 Michael D. Keller1
Michael D. Keller1 Maureen Petersen2 Peck Ong3 Joseph Church3 Kimberly Risma4 Jon Burham5 Ashish Jain6 E. Richard Stiehm7
Maureen Petersen2 Peck Ong3 Joseph Church3 Kimberly Risma4 Jon Burham5 Ashish Jain6 E. Richard Stiehm7 Eric P. Hanson8 Gulbu Uzel9
Eric P. Hanson8 Gulbu Uzel9 Matthew A. Deardorff10
Matthew A. Deardorff10 Jordan S. Orange1*
Jordan S. Orange1*- 1 Division of Immunology, Children’s Hospital of Philadelphia, Philadelphia, PA, USA
- 2 Division of Allergy and Immunology, Walter Reed National Military Medical Center, Bethesda, MD, USA
- 3 Division of Clinical Immunology and Allergy, Children’s Hospital of Los Angeles, Los Angeles, CA, USA
- 4 Division of Allergy and Immunology, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH, USA
- 5 Division of Rheumatology, Children’s Hospital of Philadelphia, Philadelphia, PA, USA
- 6 Laboratory of Host Defenses, Clinical Immunology Unit, National Institute of Allergy and Infectious Diseases, Bethesda, MD, USA
- 7 Divisions of Allergy, Immunology, and Rheumatology, Department of Pediatrics, David Geffen School of Medicine at University of California, Los Angeles, Los Angeles, CA, USA.
- 8 Immunoregulatory Section, National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health, Bethesda, MD, USA
- 9 Immunopathogenesis Section, Laboratory of Clinical Infectious Diseases, National Institute of Allergy and Infectious Diseases, Bethesda, MD, USA
- 10 Division of Genetics, Children’s Hospital of Philadelphia, Philadelphia, PA, USA
Ectodermal dysplasias (ED) are uncommon genetic disorders resulting in abnormalities in ectodermally derived structures. Many ED-associated genes have been described, of which ectodysplasin-A (EDA) is one of the more common. The NF-κB essential modulator (NEMO encoded by the IKBKG gene) is unique in that mutations result in severe humoral and cellular immunologic defects in addition to ED. We describe three unrelated kindreds with defects in both EDA and IKBKG resulting from X-chromosome crossover. This demonstrates the importance of thorough immunologic consideration of patients with ED even when an EDA etiology is confirmed, and raises the possibility of a specific phenotype arising from coincident mutations in EDA and IKBKG.
Introduction
Ectodermal dysplasias (ED) comprise over 150 heterogeneous phenotypes in which the formation of structures arising from the ectodermal cell layer is impaired to some degree. Several causative gene pathways have been identified, including proteins involved in gap junctions, cell adhesion, ectoderm-specific transcription factors, and members of the TNF/TNFr superfamily (Itin and Fistarol, 2004). Importantly, while infections have been reported to occur at increased incidence in ED (Clarke et al., 1987), most forms do not impact the immune system.
Several of the better-defined causes of ED result from mutations in the ectodysplasin-A (EDA) signaling pathway. EDA is a membrane-bound signaling molecule of the TNF superfamily and via interaction with its EDA-receptor (EDAR, a TNF superfamily receptor) plays a role in signaling between ectodermal and mesodermal cell layers during embryogenesis (Mikkola and Thesleff, 2003). Defects in this pathway result in hypohidrotic ectodermal dysplasia (HED), characterized by absent sweat glands, hypodontia/oligodontia, hypotrichosis, eczema, and distinctive facial features including frontal bossing and a depressed nasal bridge. Approximately 80% of cases of HED are caused by mutations in the gene EDA (OMIM 305100, XLHED, ectodermal dysplasia, type 1; ED1) on the X-chromosome, while a smaller subset of cases is caused by mutations in the EDAR, the adapter protein EDARADD, or WNT10A (Chassaing et al., 2010; Cluzeau et al., 2011). Defects in these latter genes can manifest with autosomal recessive (ectodermal dysplasia anhidrotic; EDA; OMIM 224900) and autosomal dominant forms (ectodermal dysplasia type 3; ED3; OMIM 129490).
Although EDA pathway defects have no discernable affect on the immune system, hypomorphic mutations in the NF-κB essential modulator (NEMO) encoded by the X-linked IKBKG gene, result in HED with immunodeficiency (HED-ID or EDAXID, OMIM 300291). NEMO is an integral component of the inhibitor of NF-κB (IκB)-kinase (IKK) complex, which affects the phosphorylation of IκB, and thereby allows nuclear translocation of NF-κB (Doffinger et al., 2001; Orange et al., 2005). HED-ID is characterized by exquisite susceptibility to mycobacteria, pyogenic bacteria, and herpesviruses due to the essential role played by NF-κB signaling in both innate and adaptive immunity (Orange et al., 2005; Hanson et al., 2008). The immunologic phenotype of HED-ID is therefore diverse and highly mutation dependent, having the potential to impact Toll-like receptor (TLR) function, immunoglobulin class switch recombination, and T-cell function. Of note, NF-κB is an essential downstream component of the EDA, RANK-ligand, and VEGFR-3 pathways, and accordingly, observed phenotypes in HED-ID variably include ectodermal dysplasia, osteoclast abnormalities, and lymphedema (Orange et al., 2005; Hanson et al., 2008), as mutations of these individual genes result in these specific isolated phenotypes (Kere et al., 1996; Hughes et al., 2000; Karkainnen et al., 2000). Heterozygous null mutations or large deletions of IKBKG in females are associated with the rare neurocutaneous disorder incontinentia pigmenti (IP). Unlike the hypomorphic mutations associated with HED-ID, these larger defects are lethal to male offspring in utero (Dupuis-Girod et al., 2002; Fusco et al., 2008).
EDA is located on Xq12-q13, and the prevalence of HED is estimated to affect 1 in 100,000 newborns (Clarke, 1987). IKBKG is located on Xq28, and HED-ID occurs in approximately 1 in 250,000 newborns (Orange et al., 2004). Here, we describe three unrelated patients with HED-ID, who were found to have mutations in both EDA and IKBKG thus raising true diagnostic challenges when considering the etiology of HED.
Materials and Methods
Patient Data
Clinical information was obtained via chart review after IRB approval from the host institutions.
Gene Sequence Analysis
Genomic DNA and cDNA were prepared from patient leukocytes. Bi-directional DNA gene sequencing of all EDA and IKBKG exons was performed with genomic DNA from the patients as previously described (Orange et al., 2004), and targeted gene sequencing was subsequently performed to assess mutation status in relatives. Reference sequences used for this work were: EDA (NM_001399) and IKBKG (NM_001099856).
Immunologic Assays
Serum immunoglobulin concentrations (determined by nephelometry), antibody titers, and leukocyte enumeration were measured in the Children’s Hospital Clinical, or other CLIA-certified Laboratories and were compared with laboratory specific, age-related normal values.
Results
Case 1
The proband was a 2-year-old male with failure to thrive and recurrent infections (Table 1). He developed feeding intolerance, diarrhea, and failure to thrive in the first months of life, which was considered to be a result of food allergy. This did not improve, however, in response to a strict elemental diet. Beginning at 5 months of life, he experienced recurrent infections, including lymphadenitis and bacteremia with Staphylococcus, Streptococcus species, and Haemophilus influenzae. He also developed 5 weeks of persistent rash after receiving the live Varicella virus vaccine that required treatment with varicella immunoglobulin.
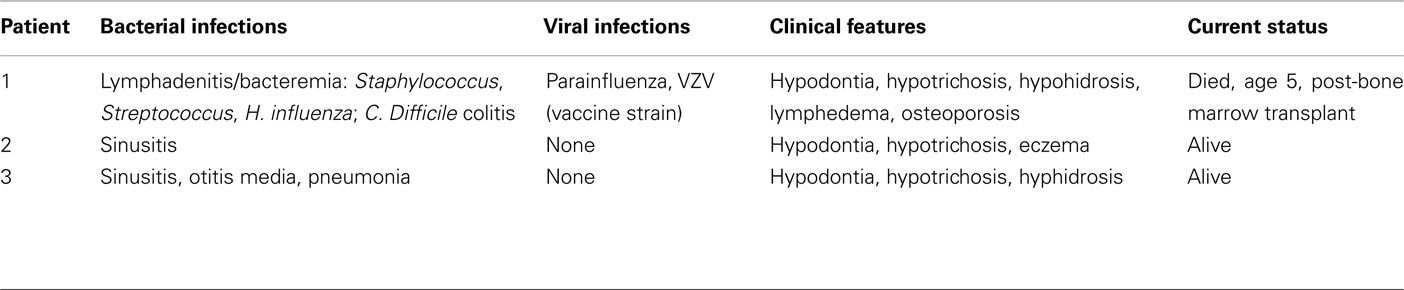
Table 1. Clinical characteristics of patients.
The family history was notable for a maternal uncle (Figure 1A, I5) who died at 8 months of age due to lymphangiectasia and dehydration. There was also a maternal first cousin who had granulomatous colitis, ectodermal abnormalities and died at 3 years of age because of E. coli bacteremia (I7). Several other maternal relatives had milder findings suggestive of ectodermal dysplasia, which mainly comprised of dental abnormalities (missing teeth, enamel defects).
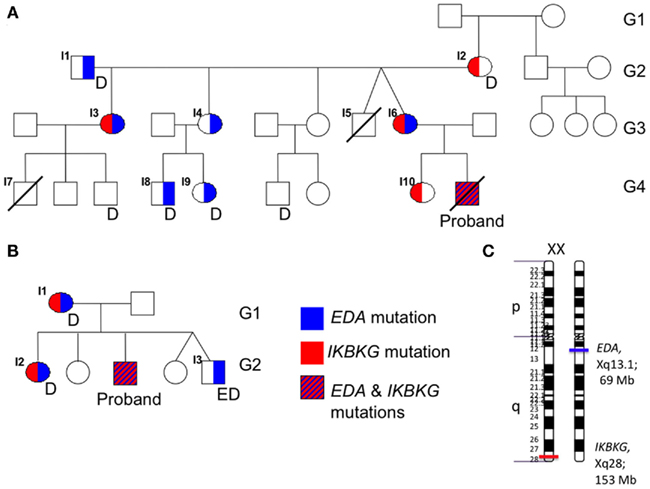
Figure 1. Family trees demonstrate the inheritance of EDA (red) and IKBKG (blue) mutations. (A) In the family of patient #1, a male maternal cousin of patient #1 (I7) had died with a diagnosis of granulomatous colitis. Multiple members also demonstrated dental abnormalities (D). (B) In the family of patient #2, the brother (I5) had features of ectodermal dysplasia (ED). No family history was available for family 3. (C) The respective positions of EDA and IKBKG on the long arm of the X-chromosome are depicted.
The proband’s physical findings included sparse hair, hypodontia with conical teeth, and edema of the lower extremities and scrotum (Figure 2). Radiologic studies were suggestive of osteopenia (Figure 3), and a lumbar spine dual X-ray absorptiometry scan showed low normal bone mass for age (Z-score = −1.0). Immunologic evaluation identified normal numbers of T-cells, B-cells, and NK cells (Table 2), as well as lymphocyte proliferation in response to mitogens phytohemagglutinin, concanavalin A, and pokeweed that were comparable to control donors. Quantitative immunoglobulin levels defined hypogammaglobulinemia and elevated IgM, along with vaccine-antigen specific IgG titers (tetanus, diphtheria, H. influenzae, Streptococcus pneumoniae, and two of three polio strains) that were below protective ranges despite his having received these vaccines. Analysis of B-cell subsets demonstrated naïve B-cells, and decreased switched memory B-cells (Table 2). TLR induced-TNF production (especially TLR7 and TLR9) and TNF-induced IκB degradation were impaired (not shown).
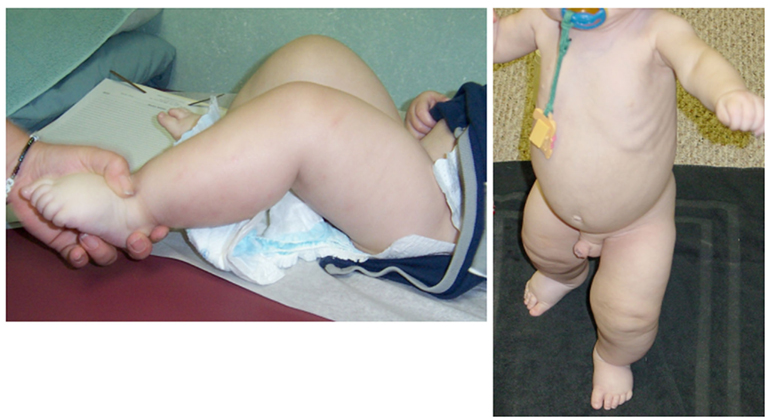
Figure 2. Lower extremity edema in patient #1.
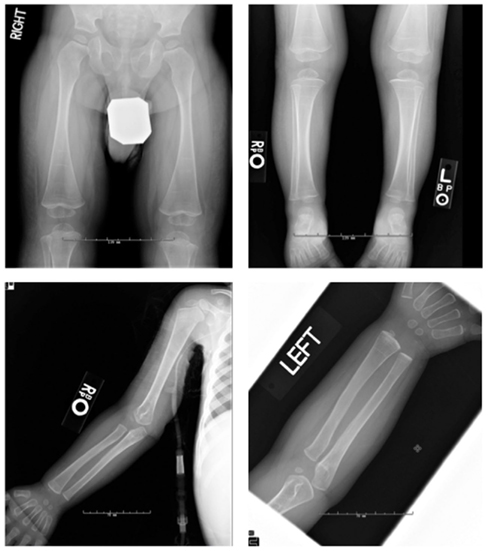
Figure 3. Radiographs of patient #1 demonstrated osteopenia, multiple growth recovery lines, and diminished tubulation of long bones. DEXA showed lumbar spine L1–4 bone mineral density of 0.412 g/cm2, Z-score = −1.0.
Due to the presence of ED, the patient’s EDA gene was sequenced. A c.458G>A substitution in exon 3 was identified, predicting an arginine to histidine missense mutation at amino acid position 153 (p.R153H). No similar mutation was found in several publically available databases of single nucleotide polymorphisms (data not shown). This region was previously described to correspond to the furin domain of EDA and thus the mutation was predicted to be impactful (Schneider et al., 2001). Sequence analysis of the IKBKG gene was performed as well due to evidence for immunodeficiency and identified a nucleotide substitution (c.518+5G>A or IVS6+5G>A), previously described to result in abnormal splicing with generation of a NEMO protein excluding the region corresponding to three exons (Orstavik et al., 2006). This mutation was also associated with the clinical syndrome of HED-ID. Analysis of NEMO protein expression in the proband’s PBMC by western blot confirmed abnormally sized protein consistent with the published report of this mutation (not shown; Orstavik et al., 2006).
DNA from the proband’s family members were subsequently analyzed for sequence of their EDA and IKBKG genes (Figure 1A). This demonstrated the presence of the mutant EDA and IKBKG genes in the maternal grandfather (I1) and grandmother (I2), respectively. Two of the daughters (I3, I6) inherited both of the mutant genes. One of these was the proband’s mother (I6), while the other daughter (I3) had a child with ectodermal dysplasia (I7) who died from bacteremia at 3 years of age. While material for gene sequencing was not available from the deceased cousin, a recombination event must have occurred in the proband’s mother. While it was not clear if the presence of the two mutant genes resulted in an exacerbated phenotype in the proband, examination of the two males possessing only the EDA mutation (I8, I9) was performed. While both had mild dental abnormalities, neither had florid HED or history suggestive of immunodeficiency. Finally, the proband’s sister (I10), who is unaffected, was found to carry the mutation in IKBKG, but no mutation in EDA. Thus, in this first family, while the EDA mutation was likely to have at least some affect, the most clinically relevant inheritance was of the IKBKG mutation. Given the family members available for sequence analysis it is not possible to conclude if the proband had a more severe presentation due to the inheritance of the two mutant genes.
After he was diagnosed with HED-ID the patient was treated with intravenous immunoglobulin and prophylactic antibiotics. After his health improved, at age 3.5 years of age he was treated with a matched-unrelated bone marrow transplantation. The details of this are reported elsewhere (Fish et al., 2009), but it was unsuccessful and he died from Parainfluenza infection following engraftment failure.
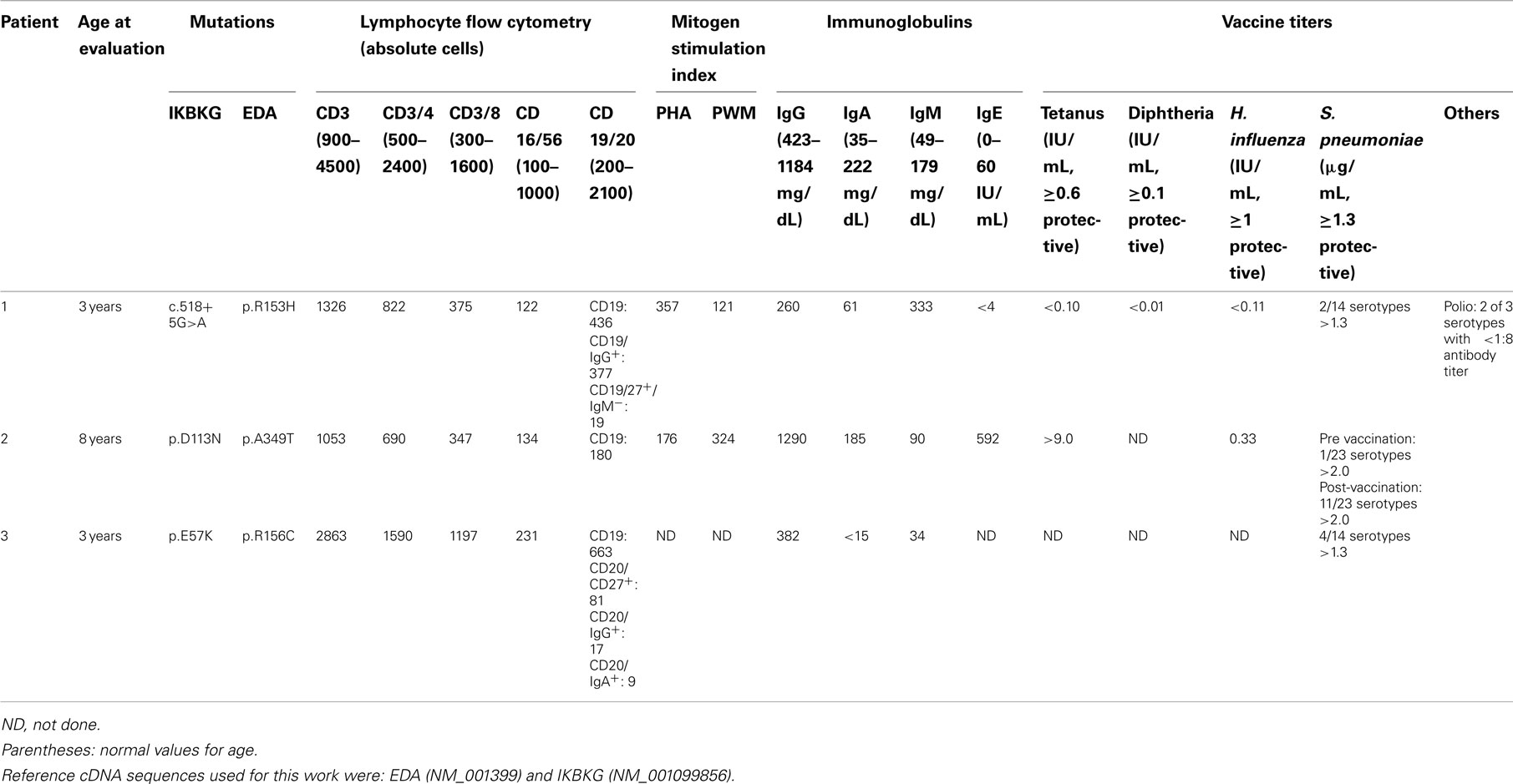
Table 2. Immunologic characteristics of patients.
Case 2
The proband was diagnosed with HED as an 8-year-old male. He had a history of severe generalized eczema and ectodermal dysplasia as characterized by severe hypodontia, dystrophic nails and hypotrichosis. He had a relatively benign history of infections including only sinusitis. There was a history of dental abnormalities in both the proband’s mother (Figure 1B, I1), 15-year-old sister (I2), and 5-year-old brother (I3), who also had sparse hair and eczema. There was no history in the family of recurrent infections or premature deaths, and the proband’s siblings and maternal uncles were healthy.
Due to his history of sinusitis and eczema, immunologic studies of the proband were performed and demonstrated normal quantitative immunoglobulins, elevated IgE, and protective antibody responses to his previous tetanus and H. influenzae vaccinations. Titers to S. pneumoniae serotypes were initially poor, but boosted appropriately following re-vaccination. Lymphocyte subsets were within normal ranges as were proliferative responses to phytohemagglutinin and pokeweed (Table 2).
Due to the presence of ectodermal dysplasia, the EDA gene was sequenced in the proband and analysis demonstrated a hemizygous c.1045G>A substitution, resulting in a predicted p.A349T missense mutation. This mutation has been previously described (Schneider et al., 2001), and corresponds to the TNF homology domain. Sequencing of the IKBKG gene was also performed due to history of sinusitis, and identified a hemizygous c.337G>A nucleotide substitution, resulting in a predicted p.D113N missense mutation, which has been previously described to be associated with immunodeficiency without causing HED (Salt et al., 2008). Heterozygous mutations in both EDA and IKBKG were found in his mother and eldest sister, while his brother was found to have the hemizygous EDA mutation only (Figure 1B). While the distinct origins of these two mutations was not identified as in the first case, the proband’s brother had only the EDA mutation suggesting a recombination event unique to the proband. The proband’s mild history of infection has continued and his immune status continues to be monitored.
Case 3
The proband was an adopted male with hypotrichosis, hypohidrosis, and hypodontia born to a mother of Italian descent. He had a history of recurrent sinusitis, otitis media, and an episode of lobar pneumonia requiring hospitalization at age 11 months. Due to his history of infections, immunologic laboratory studies were performed when he was 3.5 years old, which identified decreased IgG, and IgA compared to normal values for the patient’s age (Table 2). Analysis of lymphocyte subsets identified decreased CD4 Memory T-cells (absolute CD3+/CD4+/CD45RO: 169 cells/μL, normal: 203–975) and decreased CD8 Memory T-cells (absolute CD3+/CD8+/CD45RO: 37 cells/μL, normal: 34–309). Pneumococcal titers were non-protective (<1.3 μg) for 10/14 serotypes checked randomly at 3.5 years.
Because of the presence of ectodermal dysplasia, initially EDA was sequenced, and demonstrated a hemizygous c.466C>T substitution predicting a p.R156C missense mutation, which has been previously described in ED (Schneider et al., 2001). Due to the proband’s history of infections and immune abnormalities, his IKBKG gene was also sequenced and analysis identified a c.169G>A substitution, predicted to cause a p.E57K missense mutation. This mutation has been previously reported in association with IP (Fusco et al., 2004; Gautheron et al., 2010). Due to the proband having been adopted, additional biological family members were not available for sequence analyses.
The proband was treated with immunoglobulin replacement and prophylactic azithromycin against mycobacterial infections. He has developed normally and has not experienced any significant infectious episodes.
Discussion
We describe three patients with mutations in both EDA and IKBKG genes who have had features that could be attributed to one or the other gene abnormalities. In each of the three families there were additional family members with related features suggesting the influence of the individual gene mutations. This series emphasizes the importance of ascertaining the complete historical and family details and specifically considering distinct features that might suggest gene mutations with overlapping clinical impact. In this case both EDA and IKBKG gene mutations could be conceived to contribute to the clinical presentations of the patients. EDA mutation, however, should not account for the immunologic and infectious features presented here. Importantly, in X-linked disorders the possibility for males affected by a second clinically overlapping disorder resulting from a distinct alteration of the X-chromosome should be considered.
The immunologic and ED phenotypes in HED-ID are highly mutation dependent, and the associations of specific mutations with particular phenotypes have been reported previously (Cluzeau et al., 2011). In particular there are specific IKBKG mutations that do not cause ED (Nihues et al., 2004; Orange et al., 2004; Hanson et al., 2008). Presently this accounts for one-quarter of patients diagnosed with a clinically impactful IKBKG mutation. In general mothers carrying an IKBKG mutation do not demonstrate features of ED, but rare mutations associated with an IP phenotype have been observed (Dupuis-Girod et al., 2002; Fusco et al., 2008). These are considered to be quite rare, however, as most IP-causing mutations of IKBKG in women are incompatible with survival of males inheriting a single copy of the mutant-containing X-chromosome (Smahi et al., 2000).
In contrast to the presence or absence of ED, all males with IKBKG mutation appear to possess at least some variety of immunodeficiency. The immunodeficiency, however, is quite variable and may result from defects in signaling through the T-cell receptor, B-cell receptor, NK cell receptors, TLRs, CD40, as well as other TNF superfamily receptors, all of which are upstream of NF-κB. Some IKBKG mutations are more likely to affect particular immune functions; for example, some typically cause hypogammaglobulinemia, while others (particularly impacting the NEMO leucine zipper domain) often do not (Hanson et al., 2008). Some mutations are associated with T-cell defects, though selective memory defects have been mostly attributed to mutations in the downstream protein Inhibitor of kB (IKBα; Courtois et al., 2003). Mature/memory T-cells have been shown to depend on signaling through the IKK complex (Schmidt-Supprian et al., 2003), which may explain the low memory T-cell phenotype observed in these patients.
While conceivable, phenotypic heterogeneity attributable to particular gene mutations in EDA-related HED has not been found (Monreal et al., 1998; Schneider et al., 2001; Vincent et al., 2001). Thus it is unclear if and how the EDA variants identified in the patients we describe affect their phenotype. In the second case, it is quite likely that the EDA mutation has imparted the HED, since this particular IKBKG mutation has been reported previously to not cause HED. However, he did not seem to have a significant immunologic phenotype, with sinusitis being his only infectious problem. It was previously reported that D113N mutations in IKBKG may impart susceptibility to opportunistic infections with Pneumocystis or herpesviruses, but the mutations does not seem to impart susceptibility to encapsulated bacteria (Hanson et al., 2008). The same mutation has further been defined to cause a T-cell functional defect (Salt et al., 2008), which was not specifically evaluated in our patient. His minimal infectious history may reflect heterogeneity in the immunologic phenotype of this mutation, as few patients have been described. In the other two cases, individual or combined contributions of the EDA and IKBKG mutations are less clear. With the IKBKG mutation found in the first case, severe immunodeficiency and HED have been previously reported (Orstavik et al., 2006). The mild appearance of ED-related findings in family members who possessed only the EDA mutation (Family 1) suggests it was of minimal impact. The mutation described for EDA in this case had not previously been reported and would be predicted to only result in a minor charge variation via a single amino acid change. It is unclear, however, if inheritance of both mutations on a single chromosome resulted in an exacerbated clinical phenotype in the proband. Sequence analysis of the deceased cousin or uncle might have provided some clarity, but these samples were not available. It is worth considering, however, if this particular occurrence might have impacted the negative outcome of the hematopoietic stem cell transplantation via graft failure because of some unusual effect on the bone marrow stromal/physical environment. With regards to the third case, the EDA mutation was classic for being associated with disease, and the IKBKG mutation was associated with IP, but not HED-ID (Fusco et al., 2004; Gautheron et al., 2010). The presence of immunodeficiency, however, implies the impact of the latter while its contribution to ED or the influence of the combined mutations is unclear for the patient. Thus in all three of the probands, it is difficult to attribute all of the clinical impact to one of the two mutant genes, and it would appear likely that some affect is derived from both. What remains unclear is if combined EDA and IKBKG mutations are more severe than IKBKG alone. The first case suggests this might be valid, but cannot be proven at present. It does, however, suggest the possibility for a specific disease phenotype resulting from the coincidence of mutations. Given the experience regarding Patient 1, it may be feasible to evaluate for this genotype in considering hematopoietic stem cell transplantation.
While we report three unrelated cases, the concurrence of these mutations is not expected to be common. The prevalence of EDA-related HED is estimated to occur in less than 1 in 100,000 newborns (Clarke, 1987) and IKBKG-related HED-ID has also been estimated to occur in approximately 1 in 250,000 newborns (Orange et al., 2004). Thus the estimated chance of their occurrence in the same patient would be 1 in 25 billion. Were this to be the case, however, we would effectively be reporting all of the existing individuals with this particular genotype.
Due to the theoretical rarity of the diagnoses, one might consider the possibility that the genetic events, i.e., mutations of both EDA and IKBKG are linked. Although they are both genes on the X-chromosome, further assessment suggests that the co-occurrence exists by chance. This estimate is based upon data demonstrating a 1.19-cM recombination frequency per megabase of the human X-chromosome (Kong PMID: 12053178, Coop PMID: 18239090). EDA is located at Xq13.1 at a genomic position ∼69 Mb and IKBKG is on Xq28at ∼153 Mb, a separation of 84 Mb (Figure 1C). This would predict a 70% chance of recombination, well above 50% indicative of random segregation. Thus, while not linked per se, the coincidence is somewhat more likely than would be predicted for distant loci. Therefore, there are likely to be other patients possessing both EDA and IKBKG mutations in need of more definitive molecular diagnosis.
This work emphasizes that the presence of a mutation in EDA does not necessarily exclude the presence of a more serious defect in IKBKG. Early diagnosis may allow prevention of life-threatening infections via prophylactic antibiotics and immunoglobulin replacement. Though most causes of ectodermal dysplasia are not associated with immune defects, a high index of suspicion is warranted with HED for the possibility of mutations in IKBKG resulting in HED-ID. A thorough family history should be performed in pursuit of clues suggesting immunodeficiency, especially in maternally related males. Immunologic screening should be considered for all children with HED, and a thorough workup is essential if there is any abnormal pattern of infections.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Linda Monaco-Shawver, Pinaki Banerjee, Sherry Bale, David Adams, Raquel Deering, and Veronique Weinstein for their assistance in this study. Supported by National Institutes of Health (NIH) AI079731 (Jordan S. Orange).
References
Chassaing, N., Cluzeau, C., Bal, E., Guigue, P., Vincent, M. C., Viot, G., Ginisty, D., Munnich, A., Smahi, A., and Calvas, P. (2010). Mutations in ERARADD account for a small proportion of hypohidrotic ectodermal dysplasia cases. Br. J. Dermatol. 162, 1044–1048.
Clarke, A., Phillips, D. I., Brown, R., and Harper, P. S. (1987). Clinical aspects of X-linked hypohidrotic ectodermal dysplasia. Arch. Dis. Child. 62, 989–996.
Cluzeau, C., Rabia, S. H., Jambou, M., Mansour, S., Guigue, P., Masmoudi, S., Bal, E., Chassaing, N., Vincent, M. C., Viot, G., Clauss, F., Maniere, M. C., Toupenay, S., Le Merrer, M., Lyonnet, S., Cormier-Daire, V., Amiel, J., Faivre, L., de Prost, Y., Munnich, A., Bonnefont, J. P., Bodemer, C., and Smahi, A. (2011). Only four genes (EDA, EDAR, EDARADD, and WNT10A) account for 90% of hypohidrotic/anhidrotic ectodermal dysplasia cases. Hum. Mutat. 32, 70–77.
Courtois, G., Smahi, A., Reichenbach, J., Doffinger, R., Cancrini, C., Bonnet, M., Puel, A., Chable-Bessia, C., Yamaoka, S., Feinberg, J., Dupuis-Girod, S., Bodemer, C., Livadiotti, S., Novelli, F., Rossi, P., Fischer, A., Israel, A., Munnich, A., Le Deist, F., and Casanova, J. L. (2003). A hypermorphic IKBa mutation is associated with autosomal dominant anhidrotic ectodermal dysplasia and T cell immunodeficiency. J. Clin. Invest. 112, 1108–1115.
Doffinger, R., Smahi, A., Bessia, C., Geissmann, F., Feinberg, J., Durandy, A., Bodemer, C., Kenwrick, S., Dupuis-Girod, S., Blanche, S., Wood, P., Rabia, S. H., Headon, D. J., Overbeek, P. A., De Deist, F., Holland, S. M., Belani, K., Kumararatne, D. S., Fischer, A., Shapiro, R., Conley, M. E., Reimund, E., Kalhoff, H., Abinun, M., Munnich, A., Israel, A., Courtois, G., and Casanova, J. L. (2001). X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kB signaling. Nat. Genet. 27, 277–285.
Dupuis-Girod, S., Corradini, N., Rabia, S. H., Fournet, J. C., Faivre, L., Le Deist, F., Durand, P., Doffinger, R., Smahi, A., Israel, A., Courtois, G., Brousse, N., Blanche, S., Munnich, A., Fischer, A., Casanova, J. L., and Bodemer, C. (2002). Osteopetrosis, lymphedema, anhidrotic ectodermal dysplasia, and immunodeficiency in a boy and incontinentia pigmenti in his mother. Pediatrics 109, e97.
Fish, J. D., Duerst, R. E., Gelfand, E. W., Orange, J. S., and Bunin, N. (2009). Challenges in the use of allogeneic hematopoietic SCT for ectodermal dysplasia with immune deficiency. Bone Marrow Transplant. 43, 217–221.
Fusco, F., Bardaro, T., Fimiani, G., Mercadante, V., Miano, M. G., Falco, G., Israel, A., Courtois, G., D’Urso, M., and Ursini, M. V. (2004). Molecular analysis of the genetic defect in a large cohort of IP patients and identification of novel NEMO mutations interfering with NF-kB activation. Hum. Mol. Genet. 13, 1763–1773.
Fusco, F., Pescatore, A., Bal, E., Ghoul, A., Paciolla, M., Lioi, M. B., D’Urso, M., Rabia, S. H., Bodermer, C., Bonnefont, J. P., Munnich, A., Miano, M. G., Smahi, A., and Ursini, M. V. (2008). Alterations of the IKBKB locus and diseases: an update and a report of 13 novel mutations. Hum. Mutat. 29, 595–604.
Gautheron, J., Pescatore, A., Fusco, F., Esposito, E., Yamaoka, S., Agou, F., Ursini, M. V., and Courtois, G. (2010). Identification of a new NEMO/TRAF6 interface affected in incontinentia pigmenti pathology. Hum. Mol. Genet. 19, 3138–3149.
Hanson, E. P., Monaco-Shawver, L., Solt, L. A., Madge, L. A., Banerjee, P. P., May, M. J., and Orange, J. S. (2008). Hypomorphic nuclear factor KB essential modulator mutation database and reconstitution system identifies phenotypic and immunologic diversity. J. Allergy Clin. Immunol. 122, 1169–1177.
Hughes, A. E., Ralston, S. H., Marken, J., Bell, C., MacPherson, H., Wallace, R. G., van Hul, W., Whyte, M. P., Nakatuka, K., Hovy, L., and Anderson, D. M. (2000). Mutations in TNFRSF11A, affecting the signal peptide of RANK, cause familial expansile osteolysis. Nat. Genet. 24, 45–48.
Itin, P. H., and Fistarol, S. K. (2004). Ectodermal dysplasias. Am. J. Med. Genet. C Semin. Med. Genet. 131, 45–51.
Karkainnen, M. J., Ferrell, R. E., Lawrence, E. C., Kimak, M. A., Levinson, K. L., McTigue, M. A., Alitalo, K., and Finegold, D. N. (2000). Missense mutations interfere with VEGFR-3 signaling in primary lymphedema. Nat. Genet. 25, 153–159.
Kere, J., Srivastava, A. K., Montonen, O., Zonana, J., Thomas, N., Ferguson, B., Munoz, F., Morgan, D., Clarke, A., Baybayan, P., Chen, E. Y., Ezer, S., Saarialho-Kere, U., de la Chapelle, A., and Schlessinger, D. (1996). X-linked anhidrotic (hypohidrotic) ectodermal dysplasia is caused by a mutation in a novel transmembrane protein. Nat. Genet. 13, 409–416.
Mikkola, M. L., and Thesleff, I. (2003). Ectodysplasin signaling in development. Cytokine Growth Factor Rev. 14, 211–224.
Monreal, A. W., Zohana, J., and Ferguson, B. (1998). Identification of a new splice form of the EDA gene permits detection of nearly all X-linked hypohidrotic ectodermal dysplasia mutations. Am. J. Hum. Genet. 63, 380–389.
Nihues, T., Reichenbach, J., Neubert, J., Gudowius, S., Puel, S., Horneff, G., Lainka, E., Dirksen, U., Schroten, H., Doffinger, R., Casanova, J. L., and Wahn, V. (2004). Nuclear factor kB essential modulator-deficient child with immunodeficiency yet without anhidrotic ectodermal dysplasia. J. Allergy Clin. Immunol. 114, 1456–1462.
Orange, J. S., Jain, A., Ballas, Z. K., Schneider, L. C., Geha, R. S., and Bonilla, F. A. (2004). The presentation and natural history of immunodeficiency caused by nuclear factor KB essential modulator. J. Allergy Clin. Immunol. 113, 725–733.
Orange, J. S., Levy, O., and Geha, R. S. (2005). Human disease resulting from gene mutations that interfere with appropriate nuclear factor-KB activation. Immunol. Rev. 203, 21–37.
Orstavik, K. H., Kristiansen, M., Knudsen, G. P., Storhaug, K., Vege, A., Eiklid, K., Abrahamsen, T. G., Smahi, A., and Steen-Johnsen, J. (2006). Novel splicing mutation in the NEMO (IKK-gamma) gene with severe immunodeficiency and heterogeneity of X-chromosome inactivation. Am. J. Med. Genet. A 140, 31–39.
Salt, B. H., Niemela, J. E., Pandey, R., Hanson, E. P., Deering, R. P., Quinones, R., Jain, A., Orange, J. S., and Gelfand, E. W. (2008). IKBKG (NEMO) mutation can be associated with opportunistic infection without impairing TLR function. J. Allergy Clin. Immunol. 121, 976–982.
Schmidt-Supprian, M., Courtois, G., Tian, J., Coyle, A. J., Israel, A., Rajewsky, K., and Pasparakis, M. (2003). Mature T cells depend on signaling through the IKK Complex. Immunity 19, 377–389.
Schneider, P., Street, S. L., Gaide, O., Hertig, S., Tardivel, A., Tschopp, J., Runkel, L., Alevizopoulos, K., Ferguson, B. M., and Zonana, J. (2001). Mutations leading to X-linked hypohidrotic ectodermal dysplasia affect three major functional domains in the tumor necrosis factor family member ectodysplasin-A. J. Biol. Chem. 276, 18819–18827.
Smahi, A., Courtois, G., Vabres, P., Yamaoka, S., Heuertz, S., Munnich, A., Israel, A., Heiss, N. S., Klauck, S. M., Kioschis, P., Wiemann, S., Poustka, A., Esposito, T., Bardaro, T., Gianfrancesco, F., Ciccodicola, A., D’Urso, M., Woffendin, H., Jakins, T., Donnai, D., Stewart, H., Kenwrick, S. J., Aradhya, S., Yamagata, T., Levy, M., Lewis, R. A., and Nelson, D. L. (2000). Genomic rearrangement in NEMO impairs NF-kB activation and is a cause of incontinentia pigmenti. Nature 405, 466–472.
Keywords: NEMO, EDA, ectodermal dysplasia, immunodeficiency
Citation: Keller MD, Petersen M, Ong P, Church J, Risma K, Burham J, Jain A, Stiehm ER, Hanson EP, Uzel G, Deardorff MA and Orange JS (2011) Hypohidrotic ectodermal dysplasia and immunodeficiency with coincident NEMO and EDA mutations. Front. Immun. 2:61. doi: 10.3389/fimmu.2011.00061
Received: 15 September 2011; Paper pending published: 03 October 2011;
Accepted: 20 October 2011; Published online: 08 November 2011.
Edited by:
Sergio Rosenzweig, National Institutes of Health, USAReviewed by:
Sergio Rosenzweig, National Institutes of Health, USALaurence E. Cheng, University of California San Francisco, USA
Elena Perez, University of South Florida, USA
Copyright: © 2011 Keller, Petersen, Ong, Church, Risma, Burham, Jain, Stiehm, Hanson, Uzel, Deardorff and Orange. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Jordan S. Orange, Abramson Research Building Room 909, Children’s Hospital of Philadelphia, Philadelphia, PA 19104, USA. e-mail:b3JhbmdlQG1haWwubWVkLnVwZW5uLmVkdQ==