a phenomenon that is crucial not only for bactericidal activities, but also for extracellular matrix degradation during trans-tissular migration, the molecular actors of this process could then be essential effectors of the inflammation response.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The vector encoding Cathepsin-D-mCherry was a kind gift from Dr. François Darchen (Paris, France); Raw264.7 cells stably expressing actin-GFP were provided by Dr. Sergio Grinstein (Toronto, Canada). We thank Dr. Joly for critical reading of our manuscript, Dr. Charrière and Mr. Romain Guiet for helpful discussions during the advancement of this work. A. Labrousse was funded by the University of Toulouse; this work was supported by Assocation pour la Recherche contre le Cancer (SSFI 20101201733) and Agence National de la Recherche (2010 Midi 01301: Migreflame).
Supplementary Material
The Movies 1–3 for this article can be found online at http://www.frontiersin.org/Inflammation/10.3389/fimmu.2011.00051/abstract
Movie S1. RAW267.4 RAW267.4 macrophage stably expressing GFP-actin undergoes frustrated phagocytosis on micro-patterned immune complexes. The cell makes contact with the substratum at the top right corner of the field and form two successive frustrated phagosomes toward the left in the lower part of the field. Accelerated 2 times.
Movie S2. RAW267.4 macrophage stably expressing GFP-actin undergoes frustrated phagocytosis on micro-patterned immune complexes. This cell rapidly forms two frustrated phagosomes, which seem to be linked by a dynamic actin structure. Accelerated 2 times.
Movie S3. RAW267.4 macrophage stably co-expressing GFP-actin (left panel) and CathepsinD-mCherry (right panel). Images of GFP and mCherry fluorescence were taken every 10 sec to avoid bleaching, and 250 ms, respectively. Arrows show examples of tracked lysosomes. Accelerated 4 times.
References
Astarie-Dequeker, C., Carreno, S., Cougoule, C., and Maridonneau-Parini, I. (2002). The protein tyrosine kinase Hck is located on lysosomal vesicles that are physically and functionally distinct from CD63-positive lysosomes in human macrophages. J. Cell Sci. 115, 81–89.
Bainton, D. F., Takemura, R., Stenberg, P. E., and Werb, Z. (1989). Rapid fragmentation and reorganization of Golgi membranes during frustrated phagocytosis of immobile immune complexes by macrophages. Am. J. Pathol. 134, 15–26.
Barral, D. C., and Seabra, M. C. (2004). The melanosome as a model to study organelle motility in mammals. Pigment Cell Res. 17, 111–118.
Claus, V., Jahraus, A., Tjelle, T., Berg, T., Kirschke, H., Faulstich, H., and Griffiths, G. (1998). Lysosomal enzyme trafficking between phagosomes, endosomes, and lysosomes in J774 macrophages. Enrichment of cathepsin H in early endosomes. J. Biol. Chem. 273, 9842–9851.
Cougoule, C., Carreno, S., Castandet, J., Labrousse, A., Astarie-Dequeker, C., Poincloux, R., Le Cabec, V., and Maridonneau-Parini, I. (2005). Activation of the lysosome-associated p61Hck isoform triggers the biogenesis of podosomes. Traffic 6, 682–694.
Cougoule, C., Le Cabec, V., Poincloux, R., Al Saati, T., Mege, J. L., Tabouret, G., Lowell, C. A., Laviolette-Malirat, N., and Maridonneau-Parini, I. (2010). Three-dimensional migration of macrophages requires Hck for podosome organization and extracellular matrix proteolysis. Blood 115, 1444–1452.
Desjardins, M. (1995). Biogenesis of phagolysosomes: the “kiss and run” hypothesis. Trends Cell Biol. 5, 183–186.
Eng, E. W., Bettio, A., Ibrahim, J., and Harrison, R. E. (2007). MTOC reorientation occurs during FcgammaR-mediated phagocytosis in macrophages. Mol. Biol. Cell 18, 2389–2399.
Falcon-Perez, J. M., Nazarian, R., Sabatti, C., and Dell’angelica, E. C. (2005). Distribution and dynamics of Lamp1-containing endocytic organelles in fibroblasts deficient in BLOC-3. J. Cell Sci. 118, 5243–5255.
Guiet, R., Poincloux, R., Castandet, J., Marois, L., Labrousse, A., Le Cabec, V., and Maridonneau-Parini, I. (2008). Hematopoietic cell kinase (Hck) isoforms and phagocyte duties – from signaling and actin reorganization to migration and phagocytosis. Eur. J. Cell Biol. 87, 527–542.
Harrison, R. E., Bucci, C., Vieira, O. V., Schroer, T. A., and Grinstein, S. (2003). Phagosomes fuse with late endosomes and/or lysosomes by extension of membrane protrusions along microtubules: role of Rab7 and RILP. Mol. Cell. Biol. 23, 6494–6506.
Heiple, J. M., Wright, S. D., Allen, N. S., and Silverstein, S. C. (1990). Macrophages form circular zones of very close apposition to IgG-coated surfaces. Cell Motil. Cytoskeleton 15, 260–270.
Holt, O. J., Gallo, F., and Griffiths, G. M. (2006). Regulating secretory lysosomes. J. Biochem. 140, 7–12.
Huynh, K. K., Eskelinen, E. L., Scott, C. C., Malevanets, A., Saftig, P., and Grinstein, S. (2007). LAMP proteins are required for fusion of lysosomes with phagosomes. EMBO J. 26, 313–324.
Kjeken, R., Egeberg, M., Habermann, A., Kuehnel, M., Peyron, P., Floetenmeyer, M., Walther, P., Jahraus, A., Defacque, H., Kuznetsov, S. A., and Griffiths, G. (2004). Fusion between phagosomes, early and late endosomes: a role for actin in fusion between late, but not early endocytic organelles. Mol. Biol. Cell 15, 345–358.
Knapp, P. E., and Swanson, J. A. (1990). Plasticity of the tubular lysosomal compartment in macrophages. J. Cell Sci. 95(Pt 3), 433–439.
Kumar, Y., and Valdivia, R. H. (2009). Leading a sheltered life: intracellular pathogens and maintenance of vacuolar compartments. Cell Host Microbe 5, 593–601.
Labernadie, A., Thibault, C., Vieu, C., Maridonneau-Parini, I., and Charriere, G. M. (2010). Dynamics of podosome stiffness revealed by atomic force microscopy. Proc. Natl. Acad. Sci. U.S.A. 107, 21016–21021.
Liebl, D., and Griffiths, G. (2009). Transient assembly of F-actin by phagosomes delays phagosome fusion with lysosomes in cargo-overloaded macrophages. J. Cell Sci. 122, 2935–2945.
Linder, S., and Aepfelbacher, M. (2003). Podosomes: adhesion hot-spots of invasive cells. Trends Cell Biol. 13, 376–385.
Linder, S., Wiesner, C., and Himmel, M. (2010). Degrading devices: invadosomes in proteolytic cell invasion. Annu. Rev. Cell Dev. Biol. 27, 24.1–24.27.
Luzio, J. P., Pryor, P. R., and Bright, N. A. (2007). Lysosomes: fusion and function. Nat. Rev. Mol. Cell Biol. 8, 622–632.
Newman, S. L., Mikus, L. K., and Tucci, M. A. (1991). Differential requirements for cellular cytoskeleton in human macrophage complement receptor- and Fc receptor-mediated phagocytosis. J. Immunol. 146, 967–974.
Rabinowitz, S., Horstmann, H., Gordon, S., and Griffiths, G. (1992). Immunocytochemical characterization of the endocytic and phagolysosomal compartments in peritoneal macrophages. J. Cell Biol. 116, 95–112.
Reddy, A., Caler, E. V., and Andrews, N. W. (2001). Plasma membrane repair is mediated by Ca(2(+)-regulated exocytosis of lysosomes. Cell 106, 157–169.
Ross, J. L., Shuman, H., Holzbaur, E. L., and Goldman, Y. E. (2008). Kinesin and dynein-dynactin at intersecting microtubules: motor density affects dynein function. Biophys. J. 94, 3115–3125.
Saltel, F., Chabadel, A., Bonnelye, E., and Jurdic, P. (2008). Actin cytoskeletal organisation in osteoclasts: a model to decipher transmigration and matrix degradation. Eur. J. Cell Biol. 87, 459–468.
Schroeder, H. W. III, Mitchell, C., Shuman, H., Holzbaur, E. L., and Goldman, Y. E. (2010). Motor number controls cargo switching at actin-microtubule intersections in vitro. Curr. Biol. 20, 687–696.
Stinchcombe, J. C., Page, L. J., and Griffiths, G. M. (2000). Secretory lysosome biogenesis in cytotoxic T lymphocytes from normal and Chediak Higashi syndrome patients. Traffic 1, 435–444.
Takemura, R., Stenberg, P. E., Bainton, D. F., and Werb, Z. (1986). Rapid redistribution of clathrin onto macrophage plasma membranes in response to Fc receptor-ligand interaction during frustrated phagocytosis. J. Cell Biol. 102, 55–69.
Touret, N., Paroutis, P., Terebiznik, M., Harrison, R. E., Trombetta, S., Pypaert, M., Chow, A., Jiang, A., Shaw, J., Yip, C., Moore, H. P., Van Der Wel, N., Houben, D., Peters, P. J., De Chastellier, C., Mellman, I., and Grinstein, S. (2005). Quantitative and dynamic assessment of the contribution of the ER to phagosome formation. Cell 123, 157–170.
Tu, C., Ortega-Cava, C. F., Chen, G., Fernandes, N. D., Cavallo-Medved, D., Sloane, B. F., Band, V., and Band, H. (2008). Lysosomal cathepsin B participates in the podosome-mediated extracellular matrix degradation and invasion via secreted lysosomes in v-Src fibroblasts. Cancer Res. 68, 9147–9156.
Van Goethem, E., Guiet, R., Balor, S., Charrière, G. M., Poincloux, R., Labrousse, A., Maridonneau-Parini, I., and Le Cabec, V. (2011). Macrophage podosomes go 3D. Eur J. Cell. Biol. 90, 224–236.
Van Goethem, E., Poincloux, R., Gauffre, F., Maridonneau-Parini, I., and Le Cabec, V. (2010). Matrix architecture dictates three-dimensional migration modes of human macrophages: differential involvement of proteases and podosome-like structures. J. Immunol. 184, 1049–1061.
Verollet, C., Zhang, Y. M., Le Cabec, V., Mazzolini, J., Charriere, G., Labrousse, A., Bouchet, J., Medina, I., Biessen, E., Niedergang, F., Benichou, S., and Maridonneau-Parini, I. (2010). HIV-1 Nef triggers macrophage fusion in a p61Hck- and protease-dependent manner. J. Immunol. 184, 7030–7039.
Vincent, C., Maridonneau-Parini, I., Le Clainche, C., Gounon, P., and Labrousse, A. (2007). Activation of p61Hck triggers WASp- and Arp2/3-dependent actin-comet tail biogenesis and accelerates lysosomes. J. Biol. Chem. 282, 19565–19574.
Wiesner, C., Faix, J., Himmel, M., Bentzien, F., and Linder, S. (2010). KIF5B and KIF3A/KIF3B kinesins drive MT1-MMP surface exposure, CD44 shedding, and extracellular matrix degradation in primary macrophages. Blood 116, 1559–1569.
Wright, S. D., and Silverstein, S. C. (1984). Phagocytosing macrophages exclude proteins from the zones of contact with opsonized targets. Nature 309, 359–361.
Wu, M., Baumgart, T., Hammond, S., Holowka, D., and Baird, B. (2007). Differential targeting of secretory lysosomes and recycling endosomes in mast cells revealed by patterned antigen arrays. J. Cell Sci. 120, 3147–3154.
Appendix
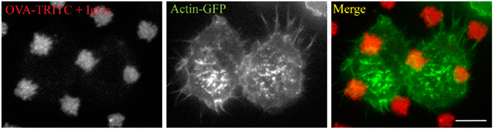
Figure A1. Fc-receptors are necessary to trigger frustrated phagocytosis. RAW267.4 macrophage stably expressing GFP-actin (green) were pre-treated with anti-CD16/CD32 blocking antibodies, layered on micro-patterned immune complexes (red) for 15 minutes before being fixed.
Keywords: lysosome, frustrated phagocytosis, macrophages, micro-patterned immune complexes
Citation: Labrousse AM, Meunier E, Record J, Labernadie A, Beduer A, Vieu C, Ben Safta T and Maridonneau-Parini I (2011) Frustrated phagocytosis on micro-patterned immune complexes to characterize lysosome movements in live macrophages. Front. Immun. 2:51. doi: 10.3389/fimmu.2011.00051
Received: 04 July 2011; Paper pending published: 25 July 2011;
Accepted: 19 September 2011; Published online: 12 October 2011.
Edited by:
Patrizia Rovere Querini, Fondazione Centro San Raffaele del Monte Tabor and Vita-Salute University, ItalyReviewed by:
Pablo Pelegrin, Hospital Universitario Virgen Arrixaca, SpainPatrizia Rovere Querini, Fondazione Centro San Raffaele del Monte Tabor and Vita-Salute University, Italy
Copyright: © 2011 Labrousse, Meunier, Record, Labernadie, Beduer, Vieu, Ben Safta and Maridonneau-Parini. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Isabelle Maridonneau-Parini, UMR5089, CNRS, Institut de Pharmacologie et de Biologie Structurale, 205 route de Narbonne, 31077 Toulouse Cedex, France. e-mail: isabelle.maridonneau-parini@ ipbs.fr
† Etienne Meunier and Julien Record have contributed equally to this work.