 Aisling Kennedy1,2
Ursula Fearon2,3
Douglas J. Veale2,3
Aisling Kennedy1,2
Ursula Fearon2,3
Douglas J. Veale2,3 Catherine Godson1,2*
Catherine Godson1,2*- 1 School of Medicine and Medical Sciences, University College Dublin Conway Institute, Dublin, Ireland
- 2 University College Dublin, Dublin, Ireland
- 3 Dublin Academic Medical Centre, St. Vincent’s University Hospital, Dublin, Ireland
Synovial macrophages are one of the resident cell types in synovial tissue and while they remain relatively quiescent in the healthy joint, they become activated in the inflamed joint and, along with infiltrating monocytes/macrophages, regulate secretion of pro-inflammatory cytokines and enzymes involved in driving the inflammatory response and joint destruction. Synovial macrophages are positioned throughout the sub-lining layer and lining layer at the cartilage–pannus junction and mediate articular destruction. Sub-lining macrophages are now also considered as the most reliable biomarker for disease severity and response to therapy in rheumatoid arthritis (RA). There is a growing understanding of the molecular drivers of inflammation and an appreciation that the resolution of inflammation is an active process rather than a passive return to homeostasis, and this has implications for our understanding of the role of macrophages in inflammation. Macrophage phenotype determines the cytokine secretion profile and tissue destruction capabilities of these cells. Whereas inflammatory synovial macrophages have not yet been classified into one phenotype or another it is widely known that TNFα and IL-l, characteristically released by M1 macrophages, are abundant in RA while IL-10 activity, characteristic of M2 macrophages, is somewhat diminished. Here we will briefly review our current understanding of macrophages and macrophage polarization in RA as well as the elements implicated in controlling polarization, such as cytokines and transcription factors like NFκB, IRFs and NR4A, and pro-resolving factors, such as LXA4 and other lipid mediators which may promote a non-inflammatory, pro-resolving phenotype, and may represent a novel therapeutic paradigm.
Introduction
Macrophages (Mɸ) are one of the resident cell types in synovial tissue, along with fibroblasts. While quiescent in health, Mɸ become activated in the inflamed joint, where they make up around 30–40% of the cellular content, and regulate secretion of pro-inflammatory cytokines and enzymes involved in driving the inflammatory response and joint destruction (Firestein and Zvaifler, 1990). Their position throughout the sub-lining layer and lining layer at the cartilage–pannus junction facilitates their role mediating articular destruction. It is estimated that rheumatoid arthritis (RA) and psoriatic arthritis (PsA) each affects approximately 1% of the population (Firestein, 2003; Gladman, 2009), leading to patient pain and disability as well as contributing to a great economic burden in terms of lost working days and patient health services (Cooper, 2000) and therefore is an area of intense investigation.
As our understanding of inflammation progresses, including the recent concept that resolution of inflammation is an active process rather than a passive return to homeostasis, the role of Mɸ is increasingly appreciated. The inability to resolve acute inflammation may lead to a chronic inflammatory state. Depending on their phenotype, Mɸ can secrete either pro- or anti-inflammatory cytokines and mediate matrix destruction or deposition. Synovial Mɸ participate in many of the events driving inflammation including the stimulation of angiogenesis, leukocyte and lymphocyte recruitment, fibroblast proliferation, and protease secretion leading to eventual joint destruction (Burmester et al., 1997; Vallejo et al., 2003; Abeles and Pillinger, 2006). While RA and PsA are considered more inflammatory than osteoarthritis (OA), it can still contain an inflammatory component, of which Mɸ play a large part. In all of these conditions Mɸ derived mediators can drive inflammation and cartilage destruction. Depletion of Mɸ from OA synovial cell cultures significantly reduced TNFα and IL-1β levels. Depletion of Mɸ from both RA and OA synovial cell cultures leads to reduced synovial fibroblast responses such as cytokine and MMP production (Janusz and Hare, 1993; Bondeson et al., 2010). Both macrophages and fibroblasts display an activated cell phenotype with increased cell surface expression of HLA-DR and leukocyte adhesion molecules (Athanasou et al., 1988; Alvaro-Gracia et al., 1990) participating in T-cell activation. Interaction of Mɸ with T-cells potentiates the expression of several pro-inflammatory mediators such as IL-1α and β and MMPs (McInnes et al., 2000).
Important pro-inflammatory cytokines like TNFα and IL-l are abundant in the inflamed synovium and are characteristically released by classically activated (M1) Mɸ. These cytokines are central to joint destruction. The importance of Mɸ in driving the inflammatory response has been highlighted by several quantitative microscopic studies, where they have shown that Mɸ number; correlates with disease activity (Tak et al., 1997), has potential use as a biomarker for disease (Kruithof et al., 2006; Bresnihan et al., 2009) and declines in response to therapy (Goedkoop et al., 2004; Canete et al., 2010). Mɸ can induce angiogenesis (Leibovich et al., 1987), and hypoxia, a prominent feature of the inflamed joint, promotes the survival of monocytes/macrophages and induces their anaerobic adaptations including glycolysis (Roiniotis et al., 2009).
It is long appreciated that Mɸ play an important role in the pathogenesis of arthritis and this observation was supported by studies showing that the number of Mɸ was increased in clinically affected joints compared to non-affected joints (Kraan et al., 1998). Several studies also linked the number of synovial Mɸ to inflammatory cytokine production joint destruction (Mulherin et al., 1996). As the search for a reliable biomarker in RA continued, the role of Mɸ was again highlighted. The culmination of this work has led to sub-lining CD68 positive synovial Mɸ currently being the only validated biomarker for disease severity (Tak et al., 1997) and response to therapy in arthritis (Haringman et al., 2005), further confirming their importance in the pathogenesis of this disease, a finding which is independent of treatment type (Haringman et al., 2005; Thurlings et al., 2008). Considering the similarities between synovial inflammation in RA and PsA, Mɸ have also been proposed as a biomarker for response to therapy in PsA. Several studies have concluded that Mɸ number is decreased in PsA synovial tissue following therapy (Goedkoop et al., 2004; Kruithof et al., 2006; Canete et al., 2010).
Activation of Synovial Macrophages
Besides the abundant pro-inflammatory cytokines and chemokines present in inflamed synovial tissue, activation, and survival of Mɸ can be achieved through acetylation or de-acetylation of histones. Downstream effects of TNFα and other molecules results in the induction of histone acetyltransferase (HAT) activity in Mɸ which causes acetylation of histones and subsequent modulation of transcriptional activity. HAT activity is counteracted by histone deacetylases (HDAC). Two recent studies have found evidence of depressed HDAC activity in RA, particularly in synovial macrophages and fibroblasts. The ratio of HDAC:HAT activity was significantly lower in RA synovial tissue compare to healthy controls. In combination with this, HDAC inhibition decreases IL-10 production from whole tissue synovial explants cultures, indicating a negative effect on anti-inflammatory pathways, which would lead us to believe that a lack of HDAC may contribute to perpetuation of inflammation (Huber et al., 2007; Grabiec et al., 2008, 2010). Despite this, HDAC inhibition is showing promise for inflammatory diseases. HDAC inhibitors reduced IL-6 production from TNFα stimulated Mɸ and induced apoptosis of RA synovial fluid (SF) Mɸ, even in the presence of a pro-inflammatory stimulus (Grabiec et al., 2010). This is of interest considering the ability of synovial cells and infiltrating cells to evade apoptosis during joint inflammation contributing to synovial hypercellularity (Salmon et al., 1997; Perlman et al., 2001). The potential use of HDAC inhibitors has been further promoted by their success in suppressing synovial inflammation and cartilage destruction in a CIA mouse model (Nasu et al., 2008).
Toll like receptors (TLR) are pattern recognition receptors that mediate response to infection. However, it is becoming apparent that some of these receptors may become activated by non-infectious agents from within the body and may therefore play a role in autoimmune conditions such as RA. Engagement of TLRs induces signaling through a well defined pathway involving MyD88 that leads to transcriptional activation (Joosten et al., 2003). TLR2 and TLR4 appear to be particularly associated with RA. TLR knockout and arthritis mouse models, or a combination of both, have highlighted the position of TLRs in the pathogenesis of arthritis. In a model of spontaneous arthritis due to IL-1 receptor antagonist knockout, simultaneous knockout of TLR4 attenuated inflammation while TLR2 knockout produced a more severe arthritis. Knockout of TLR9 had no effect (Abdollahi-Roodsaz et al., 2008). This clearly indicates a potential benefit for TLR4 antagonism in RA. However the role of TLR2 seems less defined as other studies have shown that knockdown of TLR2 produces beneficial effects in arthritis (Joosten et al., 2003). Further to this, many TLR ligands have been identified in synovial inflammation (Okamura et al., 2001; Park et al., 2004). Acute serum amyloid A (SAA), which is significantly upregulated in arthritis and propagates pro-inflammatory effects similar to TNFα (O’Hara et al., 2000; Mullan et al., 2006; Connolly et al., 2011), is a functional ligand for TLR2 and may contribute to the deleterious effects of SAA in arthritis (Cheng et al., 2008). RA Mɸ are more responsive to stimulation than Mɸ from other forms of inflammatory arthritis, despite no difference in Mɸ number (Huang et al., 2007). Therefore, engagement of TLR2 and 4 may contribute to Mɸ activation and a sustained Mɸ response in RA.
Rheumatoid factor (RF) is one of the diagnostic criteria for RA and can help to distinguish RA from similar arthropathies like PsA. Classification of RA as an autoimmune disease came initially from the discovery of IgG auto-antibodies in the blood of patients (Waaler, 1940; Franklin et al., 1957). RF is mostly IgM-RF, but IgG-RF and IgA-RF can also be detected in some patients. The cellular receptors for IgG are the Fcγ receptors, FCγRI (CD64), FcγRII (CD32), and FcγRIII (CD16). All three receptors are expressed on synovial Mɸ (Laurent et al., 2011) as well as lymphocytes. FcγRIII has been demonstrated to play a role in the development of arthritis through animal models. Mice deficient in FcγRIII are protected from the development of collagen induced arthritis without alteration of their humoral response, and therefore the protection is not due to alterations in T-cell responses (Ståhl et al., 2002; Andrén et al., 2006). Polymorphisms in Fcγ receptors are associated with incidence of RA as well as response to therapy (Morgan et al., 2006; Canete et al., 2009; Thabet et al., 2009; Morales-Lara et al., 2010).
Are Synovial Macrophages Polarized?
In the immune system Mɸ are effective antigen presenting cells with phagocytic activity which respond to lymphocyte derived cytokines. However, the responses elicited by Mɸ are variable and depend entirely on the tissue environment. We now know these responses can be either pro- or anti-inflammatory. Dedicated reviews on this topic discuss in more detail the cytokines and chemokines involved in promoting one phenotype over another (Mantovani et al., 2004; Murray and Wynn, 2011) but an overview of the main components are outlined in Figure 1. Classically activated M1 Mɸ have a pro-inflammatory phenotype, producing high levels of TNFα, IL-1, IL-6, IL-12, IL-23, reactive oxygen species, and low levels of IL-10. Alternatively activated Mɸ, of which there are three subsets (Mantovani et al., 2004; Martinez et al., 2008), display and anti-inflammatory phenotype, producing high levels of IL-10, IL-1 receptor antagonist, decoy IL-1RII, TGFβ, and low levels of IL-12. Both types are necessary for correct resolution of inflammation. An interesting, and potentially useful, property of these Mɸ is that they remain plastic and polarization into one phenotype does preclude re-polarization (Stout et al., 2005). Therefore, if we could elucidate the exact pathways and transcription factors involved in promoting one phenotype over the other in vivo, this system could be exploited for therapeutic gain.
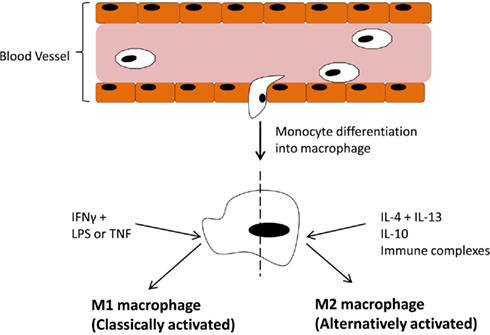
Figure 1. Blood-borne monocytes exit the blood stream and differentiate into macrophages. The macrophage response depends on the stimulus provided by the microenvironment. IFNγ along with LPS or TNFα drive polarization of M1 (classically activated) macrophages which participate in pro-inflammatory activities. On the other hand, IL-4 + IL-13, IL-10, or immune complexes drive M2 (alternatively activated) macrophages, which participate in anti-inflammatory responses.
There appears to be a lack of evidence for Mɸ polarization in either direction in the inflamed joint. It has been suggested that spondyloarthropathies such as PsA display a more M2 profile compared to RA patients and that M1 mediators correlate with joint inflammation in RA (Vandooren et al., 2009). However, in general, most studies of Mɸ in arthritis focus on important Mɸ functions and not polarization. The mediators that can control Mɸ polarization are indeed present in the synovium and some show potential as therapeutic targets. Synovial lining layer thickness is greater in RA, compared to PsA or healthy control subjects, which is associated with an increase in synovial Mɸ and fibroblasts. PsA patients tend to have less lining layer Mɸ than RA patients. This has been observed in many comparative studies. Danning et al. (2000) also found similar levels of IL-10 in RA and PsA synovium, despite the difference in synovial lining layer Mɸ numbers, however levels were described as being quite low. It is difficult to determine if this lack of IL-10 is a contributor to or consequence of the overwhelming inflammation in the joint. A study by Mottonen et al. (1998) found that 68% of Mɸ isolated from RA SF were CD86 positive and that SF Mɸ can take on a dendritic cell phenotype when exposed to a combination of IL-4 and GM-CSF and that these cells were more effective at activating T-cells than control or TNFα stimulated Mɸ. The effects of IL-4 + GM-CSF were mediated through CD86, a marker of classically activated Mɸ. IL-10 was able to inhibit the observed effects with IL-4 + GM-CSF as it downregulated the expression of CD86, as well as CD-40 and HLA-DR which also participate in Mɸ mediated T-cell activation. This is consistent with the classification that M2c Mɸ, which are driven by IL-10 are involved in suppression of the immune response (Mantovani et al., 2004). These results may appear confusing as IL-4 along with IL-13 drive the M2a or alternative Mɸ phenotype which should be involved in anti-inflammatory responses. However GM-CSF drives the M1 phenotype in monocyte derived macrophages so this may be the driving force for inflammatory responses in these experiments.
What Regulators in Synovial Inflammation Could Potentially Influence Macrophage Polarization?
Figure 1 has outlined the cytokines and regulators that promote M1 or M2 polarization. The extent of expression of these factors in the joint varies. The M1 Mɸ phenotype is induced by interferon-γ (IFNγ) in combination with either lipopolysaccharide (LPS) or TNFα. IFNγ is highly expressed in RA synovial tissue and its levels significantly correlate with disease severity (Milman et al., 2010). Exposure to INFγ increases the response of Mɸ exposed to other stimuli by either upregulating pro-inflammatory cytokines, like TNFα, or downregulating anti-inflammatory cytokines, like IL-10 (Erwig et al., 1998; Wallet et al., 2010). TNFα is a master cytokine in inflammation and as such is a potent inducer of other pro-inflammatory cytokines (Nawroth et al., 1986; Butler et al., 1995), is chemotactic for leukocytes, is a potent inducer of angiogenesis (Leibovich et al., 1987), stimulates adhesion molecule expression in SFC in vitro (Marlor et al., 1992), and lymphoid migration into inflamed synovial tissue in vivo (Wahid et al., 2000). Within the inflamed joint macrophages, fibroblasts, lymphocytes, and endothelial cells produce TNFα. An important role for TNFα in arthritis was confirmed by studies which showed its potential to degrade both cartilage (Dayer et al., 1985) and bone (Bertolini et al., 1986). Further rationale for the involvement of TNFα in the progression of inflammatory arthritis was provided when transgenic mice expressing a modified human TNFα gene spontaneously developed arthritis which exhibited increased human TNFα protein, joint inflammation, bone erosion, and cartilage destruction. In this study, antibodies specific for human, but not mouse TNFα reduced disease severity (Keffer et al., 1991). In subsequent studies administration of a monoclonal antibody to TNFα ameliorated inflammation and joint damage after disease onset in a CIA model of arthritis (Williams et al., 1992). TNFα cytokine targeted therapies have now been developed for inflammatory arthritis. The first clinical trial was undertaken in the UK in 1992 and demonstrated that targeted biologic therapy decreased serum IL-6 levels, swollen joint numbers and levels of the acute phase proteins CRP and A-SAA which are markers of inflammation (Elliott et al., 1993). Alternatively, anti-inflammatory and M2 polarizing cytokines like IL-10 are lowly expressed in arthritis as its signaling is blocked during FCγ receptor ligation (Ji et al., 2003), and treatment with the pro-resolving mediator annexin A1 stimulates release of IL-10 (Ferlazzo et al., 2003). Treatment of PBMC with IL-10 caused a change in the ratio of Th17:Treg cells in favor of Treg cells and decreased production of the pro-inflammatory cytokine IL-17 (Heo et al., 2010). Animal models of arthritis have also demonstrated how treatment with IL-10 can suppress the development and progression of joint inflammation, even in established disease (Walmsley et al., 1996; Whalen et al., 1999; Mauri et al., 2003).
The cytokines involved in promoting polarization are well defined, however less is known about which transcription factors are utilized to induce polarization. IRF5 (interferon regulatory factor 5) has been implicated in driving the M1 phenotype as well as actively suppressing M2 polarization and driving Th1 and Th17 responses (Krausgruber et al., 2011). While the study by Krausgruber et al. (2011) was not performed in synovial Mɸ, animal studies suggest that inflammation in RA is driven by Th1 cytokines such as IFNγ, which is upregulated early in the disease process (Miltenburg et al., 1992; Schulze-Koops and Kalden, 2001) and a rapid growth in interest in the Th17 pathway and indeed IL-17 itself in the last few years would suggest that this would warrant investigation in the inflamed joint. Recent reports confirm that alterations in the IRF5 gene confers susceptibility to RA (Dieguez-Gonzalez et al., 2008; Han et al., 2009; Dawidowicz et al., 2011) as well as many related illnesses such as inflammatory bowel disease, Sjogrens syndrome, and systemic lupus erythematosus (Dideberg et al., 2007; Graham et al., 2007; Miceli-Richard et al., 2007). Other transcription factors in the IRF family, like IRF3 (Biswas et al., 2006) and IRF4 (Satoh et al., 2010) have been implicated in promoting Mɸ polarization in other disease settings, and IRF family members contribute to determination of dendritic cell fate (Tamura et al., 2005). These findings make the IRF family attractive candidates to study in the context of Mɸ’s in arthritis.
NR4A is part of the orphan nuclear receptor superfamily which have roles in lipid metabolism and inflammation (Desreumaux et al., 2001; Oosterveer et al., 2010; Hong et al., 2011). Receptors in the same superfamily as NR4A are downregulated in arthritic tissue and their activation appears to play a role in inhibiting disease progression (Bonnelye et al., 2008; Park et al., 2010). However members of the NR4A subfamily appear to have less clearly defined effects to the anti-inflammatory family members liver X receptor and peroxisome-proliferator-activator receptor and drive inflammation in human synovial tissue (Murphy et al., 2001). The role of NR4A receptors specifically in Mɸ polarization has not yet been elucidated, however, any role for NR4A in Mɸ polarization would be an interesting finding due to the modulation of NR4A by both dexamethasone and methotrexate, which are effective treatments for joint inflammation in some patients. NR4A receptors can also activate NFκB in murine Mɸ (Pei et al., 2006) where it promotes transcription of pro-inflammatory genes. In arthritis, and inflammation in general, NFκB can be considered a master transcription factor as it is utilized by many ligand–receptor complexes to modulate gene transcription. TNFα and IL-1β which are abundant in the inflamed joint employ this transcription factor and in turn can be regulated by it. NFκB activation has been detected prior to the clinical onset of arthritis in animal models (Tsao et al., 1997; Han et al., 1998) and the NFκB pathway has been directly targeted as a treatment method for RA confirming its essential role in the pathogenesis of this disease (Wakamatsu et al., 2005). Immunohistochemical staining has confirmed nuclear expression of NFκB subunits in synovial Mɸ (Handel et al., 1995). The NFκB family consists of five proteins; p50, p52, RelA (p65), RelB, and c-Rel. These proteins form homo or heterodimers to determine gene transcription. RelA, RelB, and c-Rel contain a transcriptional activation domain and therefore upregulate gene expression, however p50 and p52 do not contain the transcriptional activation domain and homodimers of these proteins can sometimes have a repressing function (Bohuslav et al., 1998). NFκB p50 activation has been linked to promoting M2 polarizing genes in Mɸ (Porta et al., 2009). This study found that knockout of the NFκB p50 subunit prevented the development of tolerance in LPS challenged Mɸ by restoring M1 mediators and inhibiting M2 cytokines. Similarly, Saccani et al. (2006) found that accumulation of the p50 homodimer occurred in the nuclei of tumor associated Mɸ and that these Mɸ expressed an M2 phenotype. Therefore due to the prominence of the NFκB pathway in RA it remains an interesting candidate for influencing Mɸ polarization. A summary of all factors discussed here are outlined in Table 1.
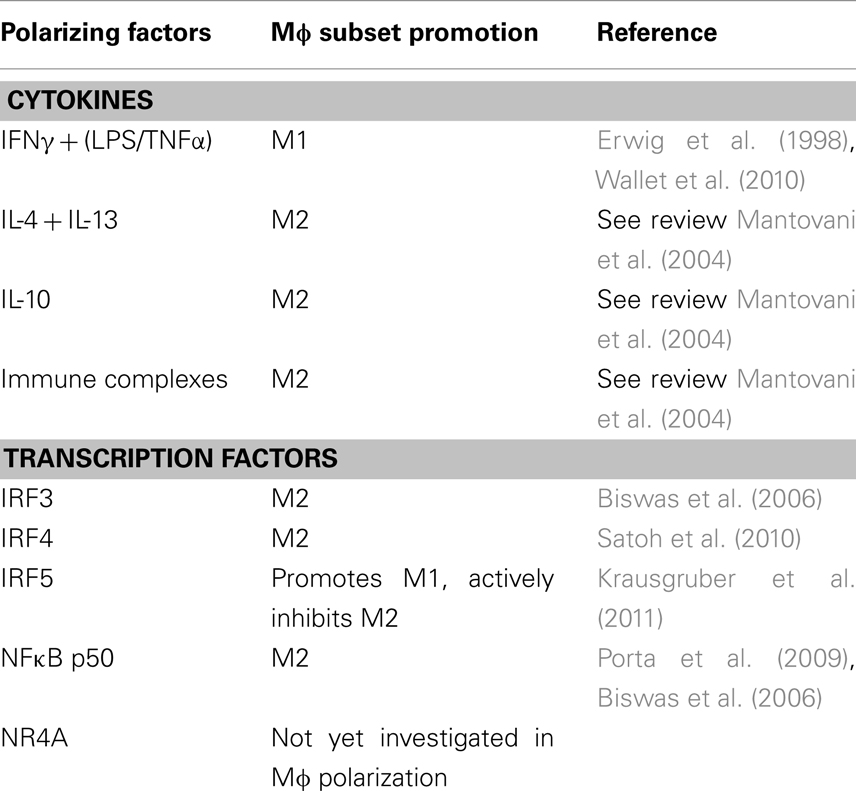
Table 1. Description of the regulators in synovial inflammation that could potentially influence macrophage polarization.
Macrophages and Resolution of Inflammation
There is a growing understanding of the molecular drivers of inflammation and an appreciation that the resolution of inflammation is an active process rather than a passive return to homeostasis. Endogenously produced mediators that actively promote the resolution of inflammation are now under investigation for their therapeutic use. These are molecules such as lipoxins, resolvins, protectins, and annexins. Lipoxin A4 (LXA4) is an eicosanoid produced by the transcellular metabolism of arachidonic acid by 15/5- or 5/12-lipoxinagese (Serhan et al., 1984). Its biosynthesis is co-incident with the resolution phase of inflammation and many of its bioactions are mediated through ligation of its receptor, ALX/FPR2 (Fiore et al., 1994). LXA4 is produced in inflamed synovial tissue (Thomas et al., 1995) where it can downregulate pro-inflammatory activities of activated fibroblasts and upregulate anti-inflammatory activities, even in the presence of a pro-inflammatory stimulus which acts through the same receptor (Sodin-Semrl et al., 2004; Kronke et al., 2009; Chan and Moore, 2010). In other disease models, LXA4 has been shown to induce anti-inflammatory/pro-resolving actions such as inhibition of neutrophil recruitment and activation (Filep et al., 1999), regulation of NFκB activation (Decker et al., 2009), and the clearance of apoptotic cells by Mɸ (Godson et al., 2000). Neutrophils are the first effector cells at the site of inflammation. Once these cells have carried out their functions in regard to host defense they are programmed to die by apoptosis. Resolution of inflammation and return to homeostasis involves phagocytosis of apoptotic neutrophils to prevent the persistence to necrosis and leakage of cellular contents, which may itself begin an inflammatory reaction. Despite the lack of apoptosis occurring in all cell types in the inflamed synovium, resident synovial Mɸ retain the capacity to phagocytose apoptotic cells, even at an early timepoint after arthritis induction (van Lent et al., 2001). If normal apoptosis and phagocytosis could be induced in the inflamed synovium, possibly by native LXA4 or its stable analogs, this process may trigger a normal resolution of inflammation.
In order to encourage phagocytosis, apoptotic cells release mediators that attract phagocytes toward them, essentially flagging themselves for engulfment. One such mediator is the anti-inflammatory compound annexin A1 (Arur et al., 2003; Scannell et al., 2007). Annexin A1 is a 37-kDa protein of the annexin superfamily where all family members contain a similar core region and a distinct N-terminal region which confers specificity of function. Generally annexin A1 is localized to the cytoplasm where, upon stimulation, it is mobilized to the cell membrane and secreted (for an extensive review on annexin A1 see Perretti and Dalli, 2009). Interestingly annexin A1 also signals through ALX/FPR2, the same receptor utilized by LXA4 and SAA. Annexin A1 is widely expressed in many cell types including Mɸ. Immunohistochemical analysis has demonstrated an increased expression of annexin A1 in the RA synovial lining layer macrophages and fibroblasts compared to OA and normal joints. This may, however, be due to the increased lining layer thickness in this condition (Goulding et al., 1995) as other studies have shown decreased binding of annexin A1 to several cell types in RA (Goulding et al., 1992; Sampey et al., 2000). Glucocorticoid stimulation causes annexin A1 mobilization to the cell surface and secretion where it mediates glucocorticoid induced anti-inflammatory effects. This is of particular interest in arthritis as glucocorticoid therapy is one of the current treatments for this condition (Flower, 1988; Podgorski et al., 1992; Yang et al., 1998, 1999; Maderna et al., 2005). However, as is increasingly the case for many mediators, the role of annexin A1 may not be as unambiguous as initially described and it may also potentiate pro-inflammatory actions in arthritis. An investigation by Tagoe et al. (2008) has revealed synergistic actions with TNFα and annexin A1 in terms of MMP production from synovial fibroblast cells. They saw firstly that TNFα can induce expression of endogenous annexin A1 and secondly that TNFα along with the annexin A1 mimetic peptide Ac2-26 enhanced secretion of MMP-1 which was dependent on FPR2/ALX, Erk, Jnk, and NFκB (Tagoe et al., 2008). As mentioned, this study was not performed in synovial Mɸ, but as they have similar actions to synovial fibroblasts, the same results may be produced by these cells once investigated. Further to this, it has also been shown that administration of human recombinant annexin A1 during the immunization phase of the collagen induced arthritis model perpetuated the development of the signs and symptoms of arthritis. This may have been due to the increased T-cell activation and skewing toward a Th1 phenotype by annexin A1 acting through FPR2/ALX (D’Acquisto et al., 2007). T-cells from RA patients 48 h post steroid therapy demonstrated depressed expression of annexin A1 (D’Acquisto et al., 2008) further lending support to the possibility that annexin A1 may also mediate pro-inflammatory actions. However we must be careful to acknowledge the actions of specific cleavage products from full length annexin. One such cleavage product has been identified as causing neutrophil extravasation, an important event in inflammation, where other truncated forms of annexin cannot (Williams et al., 2010).
Conclusion
In the study of inflammation and our efforts to promote its normal resolution, Mɸ remain to the fore of our interest. In the inflamed joint, Mɸ will continue to be a focal point for therapeutic intervention which, currently, centers around cytokine blockade but now has the possibility of extending into Mɸ re-programming. This remains an interesting and a yet to be fully explored option in terms of treatment for synovial inflammation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abdollahi-Roodsaz, S., Joosten, L. A. B., Koenders, M. I., Devesa, I., Roelofs, M. F., Radstake, T. R. D. J., Heuvelmans-Jacobs, M., Akira, S., Nicklin, M. J. H., Ribeiro-Dias, F. T., and Van Den Berg, W. B. (2008). Stimulation of TLR2 and TLR4 differentially skews the balance of T cells in a mouse model of arthritis. J. Clin. Invest. 118, 205–216.
Abeles, A., and Pillinger, M. (2006). The role of the synovial fibroblast in rheumatoid arthritis: cartilage destruction and the regulation of matrix metalloproteinases. Bull. NYU. Hosp. Jt. Dis. 24, 20–24.
Alvaro-Gracia, J. M., Zvaifler, N. J., and Firestein, G. S. (1990). Cytokines in chronic inflammatory arthritis. V. Mutual antagonism between interferon-gamma and tumor necrosis factor-alpha on HLA-DR expression, proliferation, collagenase production, and granulocyte macrophage colony-stimulating factor production by rheumatoid arthritis synoviocytes. J. Clin. Invest. 86, 1790–1798.
Andrén, M., Xiang, Z., Nilsson, G., and Kleinau, S. (2006). FcγRIII-expressing macrophages are essential for development of collagen-induced arthritis. Scand. J. Immunol. 63, 282–289.
Arur, S., Uche, U. E., Rezaul, K., Fong, M., Scranton, V., Cowan, A. E., Mohler, W., and Han, D. K. (2003). Annexin I is an endogenous ligand that mediates apoptotic cell engulfment. Dev. Cell 4, 587–598.
Athanasou, N., Quinn, J., Heryet, A., Puddle, B., Woods, C., and Mcgee, J. (1988). The immunohistology of synovial lining cells in normal and inflamed synovium. J. Pathol. 155, 133–142.
Bertolini, D. R., Nedwin, G. E., Bringman, T. S., Smith, D. D., and Mundy, G. R. (1986). Stimulation of bone resorption and inhibition of bone formation in vitro by human tumour necrosis factors. Nature 319, 516–518.
Biswas, S. K., Gangi, L., Paul, S., Schioppa, T., Saccani, A., Sironi, M., Bottazzi, B., Doni, A., Vincenzo, B., Pasqualini, F., Vago, L., Nebuloni, M., Mantovani, A., and Sica, A. (2006). A distinct and unique transcriptional program expressed by tumor-associated macrophages (defective NF-kB and enhanced IRF-3/STAT1 activation). Blood 107, 2112–2122.
Bohuslav, J., Kravchenko, V. V., Parry, G. C., Erlich, J. H., Gerondakis, S., Mackman, N., and Ulevitch, R. J. (1998). Regulation of an essential innate immune response by the p50 subunit of NF-kappaB. J. Clin. Invest. 102, 1645–1652.
Bondeson, J., Blom, A. B., Wainwright, S., Hughes, C., Caterson, B., and Van Den Berg, W. B. (2010). The role of synovial macrophages and macrophage-produced mediators in driving inflammatory and destructive responses in osteoarthritis. Arthritis Rheum. 62, 647–657.
Bonnelye, E., Laurin, N., Jurdic, P., Hart, D. A., and Aubin, J. E. (2008). Estrogen receptor-related receptor-alpha (ERR-alpha) is dysregulated in inflammatory arthritis. Rheumatology 47, 1785–1791.
Bresnihan, B., Pontifex, E., Thurlings, R. M., Vinkenoog, M., El-Gabalawy, H., Fearon, U., Fitzgerald, O., Gerlag, D. M., Rooney, T., Van De Sande, M. G., Veale, D., Vos, K., and Tak, P.-P. (2009). Synovial tissue sublining CD68 expression is a biomarker of therapeutic response in rheumatoid arthritis clinical trials: consistency across centers. J. Rheumatol. 36, 1800–1802.
Burmester, G., Stuhlmuller, B., Keyszer, G., and Kinne, R. (1997). Mononuclear phagocytes and rheumatoid synovitis. Mastermind or workhorse in arthritis? Arthritis Rheum. 40, 5–18.
Butler, D. M., Maini, R. N., Feldmann, M., and Brennan, F. M. (1995). Modulation of proinflammatory cytokine release in rheumatoid synovial membrane cell cultures. Comparison of monoclonal anti TNF-alpha antibody with the interleukin-1 receptor antagonist. Eur. Cytokine Netw. 6, 225–230.
Canete, J. D., Celis, R., Hernandez, M. V., Pablos, J. L., and Sanmarti, R. (2010). Synovial immunopathological changes associated with successful abatacept therapy in a case of severe refractory psoriatic arthritis. Ann. Rheum. Dis. 69, 935–936.
Canete, J. D., Suarez, B., Hernandez, M. V., Sanmarti, R., Rego, I., Celis, R., Moll, C., Pinto, J. A., Blanco, F. J., and Lozano, F. (2009). Influence of variants of Fc-gamma receptors IIA and IIIA on the American college of rheumatology and European league against rheumatism responses to anti-tumour necrosis factor-alpha therapy in rheumatoid arthritis. Ann. Rheum. Dis. 68, 1547–1552.
Chan, M. M.-Y., and Moore, A. R. (2010). Resolution of inflammation in murine autoimmune arthritis is disrupted by cyclooxygenase-2 inhibition and restored by prostaglandin E2-mediated lipoxin A4 production. J. Immunol. 184, 6418–6426.
Cheng, N., He, R., Tian, J., Ye, P. P., and Ye, R. D. (2008). Cutting edge: TLR2 is a functional receptor for acute-phase serum amyloid A. J. Immunol. 181, 22–26.
Connolly, M., Veale, D. J., and Fearon, U. (2011). Acute serum amyloid A regulates cytoskeletal rearrangement, cell matrix interactions and promotes cell migration in rheumatoid arthritis. Ann. Rheum. Dis. 70, 1296–1303.
Cooper, N. J. (2000). Economic burden of rheumatoid arthritis: a systematic review. Rheumatology 39, 28–33.
D’Acquisto, F., Merghani, A., Lecona, E., Rosignoli, G., Raza, K., Buckley, C. D., Flower, R. J., and Perretti, M. (2007). Annexin-1 modulates T-cell activation and differentiation. Blood 109, 1095–1102.
D’Acquisto, F., Paschalidis, N., Raza, K., Buckley, C. D., Flower, R. J., and Perretti, M. (2008). Glucocorticoid treatment inhibits annexin-1 expression in rheumatoid arthritis CD4+ T cells. Rheumatology 47, 636–639.
Danning, C. L., Illei, G. G., Hitchon, C., Greer, M. R., Boumpas, D. T., and Mcinnes, I. B. (2000). Macrophage-derived cytokine and nuclear factor kappaB p65 expression in synovial membrane and skin of patients with psoriatic arthritis. Arthritis Rheum. 43, 1244–1256.
Dawidowicz, K., Allanore, Y., Guedj, M., Pierlot, C., Bombardieri, S., Balsa, A., Westhovens, R., Barrera, P., Alves, H., Teixeira, V. H., Petit-Teixeira, E., Van De Putte, L., Van Riel, P., Prum, B., Bardin, T., Meyer, O., Cornelis, F., Dieude, P., and Ecraf, F. (2011). The interferon regulatory factor 5 gene confers susceptibility to rheumatoid arthritis and influences its erosive phenotype. Ann. Rheum. Dis. 70, 117–121.
Dayer, J. M., Beutler, B., and Cerami, A. (1985). Cachectin/tumor necrosis factor stimulates collagenase and prostaglandin E2 production by human synovial cells and dermal fibroblasts. J. Exp. Med. 162, 2163–2168.
Decker, Y., Mcbean, G., and Godson, C. (2009). Lipoxin A4 inhibits IL-1b-induced IL-8 and ICAM-1 expression in 1321N1 human astrocytoma cells. Am. J. Physiol. Cell Physiol. 296, C1420–C1427.
Desreumaux, P., Dubuquoy, L., Nutten, S., Peuchmaur, M., Englaro, W., Schoonjans, K., Derijard, B., Desvergne, B., Wahli, W., Chambon, P., Leibowitz, M. D., Colombel, J.-F. D. R., and Auwerx, J. (2001). Attenuation of colon inflammation through activators of the retinoid X receptor (Rxr)/peroxisome proliferator-activated receptor g (Pparg) heterodimer. J. Exp. Med. 193, 827–838.
Dideberg, V., Kristjansdottir, G., Milani, L., Libioulle, C., Sigurdsson, S., Louis, E., Wiman, A.-C., Vermeire, S., Rutgeerts, P., Belaiche, J., Franchimont, D., Van Gossum, A., Bours, V., and Syvanen, A.-C. (2007). An insertion/deletion polymorphism in the interferon regulatory factor 5 (IRF5) gene confers risk of inflammatory bowel diseases. Hum. Mol. Genet. 16, 3008–3016.
Dieguez-Gonzalez, R., Calaza, M., Perez-Pampin, E., De La Serna, A. R., Fernandez-Gutierrez, B., Castañeda, S., Largo, R., Joven, B., Narvaez, J., Navarro, F., Marenco, J. L., Vicario, J. L., Blanco, F. J., Fernandez-Lopez, J. C., Caliz, R., Collado-Escobar, M. D., Carreño, L., Lopez-Longo, J., Cañete, J. D., Gomez-Reino, J. J., and Gonzalez, A. (2008). Association of interferon regulatory factor 5 haplotypes, similar to that found in systemic lupus erythematosus, in a large subgroup of patients with rheumatoid arthritis. Arthritis Rheum. 58, 1264–1274.
Elliott, M. J., Maini, R. N., Feldmann, M., Long-Fox, A., Charles, P., Katsikis, P., Brennan, F. M., Walker, J., Bijl, H., Ghrayeb, J., and Woody, J. N. (1993). Treatment of rheumatoid arthritis with chimeric monoclonal antibodies to tumor necrosis factor α. Arthritis Rheum. 36, 1681–1690.
Erwig, L.-P., Kluth, D. C., Walsh, G. M., and Rees, A. J. (1998). Initial cytokine exposure determines function of macrophages and renders them unresponsive to other cytokines. J. Immunol. 161, 1983–1988.
Ferlazzo, V., D’agostino, P., Milano, S., Caruso, R., Feo, S., Cillari, E., and Parente, L. (2003). Anti-inflammatory effects of annexin-1: stimulation of IL-10 release and inhibition of nitric oxide synthesis. Int. Immunopharmacol. 3, 1363–1369.
Filep, J. N. G., Zouki, C., Petasis, N. A., Hachicha, M., and Serhan, C. N. (1999). Anti-inflammatory actions of lipoxin A4 stable analogs are demonstrable in human whole blood: modulation of leukocyte adhesion molecules and inhibition of neutrophil-endothelial interactions. Blood 94, 4132–4142.
Fiore, S., Maddox, J. F., Perez, H. D., and Serhan, C. N. (1994). Identification of a human cDNA encoding a functional high affinity lipoxin A4 receptor. J. Exp. Med. 180, 253–260.
Firestein, G. S., and Zvaifler, N. J. (1990). How important are T cells in chronic rheumatoid synovitis? Arthritis Rheum. 33, 768–773.
Flower, R. J. (1988). Eleventh Gaddum memorial lecture. Lipocortin and the mechanism of action of the glucocorticoids. Br. J. Pharmacol. 94, 987–1015.
Franklin, E. C., Holman, H. R., Muller-Eberhard, H. J., and Kunkel, H. G. (1957). An unusual protein component of high molecular weight in the serum of certain patients with rheumatoid arthritis. J. Exp. Med. 105, 425–438.
Godson, C., Mitchell, S., Harvey, K., Petasis, N. A., Hogg, N., and Brady, H. R. (2000). Cutting edge: lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J. Immunol. 164, 1663–1667.
Goedkoop, A. Y., Kraan, M. C., Teunissen, M. B. M., Picavet, D. I., De Rie, M. A., Bos, J. D., and Tak, P. P. (2004). Early effects of tumour necrosis factor {alpha} blockade on skin and synovial tissue in patients with active psoriasis and psoriatic arthritis. Ann. Rheum. Dis. 63, 769–773.
Goulding, N. J., Dixey, J., Morand, E. F., Dodds, R. A., Wilkinson, L. S., Pitsillides, A. A., and Edwards, J. C. (1995). Differential distribution of annexins-I, -II, -IV, and -VI in synovium. Ann. Rheum. Dis. 54, 841–845.
Goulding, N. J., Jefferiss, C. M., Pan, L., Rigby, W. F., and Guyre, P. M. (1992). Specific binding of lipocortin-1 (annexin I) to monocytes and neutrophils is decreased in rheumatoid arthritis. Arthritis Rheum. 35, 1395–1397.
Grabiec, A. M., Krausz, S., De Jager, W., Burakowski, T., Groot, D., Sanders, M. E., Prakken, B. J., Maslinski, W., Eldering, E., Tak, P. P., and Reedquist, K. A. (2010). Histone deacetylase inhibitors suppress inflammatory activation of rheumatoid arthritis patient synovial macrophages and tissue. J. Immunol. 184, 2718–2728.
Grabiec, A. M., Tak, P. P., and Reedquist, K. A. (2008). Targeting histone deacetylase activity in rheumatoid arthritis and asthma as prototypes of inflammatory disease: should we keep our HATs on? Arthritis Res. Ther. 10, 226.
Graham, R. R., Kyogoku, C., Sigurdsson, S., Vlasova, I. A., Davies, L. R. L., Baechler, E. C., Plenge, R. M., Koeuth, T., Ortmann, W. A., Hom, G., Bauer, J. W., Gillett, C., Burtt, N., Cunninghame Graham, D. S., Onofrio, R., Petri, M., Gunnarsson, I., Svenungsson, E., Ronnblom, L., Nordmark, G., Gregersen, P. K., Moser, K., Gaffney, P. M., Criswell, L. A., Vyse, T. J., Syvanen, A.-C., Bohjanen, P. R., Daly, M. J., Behrens, T. W., and Altshuler, D. (2007). Three functional variants of IFN regulatory factor 5 (IRF5) define risk and protective haplotypes for human lupus. Proc. Natl. Acad. Sci. 104, 6758–6763.
Han, S. W., Lee, W. K., Kwon, K. T., Lee, B. K., Nam, E. J., and Kim, G. W. (2009). Association of polymorphisms in interferon regulatory factor 5 gene with rheumatoid arthritis: a metaanalysis. J. Rheumatol. 36, 693–697.
Han, Z., Boyle, D. L., Manning, A. M., and Firestein, G. S. (1998). AP-1 and NF-kB regulation in rheumatoid arthritis and murine collagen-induced arthritis. Autoimmunity 28, 197–208.
Handel, M. L., Mcmorrow, L. B., and Gravallese, E. M. (1995). Nuclear factor–kB in rheumatoid synovium. Localization of P50 and P65. Arthritis Rheum. 38, 1762–1770.
Haringman, J. J., Gerlag, D. M., Zwinderman, A. H., Smeets, T. J. M., Kraan, M. C., Baeten, D., Mcinnes, I. B., Bresnihan, B., and Tak, P. P. (2005). Synovial tissue macrophages: a sensitive biomarker for response to treatment in patients with rheumatoid arthritis. Ann. Rheum. Dis. 64, 834–838.
Heo, Y.-J., Joo, Y.-B., Oh, H.-J., Park, M.-K., Heo, Y.-M., Cho, M.-L., Kwok, S.-K., Ju, J.-H., Park, K.-S., Cho, S. G., Park, S.-H., Kim, H.-Y., and Min, J.-K. (2010). IL-10 suppresses Th17 cells and promotes regulatory T cells in the CD4+ T cell population of rheumatoid arthritis patients. Immunol. Lett. 127, 150–156.
Hong, C., Walczak, R., Dhamko, H., Bradley, M. N., Marathe, C., Boyadjian, R., Salazar, J. V., and Tontonoz, P. (2011). Constitutive activation of LXR in macrophages regulates metabolic and inflammatory gene expression: identification of ARL7 as a direct target. J. Lipid Res. 52, 531–539.
Huang, Q., Ma, Y., Adebayo, A., and Pope, R. M. (2007). Increased macrophage activation mediated through toll-like receptors in rheumatoid arthritis. Arthritis Rheum. 56, 2192–2201.
Huber, L. C., Brock, M., Hemmatazad, H., Giger, O. T., Moritz, F., Trenkmann, M., Distler, J. H. W., Gay, R. E., Kolling, C., Moch, H., Michel, B. A., Gay, S., Distler, O., and Jüngel, A. (2007). Histone deacetylase/acetylase activity in total synovial tissue derived from rheumatoid arthritis and osteoarthritis patients. Arthritis Rheum. 56, 1087–1093.
Janusz, M., and Hare, M. (1993). Cartilage degradation by cocultures of transformed macrophage and fibroblast cell lines. A model of metalloproteinase-mediated connective tissue degradation. J. Immunol. 150, 1922–1931.
Ji, J.-D., Tassiulas, I., Park-Min, K.-H., Aydin, A., Mecklenbrauker, I., Tarakhovsky, A., Pricop, L., Salmon, J. E., and Ivashkiv, L. B. (2003). Inhibition of interleukin 10 signaling after Fc receptor ligation and during rheumatoid arthritis. J. Exp. Med. 197, 1573–1583.
Joosten, L. A. B., Koenders, M. I., Smeets, R. L., Heuvelmans-Jacobs, M., Helsen, M. M. A., Takeda, K., Akira, S., Lubberts, E., Van De Loo, F. A. J., and Van Den Berg, W. B. (2003). Toll-like receptor 2 pathway drives streptococcal cell wall-induced joint inflammation: critical role of myeloid differentiation factor 88. J. Immunol. 171, 6145–6153.
Keffer, J., Probert, L., Cazlaris, H., Georgopoulos, S., Kaslaris, E., Kioussis, D., and Kollias, G. (1991). Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J. 10, 4025–4031.
Kraan, M. C., Versendaal, H., Jonker, M., Bresnihan, B., Post, W. J., T Hart, B. A., Breedveld, F. C., and Tak, P. P. (1998). Asymptomatic synovitis precedes clinically manifest arthritis. Arthritis Rheum. 41, 1481–1488.
Krausgruber, T., Blazek, K., Smallie, T., Alzabin, S., Lockstone, H., Sahgal, N., Hussell, T., Feldmann, M., and Udalova, I. A. (2011). IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat. Immunol. 12, 231–238.
Kronke, G., Katzenbeisser, J., Uderhardt, S., Zaiss, M. M., Scholtysek, C., Schabbauer, G., Zarbock, A., Koenders, M. I., Axmann, R., Zwerina, J., Baenckler, H. W., Van Den Berg, W., Voll, R. E., Kuhn, H., Joosten, L. A. B., and Schett, G. (2009). 12/15-Lipoxygenase counteracts inflammation and tissue damage in arthritis. J. Immunol. 183, 3383–3389.
Kruithof, E., Rycke, L. D., Vandooren, B., Keyser, F. D., Fitzgerald, O., Mcinnes, I., Tak, P. P., Bresnihan, B., Veys, E. M., Baeten, D., OMERACT Special Interest Group on Synovial Analysis in Clinical Trials. (2006). Identification of synovial biomarkers of response to experimental treatment in early-phase clinical trials in spondylarthritis. Arthritis Rheum. 54, 1795–1804.
Laurent, L., Clavel, C., Lemaire, O., Anquetil, F., Cornillet, M., Zabraniecki, L., Nogueira, L., Fournie, B., Serre, G., and Sebbag, M. (2011). Fc-gamma receptor profile of monocytes and macrophages from rheumatoid arthritis patients and their response to immune complexes formed with autoantibodies to citrullinated proteins. Ann. Rheum. Dis. 70, 1052–1059.
Leibovich, S. J., Polverini, P. J., Shepard, H. M., Wiseman, D. M., Shively, V., and Nuseir, N. (1987). Macrophage-induced angiogenesis is mediated by tumour necrosis factor-[alpha]. Nature 329, 630–632.
Maderna, P., Yona, S., Perretti, M., and Godson, C. (2005). Modulation of phagocytosis of apoptotic neutrophils by supernatant from dexamethasone-treated macrophages and annexin-derived peptide Ac2–26. J. Immunol. 174, 3727–3733.
Mantovani, A., Sica, A., Sozzani, S., Allavena, P., Vecchi, A., and Locati, M. (2004). The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 25, 677–686.
Marlor, C., Webb, D., Bombara, M., Greve, J., and Blue, M. (1992). Expression of vascular cell adhesion molecule-1 in fibroblast like synoviocytes after stimulation with tumor necrosis factor. Am. J. Pathol. 140, 1055–1060.
Martinez, F. O., Sica, A., Mantovani, A., and Locati, M. (2008). Macrophage activation and polarization. Front. Biosci. 13, 453–461.
Mauri, C., Gray, D., Mushtaq, N., and Londei, M. (2003). Prevention of arthritis by interleukin 10-producing B cells. J. Exp. Med. 197, 489–501.
McInnes, I. B., Leung, B. P., and Liew, F. Y. (2000). Cell-cell interactions in synovitis: interactions between T lymphocytes and synovial cells. Arthritis Res. 2, 374–378.
Miceli-Richard, C., Comets, E., Loiseau, P., Puechal, X., Hachulla, E., and Mariette, X. (2007). Association of an IRF5 gene functional polymorphism with Sjögren’s syndrome. Arthritis Rheum. 56, 3989–3994.
Milman, N., Karsh, J., and Booth, R. A. (2010). Correlation of a multi-cytokine panel with clinical disease activity in patients with rheumatoid arthritis. Clin. Biochem. 43, 1309–1314.
Miltenburg, A. M. M., Van Laar, J. M., De Kniper, M. R., Daha, M. R., and Breedveld, F. C. (1992). T cells cloned from human rheumatoid synovial membrane functionally represent the Th1 subset. Scand. J. Immunol. 35, 603–610.
Morales-Lara, M., Conesa-Zamora, P., Garcia-Simon, M., Pedrero, F., Santaclara, V., Perez-Guillermo, M., and Soriano-Navarro, E. (2010). Association between the FCGR3A V158F polymorphism and the clinical response to infliximab in rheumatoid arthritis and spondyloarthritis patients. Scand. J. Rheumatol. 39, 518–520.
Morgan, A. W., Barrett, J. H., Griffiths, B., Subramanian, D., Robinson, J. I., Keyte, V. H., Ali, M., Jones, E. A., Old, R. W., Ponchel, F., Boylston, A. W., Situnayake, R. D., Markham, A. F., Emery, P., and Isaacs, J. D. (2006). Analysis of Fcgamma receptor haplotypes in rheumatoid arthritis: FCGR3A remains a major susceptibility gene at this locus, with an additional contribution from FCGR3B. Arthritis Res. Ther. 8, R5.
Mottonen, M., Isomaki, P., Saario, R., Toivanen, P., Punnonen, J., and Lassila, O. (1998). Interleukin-10 inhibits the capacity of synovial macrophages to function as antigen-presenting cells. Rheumatology 37, 1207–1214.
Mulherin, D., Fitzgerald, O., and Bresnihan, B. (1996). Synovial tissue macrophage populations and articular damage in rheumatoid arthritis. Arthritis Rheum. 39, 115–124.
Mullan, R. H., Bresnihan, B., Golden-Mason, L., Markham, T., O’hara, R., Fitzgerald, O., Veale, D. J., and Fearon, U. (2006). Acute-phase serum amyloid A stimulation of angiogenesis, leukocyte recruitment, and matrix degradation in rheumatoid arthritis through an NF-kappaB-dependent signal transduction pathway. Arthritis Rheum. 54, 105–114.
Murphy, E. P., Mcevoy, A., Conneely, O. M., Bresnihan, B., and Fitzgerald, O. (2001). Involvement of the nuclear orphan receptor NURR1 in the regulation of corticotropin-releasing hormone expression and actions in human inflammatory arthritis. Arthritis Rheum. 44, 782–793.
Murray, P. J., and Wynn, T. A. (2011). Obstacles and opportunities for understanding macrophage polarization. J. Leukoc. Biol. 89, 557–563.
Nasu, Y., Nishida, K., Miyazawa, S., Komiyama, T., Kadota, Y., Abe, N., Yoshida, A., Hirohata, S., Ohtsuka, A., and Ozaki, T. (2008). Trichostatin A, a histone deacetylase inhibitor, suppresses synovial inflammation and subsequent cartilage destruction in a collagen antibody-induced arthritis mouse model. Osteoarthr. Cartil. 16, 723–732.
Nawroth, P., Bank, I., Handley, D., Cassimeris, J., Chess, L., and Stern, D. (1986). Tumor necrosis factor/cachectin interacts with endothelial cell receptors to induce release of interleukin 1. J. Exp. Med. 163, 1363–1375.
O’Hara, R., Murphy, E. P., Whitehead, A. S., Fitzgerald, O., and Bresnihan, B. (2000). Acute-phase serum amyloid A production by rheumatoid arthritis synovial tissue. Arthritis Res. Ther. 2, 142–144.
Okamura, Y., Watari, M., Jerud, E. S., Young, D. W., Ishizaka, S. T., Rose, J., Chow, J. C., and Strauss, J. F. (2001). The extra domain A of fibronectin activates toll-like receptor 4. J. Biol. Chem. 276, 10229–10233.
Oosterveer, M. H., Grefhorst, A., Groen, A. K., and Kuipers, F. (2010). The liver X receptor: control of cellular lipid homeostasis and beyond: implications for drug design. Prog. Lipid Res. 49, 343–352.
Park, J. S., Svetkauskaite, D., He, Q., Kim, J.-Y., Strassheim, D., Ishizaka, A., and Abraham, E. (2004). Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J. Biol. Chem. 279, 7370–7377.
Park, M.-C., Kwon, Y.-J., Chung, S.-J., Park, Y.-B., and Lee, S.-K. (2010). Liver X receptor agonist prevents the evolution of collagen-induced arthritis in mice. Rheumatology 49, 882–890.
Pei, L., Castrillo, A., and Tontonoz, P. (2006). Regulation of macrophage inflammatory gene expression by the orphan nuclear receptor Nur77. Mol. Endocrinol. 20, 786–794.
Perlman, H., Pagliari, L. J., Liu, H., Koch, A. E., Haines, G. K., and Pope, R. M. (2001). Rheumatoid arthritis synovial macrophages express the Fas-associated death domain–like interleukin-1β–converting enzyme–inhibitory protein and are refractory to Fas-mediated apoptosis. Arthritis Rheum. 44, 21–30.
Perretti, M., and Dalli, J. (2009). Exploiting the annexin A1 pathway for the development of novel anti-inflammatory therapeutics. Br. J. Pharmacol. 158, 936–946.
Podgorski, M. R., Goulding, N. J., Hall, N. D., Flower, R. J., and Maddison, P. J. (1992). Autoantibodies to lipocortin-1 are associated with impaired glucocorticoid responsiveness in rheumatoid arthritis. J. Rheumatol. 19, 1668–1671.
Porta, C., Rimoldi, M., Raes, G., Brys, L., Ghezzi, P., Di Liberto, D., Dieli, F., Ghisletti, S., Natoli, G., De Baetselier, P., Mantovani, A., and Sica, A. (2009). Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor kB. Proc. Natl. Acad. Sci. 106, 14978–14983.
Roiniotis, J., Dinh, H., Masendycz, P., Turner, A., Elsegood, C. L., Scholz, G. M., and Hamilton, J. A. (2009). Hypoxia prolongs monocyte/macrophage survival and enhanced glycolysis is associated with their maturation under aerobic conditions. J. Immunol. 182, 7974–7981.
Saccani, A., Schioppa, T., Porta, C., Biswas, S. K., Nebuloni, M., Vago, L., Bottazzi, B., Colombo, M. P., Mantovani, A., and Sica, A. (2006). p50 Nuclear factor-kB overexpression in tumor-associated macrophages inhibits M1 inflammatory responses and antitumor resistance. Cancer Res. 66, 11432–11440.
Salmon, M., Scheel-Toellner, D., Huissoon, A. P., Pilling, D., Shamsadeen, N., Hyde, H., D’angeac, A. D., Bacon, P. A., Emery, P., and Akbar, A. N. (1997). Inhibition of T cell apoptosis in the rheumatoid synovium. J. Clin. Invest. 99, 439–446.
Sampey, A. V., Hutchinson, P., and Morand, E. F. (2000). Annexin I surface binding sites and their regulation on human fibroblast-like synoviocytes. Arthritis Rheum. 43, 2537–2542.
Satoh, T., Takeuchi, O., Vandenbon, A., Yasuda, K., Tanaka, Y., Kumagai, Y., Miyake, T., Matsushita, K., Okazaki, T., Saitoh, T., Honma, K., Matsuyama, T., Yui, K., Tsujimura, T., Standley, D. M., Nakanishi, K., Nakai, K., and Akira, S. (2010). The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat. Immunol. 11, 936–944.
Scannell, M., Flanagan, M. B., Destefani, A., Wynne, K. J., Cagney, G., Godson, C., and Maderna, P. (2007). Annexin-1 and peptide derivatives are released by apoptotic cells and stimulate phagocytosis of apoptotic neutrophils by macrophages. J. Immunol. 178, 4595–4605.
Schulze-Koops, H., and Kalden, J. R. (2001). The balance of Th1/Th2 cytokines in rheumatoid arthritis. Best Pract. Res. Clin. Rheumatol. 15, 677–691.
Serhan, C. N., Hamberg, M., and Samuelsson, B. (1984). Lipoxins: novel series of biologically active compounds formed from arachidonic acid in human leukocytes. Proc. Natl. Acad. Sci. 81, 5335–5339.
Sodin-Semrl, S., Spagnolo, A., Mikus, R., Barbaro, B., Varga, J., and Fiore, S. (2004). Opposing regulation of interleukin-8 and NF-kappaB responses by lipoxin A4 and serum amyloid A via the common lipoxin A receptor. Int. J. Immunopathol. Pharmacol. 17, 145–156.
Ståhl, T. D. D., Andrén, M., Martinsson, P., Verbeek, J. S., and Kleinau, S. (2002). Expression of FcγRIII is required for development of collagen-induced arthritis. Eur. J. Immunol. 32, 2915–2922.
Stout, R. D., Jiang, C., Matta, B., Tietzel, I., Watkins, S. K., and Suttles, J. (2005). Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J. Immunol. 175, 342–349.
Tagoe, C. E., Marjanovic, N., Park, J. Y., Chan, E. S., Abeles, A. M., Attur, M., Abramson, S. B., and Pillinger, M. H. (2008). Annexin-1 mediates TNF-a stimulated matrix metalloproteinase secretion from rheumatoid arthritis synovial fibroblasts. J. Immunol. 181, 2813–2820.
Tak, P. P., Smeets, T. J. M., Daha, M. R., Kluin, P. M., Meijers, K. A. E., Brand, R., Meinders, A. E., and Breedveld, F. C. (1997). Analysis of the synovial cell infiltrate in early rheumatoid synovial tissue in relation to local disease activity. Arthritis Rheum. 40, 217–225.
Tamura, T., Tailor, P., Yamaoka, K., Kong, H. J., Tsujimura, H., Oâ’shea, J. J., Singh, H., and Ozato, K. (2005). IFN regulatory factor-4 and -8 govern dendritic cell subset development and their functional diversity. J. Immunol. 174, 2573–2581.
Thabet, M. M., Huizinga, T. W. J., Marques, R. B., Stoeken-Rijsbergen, G., Bakker, A. M., Kurreeman, F. A., White, S. J., Toes, R. E. M., and Van Der Helm-Van Mil, A. H. M. (2009). Contribution of Fcgamma receptor IIIA gene 158V/F polymorphism and copy number variation to the risk of ACPA-positive rheumatoid arthritis. Ann. Rheum. Dis. 68, 1775–1780.
Thomas, E., Leroux, J. L., Blotman, F., and Chavis, C. (1995). Conversion of endogenous arachidonic acid to 5,15-diHETE and lipoxins by polymorphonuclear cells from patients with rheumatoid arthritis. Inflamm. Res. 44, 121–124.
Thurlings, R. M., Vos, K., Wijbrandts, C. A., Zwinderman, A. H., Gerlag, D. M., and Tak, P. P. (2008). Synovial tissue response to rituximab: mechanism of action and identification of biomarkers of response. Ann. Rheum. Dis. 67, 917–925.
Tsao, P. W., Suzuki, T., Totsuka, R., Murata, T., Takagi, T., Ohmachi, Y., Fujimura, H., and Takata, I. (1997). The effect of dexamethasone on the expression of activated NF-[kappa]B in adjuvant arthritis. Clin. Immunol. Immunopathol. 83, 173–178.
Vallejo, A. N., Yang, H., Klimiuk, P. A., Weyand, C. M., and Goronzy, J. J. (2003). Synoviocyte-mediated expansion of inflammatory T cells in rheumatoid synovitis is dependent on CD47-thrombospondin 1 interaction. J. Immunol. 171, 1732–1740.
van Lent, P. L. E. M., Licht, R., Dijkman, H., Holthuysen, A. E. M., Berden, J. H. M., and Van Den Berg, W. B. (2001). Uptake of apoptotic leukocytes by synovial lining macrophages inhibits immune complex–mediated arthritis. J. Leukoc. Biol. 70, 708–714.
Vandooren, B., Noordenbos, T., Ambarus, C., Krausz, S., Cantaert, T., Yeremenko, N., Boumans, M., Lutter, R., Tak, P. P., and Baeten, D. (2009). Absence of a classically activated macrophage cytokine signature in peripheral spondylarthritis, including psoriatic arthritis. Arthritis Rheum. 60, 966–975.
Waaler, E. (1940). On the occurrence of a factor in human serum activating the specific agglutintion of sheep blood corpuscles. APMIS 115, 422–438.
Wahid, S., Blades, M. C., Lord, D. D., Brown, I., Blake, G., Yanni, G., Haskard, D. O., Panayi, G. S., and Pitzalis, C. (2000). Tumour necrosis factor-alpha (TNF-alpha) enhances lymphocyte migration into rheumatoid synovial tissue transplanted into severe combined immunodeficient (SCID) mice. Clin. Exp. Immunol. 122, 133–142.
Wakamatsu, K., Nanki, T., Miyasaka, N., Umezawa, K., and Kubota, T. (2005). Effect of a small molecule inhibitor of nuclear factor-κB nuclear translocation in a murine model of arthritis and cultured human synovial cells. Arthritis Res. Ther. 7, R1348–R1359.
Wallet, M. A., Wallet, S. M., Guiulfo, G., Sleasman, J. W., and Goodenow, M. M. (2010). IFN-gamma primes macrophages for inflammatory activation by high molecular weight hyaluronan. Cell. Immunol. 262, 84–88.
Walmsley, M., Katsikis, P. D., Abney, E., Parry, S., Williams, R. O., Maini, R. N., and Feldmann, M. (1996). Interleukin-10 inhibition of the progression of established collagen-induced arthritis. Arthritis Rheum. 39, 495–503.
Whalen, J. D., Lechman, E. L., Carlos, C. A., Weiss, K., Kovesdi, I., Glorioso, J. C., Robbins, P. D., and Evans, C. H. (1999). Adenoviral transfer of the viral IL-10 Gene periarticularly to mouse paws suppresses development of collagen-induced arthritis in both injected and uninjected paws. J. Immunol. 162, 3625–3632.
Williams, R. O., Feldmann, M., and Maini, R. N. (1992). Anti-tumor necrosis factor ameliorates joint disease in murine collagen-induced arthritis. Proc. Natl. Acad. Sci. 89, 9784–9788.
Williams, S. L., Milne, I. R., Bagley, C. J., Gamble, J. R., Vadas, M. A., Pitson, S. M., and Khew-Goodall, Y. (2010). A proinflammatory role for proteolytically cleaved annexin A1 in neutrophil transendothelial migration. J. Immunol. 185, 3057–3063.
Yang, Y., Hutchinson, P., and Morand, E. F. (1999). Inhibitory effect of annexin I on synovial inflammation in rat adjuvant arthritis. Arthritis Rheum. 42, 1538–1544.
Keywords: macrophage, arthritis, inflammation
Citation: Kennedy A, Fearon U, Veale DJ and Godson C (2011) Macrophages in synovial inflammation. Front. Immun. 2:52. doi: 10.3389/fimmu.2011.00052
Received: 30 June 2011; Paper pending published: 26 July 2011;
Accepted: 19 September 2011; Published online: 10 October 2011.
Edited by:
Amiram Ariel, University of Haifa, IsraelReviewed by:
Fulvio D’Acquisto, Queen Mary University of London, UKFrancesco Saverio Di Giovine, University of Sheffield, UK
Copyright: © 2011 Kennedy, Fearon, Veale and Godson. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Catherine Godson, School of Medicine and Medical Sciences, University College Dublin Conway Institute, Belfield, Dublin 4, Ireland. e-mail:Y2F0aGVyaW5lLmdvZHNvbkB1Y2QuaWU=