
 S. Courtney Frasch
S. Courtney Frasch
- Division of Cell Biology, Department of Pediatrics, National Jewish Health, Denver, CO, USA
A critical function of macrophages within the inflammatory milieu is the removal of dying cells by a specialized phagocytic process called efferocytosis (“to carry to the grave”). Through specific receptor engagement and induction of downstream signaling, efferocytosing macrophages promote resolution of inflammation by (i) efficiently engulfing dying cells, thus avoiding cellular disruption and release of inflammatory contents, and (ii) producing anti-inflammatory mediators such as IL-10 and TGF-β that dampen pro-inflammatory responses. Evidence suggests that plasticity in macrophage programming, in response to changing environmental cues, modulates efferocytic capability. Essential to programming for enhanced efferocytosis is activation of the nuclear receptors PPARγ, PPARδ, LXR, and possibly RXRα. Additionally, a number of signals in the inflammatory milieu, including those from dying cells themselves, can influence efferocytic efficacy either by acting as immediate inhibitors/enhancers or by altering macrophage programming for longer-term effects. Importantly, sustained inflammatory programming of macrophages can lead to defective apoptotic cell clearance and is associated with development of autoimmunity and other chronic inflammatory disorders. This review summarizes the current knowledge of the multiple factors that modulate macrophage efferocytic ability and highlights emerging therapeutic targets with significant potential for limiting chronic inflammation.
Introduction
Efferocytosis, or engulfment of apoptotic cells by macrophages is an essential process with roles in tissue homeostasis, embryologic development, immunity, and resolution of inflammation, the latter, the focus of this review. Apoptotic cells exhibit surface changes, especially exposure of the plasma membrane inner leaflet phospholipid phosphatidylserine (PS) or its oxidized forms (Fadok et al., 1992; Greenberg et al., 2006), that distinguish them from viable cells and allow recognition by a multiplicity of macrophage efferocytic receptors (Table 1). Other surface ligands on apoptotic cells, e.g., calreticulin and deposited complement, are reviewed elsewhere (Henson and Bratton, 2009). In many instances, factors in plasma (or serum), often produced by macrophages themselves, are utilized as bridge molecules to couple apoptotic cells to the macrophage receptors (Table 1). Engagement of efferocytic receptors initiates signaling events modulated by two main complexes, CrkII/ELMO/Dock180 (Gumienny et al., 2001) or ABCA1/GULP (Kinchen et al., 2005), both resulting in activation of Rac1, which initiates cytoskeletal rearrangement and subsequent engulfment. Rac1 and RhoA, two small Rho GTPases, have opposing roles in regulating efferocytosis; Rac1 enhances, while RhoA inhibits the process (Leverrier and Ridley, 2001; Nakaya et al., 2006). Thus, the relative balance between them plays a key role in determining macrophage efferocytic ability, and imbalance favoring active RhoA can lead to defective clearance.
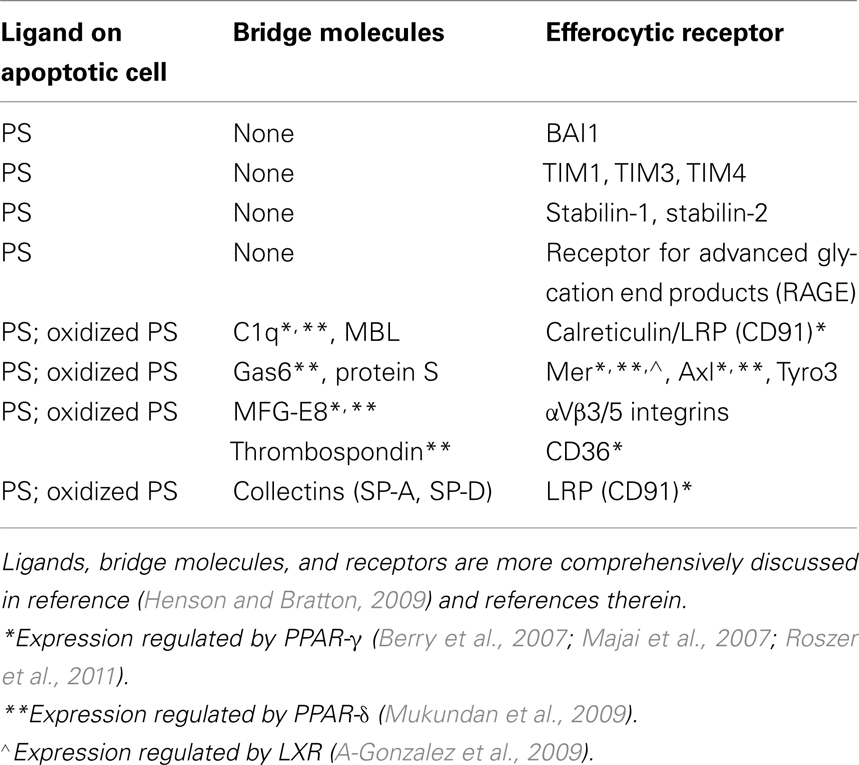
Table 1. Phosphatidylserine, bridge molecules, and receptors for apoptotic cells including those known to be modulated by nuclear receptor signaling.
Macrophages subserve key roles during inflammation and its resolution. As innate immune sentinels, resident tissue macrophages detect and interpret signals indicating tissue injury or pathogen infiltration and initiate responses through release of cytokines and other mediators. Consequently, leukocyte recruitment ensues, including blood monocytes that differentiate into macrophages. Changing cues in the inflammatory milieu alter macrophage programming and modulate various endocytic functions. Phagocytosis and macropinocytosis are essential for removal of infectious agents, antigen presentation, and activation of adaptive immune responses, while efferocytosis can help initiate restoration of tissue structure and function. Mechanistically, efferocytosis resembles stimulated macropinocytosis and is morphologically distinct from classic forms of phagocytosis (Ogden et al., 2001; Henson and Bratton, 2009). Due to differences in recognition mechanisms, engagement, and signaling pathways, it is not surprising that environmental factors have differential effects on the various forms of endocytosis (Gratchev et al., 2006; Feng et al., 2011).
Apoptotic cell recognition and efferocytosis by macrophages has a profound influence on resolution of inflammation, largely through secretion of anti-inflammatory cytokines, such as TGF-β and IL-10 that inhibit inflammatory mediator production (Voll et al., 1997; Fadok et al., 1998). Thus, macrophages responding to PS on apoptotic cells have been shown to accelerate resolution of LPS-induced lung inflammation in a TGF-β dependent manner (Huynh et al., 2002), and infusion of PS liposomes attenuated inflammation in murine skin edema and myocardial infarction models (Ramos et al., 2007; Harel-Adar et al., 2011). Additionally, local injection of PS liposomes decreased the amplitude of a CD4+ T cell response (Hoffmann et al., 2005) indicating that apoptotic cell signaling can modulate adaptive immune responses. Finally, efferocytosis of dying cells prevents their deterioration and release of phlogistic intracellular contents that can contribute to inflammation and autoimmunity. Given the impact of efferocytosis on suppression of inflammation, restoration of tissue homeostasis, and shaping the immune response, this review will focus on factors in the tissue environment that modulate the ability of macrophages to carry out these functions.
Section 1: Macrophage Programming and Modulation of Efferocytosis
The increasingly complex list of so-called macrophage “phenotypes” have been comprehensively reviewed elsewhere (Benoit et al., 2008; Mosser and Zhang, 2008; Martinez et al., 2009) The concept of phenotypes, while useful, is oversimplified, because macrophages exhibit substantial plasticity, with markers and functions readily altered by external signals (Gratchev et al., 2006; Fernandez-Boyanapalli et al., 2009). Accordingly, we argue that it is important to address the functional attributes of macrophages under study or discussion, leading us to focus, here, on those associated with differential capacity for efferocytosis. In general terms, “classically activated” or M1 macrophages demonstrate diminished efferocytosis, increased phagocytosis of foreign organisms, and enhanced production of pro-inflammatory cytokines, reactive oxygen species (ROS), and nitric oxide (NO; Benoit et al., 2008; Figure 1). This M1 programming state results from stimulation with LPS and the innate cytokine IFNγ, or can be elicited by IFNα/β-inducing TLR agonists (Mosser and Zhang, 2008), leading to inhibition of efferocytosis in part through TNFα and/or oxidant-mediated mechanisms (McPhillips et al., 2007; Michlewska et al., 2009). Because M1 macrophages are also associated with Th1 T cell polarization, they exhibit protection against bacterial infection. Such macrophages are evident early in the course of infection or injury, e.g., peritonitis (Fernandez-Boyanapalli et al., 2010a) and are designated in this review as inflammatory, “efferocytic-low” macrophages.
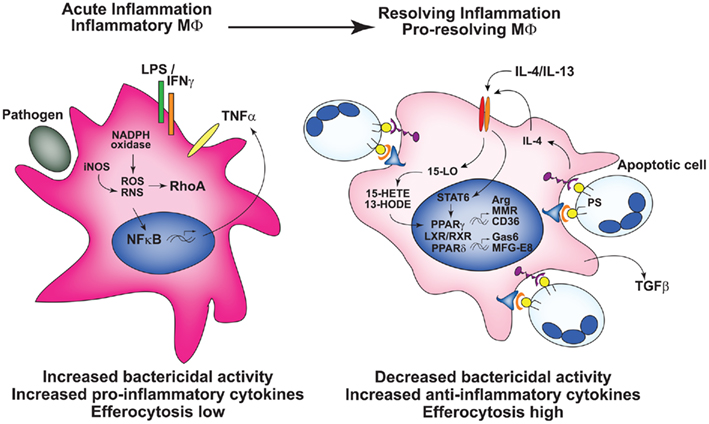
Figure 1. Macrophage programming either in vitro or in vivo determines efferocytic capacity. Left: Inflammatory macrophages, e.g., generated by stimuli such as LPS + IFNγ have heightened bactericidal activity and production of pro-inflammatory mediators, and are programmed poorly for efferocytosis. Right: Following activation of the nuclear receptors, pro-resolving, or resolution-phase macrophages are programmed for heightened efferocytosis, with increased expression of receptors and bridge molecules required for the recognition of apoptotic cells, and the production of anti-inflammatory cytokines. In vivo, over the course of acute inflammation macrophage programming and function transitions from that of inflammatory macrophages to resolution-phase macrophages.
On the opposite end of the spectrum, various “alternatively activated” or M2 macrophages can be elicited by IL-4 and IL-13 (M2a), a combination of TLR agonists and immune complexes (M2b), glucocorticoids and IL-10 (M2c), M-CSF, or TGF-β (Xu et al., 2006; Benoit et al., 2008; Mosser and Zhang, 2008; Martinez et al., 2009). Collectively, these macrophages exhibit increased expression and/or activity of the nuclear receptors, PPARγ and PPARδ, essential to their acquisition of “alternative activation” (Bouhlel et al., 2007; Odegaard et al., 2007). They often display increased levels of arginase, certain receptors (e.g., the macrophage mannose receptor) and anti-inflammatory cytokines, whereas production of pro-inflammatory cytokines, RNS, and ROS are downregulated (Benoit et al., 2008; Olefsky and Glass, 2010; Figure 1). The majority of these “alternative activation” states have been associated with enhanced efferocytosis of apoptotic cells (and likely also necrotic cells and cellular debris) supporting a role in resolution of inflammation. As such, these macrophages are seen later during the resolution phase of inflammation, and henceforth, are called pro-resolving, or “efferocytic-high” (Bystrom et al., 2008; Fernandez-Boyanapalli et al., 2010a; Schif-Zuck et al., 2011). Whether the in vivo shift from inflammatory to pro-resolving state is attributable to recruitment, or expansion of different macrophage populations within the milieu at differing stages of inflammation (Jenkins et al., 2011), or rather represents given macrophages responding to the changing milieu with a switch in programming (Bystrom et al., 2008), or a combination, remains an important, and largely unanswered question for most inflammatory processes. Importantly, two additional caveats deserve mention: (i) much of the literature is based on programming of murine macrophages which likely differs from human, and (ii) cultured macrophages (e.g., M-CSF-treated human monocyte-derived macrophages or murine bone marrow macrophages) are programmed during culture with substantial influences on subsequent responses (Fernandez-Boyanapalli et al., 2009).
Macrophage Programming for Enhanced Efferocytosis and Anti-Inflammatory Consequences
Of the pro-resolving programming states, those elicited by IL-4 and IL-13 are the most thoroughly studied in relation to enhanced efferocytic capacity. IL-4 and IL-13 increase expression and activity of the nuclear receptor PPARγ via STAT6 (Welch et al., 2003; Berry et al., 2007; Szanto et al., 2010). IL-4 also induces production of potential PPARγ-activating ligands, 13-HODE and15-HETE through 15-lipoxygenase activity (Huang et al., 1999). Macrophage PPARγ activation, in turn, has three consequences relevant to this review: (i) “alternative activation” with increased efferocytic surface receptors (Table 1) and secretion of the bridge molecule adiponectin; (ii) enhanced efferocytic capability; and (iii) suppression of inflammation. For some macrophage populations, IL-4/IL-13-induced PPARγ signaling enhances efferocytosis specifically (Fernandez-Boyanapalli et al., 2009), while in others, it non-specifically enhances other phagocytic functions: e.g., uptake of opsonized cells (Aronoff et al., 2004), parasitized RBCs (Serghides and Kain, 2001), and yeast (Gales et al., 2010). An overall increase in phagocytic ability, especially for fungal and parasitic pathogens, is likely associated with PPARγ-mediated upregulation of fungal and parasitic recognition receptors and related to the role of “alternatively activated” macrophages in immunity against Th2 response-inducing pathogens (Raes et al., 2005; Gales et al., 2010).
IL-4/IL-13 also enhanced macrophage PPARδ expression, and expression and release of bridge molecules (Table 1), and acquisition of anti-inflammatory functions (Kang et al., 2008). Both these PPARs are known to heterodimerize with other nuclear receptors to exert these actions, and accordingly, roles for LXR and RXRα in enhanced efferocytosis have been demonstrated (A-Gonzalez et al., 2009; Mukundan et al., 2009; Rebe et al., 2009; Roszer et al., 2011). Direct connections between IL-4/IL-13 and LXR and RXRα are still to be determined.
IL-4 also increases expression of the efferocytic receptors, stabilin-1 and stabilin-2, although connections with nuclear receptor signaling have not been made (Park et al., 2009). Efferocytic programming of macrophages by cytokines, such as M-CSF, IL-10, and TGF-β, are described but less understood. Likewise, pathways for the expression of other apoptotic cell receptors, and indeed the differences in the repertoire of receptors utilized by macrophages in different tissues/milieus is poorly defined (Henson and Bratton, 2009).
An important and emerging concept is that macrophage recognition of apoptotic cells themselves can reinforce signaling pathways that shift their programming toward enhanced efferocytic ability in a feedforward manner (Figure 1): e.g., apoptotic cell-induced PPARγ, PPARδ, and LXR activation results in enhanced CD36 and Mer expression and secretion of efferocytic bridge molecules (A-Gonzalez et al., 2009; Mukundan et al., 2009; Roszer et al., 2011). One mechanism by which apoptotic cells enhance efferocytic programming is through PS-dependent induction of IL-4 signaling to upregulate PPARγ (Fernandez-Boyanapalli et al., 2009). Autocrine stimulation by TGF-β produced in response to apoptotic cell recognition may similarly enhance PPARγ expression (Freire-de-Lima et al., 2006).
Suppression of inflammation also results from activation of these signaling pathways. Stimulation of PPARγ, PPARδ, and LXR, RXRα are associated with reduced production of pro-inflammatory mediators (Ghisletti et al., 2007; Mukundan et al., 2009; Roszer et al., 2011). For example, PPARγ and LXR associate with the co-repressor complex NCoR inhibiting its removal from transcriptional binding sites driven by inflammatory cytokines. Sustained NCoR occupation inhibits NF-κB down-regulating transcription of its target genes, e.g., TNFα and IL-1β (Pascual et al., 2005; Ghisletti et al., 2007; Jennewein et al., 2008). Recognition of apoptotic cells also down-regulates pro-inflammatory mediators through PS-induced stimulation of TGF-β production (Huynh et al., 2002; Freire-de-Lima et al., 2006). Observations such as these reinforce the concept of plasticity in macrophage programming during the course of an inflammatory response with early induction of protective properties and later development of restorative activities to promote resolution (Figure 1). Though the focus here is the role of efferocytic macrophages in the resolution of acute inflammation, it should also be noted that “pro-resolving” macrophages ameliorate chronic inflammation (Wang et al., 2007; Gordon and Martinez, 2010). Alternatively, depending on context, consequences of such programming may be detrimental. For instance, sustained production of TGFβ may lead to over-exuberant fibrotic responses, and there is increasing evidence that the cultivation of immunosuppressive tumor-associated macrophages promote the growth, invasion, and immune evasion of tumors (Martinez et al., 2009; Sica, 2010). While important, these downstream consequences are beyond the scope of this review and the reader is directed to recent references on the subject.
Mechanisms by Which Inflammatory Macrophage Programming Decreases Efferocytic Capacity
In the other direction, suppression of efferocytosis, but not other forms of phagocytosis, is likely mediated both by early acting inhibitors (below) as well as inflammatory macrophage programming (Michlewska et al., 2009; Feng et al., 2011). This suggests effects on pathways that are unique to the recognition and uptake of apoptotic cells. Evidence for programming-related suppression of efferocytosis stems from studies of macrophages stimulated with LPS demonstrating decreased expression of PPARγ (Welch et al., 2003), and decreased transcription and serum levels of efferocytosis-associated bridge molecules Gas6 and MFG-E8 (Komura et al., 2009; Feng et al., 2011). Likewise, LPS induces the transcription factor IRF5, which suppresses expression of efferocytic receptors CD36 and CD14 along with “alternative activation” markers as it up-regulates pro-inflammatory cytokine production (Krausgruber et al., 2011). Thus, the balance between expression of IRF5 and IRF4, the latter driven by IL-4, governs “alternative activation” marker expression (El Chartouni et al., 2010) and likely plays a role in determining macrophage efferocytic capacity. Interestingly, stimulation of macrophages with the other “classical activation” stimulus IFNγ by itself enhances phagocytosis of both apoptotic cells and IgG opsonized targets in a NO-dependent manner involving Rac activation (Fernandez-Boyanapalli et al., 2010b) suggesting further complexity in the regulation of efferocytosis.
Section 2: Early Acting Enhancers and Inhibitors in the Inflammatory Milieu that Modulate Efferocytosis
In addition to macrophage programming modulators, the inflammatory milieu contains numerous serum and cell-derived factors that have immediate (or near immediate) early effects on efferocytosis (Figure 2) separate from the slower and more prolonged consequences of macrophage programming. Many of these early acting effectors have been found to shift the balance between Rho A activation (inhibitory) and Rac 1 activation (enhancing; Figure 2).
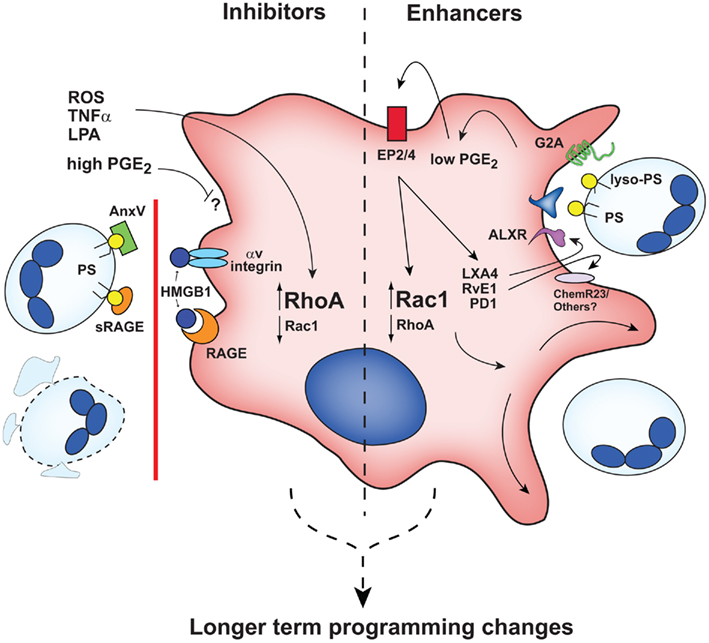
Figure 2. Early acting inhibitors and enhancers influence efferocytic capacity of macrophages. Rho/Rac balance is altered by inhibitors (left) and enhancers (right). ROS, reactive oxygen species; AnxV, annexin A5; other abbreviations are defined in the text. It is likely that early acting agents also contribute to longer-term macrophage programming as illustrated in Figure 1.
Early Acting Inhibitors
Previous studies demonstrated that LPS-mediated suppression of efferocytosis was TNFα dependent (Michlewska et al., 2009; Feng et al., 2011) and that short-term exposure to TNFα inhibited macrophage efferocytosis in an oxidant-dependent manner involving RhoA activation (McPhillips et al., 2007; Moon et al., 2010). Similarly, lysophosphatidic acid (LPA) inhibits efferocytosis through activation of RhoA (Morimoto et al., 2006). Other inhibitors of efferocytosis in the inflammatory milieu act by blocking macrophage recognition of apoptotic cells. These include high mobility group box protein 1 (HMGB1), soluble receptor for advanced glycation end products (RAGE), and annexin A5. HMGB1 binds various macrophage receptors: its binding to RAGE blocks its recognition of PS, and binding to αV integrins blocks interactions with the bridge molecule, MFG-E8 (Table 1; Banerjee et al., 2010; Friggeri et al., 2010, 2011; He et al., 2011). HMGB1 binding to RAGE also reinforces an inflammatory macrophage state by stimulating NF-κB and enhancing pro-inflammatory cytokine production (Qin et al., 2009). Alternatively, masking of PS on apoptotic cells also impairs efferocytosis: soluble RAGE (He et al., 2011) and annexin A5 (unlike annexin A1, see below; Kenis et al., 2006) block macrophage recognition of apoptotic cells by this mechanism. Collectively, these signals downregulate efferocytosis and are likely to reinforce properties of inflammatory macrophages.
Early Acting Enhancers
Early acting enhancers are defined operationally as agents that rapidly augment the capacity and/or the capability of macrophages to engulf apoptotic cells, often by increasing Rac 1 activity (Figure 2). Such factors are found on apoptotic cells and in the milieu, and many are made and released by macrophages themselves. Most enhancers identified to date appear to be modified lipids. A distinguishing feature is their inability to drive efferocytosis on their own (Frasch et al., 2011), i.e., they act by enhancing existing stimuli. By contrast, PS or oxidized PS (Table 1), termed inducers, drive macrophages to take up even viable cells when they are inserted into the target cell membrane (Fadok et al., 1992; Greenberg et al., 2006).
A modified phosphatidylserine species, lysophosphatidylserine (lyso-PS), is an early acting enhancer produced in activated and apoptosing neutrophils through an NADPH-oxidase dependent pathway. In sterile peritonitis, lyso-PS is localized to the neutrophil surface with its accumulation peaking at a time immediately preceding rapid neutrophil clearance by macrophages (Frasch et al., 2008). Lyso-PS acts through the macrophage G2A receptor stimulating the production of prostaglandin E2 (PGE2) leading to cAMP and PKA-dependent augmentation of Rac1 activity. While it has previously been reported that PGE2 inhibits efferocytosis, this effect appears to be concentration dependent; concentrations less than or equal to 1 nM in fact enhance efferocytosis, whereas concentrations greater than 10 nM are inhibitory (Rossi et al., 1998; Frasch et al., 2011). Macrophage production of PGE2 following efferocytosis (Voll et al., 1997; Fadok et al., 1998) has been demonstrated to impair anti-microbial functions of macrophages (Aronoff et al., 2004; Medeiros et al., 2009), and whether this PGE2 ultimately enhances or inhibits subsequent efferocytosis is unknown and is likely to be context and concentration dependent.
Lipoxins are a class of pro-resolving eicosanoids derived from arachidonic acid that are produced and released by macrophages during the resolution phase of inflammation via lipoxygenase enzymes (Serhan et al., 2008). Lipoxin A4 is reported to enhance efferocytosis in vitro (Godson et al., 2000) and in vivo (Mitchell et al., 2002) by acting through the macrophage ALX receptor (ALXR), which is also utilized by the PS-recognizing bridge molecule, annexin A1 (Scannell et al., 2007; Maderna et al., 2010). Several other pro-resolution lipids derived from essential omega-3 fatty acids, have also been identified, including resolvins, protectins (PD1), and maresins, all of which enhance macrophage phagocytosis of pathogens, particles, and apoptotic cells (Schwab et al., 2007; Serhan et al., 2008). How these pro-resolving lipids enhance efferocytosis, the mechanisms and precise receptors involved, are not well understood. For instance, resolvin E1 (RvE1), but not the related RvD1, exerts its effects in part via the chimerin receptor, ChemR23. Importantly, decreased production of pro-inflammatory cytokines, perhaps as a result of efferocytosis, have been observed with these pro-resolving lipids (Schwab et al., 2007). Interestingly, the transition toward production of these pro-resolving lipids (lipoxins, resolvins, and protectins) is mediated through PGE2 (Serhan et al., 2008).
In addition to the short-term influences of enhancers or inhibitors, these factors are likely to have downstream contributions in shaping subsequent macrophage programming. For example, since apoptotic cell recognition and efferocytosis enhances the macrophage programming transition from inflammatory to pro-resolving, influencing this initial process may thwart this transition (inhibitors), or speed, reinforce, and/or sustain it (enhancers). For example, PGE2 and subsequent cAMP production appear to be pivotal to programming inflammatory macrophages to a pro-resolving phenotype under some conditions (Bystrom et al., 2008). As such, early signals, e.g., lyso-PS, PGE2, and cAMP, and possibly the other pro-resolving lipids, may contribute collaboratively to initiate the transition from inflammatory macrophage to resolving macrophage by carefully orchestrated signaling.
Section 3: Consequences of Decreased Efferocytosis and Prolonged Inflammatory Programming
Defective apoptotic cell clearance is associated with many autoimmune and chronic inflammatory disorders including SLE (Koh et al., 2000; Gaipl et al., 2007), type I diabetes (Haskins et al., 2003), chronic obstructive pulmonary disease (Hodge et al., 2008), and cardiovascular disease (Li et al., 2009). Mechanistically, disintegration of uncleared late apoptotic cells releases toxic intracellular contents that spur inflammation (e.g., HMGB1) and provide epitopes (e.g., nuclear DNA) for autoantibody production, a hallmark of autoimmune disorders such as SLE (Ma et al., 2010). As a consequence, the resulting increased production of pro-inflammatory cytokines (e.g., TNFα and IFNα; Koh et al., 2000; Haskins et al., 2003) prolongs inflammatory, efferocytic-low macrophage programming. Similarly, polymorphisms resulting in over-expression of the M1-associated transcription factor IRF5 are associated with many inflammatory diseases (Dideberg et al., 2007; Dieguez-Gonzalez et al., 2008; Kristjansdottir et al., 2008).
Ineffective apoptotic cell clearance, and enhanced inflammation are also demonstrated where there are defects in “alternative activation” mechanisms. Mice with PPARγ deficient macrophages have impaired efferocytosis, decreased expression of efferocytic receptors including CD36 and Mer tyrosine kinase, increased kidney inflammation, and autoantibody production (Roszer et al., 2011). PPARδ and LXR-deficient mice also develop systemic autoimmune disease associated with defective clearance and decreased efferocytic receptor/bridge molecule expression (A-Gonzalez et al., 2009; Mukundan et al., 2009). In a murine model of chronic granulomatous disease (CGD), which exhibits chronic inflammation, and mild autoimmunity, macrophages maintain a “classically activated” phenotype, illustrated by enhanced pro-inflammatory cytokine production, impaired efferocytosis, and decreased expression of PPARγ, MMR, CD36, and CD14 (Fernandez-Boyanapalli et al., 2009, 2010a).
The above evidence suggests that defective efferocytosis in autoimmune and chronic inflammatory conditions may result from continual exaggerated inflammatory activation of macrophages and/or ineffective induction of signals associated with programming toward pro-resolving macrophages. As such, further investigation of the mechanisms leading to impaired macrophage efferocytosis in these conditions, and approaches for its reversal, may lead to the development of more effective therapeutic strategies.
Section 4: Therapeutic Strategies to Enhance Efferocytosis and Reduce Inflammation
Currently Available Therapeutics
Based on the experimental evidence, enhancement of macrophage efferocytosis, either by early acting mediators or through programming changes, may have efficacy in treating disorders linked to chronic inflammation and aberrant macrophage function. Many anti-inflammatory drugs utilized clinically likely stimulate apoptotic cell uptake, though for the most part, mechanisms are poorly understood. For example, glucocorticoids with protean effects on inflammation also enhance efferocytosis (Rhen and Cidlowski, 2005). In this regard they appear to act through a number of mechanisms: (i) induced expression of the phospholipid binding protein annexin A1 and its receptor ALXR, which also binds lipoxin A4 (Maderna et al., 2005), (ii) enhanced signaling via Mer and its ligand, protein S (McColl et al., 2009), (iii) altered Rac/Rho balance (Giles et al., 2001), and (iv) PPARγ-induced programming (Majai et al., 2007). Statins, cholesterol lowering drugs, are used with increasing frequency to treat inflammatory diseases also enhance phagocytosis of apoptotic cells. The mode of action is likely complex, but may work in part by reducing prenylation and membrane association of inhibitory RhoA (Morimoto et al., 2006), as well as by inducing programming-related changes through activation of PPARγ (Yano et al., 2007). Another class of anti-inflammatory agents known to enhance efferocytosis is the macrolide group of antibiotics. These likely alter macrophage programming as well as increase levels of bridge molecules in vivo to enhance uptake of apoptotic cells (Yamaryo et al., 2003; Hodge et al., 2008). Many of these current treatments have diverse targets, making it difficult to determine the precise role of enhanced efferocytosis in their anti-inflammatory effects. Furthermore, many have adverse side effects. Accordingly, novel therapies that more closely mimic normal resolution processes or target re-programming of macrophages to specifically enhance efferocytosis are needed.
Novel Therapeutic Targets to Enhance Efferocytosis
Potential new therapeutic targets include modulation of signaling pathways and mediators involved in both early acting events and subsequent macrophage programming. Oxidant-mediated activation of RhoA and suppression of efferocytosis, may be amenable to antioxidant treatment. In a LPS-induced lung injury model, antioxidants reduced inflammation, and improved macrophage efferocytosis by inhibiting RhoA activation (Moon et al., 2010). Similarly, antioxidant treatment in autoimmune NOD mice dampened pro-inflammatory cytokine production and repaired the macrophage efferocytic defect (Haskins et al., 2003). Another potential therapy to enhance efferocytosis could be via lipoxins, including the related aspirin triggered 15-epi-lipoxins (Spite and Serhan, 2010) that signal through ALXR. Lipoxins as well as resolvins and protectins have been shown to reduce inflammation and tissue damage in a variety of rodent models (Serhan et al., 2008). Whether, and to what degree, any of these effects are a direct result of enhanced efferocytosis remains to be determined.
Given that nuclear receptor signaling is strongly associated with enhanced efferocytosis and suppression of inflammation, PPARγ, PPARδ, and LXR agonists are natural therapeutic targets for inflammatory diseases. LXR agonists were shown to reverse defective macrophage efferocytosis and reduce disease severity in a murine model of SLE (A-Gonzalez et al., 2009). Additionally, PPARγ agonists reduced neutrophil numbers in rodent models of acute inflammation, COPD, and asthma (Belvisi and Hele, 2008; Fernandez-Boyanapalli et al., 2010a). PPARγ and PPARδ activation were also shown to decrease inflammation and disease severity in experimental autoimmune encephalomyelitis (EAE), the murine model of multiple sclerosis (Feinstein et al., 2002; Polak et al., 2005). Recently, a direct association between improved efferocytosis and treatment efficacy has been demonstrated: treatment with a PPARγ agonist resolved prolonged zymosan-induced inflammation in a mouse model of CGD, which corresponded with macrophage “alternative activation” enhanced efferocytosis, and increased TGFβ and IL-10 production (Fernandez-Boyanapalli et al., 2010a).
In vivo administration of apoptotic cells exploits their signaling to alter inflammatory programming in inflamed tissues; their stimulation of phagocytes shapes subsequent immune and inflammatory responses (Jonson et al., 2008). This has been especially successful in graft versus host disease where a shift in Th1 to Th2 T cell responses was driven by macrophage-dependent induction of tolerogenic dendritic cells (DC) and regulatory T cells (Gorgun et al., 2002; Kleinclauss et al., 2006). In EAE, infusion of autoantigen-expressing apoptotic cells induced tolerance to self antigens and reduced disease progression in a manner-dependent on macrophage-mediated regulation of DC efferocytosis (Miyake et al., 2007). The mechanisms involved in apoptotic cell infusion-induced regulation of inflammation and induction of tolerance are not well studied, however, it is likely mediated in part through apoptotic cell-induced alterations in macrophage programming. Treatment with PS liposomes, which simulate signaling by apoptotic cells, has also been effective. In sterile inflammatory models, administration of PS liposomes led to macrophage re-programming from pro-inflammatory to anti-inflammatory states. As consequences, efferocytosis was enhanced (Fernandez-Boyanapalli et al., 2009), tissue swelling and pro-inflammatory cytokine production were reduced (Ramos et al., 2007), and repair of infarct tissue was enhanced (Harel-Adar et al., 2011). As a cautionary note, however, administration of both apoptotic cells and PS liposomes into inflamed tissues, and likely to inflammatory macrophages, can also contribute (at least transiently) to exacerbated inflammation (Medan et al., 2002; Borges et al., 2009), and disabled host defense against pathogens (Medeiros et al., 2009). These observations underscore the need to better understand the signaling pathways involved in therapeutic induction of macrophage anti-inflammatory programming and enhancement of efferocytic capacity.
Conclusion
Macrophages, through their phagocytic functions and production of cytokines and mediators, profoundly shape the course of inflammation. Interpreting cues from the environment, they orchestrate early pro-inflammatory responses to pathogens and tissue injury, and ultimately produce anti-inflammatory lipids and cytokines for the active suppression of inflammation and restoration of tissue homeostasis. Their recognition and removal of apoptotic cells are crucial to these latter events, and a considerable body of data supports that their dysregulation contributes to diverse autoimmune and chronic disease states. An increasing understanding of the normal resolution mechanisms, especially those that modulate apoptotic cell clearance through the stimulation and programming of macrophages will undoubtedly bring new therapeutic strategies to the forefront and will allow for the development of more effective targeted and/or combinatorial treatments.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to acknowledge the assistance of Ms. Brenda Sebern in the preparation of the manuscript. Funding for this work came from NIH HL034303, AI058228, HL081151, GM061031, and the Chronic Granulomatous Disorder Research Trust.
References
A-Gonzalez, N., Bensinger, S. J., Hong, C., Beceiro, S., Bradley, M. N., Zelcer, N., Deniz, J., Ramirez, C., Diaz, M., Gallardo, G., De Galarreta, C. R., Salazar, J., Lopez, F., Edwards, P., Parks, J., Andujar, M., Tontonoz, P., and Castrillo, A. (2009). Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity 31, 245–258.
Aronoff, D. M., Canetti, C., and Peters-Golden, M. (2004). Prostaglandin E2 inhibits alveolar macrophage phagocytosis through an E-prostanoid 2 receptor-mediated increase in intracellular cyclic AMP. J. Immunol. 173, 559–565.
Banerjee, S., Friggeri, A., Liu, G., and Abraham, E. (2010). The C-terminal acidic tail is responsible for the inhibitory effects of HMGB1 on efferocytosis. J. Leukoc. Biol. 88, 973–979.
Belvisi, M. G., and Hele, D. J. (2008). Peroxisome proliferator-activated receptors as novel targets in lung disease. Chest 134, 152–157.
Benoit, M., Desnues, B., and Mege, J. L. (2008). Macrophage polarization in bacterial infections. J. Immunol. 181, 3733–3739.
Berry, A., Balard, P., Coste, A., Olagnier, D., Lagane, C., Authier, H., Benoit-Vical, F., Lepert, J. C., Seguela, J. P., Magnaval, J. F., Chambon, P., Metzger, D., Desvergne, B., Wahli, W., Auwerx, J., and Pipy, B. (2007). IL-13 induces expression of CD36 in human monocytes through PPARgamma activation. Eur. J. Immunol. 37, 1642–1652.
Borges, V. M., Vandivier, R. W., Mcphillips, K. A., Kench, J. A., Morimoto, K., Groshong, S. D., Richens, T. R., Graham, B. B., Muldrow, A. M., Van Heule, L., Henson, P. M., and Janssen, W. J. (2009). TNFalpha inhibits apoptotic cell clearance in the lung, exacerbating acute inflammation. Am. J. Physiol. Lung Cell Mol. Physiol. 297, L586–L595.
Bouhlel, M. A., Derudas, B., Rigamonti, E., Dievart, R., Brozek, J., Haulon, S., Zawadzki, C., Jude, B., Torpier, G., Marx, N., Staels, B., and Chinetti-Gbaguidi, G. (2007). PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 6, 137–143.
Bystrom, J., Evans, I., Newson, J., Stables, M., Toor, I., Van Rooijen, N., Crawford, M., Colville-Nash, P., Farrow, S., and Gilroy, D. W. (2008). Resolution-phase macrophages possess a unique inflammatory phenotype that is controlled by cAMP. Blood 112, 4117–4127.
Dideberg, V., Kristjansdottir, G., Milani, L., Libioulle, C., Sigurdsson, S., Louis, E., Wiman, A. C., Vermeire, S., Rutgeerts, P., Belaiche, J., Franchimont, D., Van Gossum, A., Bours, V., and Syvanen, A. C. (2007). An insertion-deletion polymorphism in the interferon regulatory factor 5 (IRF5) gene confers risk of inflammatory bowel diseases. Hum. Mol. Genet. 16, 3008–3016.
Dieguez-Gonzalez, R., Calaza, M., Perez-Pampin, E., De La Serna, A. R., Fernandez-Gutierrez, B., Castaneda, S., Largo, R., Joven, B., Narvaez, J., Navarro, F., Marenco, J. L., Vicario, J. L., Blanco, F. J., Fernandez-Lopez, J. C., Caliz, R., Collado-Escobar, M. D., Carreno, L., Lopez-Longo, J., Canete, J. D., Gomez-Reino, J. J., and Gonzalez, A. (2008). Association of interferon regulatory factor 5 haplotypes, similar to that found in systemic lupus erythematosus, in a large subgroup of patients with rheumatoid arthritis. Arthritis Rheum. 58, 1264–1274.
El Chartouni, C., Schwarzfischer, L., and Rehli, M. (2010). Interleukin-4 induced interferon regulatory factor (Irf) 4 participates in the regulation of alternative macrophage priming. Immunobiology 215, 821–825.
Fadok, V. A., Bratton, D. L., Konowal, A., Freed, P. W., Westcott, J. Y., and Henson, P. M. (1998). Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J. Clin. Invest. 101, 890–898.
Fadok, V. A., Voelker, D. R., Campbell, P. A., Cohen, J. J., Bratton, D. L., and Henson, P. M. (1992). Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol. 148, 2207–2216.
Feinstein, D. L., Galea, E., Gavrilyuk, V., Brosnan, C. F., Whitacre, C. C., Dumitrescu-Ozimek, L., Landreth, G. E., Pershadsingh, H. A., Weinberg, G., and Heneka, M. T. (2002). Peroxisome proliferator-activated receptor-gamma agonists prevent experimental autoimmune encephalomyelitis. Ann. Neurol. 51, 694–702.
Feng, X., Deng, T., Zhang, Y., Su, S., Wei, C., and Han, D. (2011). Lipopolysaccharide inhibits macrophage phagocytosis of apoptotic neutrophils by regulating the production of tumour necrosis factor and growth arrest-specific gene 6. Immunology 132, 287–295.
Fernandez-Boyanapalli, R., Frasch, S. C., Riches, D. W., Vandivier, R. W., Henson, P. M., and Bratton, D. L. (2010a). PPARgamma activation normalizes resolution of acute sterile inflammation in murine chronic granulomatous disease. Blood 116, 4512–4522.
Fernandez-Boyanapalli, R., Mcphillips, K. A., Frasch, S. C., Janssen, W. J., Dinauer, M. C., Riches, D. W., Henson, P. M., Byrne, A., and Bratton, D. L. (2010b). Impaired phagocytosis of apoptotic cells by macrophages in chronic granulomatous disease is reversed by IFN-gamma in a nitric oxide-dependent manner. J. Immunol. 185, 4030–4041.
Fernandez-Boyanapalli, R. F., Frasch, S. C., Mcphillips, K., Vandivier, R. W., Harry, B. L., Riches, D. W., Henson, P. M., and Bratton, D. L. (2009). Impaired apoptotic cell clearance in CGD due to altered macrophage programming is reversed by phosphatidylserine-dependent production of IL-4. Blood 113, 2047–2055.
Frasch, S. C., Berry, K. Z., Fernandez-Boyanapalli, R., Jin, H. S., Leslie, C., Henson, P. M., Murphy, R. C., and Bratton, D. L. (2008). NADPH oxidase-dependent generation of lysophosphatidylserine enhances clearance of activated and dying neutrophils via G2A. J. Biol. Chem. 283, 33736–33749.
Frasch, S. C., Fernandez-Boyanapalli, R. F., Berry, K. Z., Leslie, C. C., Bonventre, J. V., Murphy, R. C., Henson, P. M., and Bratton, D. L. (2011). Signaling via macrophage G2A enhances efferocytosis of dying neutrophils by augmentation of Rac activity. J. Biol. Chem. 286, 12108–12122.
Freire-de-Lima, C. G., Xiao, Y. Q., Gardai, S. J., Bratton, D. L., Schiemann, W. P., and Henson, P. M. (2006). Apoptotic cells, through transforming growth factor-beta, coordinately induce anti-inflammatory and suppress pro-inflammatory eicosanoid and NO synthesis in murine macrophages. J. Biol. Chem. 281, 38376–38384.
Friggeri, A., Banerjee, S., Biswas, S., De Freitas, A., Liu, G., Bierhaus, A., and Abraham, E. (2011). Participation of the receptor for advanced glycation end products in efferocytosis. J. Immunol. 186, 6191–6198.
Friggeri, A., Yang, Y., Banerjee, S., Park, Y. J., Liu, G., and Abraham, E. (2010). HMGB1 inhibits macrophage activity in efferocytosis through binding to the alphavbeta3-integrin. Am. J. Physiol. Cell Physiol. 299, C1267–C1276.
Gaipl, U. S., Munoz, L. E., Grossmayer, G., Lauber, K., Franz, S., Sarter, K., Voll, R. E., Winkler, T., Kuhn, A., Kalden, J., Kern, P., and Herrmann, M. (2007). Clearance deficiency and systemic lupus erythematosus (SLE). J. Autoimmun. 28, 114–121.
Gales, A., Conduche, A., Bernad, J., Lefevre, L., Olagnier, D., Beraud, M., Martin-Blondel, G., Linas, M. D., Auwerx, J., Coste, A., and Pipy, B. (2010). PPARgamma controls dectin-1 expression required for host antifungal defense against Candida albicans. PLoS Pathog. 6, e1000714. doi:10.1371/journal.ppat.1000714
Ghisletti, S., Huang, W., Ogawa, S., Pascual, G., Lin, M. E., Willson, T. M., Rosenfeld, M. G., and Glass, C. K. (2007). Parallel SUMOylation-dependent pathways mediate gene- and signal-specific transrepression by LXRs and PPARgamma. Mol. Cell 25, 57–70.
Giles, K. M., Ross, K., Rossi, A. G., Hotchin, N. A., Haslett, C., and Dransfield, I. (2001). Glucocorticoid augmentation of macrophage capacity for phagocytosis of apoptotic cells is associated with reduced p130Cas expression, loss of paxillin/pyk2 phosphorylation, and high levels of active Rac. J. Immunol. 167, 976–986.
Godson, C., Mitchell, S., Harvey, K., Petasis, N. A., Hogg, N., and Brady, H. R. (2000). Cutting edge: lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J. Immunol. 164, 1663–1667.
Gordon, S., and Martinez, F. O. (2010). Alternative activation of macrophages: mechanism and functions. Immunity 32, 593–604.
Gorgun, G., Miller, K. B., and Foss, F. M. (2002). Immunologic mechanisms of extracorporeal photochemotherapy in chronic graft-versus-host disease. Blood 100, 941–947.
Gratchev, A., Kzhyshkowska, J., Kothe, K., Muller-Molinet, I., Kannookadan, S., Utikal, J., and Goerdt, S. (2006). Mphi1 and Mphi2 can be re-polarized by Th2 or Th1 cytokines, respectively, and respond to exogenous danger signals. Immunobiology 211, 473–486.
Greenberg, M. E., Sun, M., Zhang, R., Febbraio, M., Silverstein, R., and Hazen, S. L. (2006). Oxidized phosphatidylserine-CD36 interactions play an essential role in macrophage-dependent phagocytosis of apoptotic cells. J. Exp. Med. 203, 2613–2625.
Gumienny, T. L., Brugnera, E., Tosello-Trampont, A. C., Kinchen, J. M., Haney, L. B., Nishiwaki, K., Walk, S. F., Nemergut, M. E., Macara, I. G., Francis, R., Schedl, T., Qin, Y., Van Aelst, L., Hengartner, M. O., and Ravichandran, K. S. (2001). CED-12/ELMO, a novel member of the CrkII/Dock180/Rac pathway, is required for phagocytosis and cell migration. Cell 107, 27–41.
Harel-Adar, T., Ben Mordechai, T., Amsalem, Y., Feinberg, M. S., Leor, J., and Cohen, S. (2011). Modulation of cardiac macrophages by phosphatidylserine-presenting liposomes improves infarct repair. Proc. Natl. Acad. Sci. U.S.A. 108, 1827–1832.
Haskins, K., Bradley, B., Powers, K., Fadok, V., Flores, S., Ling, X., Pugazhenthi, S., Reusch, J., and Kench, J. (2003). Oxidative stress in type 1 diabetes. Ann. N. Y. Acad. Sci. 1005, 43–54.
He, M., Kubo, H., Morimoto, K., Fujino, N., Suzuki, T., Takahasi, T., Yamada, M., Yamaya, M., Maekawa, T., Yamamoto, Y., and Yamamoto, H. (2011). Receptor for advanced glycation end products binds to phosphatidylserine and assists in the clearance of apoptotic cells. EMBO Rep. 12, 358–364.
Henson, P., and Bratton, D. (2009). “Recognition and removal of apoptotic cells,” in Phagocyte-Pathogen Interactions: Macrophages and the Host Responses to Infection, Section VI Bridging the Gap Between the Innate and Acquired Immune Response, eds D. G. Russel and S. Gordon (Washington, DC: ASM Press), 341–365.
Hodge, S., Hodge, G., Jersmann, H., Matthews, G., Ahern, J., Holmes, M., and Reynolds, P. N. (2008). Azithromycin improves macrophage phagocytic function and expression of mannose receptor in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 178, 139–148.
Hoffmann, P. R., Kench, J. A., Vondracek, A., Kruk, E., Daleke, D. L., Jordan, M., Marrack, P., Henson, P. M., and Fadok, V. A. (2005). Interaction between phosphatidylserine and the phosphatidylserine receptor inhibits immune responses in vivo. J. Immunol. 174, 1393–1404.
Huang, J. T., Welch, J. S., Ricote, M., Binder, C. J., Willson, T. M., Kelly, C., Witztum, J. L., Funk, C. D., Conrad, D., and Glass, C. K. (1999). Interleukin-4-dependent production of PPAR-gamma ligands in macrophages by 12/15-lipoxygenase. Nature 400, 378–382.
Huynh, M. L., Fadok, V. A., and Henson, P. M. (2002). Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J. Clin. Invest. 109, 41–50.
Jenkins, S. J., Ruckerl, D., Cook, P. C., Jones, L. H., Finkelman, F. D., Van Rooijen, N., Macdonald, A. S., and Allen, J. E. (2011). Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science 332, 1284–1288.
Jennewein, C., Kuhn, A. M., Schmidt, M. V., Meilladec-Jullig, V., Von Knethen, A., Gonzalez, F. J., and Brune, B. (2008). Sumoylation of peroxisome proliferator-activated receptor gamma by apoptotic cells prevents lipopolysaccharide-induced NCoR removal from kappaB binding sites mediating transrepression of proinflammatory cytokines. J. Immunol. 181, 5646–5652.
Jonson, C. O., Pihl, M., Nyholm, C., Cilio, C. M., Ludvigsson, J., and Faresjo, M. (2008). Regulatory T cell-associated activity in photopheresis-induced immune tolerance in recent onset type 1 diabetes children. Clin. Exp. Immunol. 153, 174–181.
Kang, K., Reilly, S. M., Karabacak, V., Gangl, M. R., Fitzgerald, K., Hatano, B., and Lee, C. H. (2008). Adipocyte-derived Th2 cytokines and myeloid PPARdelta regulate macrophage polarization and insulin sensitivity. Cell Metab. 7, 485–495.
Kenis, H., Van Genderen, H., Deckers, N. M., Lux, P. A., Hofstra, L., Narula, J., and Reutelingsperger, C. P. (2006). Annexin A5 inhibits engulfment through internalization of PS-expressing cell membrane patches. Exp. Cell Res. 312, 719–726.
Kinchen, J. M., Cabello, J., Klingele, D., Wong, K., Feichtinger, R., Schnabel, H., Schnabel, R., and Hengartner, M. O. (2005). Two pathways converge at CED-10 to mediate actin rearrangement and corpse removal in C. elegans. Nature 434, 93–99.
Kleinclauss, F., Perruche, S., Masson, E., De Carvalho Bittencourt, M., Biichle, S., Remy-Martin, J. P., Ferrand, C., Martin, M., Bittard, H., Chalopin, J. M., Seilles, E., Tiberghien, P., and Saas, P. (2006). Intravenous apoptotic spleen cell infusion induces a TGF-beta-dependent regulatory T-cell expansion. Cell Death Differ. 13, 41–52.
Koh, J. S., Wang, Z., and Levine, J. S. (2000). Cytokine dysregulation induced by apoptotic cells is a shared characteristic of murine lupus. J. Immunol. 165, 4190–4201.
Komura, H., Miksa, M., Wu, R., Goyert, S. M., and Wang, P. (2009). Milk fat globule epidermal growth factor-factor VIII is down-regulated in sepsis via the lipopolysaccharide-CD14 pathway. J. Immunol. 182, 581–587.
Krausgruber, T., Blazek, K., Smallie, T., Alzabin, S., Lockstone, H., Sahgal, N., Hussell, T., Feldmann, M., and Udalova, I. A. (2011). IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat. Immunol. 12, 231–238.
Kristjansdottir, G., Sandling, J. K., Bonetti, A., Roos, I. M., Milani, L., Wang, C., Gustafsdottir, S. M., Sigurdsson, S., Lundmark, A., Tienari, P. J., Koivisto, K., Elovaara, I., Pirttila, T., Reunanen, M., Peltonen, L., Saarela, J., Hillert, J., Olsson, T., Landegren, U., Alcina, A., Fernandez, O., Leyva, L., Guerrero, M., Lucas, M., Izquierdo, G., Matesanz, F., and Syvanen, A. C. (2008). Interferon regulatory factor 5 (IRF5) gene variants are associated with multiple sclerosis in three distinct populations. J. Med. Genet. 45, 362–369.
Leverrier, Y., and Ridley, A. J. (2001). Requirement for Rho GTPases and PI 3-kinases during apoptotic cell phagocytosis by macrophages. Curr. Biol. 11, 195–199.
Li, S., Sun, Y., Liang, C. P., Thorp, E. B., Han, S., Jehle, A. W., Saraswathi, V., Pridgen, B., Kanter, J. E., Li, R., Welch, C. L., Hasty, A. H., Bornfeldt, K. E., Breslow, J. L., Tabas, I., and Tall, A. R. (2009). Defective phagocytosis of apoptotic cells by macrophages in atherosclerotic lesions of ob/ob mice and reversal by a fish oil diet. Circ. Res. 105, 1072–1082.
Ma, C. Y., Jiao, Y. L., Zhang, J., Yang, Q. R., Zhang, Z. F., Shen, Y. J., Chen, Z. J., and Zhao, Y. R. (2010). Elevated plasma level of HMGB1 is associated with disease activity and combined alterations with IFN-alpha and TNF-alpha in systemic lupus erythematosus. Rheumatol. Int. 1–8.
Maderna, P., Cottell, D. C., Toivonen, T., Dufton, N., Dalli, J., Perretti, M., and Godson, C. (2010). FPR2/ALX receptor expression and internalization are critical for lipoxin A4 and annexin-derived peptide-stimulated phagocytosis. FASEB J. 24, 4240–4249.
Maderna, P., Yona, S., Perretti, M., and Godson, C. (2005). Modulation of phagocytosis of apoptotic neutrophils by supernatant from dexamethasone-treated macrophages and annexin-derived peptide Ac(2-26). J. Immunol. 174, 3727–3733.
Majai, G., Sarang, Z., Csomos, K., Zahuczky, G., and Fesus, L. (2007). PPARgamma-dependent regulation of human macrophages in phagocytosis of apoptotic cells. Eur. J. Immunol. 37, 1343–1354.
Martinez, F. O., Helming, L., and Gordon, S. (2009). Alternative activation of macrophages: an immunologic functional perspective. Annu. Rev. Immunol. 27, 451–483.
McColl, A., Bournazos, S., Franz, S., Perretti, M., Morgan, B. P., Haslett, C., and Dransfield, I. (2009). Glucocorticoids induce protein S-dependent phagocytosis of apoptotic neutrophils by human macrophages. J. Immunol. 183, 2167–2175.
McPhillips, K., Janssen, W. J., Ghosh, M., Byrne, A., Gardai, S., Remigio, L., Bratton, D. L., Kang, J. L., and Henson, P. (2007). TNF-alpha inhibits macrophage clearance of apoptotic cells via cytosolic phospholipase A2 and oxidant-dependent mechanisms. J. Immunol. 178, 8117–8126.
Medan, D., Wang, L., Yang, X., Dokka, S., Castranova, V., and Rojanasakul, Y. (2002). Induction of neutrophil apoptosis and secondary necrosis during endotoxin-induced pulmonary inflammation in mice. J. Cell. Physiol. 191, 320–326.
Medeiros, A. I., Serezani, C. H., Lee, S. P., and Peters-Golden, M. (2009). Efferocytosis impairs pulmonary macrophage and lung antibacterial function via PGE2/EP2 signaling. J. Exp. Med. 206, 61–68.
Michlewska, S., Dransfield, I., Megson, I. L., and Rossi, A. G. (2009). Macrophage phagocytosis of apoptotic neutrophils is critically regulated by the opposing actions of pro-inflammatory and anti-inflammatory agents: key role for TNF-alpha. FASEB J. 23, 844–854.
Mitchell, S., Thomas, G., Harvey, K., Cottell, D., Reville, K., Berlasconi, G., Petasis, N. A., Erwig, L., Rees, A. J., Savill, J., Brady, H. R., and Godson, C. (2002). Lipoxins, aspirin-triggered epi-lipoxins, lipoxin stable analogues, and the resolution of inflammation: stimulation of macrophage phagocytosis of apoptotic neutrophils in vivo. J. Am. Soc. Nephrol. 13, 2497–2507.
Miyake, Y., Asano, K., Kaise, H., Uemura, M., Nakayama, M., and Tanaka, M. (2007). Critical role of macrophages in the marginal zone in the suppression of immune responses to apoptotic cell-associated antigens. J. Clin. Invest. 117, 2268–2278.
Moon, C., Lee, Y. J., Park, H. J., Chong, Y. H., and Kang, J. L. (2010). N-acetylcysteine inhibits RhoA and promotes apoptotic cell clearance during intense lung inflammation. Am. J. Respir. Crit. Care Med. 181, 374–387.
Morimoto, K., Janssen, W. J., Fessler, M. B., Mcphillips, K. A., Borges, V. M., Bowler, R. P., Xiao, Y. Q., Kench, J. A., Henson, P. M., and Vandivier, R. W. (2006). Lovastatin enhances clearance of apoptotic cells (efferocytosis) with implications for chronic obstructive pulmonary disease. J. Immunol. 176, 7657–7665.
Mosser, D. M., and Zhang, X. (2008). Activation of murine macrophages. Curr. Protoc. Immunol. Chapter 14, Unit 14.12.
Mukundan, L., Odegaard, J. I., Morel, C. R., Heredia, J. E., Mwangi, J. W., Ricardo-Gonzalez, R. R., Goh, Y. P., Eagle, A. R., Dunn, S. E., Awakuni, J. U., Nguyen, K. D., Steinman, L., Michie, S. A., and Chawla, A. (2009). PPAR-delta senses and orchestrates clearance of apoptotic cells to promote tolerance. Nat. Med. 15, 1266–1272.
Nakaya, M., Tanaka, M., Okabe, Y., Hanayama, R., and Nagata, S. (2006). Opposite effects of rho family GTPases on engulfment of apoptotic cells by macrophages. J. Biol. Chem. 281, 8836–8842.
Odegaard, J. I., Ricardo-Gonzalez, R. R., Goforth, M. H., Morel, C. R., Subramanian, V., Mukundan, L., Red Eagle, A., Vats, D., Brombacher, F., Ferrante, A. W., and Chawla, A. (2007). Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature 447, 1116–1120.
Ogden, C. A., Decathelineau, A., Hoffmann, P. R., Bratton, D., Ghebrehiwet, B., Fadok, V. A., and Henson, P. M. (2001). C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J. Exp. Med. 194, 781–795.
Olefsky, J. M., and Glass, C. K. (2010). Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 72, 219–246.
Park, S. Y., Jung, M. Y., Lee, S. J., Kang, K. B., Gratchev, A., Riabov, V., Kzhyshkowska, J., and Kim, I. S. (2009). Stabilin-1 mediates phosphatidylserine-dependent clearance of cell corpses in alternatively activated macrophages. J. Cell Sci. 122, 3365–3373.
Pascual, G., Fong, A. L., Ogawa, S., Gamliel, A., Li, A. C., Perissi, V., Rose, D. W., Willson, T. M., Rosenfeld, M. G., and Glass, C. K. (2005). A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature 437, 759–763.
Polak, P. E., Kalinin, S., Dello Russo, C., Gavrilyuk, V., Sharp, A., Peters, J. M., Richardson, J., Willson, T. M., Weinberg, G., and Feinstein, D. L. (2005). Protective effects of a peroxisome proliferator-activated receptor-beta/delta agonist in experimental autoimmune encephalomyelitis. J. Neuroimmunol. 168, 65–75.
Qin, Y. H., Dai, S. M., Tang, G. S., Zhang, J., Ren, D., Wang, Z. W., and Shen, Q. (2009). HMGB1 enhances the proinflammatory activity of lipopolysaccharide by promoting the phosphorylation of MAPK p38 through receptor for advanced glycation end products. J. Immunol. 183, 6244–6250.
Raes, G., Brys, L., Dahal, B. K., Brandt, J., Grooten, J., Brombacher, F., Vanham, G., Noel, W., Bogaert, P., Boonefaes, T., Kindt, A., Van Den Bergh, R., Leenen, P. J., De Baetselier, P., and Ghassabeh, G. H. (2005). Macrophage galactose-type C-type lectins as novel markers for alternatively activated macrophages elicited by parasitic infections and allergic airway inflammation. J. Leukoc. Biol. 77, 321–327.
Ramos, G. C., Fernandes, D., Charao, C. T., Souza, D. G., Teixeira, M. M., and Assreuy, J. (2007). Apoptotic mimicry: phosphatidylserine liposomes reduce inflammation through activation of peroxisome proliferator-activated receptors (PPARs) in vivo. Br. J. Pharmacol. 151, 844–850.
Rebe, C., Raveneau, M., Chevriaux, A., Lakomy, D., Sberna, A. L., Costa, A., Bessede, G., Athias, A., Steinmetz, E., Lobaccaro, J. M., Alves, G., Menicacci, A., Vachenc, S., Solary, E., Gambert, P., and Masson, D. (2009). Induction of transglutaminase 2 by a liver X receptor/retinoic acid receptor alpha pathway increases the clearance of apoptotic cells by human macrophages. Circ. Res. 105, 393–401.
Rhen, T., and Cidlowski, J. A. (2005). Antiinflammatory action of glucocorticoids – new mechanisms for old drugs. N. Engl. J. Med. 353, 1711–1723.
Rossi, A. G., Mccutcheon, J. C., Roy, N., Chilvers, E. R., Haslett, C., and Dransfield, I. (1998). Regulation of macrophage phagocytosis of apoptotic cells by cAMP. J. Immunol. 160, 3562–3568.
Roszer, T., Menendez-Gutierrez, M. P., Lefterova, M. I., Alameda, D., Nunez, V., Lazar, M. A., Fischer, T., and Ricote, M. (2011). Autoimmune kidney disease and impaired engulfment of apoptotic cells in mice with macrophage peroxisome proliferator-activated receptor gamma or retinoid X receptor alpha deficiency. J. Immunol. 186, 621–631.
Scannell, M., Flanagan, M. B., Destefani, A., Wynne, K. J., Cagney, G., Godson, C., and Maderna, P. (2007). Annexin-1 and peptide derivatives are released by apoptotic cells and stimulate phagocytosis of apoptotic neutrophils by macrophages. J. Immunol. 178, 4595–4605.
Schif-Zuck, S., Gross, N., Assi, S., Rostoker, R., Serhan, C. N., and Ariel, A. (2011). Saturated-efferocytosis generates pro-resolving CD11b low macrophages: modulation by resolvins and glucocorticoids. Eur. J. Immunol. 41, 366–379.
Schwab, J. M., Chiang, N., Arita, M., and Serhan, C. N. (2007). Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature 447, 869–874.
Serghides, L., and Kain, K. C. (2001). Peroxisome proliferator-activated receptor gamma-retinoid X receptor agonists increase CD36-dependent phagocytosis of plasmodium falciparum-parasitized erythrocytes and decrease malaria-induced TNF-alpha secretion by monocytes/macrophages. J. Immunol. 166, 6742–6748.
Serhan, C. N., Chiang, N., and Van Dyke, T. E. (2008). Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 8, 349–361.
Sica, A. (2010). Role of tumour-associated macrophages in cancer-related inflammation. Exp. Oncol. 32, 153–158.
Spite, M., and Serhan, C. N. (2010). Novel lipid mediators promote resolution of acute inflammation: impact of aspirin and statins. Circ. Res. 107, 1170–1184.
Szanto, A., Balint, B. L., Nagy, Z. S., Barta, E., Dezso, B., Pap, A., Szeles, L., Poliska, S., Oros, M., Evans, R. M., Barak, Y., Schwabe, J., and Nagy, L. (2010). STAT6 transcription factor is a facilitator of the nuclear receptor PPARgamma-regulated gene expression in macrophages and dendritic cells. Immunity 33, 699–712.
Voll, R. E., Herrmann, M., Roth, E. A., Stach, C., Kalden, J. R., and Girkontaite, I. (1997). Immunosuppressive effects of apoptotic cells. Nature 390, 350–351.
Wang, Y., Wang, Y. P., Zheng, G., Lee, V. W., Ouyang, L., Chang, D. H., Mahajan, D., Coombs, J., Wang, Y. M., Alexander, S. I., and Harris, D. C. (2007). Ex vivo programmed macrophages ameliorate experimental chronic inflammatory renal disease. Kidney Int. 72, 290–299.
Welch, J. S., Ricote, M., Akiyama, T. E., Gonzalez, F. J., and Glass, C. K. (2003). PPARgamma and PPARdelta negatively regulate specific subsets of lipopolysaccharide and IFN-gamma target genes in macrophages. Proc. Natl. Acad. Sci. U.S.A. 100, 6712–6717.
Xu, W., Roos, A., Schlagwein, N., Woltman, A. M., Daha, M. R., and Van Kooten, C. (2006). IL-10-producing macrophages preferentially clear early apoptotic cells. Blood 107, 4930–4937.
Yamaryo, T., Oishi, K., Yoshimine, H., Tsuchihashi, Y., Matsushima, K., and Nagatake, T. (2003). Fourteen-member macrolides promote the phosphatidylserine receptor-dependent phagocytosis of apoptotic neutrophils by alveolar macrophages. Antimicrob. Agents Chemother. 47, 48–53.
Yano, M., Matsumura, T., Senokuchi, T., Ishii, N., Murata, Y., Taketa, K., Motoshima, H., Taguchi, T., Sonoda, K., Kukidome, D., Takuwa, Y., Kawada, T., Brownlee, M., Nishikawa, T., and Araki, E. (2007). Statins activate peroxisome proliferator-activated receptor gamma through extracellular signal-regulated kinase 1/2 and p38 mitogen-activated protein kinase-dependent cyclooxygenase-2 expression in macrophages. Circ. Res. 100, 1442–1451.
Keywords: macrophage, efferocytosis, inflammation, alternative activation, classical activation, apoptotic cell
Citation: Korns D, Frasch SC, Fernandez-Boyanapalli R, Henson PM and Bratton DL (2011) Modulation of macrophage efferocytosis in inflammation. Front. Immun. 2:57. doi: 10.3389/fimmu.2011.00057
Received: 07 July 2011; Paper pending published: 05 August 2011;
Accepted: 16 October 2011; Published online: 08 November 2011.
Edited by:
Amiram Ariel, University of Haifa, IsraelReviewed by:
Angelika Bierhaus, University of Heidelberg, GermanyUdo S. Gaipl, University Hospital Erlangen, Germany
Copyright: © 2011 Korns, Frasch, Fernandez-Boyanapalli, Henson and Bratton. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Donna L. Bratton, Division of Cell Biology, Department of Pediatrics, National Jewish Health, Smith Building Room A540, 1400 Jackson Street, Denver, CO 80206, USA. e-mail:YnJhdHRvbmRAbmpoZWFsdGgub3Jn