 Pushpa Pandiyan*
Pushpa Pandiyan* Lixin Zheng and Michael J. Lenardo
Lixin Zheng and Michael J. Lenardo
- Laboratory of Immunology, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD, USA
CD4+CD25+Foxp3+ T lymphocytes, known as regulatory T cells or Tregs, have been proposed to be a lineage of professional immune suppressive cells that exclusively counteract the effects of the immunoprotective “helper” and “cytotoxic” lineages of T lymphocytes. Here we discuss new concepts on the mechanisms and functions of Tregs. There are several key points we emphasize: 1. Tregs exert suppressive effects both directly on effector T cells and indirectly through antigen-presenting cells; 2. Regulation can occur through a novel mechanism of cytokine consumption to regulate as opposed to the usual mechanism of cytokine/chemokine production; 3. In cases where CD4+ effector T cells are directly inhibited by Tregs, it is chiefly through a mechanism of lymphokine withdrawal apoptosis leading to polyclonal deletion; and 4. Contrary to the current view, we discuss new evidence that Tregs, similar to other T-cells lineages, can promote protective immune responses in certain infectious contexts (Chen et al., 2011; Pandiyan et al., 2011). Although these points are at variance to varying degrees with the standard model of Treg behavior, we will recount developing findings that support these new concepts.
Introduction
The immune system maintains a delicate balance between adaptive lymphocyte responses to foreign antigens that efficiently counteract a myriad of microbial assaults, but maintain unresponsiveness or “tolerance” to a large variety of harmless self-antigens. This requires distinguishing harmless “self” (that also includes commensal microbes) from harmful “non-self” throughout the lifetime of the host. When this balance is disturbed, it can lead to inflammatory disorders, autoimmunity, and allergy. Although immunity involves panoply of different cell types, we will focus principally on the control of T lymphocytes, which serve to orchestrate most immune responses. Tolerance is achieved in part by selecting against self-reactive T lymphocytes during ontogeny in the thymus, a powerful mechanism termed as central tolerance (Gershon and Kondo, 1971; Bonomo et al., 1995; Zheng et al., 2003; Kyewski and Klein, 2006; St Clair et al., 2007). However, there are “peripheral” tolerance mechanisms that divert immune responses away from self and toward appropriate pathogens (Miller and Morahan, 1992; Arnold et al., 1993; Anderson and Chan, 2004; Anderson et al., 2005; St Clair et al., 2007; Moraes-Vasconcelos et al., 2008).
Peripheral tolerance can be achieved through multiple mechanisms affecting the survival, differentiation, and function of effector T lymphocytes (Sprent, 1995). In this review, we will focus on the programmed death of T cells which in the periphery, accounts generally for propriocidal or autoregulatory deletion of self-reactive cells upon chronic restimulation by self-antigens (deletional tolerance; Lenardo et al., 1999; Sprent and Surh, 2001). Peripheral deletion can be mediated by cell death pathways involving Fas, TNF-∝, granzymes, or passive cytokine deprivation pathways driven by pro-apoptotic members of the Bcl-2 family such as Bim (Chen et al., 1995; Stockinger, 1999; Strasser et al., 2000; Bidere et al., 2006). Peripheral deletion pathways are typically clonal in that specific engagement of antigen receptors in the T-cells activity on internal death program leading to deletion (Lenardo, 1991; Hornung, 1997). Other mechanisms of peripheral tolerance not discussed in this review include rendering self-specific cells inactive (anergy), deviating the immune system to innocuous or protective responses (immune deviation), adjusting lymphocyte responses so that available antigen levels are below the threshold of detection (clonal ignorance) or suppression of local responses by biological mediators such as CTLA-4, IL-10, and TGF-β (suppression or regulation; Schwartz, 2003; Anderson and Chan, 2004).
Recently, a great deal of experimental work has centered on a form of dominant tolerance mediated by a class of thymus-derived CD4+ CD25hi CTLA-4hi Foxp3+ T lymphocytes, known as regulatory T cells or Tregs, that appear to suppress effector T-cell function (Sakaguchi et al., 2008). There are long-standing antecedents to that concept. T lymphocytes regulate immune responses both positively and negatively. Historically, a binary paradigm, which categorized CD4+ cells as “helpers” and CD8 cells as “suppressors” was put forward, as soon as T cells could be distinguished by cell surface markers. This paradigm has had a powerful influence on the literature of immunology (Janeway et al., 1975; Damle, 1986). However, this concept lost ground when CD8 cells were found to mediate protective anti-viral cytolysis and other problems arose in the characterization of “suppressors”(Shevach, 2011). A similar binary paradigm has been reincarnated in the past decade for CD4 cells such that Th1, Th2, and Th17 cells mediate immunoprotective helper (and autoimmune) functions and CD4+CD25+Foxp3+Treg cells mediate suppressive or anti-helper functions (Zhu and Paul, 2010). The main functions of the helper cells are that Th1 cells express interleukin-2 (IL-2), interferon- (IFN)-γ, and lymphotoxin (LT) or TNF-α, Th2 cells produce IL-4 and IL-5, and Th17 cells produce IL-17A, -17F, IL-22, and TNF-α (Bettelli et al., 2007). On the other hand, Tregs express IL-10, TGF-β, CD25, and Foxp3 but do not produce inflammatory cytokines under most situations (Fontenot and Rudensky, 2005). Severe atopic autoimmune conditions in scurfy mice and immune dysregulation polyendocrinopathy X-linked (IPEX) syndrome patients with mutations in FOXP3, highlight the functional importance of Tregs in suppressing severe autoimmune reactions in mice and humans (Bennett et al., 2001).
However, less attention has been focused on the fact that immunoprotective functions are defective when FOXP3 and presumably Treg cells are lacking (Costantino et al., 2008; Coutinho and Carneiro-Sampaio, 2008). IPEX is a primary immunodeficiency because severe infections involving bacteria, viruses, and fungi such as Candida sp. affecting a variety of organ systems contribute to early mortality in IPEX patients (Moraes-Vasconcelos et al., 2008). The immunoprotective functions of Fox P3 and presumably Tregs have not been elucidated in detail. Recent research has provided important new insights into the regulator behavior of Tregs, both as suppressors and immune stimulators. Here we discuss the molecular mechanisms governing immune responses, especially T lymphocyte effector functions by Tregs, with an emphasis on polyclonal deletional tolerance (Pandiyan et al., 2007). Furthermore, we review the recent descriptions of immunoprotective functions of Tregs including evidence for functional plasticity in Tregs, depending on the immune environment (Pandiyan et al., 2011). Finally, we suggest an alternative model of immunoregulation and immunoprotection functions being distributed among all CD4+ T lymphocyte subtypes and each subset has important positive and negative immunoregulatory roles.
Tregs – Discovery and Functions
CD4+CD25+Foxp3+ Tregs [natural (n) Tregs] are thymus derived, and constitute 5–10% of peripheral CD4 T cells. They are said to be “anergic” because they do not express the IL-2 gene and proliferate poorly when stimulated alone under most conditions. The suppressive capacity of Tregs was identified in vivo by seminal experiments showing that day 3 thymectomized mice and mice depleted of Tregs succumbed to systemic autoimmunity (Sakaguchi et al., 1982, 1996). The ability of Tregs to restrain auto-aggressive immune reactions, led to the idea that these cells represented a type of specialized suppressor cells. However, other in vitro data show that addition of exogenous IL-2 and α-CD28 combined with antigen-presenting cells (APC) not only breaks the anergic state and promotes Tregs proliferation, but also largely abrogates the suppression effect (Takahashi et al., 1998; Thornton et al., 2004b). This opened the door to possible immunological roles for Tregs besides suppression, but until recently this has been largely unexplored.
The plurality of mechanisms that have been postulated by different groups to explain nTregs function in different in vitro and in vivo settings is remarkable (Shevach, 2002; Von Boehmer, 2005; Rudensky and Campbell, 2006; Tang and Bluestone, 2008). Many investigators detect immune regulatory effects by culturing a mixture of Tregs and effector T cells in vitro (Takahashi et al., 1998; Thornton and Shevach, 1998; Von Boehmer, 2005; Pandiyan et al., 2007; Tran et al., 2009a). The manner in which such mixtures are prepared and examined often leads to varied conclusions but mainly interpreted to reinforce the preconceived binary paradigm of helpers/suppressors. For example, the presence or absence of α-CD28 antibodies, APC, cell density, and types of target cells in such mixtures have led to different interpretations by different investigators (Takahashi et al., 1998, 2000; Thornton and Shevach, 1998; Pandiyan et al., 2007). Failing to take into account the experimental details, such as the target cells that are suppressed, cell density, TCR activation strength, has caused confusion and even misleading information regarding the regulatory action of Tregs (Takahashi et al., 1998, 2000; Thornton and Shevach, 1998; Tran et al., 2009b). This is illustrated in the following examples.
First, one in vitro study sought to establish that Treg cells serve as “professional” suppressor cells that shut off IL-2 gene transcription when present during the stimulation of effector T cells, ostensibly supporting the helper/anti-helper paradigm (Thornton and Shevach, 1998). In this study, however the fate (cell death) of the effector T cells was not examined. Later studies showed that consumption of IL-2 and not IL-2 production suppression by the Treg cells led to lymphokine withdrawal death of the effector cells indicating that decreased thymidine incorporation was due to death of the effector cells (Pandiyan et al., 2007). This is explained by the fact that, in this conventional proliferation assay, the indicator cells are the proliferating cells themselves and if these cells are dying, then no conclusions can be drawn regarding the “suppression” of proliferation. IL-2 is well-known to be regulated at both transcriptional and message stability levels (Zhu et al., 2010). Since the effector T cells die under these conditions, less steady-state mRNA was found, but when examined, no direct suppressive effect on the IL-2 promoter or gene transcription was found (Klein et al., 2003; Pandiyan et al., 2007). Finally, the conclusion drawn from these assays was that Treg cells were “professional suppressor cells” that are solely suppressive, but no experiments were done to assess their helper functions. The later discovery that Treg cells can promote the differentiation and cytokine production by Th17 cells in an immunoprotective setting (described below) undermines the binary concept of “professional” helpers and suppressors in the CD4 T lineages. This is important because current investigations involve injecting Treg cells as therapy for human disease based on the professional suppressor notion. However, current findings indicate that the injected Treg cells may have unpredictable effects or even exacerbate disease owing to their plasticity (see below; Zhou, 2008; Zhou et al., 2009a; Zhu and Paul, 2010; Pandiyan et al., 2011).
Similarly, in vivo assays have led different investigators to explain immunosuppressive effects by a variety of molecules such as IL-10, TGF-β, IL-35, and CTLA-4 among others. IL-10 produced by Tregs, seems to be suppressive in vivo, keeping immune responses in check at environmental interfaces of the host, such as the colon and lungs (Klein et al., 2003; Kearley et al., 2005; Rubtsov et al., 2008). However, it is puzzling that IL-10 production by Treg cells is not required for the control of T cells in vitro (Thornton and Shevach, 1998; Takahashi et al., 2000; Rubtsov et al., 2008). Similarly, CD4 effector T cells from TGF-β receptor II (RII) deficient mice have been shown to be resistant to Treg-mediated suppression in vivo. On the other hand, evidence showing that TGF-β deficient Tregs suppress T cells in in vitro assays and autoimmune colitis in vivo clearly, which seems to exclude TGF-β mediated regulation as the principle suppressive mechanism of Tregs (Shevach et al., 2001). One alternative explanation is that TGF-β RII knockout (KO) T cells are intrinsically hyperactive and less suppressible by Tregs (Wan and Flavell, 2007). TGF-β RII KO CD4 cells may produce abnormally high levels of cytokines causing them to be refractory to cytokine deprivation death. Thus, TGF-β seems unlikely to directly mediate Treg suppression. TGF-β produced by natural Tregs may convert conventional CD4 T cells into Foxp3 expressing “induced” Tregs (iTregs; Andersson et al., 2008; Pandiyan and Lenardo, 2008; Shevach et al., 2008). These iTregs have suppressive functions and may confer “infectious tolerance” since they can be induced or recruited to spread tolerance (Shevach et al., 2008). However, the exact mechanism of how they suppress is unknown. Similarly, IL-35, a novel member of the IL-12 family, also contributes to Treg-mediated infectious tolerance by converting conventional T cells into Tregs (Collison et al., 2010). However, the presence and function of IL-35 in human CD4+ Foxp3+ Tregs is controversial (Bardel et al., 2008; Chaturvedi et al., 2011; Liu et al., 2011). Moreover, EBI-3 (IL-35) KO mice do not exhibit gross autoimmunity similar to that displayed by Foxp3, IL-2 or CTLA-4 deficient mice, raising a question about its physiological role in Tregs function (Bardel et al., 2008; Chaturvedi et al., 2011; Dokmeci et al., 2011; Liu et al., 2011). Finally, although CTLA-4 was originally claimed not to be required for the suppressive mechanism of Tregs in vitro, as described below, new evidence clearly supports a role in modulating APC function which will indirectly inhibit T-cell activation under certain circumstances (Wing et al., 2008; Qureshi et al., 2011). The contradictions in these data appear to result from the classic “blind men and the elephant” effect. Different groups have examined only selected aspects of the multi-step process of suppression. Taken together, Tregs deploy different mechanisms of regulation depending on the immunological context, location, conditions of T-cell activation and differences in target cells (Shevach, 2002; Von Boehmer, 2005; Rudensky and Campbell, 2006; Tang and Bluestone, 2008).
Recently, a few mechanisms of Tregs action have become increasingly clear. We will briefly describe two negative effects on T effector responses – mediated by the high affinity IL-2 receptor (IL-2R) and the CTLA-4 molecule – and one positive effect on T effector responses mediated by the high affinity IL-2R.
Negative Effects of Treg Cells
The negative effects are mediated, not surprisingly, by two cell surface “receptors” that are expressed extraordinarily highly on Treg cells – IL-2R (CD25) and the CD152 (CTLA-4) receptors. In order to understand the immunosuppressive effect of Tregs in vitro, it is important to distinguish the direct impact on T-cells mediated by IL-2R and indirect inhibitory effects on APC mediated by CTLA-4 (Pandiyan et al., 2007; Sakaguchi and Wing, 2011; Wing et al., 2011).
IL-2 Consumption and Cytokine Deprivation Apoptosis in CD4 T Cells
A defining feature used in the initial isolation of Treg cells was a characteristically high level of expression of CD25. It was therefore surprising that TCR stimulation of Treg cells led to no production of IL-2 under most circumstances. This led us and others to hypothesize that the CD25 (IL-2R) served an important regulatory function for Treg cells. One of the most important characteristics of nTregs is the requirement of IL-2 for their suppressive functions (Scheffold et al., 2005). Scheffold and Stockinger’s groups demonstrated that Tregs consume IL-2 to exert their suppressive functions (Barthlott et al., 2003; De La Rosa et al., 2004). Starting from these observations, we investigated whether consumption of IL-2 is an actual suppressive mechanism for immune responses by Tregs. We also recognized that none of the studies on Treg suppression had determined the fate of the “suppressed” CD4 effector cells, especially in the in vitro assay systems, However, apoptosis and cell survival were well-known immune regulatory mechanisms (Lenardo et al., 1999). Therefore, we examined the direct effects of Tregs on CD4 T cells and the fate of the “suppressed” T cells (Pandiyan et al., 2007). We observed that Tregs consume a large fraction of the IL-2 that is produced by the stimulated CD4 responder cells (effector cells). Tregs produce no IL-2 themselves even though their survival depends on it. Close proximity of a large number of Treg cells deprives the effector CD4 cells of this primary growth factor causing proliferation arrest and apoptosis of both cell populations (Pandiyan et al., 2007). This effect could quantitatively explain the drop in proliferation measured by tritiated thymidine incorporation described in earlier studies (Thornton and Shevach, 1998) and did not require the presence of APCs. Thus, Tregs directly regulate the IL-2 producing Th0 effector cells through a chain of events leading to IL-2 deprivation and polyclonal deletion (PCD). The deletion is “polyclonal” at least according to in vitro experiments, because the suppression does not depend on any clonotype specificity of Tregs and target cells consistent with prior evidence that Tregs “suppression” is not antigen specific in most settings (Thornton and Shevach, 2000; Sakaguchi et al., 2008).
Cytokine deprivation happens at two levels. First, Tregs directly consume growth cytokines secreted at early phase of activation by effector T cells that are in the immediate vicinity at early phase of activation. Close juxtaposition is crucial for interception of IL-2 in its autocrine loop of production and utilization by effector T cells. Second, they cause failure of activated cells to produce cytokines at a later phase of activation by removing a critical IL-2 positive feedback (Pandiyan et al., 2007). In Th0 cells, a powerful positive feedback loop is initiated by IL-2 production during early T-cell activation, in which autocrine IL-2 induces the high affinity IL-2R expression rendering activated T cells to react more efficiently with IL-2. This leads to the production of other T-cell cytokines and proliferation. Signaling by early IL-2 is essential for later optimal cytokine production and effector functions. By consumption of IL-2, Tregs can effectively interrupt these processes by down-regulating IL-2R expression and dampening further cytokine expression and proliferation. Thus, suppression of proliferation and death due to cytokine deprivation may have a more pronounced effect on effector and effector memory T-cell populations. Further investigations elucidated the importance of TCR stimulation strength in the processes of IL-2 consumption and Treg suppression (Tran et al., 2009b). These experiments demonstrated that human Tregs consume mouse IL-2, which inhibits mouse effector T cells at mid range but not at very strong stimulation conditions. We also find that in human Tregs, consumption causes PCD under low dose α-CD3 stimulations and not at high α-CD3 concentrations (Pandiyan and Lenardo, unpublished results). However, the minimum amount of IL-2 that must be consumed to obtain PCD, how TCR signal strength affects IL-2 consumption, other cytokines that might alter the IL-2 consumption rate and the suppression processes, and how IL-2 consumption may affect APCs, remain to be investigated. It seems intuitive that low strength TCR stimulation is more reflective of an immune response to the low-affinity self-antigens in vivo and therefore, PCD may play a predominant role in such an immune context. In addition to direct cytokine consumption, whether Bim-dependent apoptosis may also be caused by TGF-β remains to be addressed (Ramesh et al., 2008; Houde et al., 2009; Sanjabi et al., 2009; Tinoco et al., 2009). However, more work is needed to address these important questions, which will facilitate our understanding of how Tregs and PCD can be therapeutically deployed.
Cytokine Deprivation Versus Other Deletion Mechanisms in CD4 T Cells
The consumption of available IL-2 by Tregs plays a key role in PCD of responder T cells but other cytokines in vitro and in vivo may be involved (Pandiyan et al., 2007). The biochemical events were PI-3 kinase inactivation, Akt/BAD dephosphorylation and Bim-dependent apoptosis. This was verified by removing the Bim apoptosis gene in effector T cells, and examining their response to suppressive effects in co-cultures with wild-type Tregs. The Bcl-2 family member Bim is a central mediator of intrinsic apoptosis pathways, orchestrating cytokine withdrawal apoptosis in various hematopoietic cells that depend on trophic cytokines for growth and survival (Hildeman et al., 2002). In particular, it mediates lymphocyte death caused by cytokine deprivation generally at the end of immune responses (Hildeman et al., 2002). Bim knockout (KO) mice suffer with lupus-like autoimmunity and kidney disease due to defective apoptosis of T and B cells (Strasser, 2005). We found that Bim-KO effector T-cells clearly resist suppression by fully active Tregs and are not impaired in late-phase cytokine production. Hence, “suppression” was abrogated by removal of an apoptosis gene in the effector T cells that were the target of wild-type Tregs. Unaffected cytokine production in Bim-KO cells implies that the late-phase cytokine suppression resulted from the death of cytokine producers. These data validate the hypothesis that Treg cells can regulate cytokine dependent homeostatic proliferation of effector T cells by killing them in an antigen non-specific manner in vivo. This was demonstrated by the ability of Treg cells to suppress inflammatory bowel disease (IBD) in a mouse model, in which the disease is partially driven by homeostatic proliferation of naïve CD4 cells in a lymphopenic environment (Powrie et al., 1994). However, Treg cells failed to suppress IBD caused by Bim-KO cells. This indicates that direct cytokine deprivation and BIM-dependent apoptosis by Tregs plays a significant role in their ability to regulate IBD.
Polyclonal deletion mediated by natural Treg cells apparently does not involve direct cytolytic mechanisms such as activation of Fas or cell contact-dependent, granzyme-mediated cell death (Takahashi et al., 2000; Pandiyan et al., 2007). We found no evidence of death mechanisms mediated by TNF receptor family members or perforin and/or granzymes. Moreover, our data showing that PCD of the CD4 T-cells occurred slowly (3–4 days after activation) argues against the direct cytolysis mechanism, which usually kills the target cells within 24 h. Our studies also support the idea that close proximity, not necessarily direct physical contact, is crucial for Treg cells to compete effectively for cytokines produced by the effector T cells or APCs and thereby block the autocrine and paracrine loops that are essential for effector T-cell survival, proliferation, and cytokine production (Scheffold et al., 2005; Busse et al., 2010). Although the exact distance required between target cells and Treg cells for efficient IL-2 consumption is currently unknown, we believe that the closer the proximity of the two cells, the more likely it will be that the IL-2Rs on the Tregs will compete better to capture the IL-2 produced by the effector T cells before it can interact with the IL-2Rs on the T effector cells themselves.
It has been shown that Treg cells can block the autoimmune disease that occurs in IL-2 KO mice (Schimpl et al., 2002). Along with our results showing that Treg cells kill IL-2 KO T cells in vitro, it raises a possibility that Treg cells can cause PCD by consuming cytokines besides IL-2. In fact, at this point it is unclear consumption of which cytokine contributes the most to the immunoregulatory properties of Tregs. Because Treg cells are themselves heavily dependent on common gamma (γc)-chain cytokines, it is likely that in the absence of IL-2, they will consume/regulate other γc-chain cytokines to gain control of activated CD4 cells (Pandiyan and Lenardo, 2008). We found that the addition of γc-chain cytokines such as IL-4, IL-15, and others to the Treg/effector T-cell co-culture can completely rescue effector T cells from apoptosis, confirming that a variety of cytokines can tip the survival balance (Pandiyan et al., 2007). Cytokine/s that drive the proliferation of the IL-2 KO CD4 T cells in the absence of IL-2, how that cytokine is regulated by Tregs, and how APCs are affected in cytokine deprived situations in vivo, are questions that remain to be answered.
Although Tregs mediate the PCD of activated effector CD4+ T cells, extensive experimentation shows they do not suppress priming or initiation of T-cell receptor (TCR) activation and/or early IL-2 production in T cells (Thornton et al., 2004a; Oberle et al., 2007; Pandiyan et al., 2007; Esquerre et al., 2008). This contradicts a tenet of Treg suppression proposed in previous studies (Thornton and Shevach, 1998). Early IFNγ and IL-2 gene expression is not affected by mouse Tregs in conventional co-cultures, so that the simplistic paradigm of helper/anti-helper competition to control early activation events fails to explain T effector cell regulation by Tregs (Pandiyan et al., 2007). This conclusion is reinforced by the fact that in human T cells, early TCR signaling events and calcium mobilization in effector T cells are unaffected by Treg cells (Esquerre et al., 2008). Further experiments have demonstrated that the proliferation (and survival) of previously activated effector T cells are susceptible to Treg suppression, indicating that Treg cells do not need to affect initial priming of mouse T cells in order to suppress them in vitro (Pandiyan et al., 2007; Esquerre et al., 2008). Moreover, cytokine deprivation and Bim regulates the survival of Tregs themselves, and, in the absence of gamma chain cytokines, they die by apoptosis (Pandiyan and Lenardo, 2008). During an immune response, early production of IL-2 drives activated T cells into proliferation and at the same time makes the proliferating T-cells totally dependent on IL-2 (Lenardo et al., 1999). Intact early activation events and normal effector T-cell proliferation are therefore essential for the IL-2 consumption made of PCD by Treg (Klein et al., 2003). Although Tregs can modulate APCs, we have found that potent Treg suppression of effector T cells by PCD occurs in the absence of APCs in vitro. Taken together, direct induction of PCD in activated and cycling T-cells plays a major role in the Treg-mediated immune suppression but not direct impairment of naïve T cells or any direct effect on early T-cell activation.
Modulation of APC by Tregs also Contributes to Suppression
A second mode of negative regulation is the effect of Tregs on APC such as dendritic cells. When Tregs are mixed with APCs and then the APCs are isolated and used to stimulate T cells, these APCs function suboptimally in many conditions. Hence, Treg effects on APC’s could indirectly affect T-cell responses. Consistent with the idea of APC regulation, CTLA-4, which is constitutively expressed on Tregs, has been found to indirectly regulate T cells by down-regulating co-stimulatory molecules such as CD80 and CD86 on APC (Wing et al., 2008). Initially, this effect was somewhat mysterious, but elegant studies showed that CTLA-4 could capture its ligands from opposing cells, by a process of trans-endocytosis. This reduces the level of CD80 and CD86 on the APCs in the immediate vicinity, which will reduce co-stimulation provided to the effector T cells (Qureshi et al., 2011). Treg-specific CTLA-4 deficiency impairs in vivo and in vitro suppressive function of Tregs (Wing et al., 2008). In vitro blocking experiments with anti-CTLA-4 antibodies give less clear-cut results because of low/transient CTLA-4 binding at the cell surface. The role of APC modulation is further supported by the studies demonstrating that Treg cells could regulate the contact and clustering between APC and T cells (Tadokoro et al., 2006; Tang et al., 2006; Qureshi et al., 2011). Thus, strong evidence now indicates that Tregs inhibit immune responses by directly blocking the survival and effector functions of CD4 cells and indirectly by down-modulating APC co-stimulation. These new observations provide a compelling mechanism, but further validation by other groups and more information about the specificity and kinetics of the CTLA-4 transcytosis will be necessary. However, CTLA-4 and IL-2-dependent suppression can be regarded as core mechanisms of Treg-mediated suppression, explained by the high levels of CTLA-4 and CD25 (IL-2R) characteristically expressed by Tregs (Sakaguchi et al., 2009).
Coordination between Deletion of T Cells and Other Mechanisms of Suppression in vivo
Consumption of cytokines and PCD of effector T cells by Tregs plays an important role in reducing the strength of the immune response in vivo (Pandiyan et al., 2007). We speculate that in the initial phase of an immune response, the effector T:Treg ratio is higher because effector T cells are proliferating faster than Tregs. However as the response progresses, Tregs consume IL-2 from activated cells, proliferate, and cause accelerated apoptosis of IL-2-dependent effector T cells. This begins to shift the effector T:Treg balance. Bim-dependent PCD of a substantial number of cytokine producing CD4 cell progenitors in the initial phase of the response inhibits further cytokine production by affecting differentiation of CD4 effector T cells into Th1, Th2, or Th17 effectors (due to poor expression of cytokines and cytokine receptors; Figure 1). These forces all act together to reduce the number of specific effector T cells, to dampen the immune response.
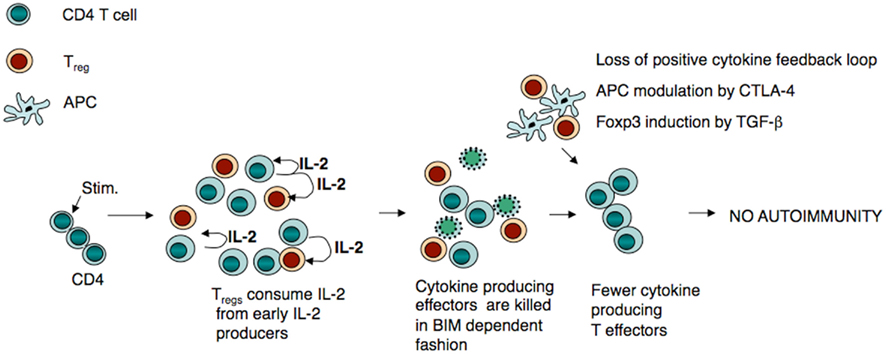
Figure 1. Treg-mediated apoptosis plays a major role in limiting Th07Th1/Th2 cell responses. Under steady-state conditions, CD4+ T cells upon priming, produce IL-2 and /or other growth factors and expand responding to these cytokines. If Tregs are present during this early phase of IL-2 production, CD4+ T cells are deprived of IL-2 leading to CD4+ T-cell death. However, cells that are not in direct/close proximity to Treg5, may remain in the system and continue to expand. Because of the low levels of IL-2 in their milieu and inactivation of APC by CTLA-4, their later effector function and cytokine production are impaired. This limits autoimmunity, perhaps under homeostatic conditions involving self-antigens.
Thus, PCD directly reduces the intensity of an immune response by tapering the T-cell population, and the diminished population of effector cells is rendered more susceptible to other suppression mechanisms (Tang and Bluestone, 2008; Figure 1). The suppressive factors could directly be produced by Tregs, or indirectly induced in other cells by Tregs. Cytokine consumption ensures loss of effector functions in the remaining effector population making them immunologically inactive and restores Treg numbers for future regulation. Even though we believe that suppression of late cytokine production is chiefly caused by the “disruption of cytokine positive feedback loop,” the effects of other postulated mechanisms could be important. Cytokines produced during effector response may further potentiate Tregs to produce other suppressive or even non-suppressive factors (see below). For example, IL-2 may trigger transcriptional events leading to the expression of IL-10, TGF-β, EBI-3, or IL-35 in Tregs (Maloy and Powrie, 2005). These suppressive molecules further reduce inflammation by inhibiting effector CD4 cells, as well as modulating APC and other inflammatory cells. Furthermore, Treg dependent Foxp3 and IL-35 induction in effector T cells may also contribute to their intrinsically impaired cytokine production at later phases (Andersson et al., 2008; Pandiyan and Lenardo, 2008; Collison et al., 2010). The specific role of each mechanism in suppressing a heterogeneous population of effector Th1, Th2, and Th17 cells in vivo remains to be seen. Deprivation of cytokines and induction of apoptosis by Tregs could differentially affect target cell functions, depending on whether the immune response is pro- or non-inflammatory, or whether the immune cells are responding to microbial infections or self-antigens. The phase (early versus late) of the immune response and the corresponding cytokine environment also likely dictate the functions of Tregs. Thus, these suppressive mechanisms may play their distinct roles in the multi-step process of immune suppression at different stages.
Are Foxp3+ CD4+ Cells Solely Devoted to Tolerance?
There is great deal of literature correlating Foxp3 expression in CD4 cells with active immune suppression. However, recent evidence indicates that Foxp3+ cells may play other immunological roles (Komatsu et al., 2009; Zhou et al., 2009b; Chen et al., 2011; Pandiyan et al., 2011). Although Foxp3+ CD4+ Tregs have suppressive capability, Foxp3 expression in CD4 cells may not always indicate suppression. Tregs may have positive immune functions and this possibility has been explored much less because of the preconception that they are simply “suppressor” cells. Transient expression of Foxp3 in a subset of activated cells or in TGF-β-induced iTregs does not necessarily mean that these cells can function like-thymus-derived suppressive “natural” Treg cells (Hoffmann et al., 2009; Huehn et al., 2009; Komatsu et al., 2009; Lahl et al., 2009; Thornton et al., 2010). Also, Foxp3 alone does not control all the elements of Treg signature phenotype (Zhou et al., 2008). The presence of Foxp3+ cells in vivo may reflect an inflammation that has induced Foxp3 expression in activated cells (iTregs) or proliferation of natural Tregs, and not necessarily associated with active suppression of immune responses. Similar to Tregs induced in vitro (iTregs), a subset of natural Tregs might represent a population with unstable Foxp3 expression in vivo. Given the plasticity of Tregs and Foxp3 expression in activated cells, care should be taken when interpreting the data from experiments involving Foxp3 positive cells in disease conditions.
Although controversial, recent accumulating evidence shows that Foxp3+ Treg cells can themselves become effector cells or produce cytokines under strong pro-inflammatory conditions (Komatsu et al., 2009; Zhou et al., 2009b; Rubtsov et al., 2010). In some instances they are even shown to exacerbate pathogenic immune responses (Zhou et al., 2009a,b). It has been observed in an experimental allergic encephalomyelitis (EAE) model, that Ag specific Tregs accumulate in brain but fail to control inflammation (Korn et al., 2007). This raises an interesting possibility that Treg functions are not limited solely to suppression but also to other effector functions, including protection against infection. Evidence to support this idea emerged with the demonstration that Tregs play an important role in controlling lethal West Nile virus (Lanteri et al., 2009). Those functions may be largely dependent on cytokine milieu or innate immune responses.
We and others have shown that under Th17 milieu, IL-2 consumption by Tregs actually leads to an enhanced proliferation and cytokine production of early Th17 cells in vitro and in the context of oral Candida infection or inflammation in vivo (Chen et al., 2011; Pandiyan et al., 2011). IL-2 has been known to inhibit Th17 differentiation. By enhancing Th17 cells, these non-suppressive Tregs reduce the fungal burden in mice (Pandiyan et al., 2011; Figure 2). Moreover, our preliminary studies show that Tregs themselves lose Foxp3 in Th17 inflammatory conditions in vitro, which also likely contributes to the loss of their suppressive properties (unpublished results). These destabilized Tregs (Ex-Tregs) are only present transiently, because in a chronic Th17 IBD model, such Ex-Tregs can regain suppressive functions and show a delayed, but strong potential to suppress Th17 inflammation (Pandiyan et al., 2011). The mechanism of reacquisition of suppressive properties by Tregs and the mechanism of the delayed suppression of Th17 cells remain to be investigated. The loss of suppressive functions in Tregs during infections might be associated with certain toll like receptor (TLR) ligands and strong responses through massive production of inflammatory cytokines (Pasare and Medzhitov, 2003). This mechanism might allow protective immune responses against microbes to proceed normally. Thus, Foxp3+ Tregs could be principally suppressive when inflammatory cytokines are low, such as during late-phase responses, which prevents immunopathology in the host. However, they may not be destined solely for suppressive functions, because they also act as helper cells to promote certain immune responses in cytokine-rich environments, such as during infections (Pandiyan et al., 2004; Zhou, 2008; Lanteri et al., 2009). Taken together, Foxp3+ cells may represent a subset of protective immune cells like T helper cells, but with elaborate suppressive capacities.
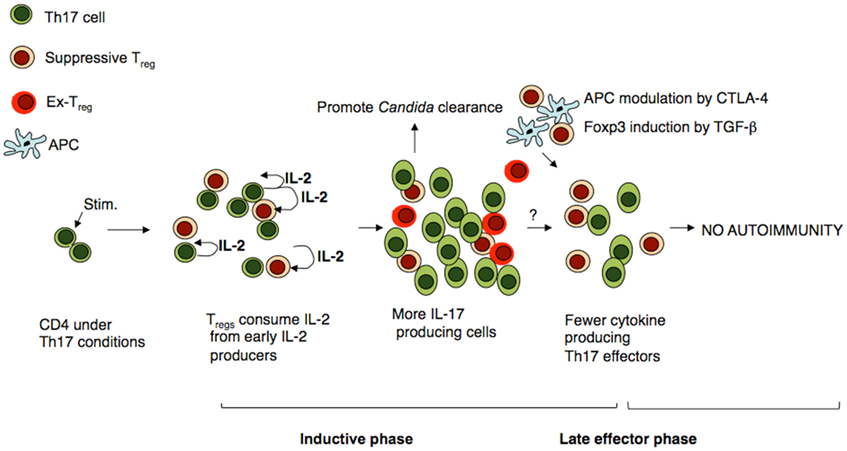
Figure 2. Treg cells promote Th17 cells at early phase and modulate inflammatory Th17 responses at later phases. Under Th17 inflammatory conditions, CD4+ T cells and expand responding to factors such as IL-21 and IL-6. Tregs do not inhibit the survival or proliferation of those cells because Th17 cells produce excess of IL-2, IL-21, IL-6, and TNF-α, all of which overcome the suppression by cytokine deprivation. Some Tregs also lose Foxp3 expression (Ex-Treg), which also likely contributes to the absence of suppression. However, Tregs enhance IL-17 production by consuming IL-2. This promotes clearance of pathogens that require protective Th17 responses. However when the cytokine levels drop at later phases, Tregs limit Th17 cells, likely by other mechanisms such as TGF-β and APC modulation by CTLA-4.
Even T helper cells such as Th1, Th2, and Th17 cells can be regarded to possess regulatory potentials. These cells along with Th22 cells and inducible regulatory T cells such as Foxp3+ iTregs and iTr35 (the inducible regulatory T-cells expressing IL-35) polarize under specific cytokine milieu along with TCR stimulation. (Yang et al., 2008; Bluestone et al., 2009; Lohr et al., 2009; Bending et al., 2011; Hirota et al., 2011). For example, under a Th1 skewing condition, IFN-γ and IL-12 activate signal transducer and activator of transcription 4 (STAT4) and STAT1, leading to the activation of T-bet, a transcription factor and a master regulator for Th1 differentiation (Ashkar et al., 2000; Glimcher and Murphy, 2000; Szabo et al., 2000). Along with Runx3 activation, a positive feedback loop forms and accelerates the expression of the Th1 hallmark cytokine IFN-γ. Meanwhile, activated T-bet and Runx3 can suppress GATA3 signaling, causing the expression of the Th2-specific IL-4 gene to be inhibited (Hegazy et al., 2010; Zhu and Paul, 2010). Inhibition of Th2 factors is critical for the maintenance of Th1 cells. On the other hand, IL-4 and IL-2 are the major cytokines that promote Th2 differentiation (Ben-Sasson et al., 1990; Le Gros et al., 1990). Through stimulating respectively STAT6 and STAT5, IL-4 (also produced by DC) and IL-2 activate GATA3, a master regulator of Th2 cell differentiation, and then promote IL-4 gene expression (Ouyang et al., 2000; Hofer et al., 2002; Tykocinski et al., 2005). Activated GATA3 also inhibits Runx3 and thereby turns off IFN-γ gene expression in order to suppress Th1 and augment Th2 differentiation (Yagi et al., 2010). This phenomenon, where one type of cytokine milieu favors a lineage of T helper cell differentiation, is always accompanied by suppression of the other lineages. (Mosmann and Coffman, 1989a,b; Paul and Seder, 1994; Seder and Paul, 1994; Zhu and Paul, 2010). This process can also be called as cross regulation. The induction versus suppression of differentiating CD4 T-cells appears to be dependent on available cytokines in the microenvironment of any given immune response. Although most of the available data indicate that inhibition of transcription factors during Th differentiation occurs in the same cell, we speculate that it is likely to occur through cytokines at intercellular level as well. For example, differentiated Th1 cells may suppress Th2 cells and vice versa. It will be interesting to study the cross regulation of well-differentiated Th1 or Th2 cells. In essence, while the differentiated helper T cells can assist in mounting appropriate immune responses, the same “helper” cells can exercise functions of suppression, at least on some differentiating T cells. Thus, most immune cells may harbor both immunosuppressive and immune promoting functions and immune suppression function may not be confined to specific subsets.
A recent example that supports this tenet is the identification of Foxp3+ suppressive macrophages (Zorro Manrique et al., 2011). Historically, macrophages are regarded as effector cells, classical APCs and one of the first responders in an immune response. Now, it has been shown that under certain conditions they can be suppressive in function. More importantly, we have also shown that even effector CD4 T cells with no Foxp3 expression can suppress CD4 T cells by consuming IL-2. These “pseudo-Tregs,” mimic nTregs in expressing CD25 and being dependent on IL-2, but being unable to produce IL-2 (Pandiyan et al., 2007). Therefore, “helpers” and “suppressors” should be regarded as relative terms; suppressive properties can reside in cells that are conventionally conceived as effector/helper cells. The dose of antigens, site of action, timing and cytokine milieu may dictate the function of the immune cells, whether “immunosuppressive” or “immune promoting.” This explains why Foxp3+ CD4+ cells regulate Th1 and Th2 functions by cytokine deprivation but promote other types of responses such as the Th17 response, which in and of themselves may regulate each other. Thus, the binary model in which Foxp3+ CD4+ cells are exclusively suppressive and other effector T helper cells function exclusively to promote immune responses, may not explain all immune functions. Ultimately, coordination between suppression of certain cells and enhancement of certain other effector cells leads to appropriate immune responses.
Conclusion
In an optimal immune response by CD4 cells, mixture of Th1, Th2, and Th17 effector cells are generated and the dominant population in a given response varies. Treg cells are probably equipped mainly to kill IL-2-dependent Th0/Th1 cells but not all the effector cells equally. Importantly, Tregs promote (and clearly do not suppress) Th17 responses during acute inflammation. Thus the function and stability of Tregs can depend largely on the immunological milieu in which they are operating. These parameters are complex and therefore it is important to identify the signals that regulate the function of Foxp3+ Tregs and other Th cells and determine how they cross regulate each other at the molecular level. Regarding the overall suppression, PCD of T-cells combined with suppression of APC by CTLA-4 and other suppressive mechanisms ensures more stringent control of different T-cells subsets. The cytokine milieu and the target effector cells that are suppressed determine the predominant mechanism employed by Tregs in a particular immune response. The temporal and spatial parameters that determine which mechanism predominates suppression by Tregs and what is the sequence in which the core mechanisms, i.e., cytokine deprivation and regulation by CTLA-4 are employed remain to be studied. Relative contribution of each of the mechanism of suppression by Tregs and when Tregs lose their suppressive properties remain to be the important questions for future investigations. When planning strategies for intervention of immune-related disorders using Tregs, cellular and cytokine networks as a whole, and not the function of a single subset of cells, should be taken into consideration. This is a fascinating area of research and warrants vigorous investigation, given its potential implications in chronic inflammation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Pushpa Pandiyan was partially supported by a fellowship from the National Research Council (NRC), National Academy of Sciences, This work was supported by the intramural research program of National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH).
References
Anderson, C. C., and Chan, W. F. (2004). Mechanisms and models of peripheral CD4 T cell self-tolerance. Front. Biosci. 9, 2947–2963.
Anderson, M. S., Venanzi, E. S., Chen, Z., Berzins, S. P., Benoist, C., and Mathis, D. (2005). The cellular mechanism of Aire control of T cell tolerance. Immunity 23, 227–239.
Andersson, J., Tran, D. Q., Pesu, M., Davidson, T. S., Ramsey, H., O’Shea, J. J., and Shevach, E. M. (2008). CD4+ FoxP3+ regulatory T cells confer infectious tolerance in a TGF-beta-dependent manner. J. Exp. Med. 205, 1975–1981.
Arnold, B., Schonrich, G., and Hammerling, G. J. (1993). Multiple levels of peripheral tolerance. Immunol. Today 14, 12–14.
Ashkar, S., Weber, G. F., Panoutsakopoulou, V., Sanchirico, M. E., Jansson, M., Zawaideh, S., Rittling, S. R., Denhardt, D. T., Glimcher, M. J., and Cantor, H. (2000). Eta-1 (osteopontin): an early component of type-1 (cell-mediated) immunity. Science 287, 860–864.
Bardel, E., Larousserie, F., Charlot-Rabiega, P., Coulomb-L’Hermine, A., and Devergne, O. (2008). Human CD4+ CD25+ Foxp3+ regulatory T cells do not constitutively express IL-35. J. Immunol. 181, 6898–6905.
Barthlott, T., Kassiotis, G., and Stockinger, B. (2003). T cell regulation as a side effect of homeostasis and competition. J. Exp. Med. 197, 451–460.
Bending, D., Newland, S., Krejci, A., Phillips, J. M., Bray, S., and Cooke, A. (2011). Epigenetic changes at Il12rb2 and Tbx21 in relation to plasticity behavior of Th17 cells. J. Immunol. 186, 3373–3382.
Bennett, C. L., Christie, J., Ramsdell, F., Brunkow, M. E., Ferguson, P. J., Whitesell, L., Kelly, T. E., Saulsbury, F. T., Chance, P. F., and Ochs, H. D. (2001). The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet. 27, 20–21.
Ben-Sasson, S. Z., Le Gros, G., Conrad, D. H., Finkelman, F. D., and Paul, W. E. (1990). IL-4 production by T cells from naive donors. IL-2 is required for IL-4 production. J. Immunol. 145, 1127–1136.
Bettelli, E., Korn, T., and Kuchroo, V. K. (2007). Th17: the third member of the effector T cell trilogy. Curr. Opin. Immunol. 19, 652–657.
Bidere, N., Su, H. C., and Lenardo, M. J. (2006). Genetic disorders of programmed cell death in the immune system. Annu. Rev. Immunol. 24, 321–352.
Bluestone, J. A., Mackay, C. R., O’Shea, J. J., and Stockinger, B. (2009). The functional plasticity of T cell subsets. Nat. Rev. Immunol. 9, 811–816.
Bonomo, A., Kehn, P. J., and Shevach, E. M. (1995). Post-thymectomy autoimmunity: abnormal T-cell homeostasis. Immunol. Today 16, 61–67.
Busse, D., De La Rosa, M., Hobiger, K., Thurley, K., Flossdorf, M., Scheffold, A., and Hofer, T. (2010). Competing feedback loops shape IL-2 signaling between helper and regulatory T lymphocytes in cellular microenvironments. Proc. Natl. Acad. Sci. U.S.A. 107, 3058–3063.
Chaturvedi, V., Collison, L. W., Guy, C. S., Workman, C. J., and Vignali, D. A. (2011). Cutting edge: human regulatory T cells require IL-35 to mediate suppression and infectious tolerance. J. Immunol. 186, 6661–6666.
Chen, Y., Haines, C. J., Gutcher, I., Hochweller, K., Blumenschein, W. M., Mcclanahan, T., Hammerling, G., Li, M. O., Cua, D. J., and Mcgeachy, M. J. (2011). Foxp3(+) regulatory T cells promote T helper 17 cell development in vivo through regulation of interleukin-2. Immunity 34, 409–421.
Chen, Y., Inobe, J., Marks, R., Gonnella, P., Kuchroo, V. K., and Weiner, H. L. (1995). Peripheral deletion of antigen-reactive T cells in oral tolerance. Nature 376, 177–180.
Collison, L. W., Chaturvedi, V., Henderson, A. L., Giacomin, P. R., Guy, C., Bankoti, J., Finkelstein, D., Forbes, K., Workman, C. J., Brown, S. A., Rehg, J. E., Jones, M. L., Ni, H. T., Artis, D., Turk, M. J., and Vignali, D. A. (2010). IL-35-mediated induction of a potent regulatory T cell population. Nat. Immunol. 11, 1093–1101.
Costantino, C. M., Baecher-Allan, C. M., and Hafler, D. A. (2008). Human regulatory T cells and autoimmunity. Eur. J. Immunol. 38, 921–924.
Coutinho, A., and Carneiro-Sampaio, M. (2008). Primary immunodeficiencies unravel critical aspects of the pathophysiology of autoimmunity and of the genetics of autoimmune disease. J. Clin. Immunol. 28(Suppl. 1), S4–S10.
De La Rosa, M., Rutz, S., Dorninger, H., and Scheffold, A. (2004). Interleukin-2 is essential for CD4+CD25+ regulatory T cell function. Eur. J. Immunol. 34, 2480–2488.
Dokmeci, E., Xu, L., Robinson, E., Golubets, K., Bottomly, K., and Herrick, C. A. (2011). EBI3 deficiency leads to diminished T helper type 1 and increased T helper type 2 mediated airway inflammation. Immunology 132, 559–566.
Esquerre, M., Tauzin, B., Guiraud, M., Muller, S., Saoudi, A., and Valitutti, S. (2008). Human regulatory T cells inhibit polarization of T helper cells toward antigen-presenting cells via a TGF-beta-dependent mechanism. Proc. Natl. Acad. Sci. U.S.A. 105, 2550–2555.
Fontenot, J. D., and Rudensky, A. Y. (2005). A well adapted regulatory contrivance: regulatory T cell development and the forkhead family transcription factor Foxp3. Nat. Immunol. 6, 331–337.
Glimcher, L. H., and Murphy, K. M. (2000). Lineage commitment in the immune system: the T helper lymphocyte grows up. Genes Dev. 14, 1693–1711.
Hegazy, A. N., Peine, M., Helmstetter, C., Panse, I., Frohlich, A., Bergthaler, A., Flatz, L., Pinschewer, D. D., Radbruch, A., and Lohning, M. (2010). Interferons direct Th2 cell reprogramming to generate a stable GATA-3(+)T-bet(+) cell subset with combined Th2 and Th1 cell functions. Immunity 32, 116–128.
Hildeman, D. A., Zhu, Y., Mitchell, T. C., Bouillet, P., Strasser, A., Kappler, J., and Marrack, P. (2002). Activated T cell death in vivo mediated by proapoptotic bcl-2 family member BIM. Immunity 16, 759–767.
Hirota, K., Duarte, J. H., Veldhoen, M., Hornsby, E., Li, Y., Cua, D. J., Ahlfors, H., Wilhelm, C., Tolaini, M., Menzel, U., Garefalaki, A., Potocnik, A. J., and Stockinger, B. (2011). Fate mapping of IL-17-producing T cells in inflammatory responses. Nat. Immunol. 12, 255–263.
Hofer, T., Nathansen, H., Lohning, M., Radbruch, A., and Heinrich, R. (2002). GATA-3 transcriptional imprinting in Th2 lymphocytes: a mathematical model. Proc. Natl. Acad. Sci. U.S.A. 99, 9364–9368.
Hoffmann, P., Boeld, T. J., Eder, R., Huehn, J., Floess, S., Wieczorek, G., Olek, S., Dietmaier, W., Andreesen, R., and Edinger, M. (2009). Loss of FOXP3 expression in natural human CD4+CD25+ regulatory T cells upon repetitive in vitro stimulation. Eur. J. Immunol. 39, 1088–1097.
Hornung, F., Zheng, L., and Lenardo, M. J. (1997). Maintenance of clonotype specificity in CD95/Apo-1/Fas-mediated apoptosis of mature T lymphocytes. J. Immunol. 159, 3816–3822.
Houde, N., Chamoux, E., Bisson, M., and Roux, S. (2009). TGF-{beta}1 induces human osteoclast apoptosis by uregulating bim. J. Biol. Chem. 284, 23397–233404.
Huehn, J., Polansky, J. K., and Hamann, A. (2009). Epigenetic control of FOXP3 expression: the key to a stable regulatory T-cell lineage? Nat. Rev. Immunol. 9, 83–89.
Janeway, C. A. Jr., Koren, H. S., and Paul, W. E. (1975). The role of thymus-derived lymphocytes in an antibody-mediated hapten-specific helper effect. Eur. J. Immunol. 5, 17–22.
Kearley, J., Barker, J. E., Robinson, D. S., and Lloyd, C. M. (2005). Resolution of airway inflammation and hyperreactivity after in vivo transfer of CD4+CD25+ regulatory T cells is interleukin 10 dependent. J. Exp. Med. 202, 1539–1547.
Klein, L., Khazaie, K., and Von Boehmer, H. (2003). In vivo dynamics of antigen-specific regulatory T cells not predicted from behavior in vitro. Proc. Natl. Acad. Sci. U.S.A. 100, 8886–8891.
Komatsu, N., Mariotti-Ferrandiz, M. E., Wang, Y., Malissen, B., Waldmann, H., and Hori, S. (2009). Heterogeneity of natural Foxp3+ T cells: a committed regulatory T-cell lineage and an uncommitted minor population retaining plasticity. Proc. Natl. Acad. Sci. U.S.A. 106, 1903–1908.
Korn, T., Anderson, A. C., Bettelli, E., and Oukka, M. (2007). The dynamics of effector T cells and Foxp3+ regulatory T cells in the promotion and regulation of autoimmune encephalomyelitis. J. Neuroimmunol. 191, 51–60.
Kyewski, B., and Klein, L. (2006). A central role for central tolerance. Annu. Rev. Immunol. 24, 571–606.
Lahl, K., Mayer, C. T., Bopp, T., Huehn, J., Loddenkemper, C., Eberl, G., Wirnsberger, G., Dornmair, K., Geffers, R., Schmitt, E., Buer, J., and Sparwasser, T. (2009). Nonfunctional regulatory T cells and defective control of Th2 cytokine production in natural scurfy mutant mice. J. Immunol. 183, 5662–5672.
Lanteri, M. C., O’Brien, K. M., Purtha, W. E., Cameron, M. J., Lund, J. M., Owen, R. E., Heitman, J. W., Custer, B., Hirschkorn, D. F., Tobler, L. H., Kiely, N., Prince, H. E., Ndhlovu, L. C., Nixon, D. F., Kamel, H. T., Kelvin, D. J., Busch, M. P., Rudensky, A. Y., Diamond, M. S., and Norris, P. J. (2009). Tregs control the development of symptomatic West Nile virus infection in humans and mice. J. Clin. Invest. 119, 3266–3277.
Le Gros, G., Ben-Sasson, S. Z., Seder, R., Finkelman, F. D., and Paul, W. E. (1990). Generation of interleukin 4 (IL-4)-producing cells in vivo and in vitro: IL-2 and IL-4 are required for in vitro generation of IL-4-producing cells. J. Exp. Med. 172, 921–929.
Lenardo, M. J. (1991). Interleukin-2 programs mouse alpha beta T lymphocytes for apoptosis. Nature 353, 858–861.
Lenardo, M., Chan, K. M., Hornung, F., Mcfarland, H., Siegel, R., Wang, J., and Zheng, L. (1999). Mature T lymphocyte apoptosis – immune regulation in a dynamic and unpredictable antigenic environment. Annu. Rev. Immunol. 17, 221–253.
Liu, F., Tong, F., He, Y., and Liu, H. (2011). Detectable expression of IL-35 in CD4+ T cells from peripheral blood of chronic hepatitis B patients. Clin. Immunol. 139, 1–5.
Lohr, J., Knoechel, B., Caretto, D., and Abbas, A. K. (2009). Balance of Th1 and Th17 effector and peripheral regulatory T cells. Microbes Infect. 11, 589–593.
Maloy, K. J., and Powrie, F. (2005). Fueling regulation: IL-2 keeps CD4+ Treg cells fit. Nat. Immunol. 6, 1071–1072.
Moraes-Vasconcelos, D., Costa-Carvalho, B. T., Torgerson, T. R., and Ochs, H. D. (2008). Primary immune deficiency disorders presenting as autoimmune diseases: IPEX and APECED. J. Clin. Immunol. 28(Suppl. 1), S11–S19.
Mosmann, T. R., and Coffman, R. L. (1989a). Heterogeneity of cytokine secretion patterns and functions of helper T cells. Adv. Immunol. 46, 111–147.
Mosmann, T. R., and Coffman, R. L. (1989b). TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu. Rev. Immunol. 7, 145–173.
Oberle, N., Eberhardt, N., Falk, C. S., Krammer, P. H., and Suri-Payer, E. (2007). Rapid suppression of cytokine transcription in human CD4+CD25 T cells by CD4+Foxp3+ regulatory T cells: independence of IL-2 consumption, TGF-beta, and various inhibitors of TCR signaling. J. Immunol. 179, 3578–3587.
Ouyang, W., Lohning, M., Gao, Z., Assenmacher, M., Ranganath, S., Radbruch, A., and Murphy, K. M. (2000). Stat6-independent GATA-3 autoactivation directs IL-4-independent Th2 development and commitment. Immunity 12, 27–37.
Pandiyan, P., Conti, H. R., Zheng, L., Peterson, A. C., Mathern, D. R., Hernandez-Santos, N., Edgerton, M., Gaffen, S. L., and Lenardo, M. J. (2011). CD4(+)CD25(+)Foxp3(+) regulatory T cells promote Th17 cells in vitro and enhance host resistance in mouse Candida albicans Th17 cell infection model. Immunity 34, 422–434.
Pandiyan, P., Gartner, D., Soezeri, O., Radbruch, A., Schulze-Osthoff, K., and Brunner-Weinzierl, M. C. (2004). CD152 (CTLA-4) determines the unequal resistance of Th1 and Th2 cells against activation-induced cell death by a mechanism requiring PI3 kinase function. J. Exp. Med. 199, 831–842.
Pandiyan, P., and Lenardo, M. J. (2008). The control of CD4+CD25+Foxp3+ regulatory T cell survival. Biol. Direct 3, 6.
Pandiyan, P., Zheng, L., Ishihara, S., Reed, J., and Lenardo, M. J. (2007). CD4(+)CD25(+)Foxp3(+) regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4(+) T cells. Nat. Immunol. 8, 1353–1362.
Pasare, C., and Medzhitov, R. (2003). Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science 299, 1033–1036.
Powrie, F., Leach, M. W., Mauze, S., Menon, S., Caddle, L. B., and Coffman, R. L. (1994). Inhibition of Th1 responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+ T cells. Immunity 1, 553–562.
Qureshi, O. S., Zheng, Y., Nakamura, K., Attridge, K., Manzotti, C., Schmidt, E. M., Baker, J., Jeffery, L. E., Kaur, S., Briggs, Z., Hou, T. Z., Futter, C. E., Anderson, G., Walker, L. S., and Sansom, D. M. (2011). Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science 332, 600–603.
Ramesh, S., Qi, X. J., Wildey, G. M., Robinson, J., Molkentin, J., Letterio, J., and Howe, P. H. (2008). TGF beta-mediated BIM expression and apoptosis are regulated through SMAD3-dependent expression of the MAPK phosphatase MKP2. EMBO Rep. 9, 990–997.
Rubtsov, Y. P., Niec, R. E., Josefowicz, S., Li, L., Darce, J., Mathis, D., Benoist, C., and Rudensky, A. Y. (2010). Stability of the regulatory T cell lineage in vivo. Science 329, 1667–1671.
Rubtsov, Y. P., Rasmussen, J. P., Chi, E. Y., Fontenot, J., Castelli, L., Ye, X., Treuting, P., Siewe, L., Roers, A., Henderson, W. R. Jr., Muller, W., and Rudensky, A. Y. (2008). Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity 28, 546–558.
Rudensky, A. Y., and Campbell, D. J. (2006). In vivo sites and cellular mechanisms of T reg cell-mediated suppression. J. Exp. Med. 203, 489–492.
Sakaguchi, S., Takahashi, T., and Nishizuka, Y. (1982). Study on cellular events in post-thymectomy autoimmune oophoritis in mice. II. Requirement of Lyt-1 cells in normal female mice for the prevention of oophoritis. J. Exp. Med. 156, 1577–1586.
Sakaguchi, S., Toda, M., Asano, M., Itoh, M., Morse, S. S., and Sakaguchi, N. (1996). T cell-mediated maintenance of natural self-tolerance: its breakdown as a possible cause of various autoimmune diseases. J. Autoimmun. 9, 211–220.
Sakaguchi, S., Wing, K., Onishi, Y., Prieto-Martin, P., and Yamaguchi, T. (2009). Regulatory T cells: how do they suppress immune responses? Int. Immunol. 21, 1105–1111.
Sakaguchi, S., Yamaguchi, T., Nomura, T., and Ono, M. (2008). Regulatory T cells and immune tolerance. Cell 133, 775–787.
Sanjabi, S., Mosaheb, M. M., and Flavell, R. A. (2009). Opposing effects of TGF-beta and IL-15 cytokines control the number of short-lived effector CD8+ T cells. Immunity 31, 131–144.
Scheffold, A., Huhn, J., and Hofer, T. (2005). Regulation of CD4+CD25+ regulatory T cell activity: it takes (IL-)two to tango. Eur. J. Immunol. 35, 1336–1341.
Schimpl, A., Berberich, I., Kneitz, B., Kramer, S., Santner-Nanan, B., Wagner, S., Wolf, M., and Hunig, T. (2002). IL-2 and autoimmune disease. Cytokine Growth Factor Rev. 13, 369–378.
Seder, R. A., and Paul, W. E. (1994). Acquisition of lymphokine-producing phenotype by CD4+ T cells. Annu. Rev. Immunol. 12, 635–673.
Shevach, E. M. (2002). CD4+ CD25+ suppressor T cells: more questions than answers. Nat. Rev. Immunol. 2, 389–400.
Shevach, E. M., Mchugh, R. S., Piccirillo, C. A., and Thornton, A. M. (2001). Control of T-cell activation by CD4+ CD25+ suppressor T cells. Immunol. Rev. 182, 58–67.
Shevach, E. M., Tran, D. Q., Davidson, T. S., and Andersson, J. (2008). The critical contribution of TGF-beta to the induction of Foxp3 expression and regulatory T cell function. Eur. J. Immunol. 38, 915–917.
Sprent, J., and Surh, C. D. (2001). Generation and maintenance of memory T cells. Curr. Opin. Immunol. 13, 248–254.
St Clair, E. W., Turka, L. A., Saxon, A., Matthews, J. B., Sayegh, M. H., Eisenbarth, G. S., and Bluestone, J. (2007). New reagents on the horizon for immune tolerance. Annu. Rev. Med. 58, 329–346.
Stockinger, B. (1999). T lymphocyte tolerance: from thymic deletion to peripheral control mechanisms. Adv. Immunol. 71, 229–265.
Strasser, A. (2005). The role of BH3-only proteins in the immune system. Nat. Rev. Immunol. 5, 189–200.
Strasser, A., Puthalakath, H., Bouillet, P., Huang, D. C., O’Connor, L., O’Reilly, L. A., Cullen, L., Cory, S., and Adams, J. M. (2000). The role of bim, a proapoptotic BH3-only member of the Bcl-2 family in cell-death control. Ann. N. Y. Acad. Sci. 917, 541–548.
Szabo, S. J., Kim, S. T., Costa, G. L., Zhang, X., Fathman, C. G., and Glimcher, L. H. (2000). A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 100, 655–669.
Tadokoro, C. E., Shakhar, G., Shen, S., Ding, Y., Lino, A. C., Maraver, A., Lafaille, J. J., and Dustin, M. L. (2006). Regulatory T cells inhibit stable contacts between CD4+ T cells and dendritic cells in vivo. J. Exp. Med. 203, 505–511.
Takahashi, T., Kuniyasu, Y., Toda, M., Sakaguchi, N., Itoh, M., Iwata, M., Shimizu, J., and Sakaguchi, S. (1998). Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state. Int. Immunol. 10, 1969–1980.
Takahashi, T., Tagami, T., Yamazaki, S., Uede, T., Shimizu, J., Sakaguchi, N., Mak, T. W., and Sakaguchi, S. (2000). Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J. Exp. Med. 192, 303–310.
Tang, Q., Adams, J. Y., Tooley, A. J., Bi, M., Fife, B. T., Serra, P., Santamaria, P., Locksley, R. M., Krummel, M. F., and Bluestone, J. A. (2006). Visualizing regulatory T cell control of autoimmune responses in nonobese diabetic mice. Nat. Immunol. 7, 83–92.
Tang, Q., and Bluestone, J. A. (2008). The Foxp3+ regulatory T cell: a jack of all trades, master of regulation. Nat. Immunol. 9, 239–244.
Thornton, A. M., Donovan, E. E., Piccirillo, C. A., and Shevach, E. M. (2004a). Cutting edge: IL-2 is critically required for the in vitro activation of CD4+CD25+ T cell suppressor function. J. Immunol. 172, 6519–6523.
Thornton, A. M., Piccirillo, C. A., and Shevach, E. M. (2004b). Activation requirements for the induction of CD4+CD25+ T cell suppressor function. Eur. J. Immunol. 34, 366–376.
Thornton, A. M., Korty, P. E., Tran, D. Q., Wohlfert, E. A., Murray, P. E., Belkaid, Y., and Shevach, E. M. (2010). Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J. Immunol. 184, 3433–3441.
Thornton, A. M., and Shevach, E. M. (1998). CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J. Exp. Med. 188, 287–296.
Thornton, A. M., and Shevach, E. M. (2000). Suppressor effector function of CD4+CD25+ immunoregulatory T cells is antigen nonspecific. J. Immunol. 164, 183–190.
Tinoco, R., Alcalde, V., Yang, Y., Sauer, K., and Zuniga, E. I. (2009). Cell-intrinsic transforming growth factor-beta signaling mediates virus-specific CD8+ T cell deletion and viral persistence in vivo. Immunity 31, 145–157.
Tran, D. Q., Andersson, J., Wang, R., Ramsey, H., Unutmaz, D., and Shevach, E. M. (2009a). GARP (LRRC32) is essential for the surface expression of latent TGF-{beta} on platelets and activated FOXP3+ regulatory T cells. J. Immunol. 182, 2929–2938.
Tran, D. Q., Glass, D. D., Uzel, G., Darnell, D. A., Spalding, C., Holland, S. M., and Shevach, E. M. (2009b). Analysis of adhesion molecules, target cells, and role of IL-2 in human FOXP3+ regulatory T cell suppressor function. J. Immunol. 182, 2929–2938.
Tykocinski, L. O., Hajkova, P., Chang, H. D., Stamm, T., Sozeri, O., Lohning, M., Hu-Li, J., Niesner, U., Kreher, S., Friedrich, B., Pannetier, C., Grutz, G., Walter, J., Paul, W. E., and Radbruch, A. (2005). A critical control element for interleukin-4 memory expression in T helper lymphocytes. J. Biol. Chem. 280, 28177–28185.
Wan, Y. Y., and Flavell, R. A. (2007). ‘Yin-Yang’ functions of transforming growth factor-beta and T regulatory cells in immune regulation. Immunol. Rev. 220, 199–213.
Wing, K., Onishi, Y., Prieto-Martin, P., Yamaguchi, T., Miyara, M., Fehervari, Z., Nomura, T., and Sakaguchi, S. (2008). CTLA-4 control over Foxp3+ regulatory T cell function. Science 322, 271–275.
Wing, K., Yamaguchi, T., and Sakaguchi, S. (2011). Cell-autonomous and -non-autonomous roles of CTLA-4 in immune regulation. Trends Immunol. 32, 428–433.
Yagi, R., Junttila, I. S., Wei, G., Urban, J. F. Jr., Zhao, K., Paul, W. E., and Zhu, J. (2010). The transcription factor GATA3 actively represses RUNX3 protein-regulated production of interferon-gamma. Immunity 32, 507–517.
Yang, X. O., Nurieva, R., Martinez, G. J., Kang, H. S., Chung, Y., Pappu, B. P., Shah, B., Chang, S. H., Schluns, K. S., Watowich, S. S., Feng, X. H., Jetten, A. M., and Dong, C. (2008). Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity 29, 44–56.
Zheng, X. X., Sanchez-Fueyo, A., Domenig, C., and Strom, T. B. (2003). The balance of deletion and regulation in allograft tolerance. Immunol. Rev. 196, 75–84.
Zhou, X., Bailey-Bucktrout, S., Jeker, L. T., and Bluestone, J. A. (2009a). Plasticity of CD4(+) FoxP3(+) T cells. Curr. Opin. Immunol. 21, 281–285.
Zhou, X., Bailey-Bucktrout, S. L., Jeker, L. T., Penaranda, C., Martinez-Llordella, M., Ashby, M., Nakayama, M., Rosenthal, W., and Bluestone, J. A. (2009b). Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat. Immunol. 10, 1000–1007.
Zhou, Z., Song, X., Li, B., and Greene, M. I. (2008). FOXP3 and its partners: structural and biochemical insights into the regulation of FOXP3 activity. Immunol. Res. 42, 19–28.
Zhu, J., and Paul, W. E. (2010). Heterogeneity and plasticity of T helper cells. Cell Res. 20, 4–12.
Zhu, P., Jiang, W., Cao, L., Yu, W., Pei, Y., Yang, X., Wan, B., Liu, J. O., Yi, Q., and Yu, L. (2010). IL-2 mRNA stabilization upon PMA stimulation is dependent on NF90-Ser647 phosphorylation by protein kinase CbetaI. J. Immunol. 185, 5140–5149.
Keywords: tregs, Th17, immune suppression mechanism, BIM cytokine deprivation death, apoptosis, regulatory T cells, Foxp3, cytokine consumption
Citation: Pandiyan P, Zheng L and Lenardo MJ (2011) The molecular mechanisms of regulatory T cell immunosuppression. Front. Immun. 2:60. doi: 10.3389/fimmu.2011.00060
Received: 13 September 2011;
Accepted: 19 October 2011;
Published online: 24 November 2011.
Edited by:
Kendall A. Smith, Weill Medical College of Cornell University, USAReviewed by:
Herman Waldmann, University of Oxford, UKChristopher E. Rudd, University of Cambridge, UK
Andrew McKenzie, Medical Research Council Laboratory of Molecular Biology, UK
Copyright: © 2011 Pandiyan, Zheng and Lenardo. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Pushpa Pandiyan, Laboratory of Immunology, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD 20892, USA. e-mail:cHBhbmRpeWFuQG5pYWlkLm5paC5nb3Y=