 Berhane Ghebrehiwet1*
Berhane Ghebrehiwet1* Jolyon Jesty1 Sulan Xu1 Rama Vinayagasundaram1 Uma Vinayagasundaram1 Yan Ji1 Alisa Valentino1 Kinga K. Hosszu1 Sally Mathew1 Kusumam Joseph2
Jolyon Jesty1 Sulan Xu1 Rama Vinayagasundaram1 Uma Vinayagasundaram1 Yan Ji1 Alisa Valentino1 Kinga K. Hosszu1 Sally Mathew1 Kusumam Joseph2 Allen P. Kaplan2
Allen P. Kaplan2 Ellinor I. B. Peerschke3
Ellinor I. B. Peerschke3
- 1 Department of Medicine, Stony Brook University, Stony Brook, NY, USA
- 2 Department of Medicine, Medical University of South Carolina, Charleston, SC, USA
- 3 Department of Pathology, Mount Sinai School of Medicine, New York, NY, USA
The endothelial cell receptor complex for kininogen (HK) comprises gC1qR, cytokeratin 1, and urokinase-type plasminogen activator receptor and is essential for activation of the kinin system that leads to bradykinin (BK) generation. Of these, gC1qR/p33 constitutes a high affinity site for HK – the BK precursor – and is therefore critical for the assembly of the kinin-generating cascade. Previous studies have identified a putative HK site within the C-terminal domain (residues 204–218) of gC1qR recognized by mAb 74.5.2. In these studies, we used information from the crystal structure of gC1qR, to engineer several deletion (Δ) mutants and test their ability to bind and/or support BK generation. While deletion of residues 204–218 (gC1qRΔ204–218), showed significantly reduced binding to HK, BK generation was not affected when tested by a sensitive bradykinin immunoassay. In fact, all of the gC1qR deletion mutants supported BK generation with the exception of gC1qRΔ154–162 and a point mutation in which Trp 233 was substituted with Gly. Binding studies also identified the existence of two additional sites at residues 144–162 and 190–202. Moreover, binding of HK to a synthetic peptide 190–202 was inhibited by mAbs 48 and 83, but not by mAb 74.5.2. Since a single residue separates domains 190–202 and 204–218, they may be part of a highly stable HK binding pocket and therefore a potential target for drug design to prevent vascular permeability and inflammation.
Introduction
The surface receptor for (gC1qR) is a ubiquitously distributed, highly anionic cellular protein of 33 kDa that binds to the globular heads of C1q (gC1q; Ghebrehiwet et al., 1994, 2001; Ghebrehiwet and Peerschke, 1998). Known alternatively as p33, p32, or hyaluronic acid binding protein 1 (HABP-1), it is a multi-ligand binding protein that is distributed in several cellular compartments, including the mitochondria, the ER, and the nucleus, in addition to the cell surface (Ghebrehiwet et al., 1994; Dedio et al., 1998; van Leuwen and O’Hare, 2001). It is its surface expression and structural basis of its function that have been the primary focus of our laboratory for the past 20 years (Ghebrehiwet et al., 1994, 2001; Ghebrehiwet and Peerschke, 1998).
The binding of HK, to gC1qR is strictly zinc dependent (Herwald et al., 1996; Joseph et al., 1996; Hasan et al., 1998). HK binds to endothelial cell (EC) surface expressed gC1qR with high affinity (Kd of 9 ± 2 nM; Herwald et al., 1996) and to isolated gC1qR with a Kd of 0.8 ± 0.7 nM (kon = 12.3 ± 5.0 × 104/M/s; koff = 0.8 ± 0.5 × 10−4/M/s; Pixley et al., 2011). The interaction between HK and cell surface expressed or isolated gC1qR is inhibited by mAb 74.5.2, which recognizes the C-terminal half of gC1qR comprising residues 204–218 (Joseph et al., 1996). The receptors, which are organized as two bimolecular complexes of gC1qR–cytokeratin 1 and urokinase-type plasminogen activator receptor (uPAR)–cytokeratin 1, are essential for the assembly and activation of the coagulation/kinin-forming cascade (Mahdi et al., 2001; Joseph et al., 2004).
The plasma kinin-forming pathway consists of three essential proteins that interact in vivo in a complex fashion once bound to a macromolecular complex formed during inflammatory response or bound to proteins along cell surfaces (Kaplan, 2004). These are coagulation factor XII [FXII (or Hageman factor, HF)], prekallikrein (PK), and high molecular weight kininogen (HK). Once FXII is activated to FXII a, it converts prekallikrein, which circulates as complex with HK, to kallikrein and the latter in turn digests HK to generate the nonapeptide BK (H-Arg-Pro-Pro-Gly-Phe-Ser-Pro-Phe-Arg-OH; Kaplan, 2004). Bradykinin, belongs to the kinin family of proinflammatory peptides, and is among the most potent vasodilator agonists known (Erdos and Sloane, 1962; Regoli and Barabe, 1980; Bhoola et al., 1992; Margolius, 1998). The affinity of HK and HKa for gC1qR is much higher [gC1qR > cytokeratin 1 > uPAR than it is for cytokeratin 1 or uPAR and thus gC1qR may be pivotal for the assembly of the kallikrein kinin system (KKS; Herwald et al., 1996; Colman et al., 1997)].
Mature gC1qR is extremely acidic with a calculated pI of 4.15 (Ghebrehiwet et al., 1994, 2002; Jiang et al., 1999). It has one Cys at residue 186 and thus does not have any intrachain-disulfide bonding. It does not dimerize by inter-chain disulfide bonding either: on SDS-PAGE, it migrates as a 33-kDa band under both reducing and non-reducing conditions (Ghebrehiwet et al., 1994). However, it behaves as a trimer on gel filtration in non-dissociating conditions (Ghebrehiwet et al., 1994) and evidence from our laboratory suggests that multimer formation is an essential process that increases the affinity of gC1qR for multivalent ligands such as C1q and HK (Ghebrehiwet and Peerschke, 1998; Ghebrehiwet et al., 2001). The non-covalent trimeric structure has since been confirmed by X-ray crystallography (Jiang et al., 1999), which reveals a donut-shaped quaternary structure with asymmetric charge distribution (Ghebrehiwet et al., 1996) with one side containing a high distribution of negatively charged residues and, which we refer to as the solution face (S face), and the other side containing a more or less neutral net charge and referred to as the membrane face (M-face; Ghebrehiwet et al., 2002). More importantly, the 3D structure of the molecule reveals the presence of several highly charged domains with potential to play a role in ligand binding and cell attachment (Jiang et al., 1999; Ghebrehiwet et al., 2002). Using this knowledge, we generated several gC1qR deletion mutants in order to confirm the previously identified HK binding site and/or identify new ones. Identification and refinement of the precise interaction sites between gC1qR and HK in turn will allow us to translate this knowledge into novel small molecule-based, peptide-based, or antibody-based diagnostic and/or therapeutic strategies to block the generation of bradykinin and other vasoactive molecules that have been shown to contribute significantly to inflammatory diseases, vascular permeability, and edema (Bossi et al., 2009).
Materials and Methods
Chemicals and Reagents
The following reagents and chemicals were purchased or obtained from the sources indicated: Dulbecco’s PBS (DPBS) with and without calcium and magnesium (Mediatech Inc., Manassas, VA, USA); Dulbecco’s modified Eagles medium (DMEM); RPMI 1640, 100× Penicillin/Streptomycin, (GIBCO, Invitrogen, Grand Island, NY, USA); heat inactivated fetal bovine serum (FBS; Hyclone, Logan, UT, USA); human serum albumin (HSA; Immuno-US, Rochester, MI, USA); p-nitrophenyl phosphate (pNPP; Pierce, Rockford, IL, USA); and Immu-Mount (Thermo Fisher, Waltham, MA, USA). Alexa-488- or Alexa-594-Streptavidin, Alexa-488- or Alexa-594-F(ab′)2, goat anti-mouse or anti-rabbit; FITC conjugated goat anti-mouse IgG F(ab′)2 or sheep anti-rabbit IgG F(ab′)2 (Invitrogen, Carlsbad, CA, USA); and alkaline phosphatase (AP) – conjugated rabbit anti-goat IgG (Pierce), and C4d-EIA test kit (Quidel, San Diego, CA, USA).
Expression of Recombinant GC1qR/p33
The strategy for the construction of a plasmid containing the full-length (mature form or wild type, WT) or truncated forms of gC1qR cDNA as well as purification of the glutathione-S-transferase (GST)–gC1qR fusion products has been described in detail in our earlier publication (Ghebrehiwet et al., 1994; Lim et al., 1996). The GST–gC1qR fusion products were then cleaved by thrombin (3.2 μg/ml) and the GST-free gC1qR proteins purified on fast protein liquid chromatography (FPLC, Pharmacia) using a Mono-Q ion exchange column. The single peak containing the gC1qR was pooled, concentrated to 1–2 mg/ml, and stored at −80°C in the presence of 50 nM PPACK (D-phenylalanyl-L-prolyl-L-arginine chloromethyl ketone) – a specific thrombin inhibitor (Sigma Aldrich).
Generation and Purification of Deletion gC1qR Mutants
The cDNA and amino acid sequences of the region of gC1qR to be mutated, were identified and selected from analysis of the crystal structure as described (Jiang et al., 1999; Ghebrehiwet et al., 2002). To generate deletions, the Stratagene QuikChange double-strand mutagenesis kit was used, using complementary sense and antisense primers that bridge the deleted region (Ghebrehiwet et al., 1994; Lim et al., 1996). Maximization of mutagenesis was achieved by bringing the melting temperature (TM) on each side of the deleted sequence to 60°C as described (Breslauer et al., 1986). Primers were extended using the PfuTurbo polymerase and the Stratagene kit, and mutant constructs were enriched by digestion of the methylated template with Dpn1, and transformed into E. coli. Final constructs were sequenced in all cases to verify sequence integrity. The fusion products were purified as described above and elsewhere (Ghebrehiwet et al., 1994; Lim et al., 1996).
Single Point Mutagenesis
A single residue mutation was performed on Trp 233, which is the only domain that projects conspicuously from the S face in the crystal structure, fairly close to the second anionic loop as described earlier (Ghebrehiwet et al., 2002). Encoded by the codon TGG, this residue was mutated by a single-base change to GGG (Gly) using double-strand mutagenesis, transformed into E. coli and the protein purified as above.
Trimer Formation
To estimate the organization and integrity of each recombinant protein, gel filtration of purified gC1qR WT as well as the various deletion mutants was carried out by analytical gel filtration on a column (1.4 cm × 30 cm) packed with Superose beads and equilibrated in 50 mM Tris–HCl buffer, pH 7.4, containing 1 mM EDTA in a manner similar to that described earlier (Ghebrehiwet et al., 1994). After adjusting the flow rate to 0.2 ml/min, the column was calibrated with two proteins having molecular weights close to those for the monomeric and trimeric forms of gC1qR, respectively. The peak elution times of these proteins – carbonic anhydrase (29 kDa) and BSA (65 kDa) – were recorded and used as the standard against which the elution time of each gC1qR protein was compared.
SDS-PAGE and Western Blot Analysis
Analyses on SDS-PAGE were performed on a 1.5-mm thick slab gels according to the method of Laemmli (1970) with samples being run unreduced or reduced and alkylated by boiling for 5 min in the presence of 0.1 M dithiothreitol and 0.2 M iodoacetamide. After electrophoresis, the gels were stained with Coomassie Brilliant Blue, destained, and dried. Samples for Western blot analysis were first run on SDS-PAGE as above then electro transferred to polyvinyl difluoride (PVDF) or nitrocellulose membranes, blocked with 5% non-fat milk containing TBST (20 mM Tris–HCl, 150 mM NaCl, and 0.05% Tween 20), and the bound proteins probed with the appropriate and specific antibodies. The bound antibodies in turn were visualized by chemiluminescence horseradish peroxidase-conjugated species-specific antibody followed by reaction with 4-chloro-1-naphthol substrate.
Cultured Cells
Human brain microvascular endothelial cells (HBr-MIVEC) were cultured in Microvascular Endothelial Cell Growth Medium-2 (EGM-2MV; Lonza, Walkersville, MD, USA) and contained 5% heat inactivated FBS. Cells grown to confluence on 2% gelatin (Type B), were first treated with 0.25% trypsin, 0.01% EDTA to dissociate cells from the gelatin matrix by incubation (30 min, 37°C) in 0.01 M Tris-buffered saline. The cells were then washed and then subcultured in DMEM. Experiments were done with cells between passages 3 and 15.
U937 cells were grown in RPMI 1640 containing 10% heat inactivated (2 h 56°C) fetal calf serum and 1% antibiotic antimycotic stock (100×) mixture consisting of penicillin G (10,000 U/ml), amphotericin B (25 μg/ml) and streptomycin sulfate (10,000 μg/ml), and maintained in a humidified 37°C incubator in an atmosphere of 95% air and 5% CO2.
Proteins and Antibodies
The purified proteins used in these studies were obtained from the following commercial sources. Single-chain or two-chain high molecular weight kininogen (HK), as well as FXII was from (Enzyme Res., South Bend, IN, USA) and monoclonal anti-HK (clone 115-21) and anti-FXII (clone 179) were a generous gift from Rebecca Rowehl (Hybridoma Core Facility, Stony Brook University), and were originally generated and characterized by Reddigari and Kaplan (1989). The various monoclonal and polyclonal antibodies to recombinant gC1qR used in these studies have been described in previous publications and represent part of the anti-gC1qR antibody databank in our laboratory (Ghebrehiwet et al., 1996).
Solid-Phase Microplate Binding Assay
The ability of the various gC1qR proteins to bind HK was assessed by solid-phase ELISA using microtiter plates (Nunc, CovaLink, NH, Denmark). Because the interactants are sticky proteins, the ELISA experiments were designed to include a measure that will exclude interaction due to “stickiness.” Briefly duplicate wells were first coated (30 min, room temp or O/N, 4°C) with 100 μl (2 μg/ml) of WT or various gC1qR mutants (ΔgC1qR) or BSA in carbonate buffer, pH 9.6 (15 mM Na2CO3 and 35 mM NaHCO3). The unbound proteins were aspirated; the wells washed 2× with TBST (20 mM Tris–HCl pH 7.5, 150 mM NaCl, and 0.05% Tween-20), and the unreacted sites blocked by incubation (30 min, room temp) with 300 μl of 1% non-fat dry milk or blotto (10 mg/ml casein in TBS, pH 8.0). After washing 2× with TBST, 100 μl each of concentrations of HK ranging from 0 to 5 μg/ml in TBSZ (TBS containing 10–50 μM Zinc) were added and incubated (30 min, room temp). The wells were then washed, 1× with TBSZ containing 0.5 M NaCl, and 2× with TBSTZ (containing 10 μM Zinc). After drying, the bound proteins were detected by sequential addition and incubation (30 min, room temp) with 100 μl of 2 μg/ml mAb anti-HK, followed by alkaline phosphatase conjugated rabbit anti-goat or goat anti-mouse IgG and pNPP (freshly dissolved in 10% diethanolamine pH 9.5, containing 5 mM MgCl2 and 0.02% NaN3). In experiments in which biotin-labeled HK was employed, the ELISA was developed using alkaline phosphatase conjugated Neutravidin followed by pNPP. The absorbance of the resulting color development in either case was measured at 405 nm using a VMax Kinetic plate reader (Molecular Devices, Menlo Park, CA, USA). The experiments were done at least three times in duplicates.
Similar protocol was used to determine the interaction between FXII and gC1qR either with gC1qR bound to the duplicate wells and the bound FXII detected by mAb anti-FXII or in the reverse order in which FXII was bound to the plate and biotinylated gC1qR mutants were bound and detected by alkaline phosphatase conjugated Neutravidin followed by pNPP.
Bradykinin Assay
The ability of gC1qR to generate bradykinin (BK) was assayed in the following manner. Duplicate microtiter plate wells were first coated (30 min, 22°C) with 1% PEG to block charged sites, then gC1qR mutants were incubated with 100 μl of a mixture (1 μg/ml each) of highly purified HK, PK in the presence of 50 μM zinc and in the presence or absence of FXII (1 μg/ml each). Samples were incubated (30 min, 22°C) and the protein in the samples was precipitated with chilled ethanol, centrifuged at 10,000 × g for 20 min, and the supernatant containing BK was lyophilized and re-suspended in the assay buffer. The amount of BK in the sample was then determined using a commercial kit from Peninsula Laboratories (Peninsula Labs, San Carlos, CA, USA).
Flow Cytometry and Immunofluorescence Analyses
For flow cytometry, the cultured cell line, U937, was used. After verifying cell viability by trypan blue exclusion (≥95%), the cells (106/ml DPBS) were preincubated (30 min, 20°C) with isotype-matched Fc fragments, followed by incubation (60 min, 20°C) with either buffer alone, or gC1qR proteins (10 μg/ml). After incubation, the cells were washed in DPBS, and further incubated with a predetermined dilution of mAb anti-gC1qR followed by another incubation with Alexa-488 conjugated and species matched IgG. The cells were then washed, fixed by suspension in 4% paraformaldehyde and then analyzed by flow cytometry. When biotin-labeled gC1qR proteins were used, then the cells were directly incubated with the proteins – thus obviating the blockade with Fc fragments – and the bound gC1qR was detected using Alexa-488 conjugated streptavidin.
Immunofluorescence studies were performed on either U937 cells or cultured HBr-MIVECs. The cells were grown on cover slips and the attached monolayer of cells reacted first with PBS containing 0.1% BSA and 1% heat inactivated human serum to block unreacted sites, followed by incubation with biotinylated gC1qR proteins and FITC conjugated secondary reagent as described above. After fixing for 10 min with 3.7% (v/v) formalin, the slides were examined by 3D imaging using deconvolution microscopy.
Solid-Phase Binding to Microplate-Fixed U937 Cells
The ability of gC1qR and its deletion mutants to bind to U937 cells was tested using a microplate assay with fixed cells as described elsewhere (Kennet, 1984; Ghebrehiwet et al., 1996). Briefly, intact U937 cells (2 × 105 cells/well) were first attached (30 min, 22°C) onto poly-L-lysine (10 μg/ml in PBS, pH 7.4) coated duplicate ELISA wells. The cells were fixed (30 min, 22°C) by addition of an equal volume of glutaraldehyde (0.5% solution in PBS), and the unreacted sites quenched with glycine–BSA (100 mM Glycine, 0.1% BSA in PBS, pH 7.4). Then, biotinylated gC1qR or deletion mutants ranging 0–5 μg/ml were added and the bound proteins detected by standard ELISA procedure as described.
Statistical Analysis
Data are presented as mean ± SEM. Student’s t-tests were performed using statistical software (Excel; Microsoft, Redmond, WA, USA). A value of p = 0.05 was considered to be a significant difference (n values represent separate experiments performed under the same conditions).
Results
Assessment of Trimer Formation
After purification to homogeneity as assessed by SDS-PAGE, the deletion proteins were subjected to analytical gel filtration to assess whether deletion of a particular domain affects the trimer formation. As shown in Table 1, three deletion mutants failed to maintain the trimeric structure of the molecule. These domains correspond to amino acid residues 74–95, 204–218, and 212–223. The N-terminal residues 74–95 are part of the αA coiled-coil region of each monomer, which forms extensive intermolecular contacts in the trimeric structure of the molecule (Jiang et al., 1999; Ghebrehiwet et al., 2002). The domain covered by residues 204–218, on the other hand, forms the bulk of the β6 structure of each monomer with residues 212–223 essentially making the entire connecting loop between the β6 and β7 and partially covers the 20-Å donut hole of the trimer (Jiang et al., 1999; Ghebrehiwet et al., 2002).
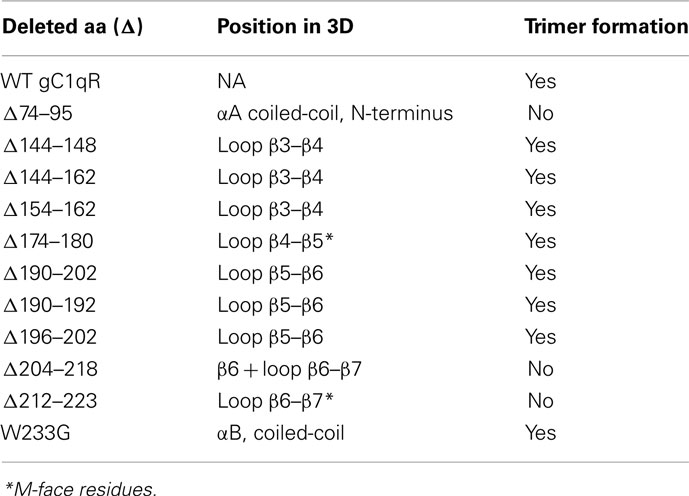
Table 1. Location of deleted residues and effect on trimerization.
Identification of gC1qR Sites for HK
As shown (Figures 1A,B), deletion of the previously identified HK site (aa 204–218) did not support HK binding. However, additional domains, whose deletion adversely affected HK binding, were also identified. These are domains covered by residues, 74–95, 144–148, 190–192, and 196–202. Furthermore, deletion of residues, which are critical for the trimeric integrity (Table 1) of the gC1qR molecule have also reduced HK binding, indicating that monomeric gC1qR does not bind HK efficiently. The relative locations of residues 144–162 (Figure 1C) and 196–2020 (Figure 1D) in the 3D structure of the trimeric molecule are provided for visual reference.
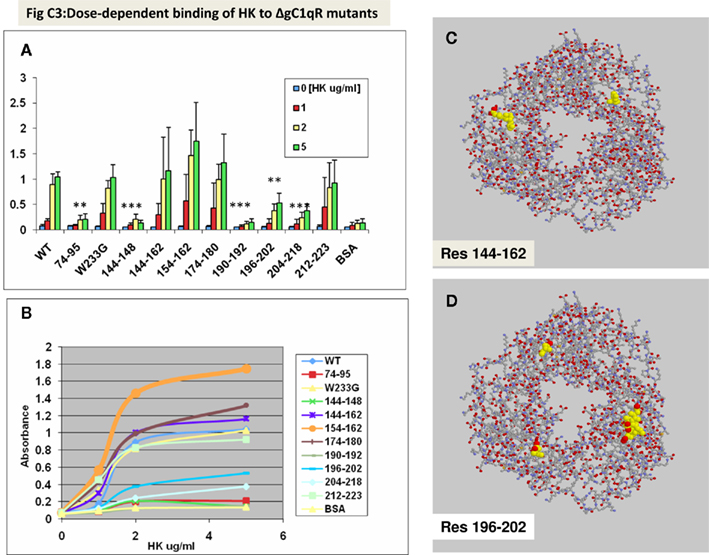
Figure 1. Binding of HK to gC1qR mutants using solid-phase microplate assay. Duplicate wells of microtiter plates were first coated with 100 μl (2 μg/ml) of wild type or gC1qR mutants (ΔgC1qR). After the unreacted sites were blocked, concentrations of HK were added as indicated. The bound HK was detected by sequential interaction with 2 μg/ml mAb anti-HK, followed by alkaline phosphatase conjugated goat anti-mouse IgG and pNPP. The absorbance of the color developed at the end of the reaction was read at 405 nm using a VMax Kinetic plate reader. Each data point (A,B) is a mean of three experiments run in duplicates and (B) represents a saturation curve of data in (A) and the location in gC1qR of residues 144–162 (C) and 196–202 (D) is provided as a reference. The significance of data is indicated as: *p ≤ 0.05; **p ≤ 0.01 and ***p ≤ 0.001.
Since a single residue separates 196–202 and residues 204–218, we hypothesized that this domain, together with residues 204–218, might be part of the HK binding pocket in gC1qR. To test this hypothesis, we generated a synthetic peptide corresponding to residues190–202 (six residues longer) and tested its ability to support HK binding. The results of this experiment showed that biotinylated HK bound to the peptide in a specific and dose-dependent manner (not shown). Further preliminary studies also showed that the interaction of HK with peptide 190–202 was not inhibited by mAb 74.5.2, which recognizes residues 204–218 (not shown). This suggested that either HK binds at multiple sites or residues 190–202 contribute residue(s) for the putative binding pocket. Using this information, we designed experiments to screen for monoclonal antibodies that could inhibit the interaction between HK and peptide 190–202. As shown in Figure 2, binding of biotinylated HK was completely blocked by mAbs 48 and 83 but not by mAbs 69 or 73.
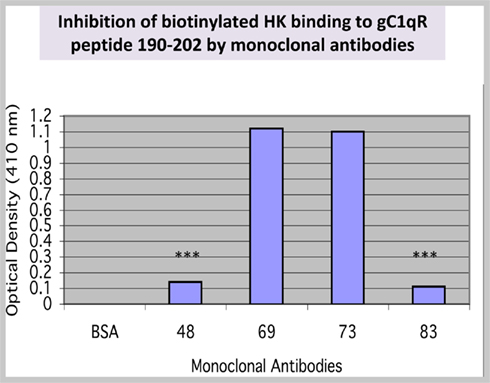
Figure 2. Binding of HK to a synthetic peptide 190–202. A peptide corresponding to residues 190–202 was synthesized and purified to ≥95% purity. Synthetic peptide (residues 190–202) was coated (50 μg/ml) onto duplicate microtiter wells. After blocking, the peptide was incubated with biotinylated HK in the presence or absence of mAbs. Control for non-specific HK binding was BSA. The data (n = 2) is a representative of experiments run in duplicates and under the same conditions. The significance of data is indicated as: *p ≤ 0.05; **p ≤ 0.01; and ***p ≤ 0.001.
Identification of gC1qR Sites for Factor XII
Previous experiments have shown that the binding of FXII to the EC surface is similar to that of HK in that it has the same requirement for zinc for binding and can compete for the same site with HK at comparable molar ratio (Reddigari et al., 1993). This observation led to the postulate that the two proteins may compete for the same receptor site on ECs. Since both HK and FXII can bind gC1qR, we examined whether they share the same or overlapping site(s) on the molecule. As shown in Figure 3, deletion of residues 204–218, which is the major binding site for HK did not affect the binding of FXII. However, several deletion mutants, including those identified as potential additional sites for HK (Figure 1) such as domains 74–95, 144–148, and 196–202 showed remarkably poor binding suggesting that the binding of HK and FXII may be to an overlapping site(s) rather than an identical site on gC1qR.
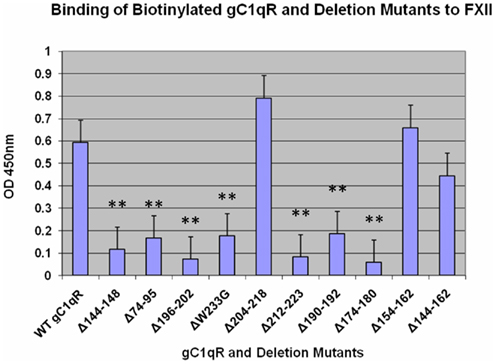
Figure 3. Binding of biotinylated gC1qR mutants to of FXII. The experimental procedures are essentially as described in the Legend to Figure 1. Each data point is a mean of three experiments run in duplicates with p values as described in the legends to Figures 1 and 2.
Assembly of Kallikrein/Kinin System and BK Generation
We have shown previously that gC1qR is capable of serving as a platform for the assembly of the KKS (Joseph et al., 2001a). Experiments were therefore designed to identify the critical site(s) for the assembly of KKS and the generation with BK. Two domains, whose deletion resulted in significantly reduced BK generation, were identified: residues 154–162, and a single point mutation W233G (not shown). Residues 154–162 are part of the disordered segment that form the connecting loop between β4 and β5 in the monomer, whereas Trp233 projects conspicuously from the trimeric structure and is at the beginning of the highly conserved sequence αB coiled-coil (Table 1).
Binding of Deletion Mutants to Cells
Although gC1qR is a membrane-associated protein, it can nonetheless be secreted into the pericellular milieu by infected cells or cells undergoing active proliferation. This soluble gC1qR can also bind to cells in a specific and dose-dependent manner (Peterson et al., 1997). When biotinylated gC1qR proteins were incubated with the fixed cells, almost all of the deletion mutants were able to bind to the cells with varying degrees and in a dose-dependent manner with the exception of gC1qRΔ144–148, gC1qRΔ174–180, gC1qRΔ190–202, and gC1qRΔ196–202, which bound poorly, suggesting that these domains might be involved in cell attachment (Figure 4). These results were further confirmed when the binding of the deletion mutants to intact U937 cells in solution was tested by flow cytometry (Figure 5). In addition, we also used deconvolution microscopy to test the binding of the deletion mutants to microvascular endothelial cells. As shown in Figure 6, both gC1qRΔ144–148 and gC1qRΔ174–180, which is on the M-face of the protein, had weak to almost no binding to ECs. Therefore these domains – in particular residues 144–148 and 174–180 – may be suitable baits to identify the cell surface molecule to which they bind.
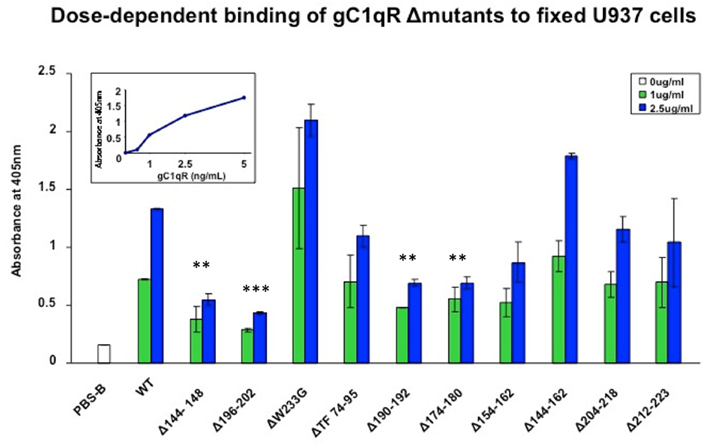
Figure 4. Dose-dependent binding of gC1qR Δmutants to fixed U937 cells. U937 cells were first attached to 96-well microtiter plates using poly-L-Lysine and fixed as described (Kennet, 1984; Ghebrehiwet et al., 1996). The cells were then reacted with biotinylated gC1qR wild type (WT) or gC1qR deletion mutants (Δ) mutants. After incubation, the plate was developed by sequential addition of alkaline phosphatase conjugated streptavidin followed by pNPP. Each data point is a mean of two experiments run in duplicates and compares the binding of WT gC1qR with gC1qRΔ mutants at two concentrations: 1 and 2.5 μg/ml. The figure in the inset is a dose-dependent binding of WT from which the optimal concentration(s) to be used was determined.
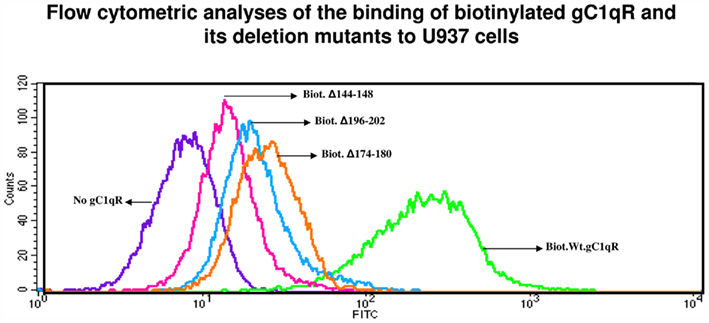
Figure 5. Flow cytometric analysis of gC1qR and deletion mutants. Biotinylated gC1qR or gC1qR mutants were first incubated with U937 cells (106/ml), and the bound proteins revealed by incubation with a predetermined dilution of Alexa-488 conjugated streptavidin, fixed by suspension in 4% paraformaldehyde and then subjected to analysis by flow cytometry. The figure is a representative of two such experiments.
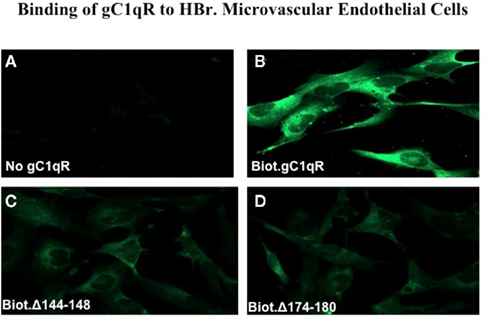
Figure 6. Immunofluorescence studies. Human Brain microvascular endothelial cells (HBr-MIVECs) were first grown on cover slips and the attached cells reacted first with PBS containing 0.1% BSA and 1% heated human serum to block unreacted sites, followed by Alexa-488 conjugated streptavidin or neutravidin. After incubation, the cells were fixed for 10 min with 3.7% (v/v) formalin, and then examined by 3D imaging using deconvolution microscopy. (A) is non-specific control and shows the image of cells reacted with Alexa-conjugated streptavidin (ACS) only. (B–D) show images of cells that were first treated with either biotinylated WT-gC1q-R, or deletion mutants 144–148 or 174–180 and then stained with ACS.
Discussion
In the absence of the 3D structure, peptide mapping, and inhibition studies were previously used to identify the binding sites for the ligands of gC1qR including C1q and HK. The availability of a detailed crystal structure (Jiang et al., 1999) of gC1qR (p32) provided a unique opportunity – and a much more realistic structural platform – to design gC1qR mutations that would permit confirmation of previously identified sites and/or identification new ones. The primary structure deduced from its cDNA shows that gC1qR is synthesized as a pre-protein of 282 residues (Ghebrehiwet et al., 1994) whereas its crystal structure reveals a donut-shaped homotrimer, with a 20-Å channel, which is partially covered by the loops connecting β6 and β7 structures, thereby reducing the channel size by half (Jiang et al., 1999). On the basis of information derived from the 3D structure, we generated and expressed 10 gC1qR deletion mutants – with regions predicted to contain either a membrane attachment site or ligand binding site. In addition, we also generated one gC1qR protein with a single mutation Trp233 to Gly (W233G) – which by virtue of its conspicuous projection from the solution face, was considered a likely target for plasma or pathogen associated ligands (Ghebrehiwet et al., 2002). Although the deleted domains are ostensibly quite large, they have been carefully selected, not only by reason of a likely functional role, but also to ensure proximity of the flanking residues in the native crystal structure (Jiang et al., 1999; Ghebrehiwet et al., 2002). The latter criterion is particularly important when considering the question whether mutant proteins fold correctly (Ghebrehiwet et al., 2002). Judging by adjacency of flanking residues in the crystal structure, all the deletions that were generated meet this requirement well. The highly purified deletion mutant proteins were then subjected to analysis using a battery of functional measurements including assessment of trimerization, specific ligand binding, general function such as bradykinin generation, which together provided a good assessment of folding and function.
On the basis of the data presented in these studies, we make the following conclusions. First, with the exception of Δ74–95, Δ204–218, and Δ212–223, all deletion mutants folded properly and formed trimers (Table 1). Interestingly, of the three that failed to form a trimer, the two domains are ligand binding sites that were identified earlier by peptide mapping and antibody inhibition studies (Ghebrehiwet et al., 1994, 2001; Ghebrehiwet and Peerschke, 1998). Residues 74–95 are part of the domain encoded by exon II (residues 78–128), which contains the C1q binding site, whereas residues 204–218 contain the HK site and are part of the domain encoded by exon V (residues193–233) – the only “self-standing” exon domain in the protein predicted to have normal folding (Ghebrehiwet et al., 2002). The N-terminal domain 74–95 is part of the αA coiled-coil region of each monomer, which forms extensive intra- and inter-monomer contacts in the trimeric structure of the molecule (Erdos and Sloane, 1962). Second, the experiments confirm that the binding site for HK is the domain covered by 204–218. The fact that mAb74.5.2, which inhibit HK binding failed (not shown) to also bind to gC1qRΔ204–218 mutant further substantiates this conclusion. However, in addition to residues 204–218, we also found that residues 190–192 and 196–202 to be critical for HK binding since deletion of these residues failed to support its binding. Furthermore, HK was able to bind to a synthetic peptide corresponding to residues 190–202 and this binding was completely inhibited by two mAbs 48 and 83, but not by mAb 74.5.2 making these antibodies useful additional tools for inhibition of ligand induced biological response. Third, whereas residues 204–218, which are recognized by mAb 74.5.2 constitute the entire β6 structure and part of the segment of the loop that connects β6 to β7, residues 190–192 and 196–202 form the entire disordered segment of the loop that connects β5 and β6 sheets in the 3D structure (Erdos and Sloane, 1962). Since the loops connecting the different β structures – in particular those connecting β6 to β7 – partially cover the donut channel, disruption of any of these residues is likely to disrupt the residues in the channel that form a tight cluster for efficient binding of mAb 74.5.2, which has been shown to bind to residues that project into the donut hole (Ghebrehiwet et al., 2002).
One of the major functions of gC1qR especially on activated or damaged ECs is its ability to bind HK with high affinity and trigger the kinin-generating pathway (Joseph et al., 2001a). In plasma, HK circulates as complex with PK and the two bind to the EC surface via either gC1qR – which is expressed in excess – or to gC1qR–cytokeratin 1 complex (Joseph et al., 2004). Bradykinin formation can then be initiated either by FXII activation – preferentially bound to the uPAR–cytokeratin complex (Joseph et al., 2004) – or by secretion of HSP-90 by activated ECs (Joseph et al., 2002). However, auto activation of FXII can also occur upon binding to gC1qR (Joseph et al., 2001b, 2009; Shariat-Madar et al., 2002). Although BK generation can be achieved in solution when HK, PK, and FXII are mixed together and left to incubate for hours, the addition of the physiological receptor, gC1qR, or kaolin accelerates this process from hours to minutes. The function of gC1qR on the cell surface is likely to be potentiated by either a conformational change upon ligand binding or the presence of other partner molecules (uPAR and cytokeratin 1). Regardless, the above experiments prove that BK can be generated in the presence of soluble gC1qR. This fact alone gives credence to the postulate that gC1qR, which is secreted into the pericellular milieu by activated or proliferating cells, can potentially induce BK generation in the presence of circulating PK, HK, and FXII, which are recruited to activated ECs (Kaplan, 2004). This is particularly relevant in infections (Staphylococcus aureus) or tumor metastasis.
In previous studies (Peterson et al., 1997), we have shown that recombinant or soluble gC1qR purified from culture supernatants of proliferating cells binds to intact U937 cells or ECs. This finding prompted us to ask two pertinent questions: why and how does gC1qR bind to intact cells? Because blockade of both gC1qR and bradykinin receptor 1 (B1R) – which is not constitutively expressed but inducible by IL-1β – abrogates BK-induced angioedema (Bossi et al., 2009), we hypothesized that secreted gC1qR could serve as a potential autocrine signal, to induce or suppresses the expression of cell surface molecules such as B1R. In order to do so however, the soluble gC1qR must bind to the cell surface through a gC1qR attachment domain presumably on the M-Face (Ghebrehiwet et al., 2002). While WT gC1qR was able to bind to U937 cells as expected, four deletion mutants were identified that failed to bind: gC1qRΔ144–148, gC1qRΔ174–180, gC1qRΔ190–192, and gC1qRΔ196–202 (Figure 4). The binding of these deletion mutants to intact cells was also either weak or negligible (Figures 5 and 6). Interestingly, domains 141–146 [(PTFDGE) contained in residues 144–148] and 191–196 (DEVGQE), have extremely high net negative charges and are not located in all three chains in the crystal structure and sequences 189–190 and 197–201 are only located in the A chain. Therefore mutants lacking part or the whole segment of the two disordered loops – 144–148 connecting β3 to β4 and 191–196 connecting β5 to β6 – are likely to be critical for ligand binding, structural stability, and/or function. What is more intriguing is that the nine aa (IKNDDGKKA) sequence which includes residues174–180 (underlined) found on the M-face of gC1qR is a highly conserved domain. With the exception of one or two amino acid substitutions (underlined), this domain is identical to sequences found in Trichomonas vaginalis G3 protein (IKNDDLKKA), Borrelia valaisiana (IKNDDGKKI), Borrelia burgdorferi B31 (IKNEDGKKV), and zebra fish (IKNDDGRKA). The relevance of this domain as a cell attachment site and to gC1qR-mediated function is currently being investigated.
In summary, the present studies not only confirm the relevance of residues 204–218 as an HK site, but also identified new sites. This in turn suggests that either HK has multiple sites or the various HK sites may each contribute residue(s) that form a ligand binding cluster in space. The gC1qR molecule plays a central role in the activation of the kinin system thereby generating not only BK, but also HKa – a cleavage product of HK – in the process. While BK is known to induce vascular permeability and to promote angiogenesis (Guo and Colman, 2005), HKa has been shown to release cytokines such as TNFα, interleukin IL-1β, IL-6, and the chemokines IL-8 and MCP-1 from human mononuclear cells, all of which contribute to inflammatory diseases (Khan et al., 2006). Inasmuch as agents that block gC1qR are likely to be useful in blocking BK generation, the gC1qR domains identified in these studies can serve as suitable templates for the design of either antibody-based or peptide-based therapeutic agents for the prevention or attenuation of vascular leakage and subsequent inflammation. This rationale is further strengthened by recent animal model studies that showed: (i) vascular permeability induced by the attack phase plasma from C1 inhibitor-deficient patients was prevented by blockade of gC1qR with mAb 74.5.2 (Bossi et al., 2009), and (ii) blockade of gC1qR – shown previously (Nguyen et al., 2000) to serve as EC and platelet receptor for protein A – inhibits adherence of S. aureus to microvascular endothelium (Sethi et al., 2011).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported in part by grants from the National Institutes of Health (R01 AI 060866 and R01 AI-084178).
References
Bhoola, K. D., Figueroa, C. D., and Worthy, K. (1992). Bioregulation of kinins: kallikrein, kininogen and kinases. Pharmacol. Rev. 44, 1–80.
Bossi, F., Fischetti, F., Regoli, D., Durigutto, P., Frossi, B., Gobeil, F. Jr., Ghebrehiwet, B., Peerschke, E. I. B., Cicardi, M., and Tedesco, F. (2009). Novel pathogenic mechanism and therapeutic approaches to angioedema associated with C1 inhibitor deficiency. J. Allergy Clin. Immunol. 124, 1303–1310.
Breslauer, K. J., Frank, R., Blocker, H., and Marky, L. A. (1986). Predicting DNA duplex stability from the base sequence. Proc. Natl. Acad. Sci. U.S.A. 83, 3746–3750.
Colman, R. W., Pixley, R. A., Najamunnisa, S., Yan, W., Wang, J., Mazar, A., and McRae, K. R. (1997). Binding of high molecular weight kininogen to human umbilical vein endothelial cells is mediated via a site within domains 2 and 3 of the urokinase receptor. J. Clin. Invest. 100, 1481–1487.
Dedio, J., Jahnen-Dechent, W., Bachmann, M., and Muller-Esterl, W. (1998). The multiligand-binding protein gC1q-R, putative C1q receptor, is a mitochondrial protein. J. Immunol. 160, 3534–3542.
Erdos, E. G., and Sloane, S. M. (1962). An enzyme in human plasma that inactivates bradykinin and kallidins. Biochem. Pharmacol. 11, 585–592.
Ghebrehiwet, B., Jesty, J., and Peerschke, E. I. B. (2002). gC1q-R/p33: structure-function predictions from the crystal structure. Immunobiology 205, 421–432.
Ghebrehiwet, B., Lim, B.-L., Kumar, R., Feng, X., and Peerschke, E. I. B. (2001). gC1q-R/p33, a member of a new class of multifunctional and multi-compartmental cellular proteins, is involved in inflammation and infection. Immunol. Rev. 180, 65–77.
Ghebrehiwet, B., Lim, B. L., Peerschke, E. I. B., Willis, A. C., and Reid, K. B. M. (1994). Isolation cDNA cloning, and overexpression of a 33-kDa cell surface glycoprotein that binds to the globular “heads” of C1q. J. Exp. Med. 179, 1809–1821.
Ghebrehiwet, B., Lu, P. D., Zhang, W., Lim, B.-L., Eggleton, P., Leigh, L. E. A., Reid, K. B. M., and Peerschke, E. I. B. (1996). Identification of functional domains on gC1q-R, a cell surface protein, which binds to the globular heads of C1q, using monoclonal antibodies and synthetic peptides. Hybridoma 15, 333–344.
Ghebrehiwet, B., and Peerschke, E. I. B. (1998). Structure and function of gC1q-R a multi-ligand binding membrane protein. Immunobiology 199, 225–238.
Guo, Y.-L., and Colman, R. W. (2005). Two faces of high-molecular-weight kininogen (HK) in angiogenesis: bradykinin turns it on and cleaved HK (HKa) turns it off. J. Thromb. Haemost. 3, 670–676
Hasan, A. A. K., Zisman, T., and Schmaier, A. H. (1998). Identification of cytokeratin 1 as a binding protein and presentation receptor for kininogens on endothelial cells. Proc. Natl. Acad. Sci. U.S.A. 95, 3615–3620.
Herwald, H., Dedio, J., Kellner, R., Loos, M., and Muller-Esterl, W. (1996). Isolation and characterization of the kininogen-binding protein p33 from endothelial cells. Identity with the gC1q receptor. J. Biol. Chem. 271, 13040–13047.
Jiang, J. Y., Zhang, K. A., and Xu, R.-M. (1999). Crystal structure of p32, a doughnut-shaped acidic mitochondrial matrix protein. Proc. Natl. Acad. Sci. U.S.A. 96, 3572–3577.
Joseph, K., Ghebrehiwet, B., and Kaplan, A. P. (2001a). Activation of the kinin-forming cascade on the surface of endothelial cells. Biochem. J. 382, 71–75.
Joseph, K., Ghebrehiwet, B., and Kaplan, A. P. (2001b). Activation of the kinin-forming cascade on the surface of endothelial cells. Biol. Chem. 382, 71–75.
Joseph, K., Ghebrehiwet, B., Peerschke, E. I. B., Reid, K. B. M., and Kaplan, A. P. (1996). Identification of the zinc-dependent endothelial cell binding protein for high molecular weight kininogen and factor XII: identity with the receptor which binds to the globular “heads” of C1q (gC1q-R). Proc. Natl. Acad. Sci. U.S.A. 93, 8552–8557.
Joseph, K., Tholanikunnel, B. G., Ghebrehiwet, B., and Kaplan, A. P. (2004). Interaction of high molecular weight binding proteins on endothelial cella. Thromb. Haemost. 91, 61–70.
Joseph, K., Tholanikunnel, B. G., and Kaplan, A. P. (2002). Heat shock protein 90 catalyzes activation of the prekallikrein-kininogen complex in the absence of factor XII. Proc. Natl. Acad. Sci. U.S.A. 99, 896–900.
Joseph, K., Tholanikunnel, B. G., and Kaplan, A. P. (2009). Factor XII-independent cleavage of high-molecular-weight kininogen by prekallikrein and inhibition by C1 inhibitor. J. Allergy Clin. Immunol. 124, 143–149.
Kaplan, A. P. (2004). “Mechanisms of bradykinin generation,” in Urticaria and Angioedema, eds M. W. Greaves and A. P. Kaplan (New York: Marcel Decker Inc.), 51–72.
Kennet, R. H. (1984). “Enzyme-linked antibody assay with cells attached with polyvinyl chloride plates,” in Monoclonal Antibodies, eds R. H. Kennet, T. J. McKearn, and K. B. Bechtol (New York: Plenum Press), 376.
Khan, M. M., Bradford, H. N., Isordia-Salas, I., Liu, Y., Wu, Y., Espinola, R. G., Ghebrehiwet, B., and Colman, R. W. (2006). High-molecular-weight kininogen fragments stimulate the secretion of cytokines and chemokines through uPAR, Mac-1, and gC1qR in monocytes. Arterioscler. Thromb. Vasc. Biol. 26, 2260–2266.
Laemmli, U. K. (1970). Cleavage of structural proteins during the assembly of the head bacteriophage T4. Nature 227, 680–685.
Lim, B.-L., Preissner, K., Ghebrehiwet, B., Leigh, L. E. A., and Reid, K. B. M. (1996). The binding protein for globular “heads” of complement C1q, gC1q-R: functional expression and characterization as a novel vitronectin binding factor. J. Biol. Chem. 271, 26739.
Mahdi, F., Shariat-Madar, Z., Todd, R. F. III, Figueroa, C. D., and Schmaier, A. H. (2001). Expression and localization of cytokeratin 1 and urokinase plasminogen activator receptor on endothelial cells. Blood 97, 2342–2350.
Margolius, H. S. (1998). Tissue kallikreins. Structure, regulation and participation in mammalian physiology and disease. Clin. Rev. Allergy Immunol. 16, 332–349.
Nguyen, T., Ghebrehiwet, B., and Peerschke, E. I. (2000). Staphylococcus aureus protein A recognizes platelet gC1qR/p33: a novel mechanism for staphylococcal interactions with platelets. Infect. Immun. 68, 2061–2068.
Peterson, K., Zhang, W., Lu, P. D., Keilbaugh, S. A., Peerschke, E. I. P., and Ghebrehiwet, B. (1997). The C1q binding membrane proteins cC1q-R and gC1q-R, are released from activated cells. Subcellular localization and immunochemical characterization. Clin. Immunol. Immunopathol. 84, 17–26.
Pixley, R. J., Espinola, R. G., Ghebrehiwet, B., Joseph, K., Kao, A., Cines, D. B., and Colman, R. W. (2011). Interaction of high molecular weight kininogen with endothelial cell binding proteins suPAR, gC1qR and cytokeratin 1 determined by plasmon resonance (BiaCore). Thromb. Haemost. 105, 1053–1059.
Reddigari, S., and Kaplan, A. P. (1989). Monoclonal antibody to human high molecular-weight kininogen recognizes its prekallikrein binding site and inhibits its coagulant activity. Blood 74, 695–702.
Reddigari, S. R., Shibayama, Y., Brunnee, T., and Kaplan, A. P. (1993). Human Hageman factor (factor XII) and high molecular kininogen compete for the same binding site on human umbilical vein endothelial cells. J. Biol. Chem. 268, 11892–11987.
Regoli, D., and Barabe, J. (1980). Pharmacology of bradykinin and related kinins. Pharmacol. Rev. 32, 1–46.
Sethi, S., Herrmann, M., Roller, J., von Müller, L., Peerschke, E. I., Ghebrehiwet, B., Bajric, I., Menger, M. D., and Laschke, M. W. (2011). Blockade of gC1qR/p33, a receptor for C1q, inhibits adherence of Staphylococcus aureus to the microvascular endothelium. Microvasc. Res. 82, 66–72.
Shariat-Madar, Z., Mahdi, F., and Schmaier, A. H. (2002). Identification and characterization of prolylcarboxypeptidase as an endothelial cell prekallikrein activator. J. Biol. Chem. 277, 17962–17969.
Keywords: gC1qR, vascular permeability, inflammation, kininogen, bradykinin
Citation: Ghebrehiwet B, Jesty J, Xu S, Vinayagasundaram R, Vinayagasundaram U, Ji Y, Valentino A, Hosszu KK, Mathew S, Joseph K, Kaplan AP and Peerschke EIB (2011) Structure–function studies using deletion mutants identify domains of gC1qR/p33 as potential therapeutic targets for vascular permeability and inflammation. Front. Immun. 2:58. doi: 10.3389/fimmu.2011.00058
Received: 04 July 2011;
Accepted: 17 October 2011;
Published online: 15 November 2011.
Edited by:
Peter F. Zipfel, Leibniz Institute for Natural Product Research and Infection Biology, GermanyReviewed by:
Mihaela Gadjeva, Harvard Medical School, USAMartin Herrmann, Universitätsklinikum Erlangen, Germany
Copyright: © 2011 Ghebrehiwet, Jesty, Xu, Vinayagasundaram, Vinayagasundaram, Ji, Valentino, Hosszu, Mathew, Joseph, Kaplan and Peerschke. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Berhane Ghebrehiwet, Health Sciences Center, Stony Brook University School of Medicine, T-16, Room 040, Stony Brook, New York, NY 11794-8161, USA. e-mail:YmVyaGFuZS5naGVicmVoaXdldEBzdG9ueWJyb29rLmVkdQ==