 Bryna E. Burrell1
Bryna E. Burrell1 Yaozhong Ding1
Yaozhong Ding1 Yumi Nakayama1 Kyung-Su Park1,2 Jiangnan Xu1 Na Yin1 and Jonathan S. Bromberg1,3,4*
Yumi Nakayama1 Kyung-Su Park1,2 Jiangnan Xu1 Na Yin1 and Jonathan S. Bromberg1,3,4*- 1 Center for Vascular and Inflammatory Diseases, University of Maryland School of Medicine, Baltimore, MD, USA
- 2 College of Medicine, The Catholic University of Korea, Seoul, South Korea
- 3 Department of Surgery, University of Maryland School of Medicine, Baltimore, MD, USA
- 4 Department of Microbiology and Immunology, University of Maryland School of Medicine, Baltimore, MD, USA
This issue of Frontiers in Immunologic Tolerance explores barriers to tolerance from a variety of views of cells, molecules, and processes of the immune system. Our laboratory has spent over a decade focused on the migration of the cells of the immune system, and dissecting the signals that determine how and where effector and suppressive regulatory T cells traffic from one site to another in order to reject or protect allografts. These studies have led us to a greater appreciation of the anatomic structure of the immune system, and the realization that the path taken by lymphocytes during the course of the immune response to implanted organs determines the final outcome. In particular, the structures, microanatomic domains, and the cells and molecules that lymphocytes encounter during their transit through blood, tissues, lymphatics, and secondary lymphoid organs are powerful determinants for whether tolerance is achieved. Thus, the understanding of complex cellular and molecular processes of tolerance will not come from “96-well plate immunology,” but from an integrated understanding of the temporal and spatial changes that occur during the response to the allograft. The study of the precise positioning and movement of cells in lymphoid organs has been difficult since it is hard to visualize cells within their three-dimensional setting; instead techniques have tended to be dominated by two-dimensional renderings, although advanced confocal and two-photon systems are changing this view. It is difficult to precisely modify key molecules and events in lymphoid organs, so that existing knockouts, transgenics, inhibitors, and activators have global and pleiotropic effects, rather than precise anatomically restricted influences. Lastly, there are no well-defined postal codes or tracking systems for leukocytes, so that while we can usually track cells from point A to point B, it is exponentially more difficult or even impossible to track them to point C and beyond. We believe this represents one of the fundamental barriers to understanding the immune system and devising therapeutic approaches that take into account anatomy and structure as major controlling principles of tolerance.
Introduction
This issue of Frontiers in Immunologic Tolerance explores barriers to tolerance from a variety of views of cells, molecules, and processes of the immune system. Our laboratory has spent over a decade focused on the migration of the cells of the immune system, and dissecting the signals that determine how and where effector and suppressive regulatory T cells traffic from one site to another in order to reject or protect allografts. These studies have led us to a greater appreciation of the anatomic structure of the immune system, and the realization that the path taken by lymphocytes during the course of the immune response to implanted organs determines the final outcome. In particular, the structures, microanatomic domains, and the cells and molecules that lymphocytes encounter during their transit through blood, tissues, lymphatics, and secondary lymphoid organs (SLOs) are powerful determinants for whether tolerance is achieved. Thus, the understanding of complex cellular and molecular processes of tolerance will not come from “96-well plate immunology,” but from an integrated understanding of the temporal and spatial changes that occur during the response to the allograft. The study of the precise positioning and movement of cells in lymphoid organs has been difficult since it is hard to visualize cells within their three-dimensional setting; instead techniques have tended to be dominated by two-dimensional renderings, although advanced confocal and two-photon systems are changing this view. It is difficult to precisely modify key molecules and events in lymphoid organs, so that existing knockouts, transgenics, inhibitors, and activators have global and pleiotropic effects, rather than precise anatomically restricted influences. Lastly, there are no well-defined postal codes or tracking systems for leukocytes, so that while we can usually track cells from point A to point B, it is exponentially more difficult or even impossible to track them to point C and beyond. We believe this represents one of the fundamental barriers to understanding the immune system and devising therapeutic approaches that take into account anatomy and structure as major controlling principles of tolerance.
Secondary Lymphoid Organs
Structure
It is first important to understand the structure, development, and regulatory mechanisms of lymphoid organs to provide the basis for understanding on how these control immunity and tolerance. The lymph node (LN) is an encapsulated, highly organized SLO (Figures 1C,D). The separate regions are the cortex, the paracortex, and the medulla. The cortex is the most outer layer and contains B cells, macrophages, and follicular dendritic cells (FDC) arranged in primary follicles. The paracortex is the next layer, which contains T cells and dendritic cells (DC). Fibroblastic reticular cells (FRC) support T cell and DC interactions in this area. The most inner layer, the medulla, consists of lymphatic tissues called medullary cords, which are separated by the lymph filled spaces of the medullary sinuses. The LN vasculature consists of high endothelial venules (HEV), the characteristic structure through which T cells and B cells enter into LN from the blood, and lymphatic vessels lined by lymphatic endothelial cells (LEC).
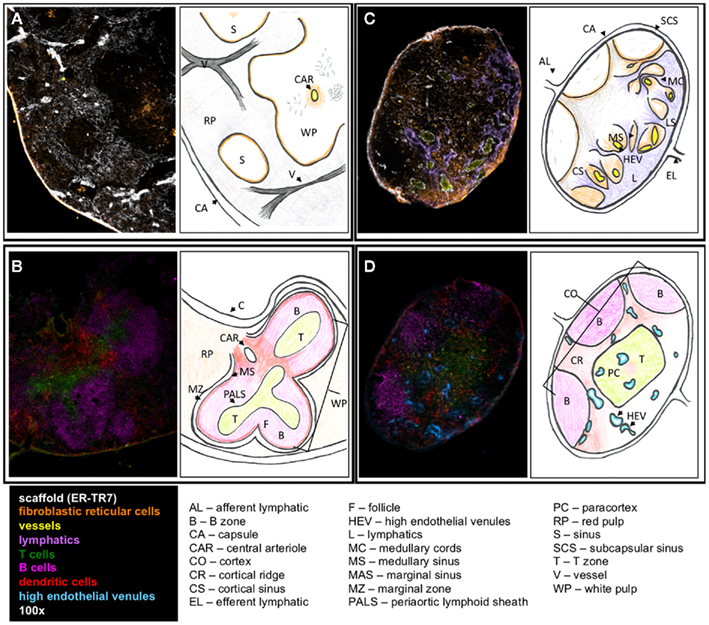
Figure 1. Structure of SLO. Fluorescent immunohistochemistry (left) and cartoons (right) illustrate the structure and cellular content of spleen (A,B) and LN (C,D). (A) The outer capsule of the spleen surrounds the red pulp, with the sinuses and white pulp embedded within. ER-TR7 fibers form a network supporting the red pulp, and FRC line the borders between the red and white pulp. A central arteriole empties into the white pulp. (B) In the white pulp, T cells populate the PALS leading into the B cell follicles. DC line the marginal sinus, enclosed by the marginal zone. (C) In the LN, the efferent lymphatics empty into the medullary sinus and travel through the medullary cords. Lymphocytes enter through the HEV, which seed the LN. (D) B cells, FDC, and macrophages lay in the cortex. The paracortex contains T cells and DC. Antigen presentation and T cell priming occur at the cortical ridge areas between B and T cell zones supported by FRC.
The spleen is surrounded by a capsule that extends many projections into the interior to form a compartmentalized structure (Figures 1A,B). There are two compartments, the red pulp and the white pulp, and a diffuse marginal zone that separates them. The red pulp is a network of sinusoids populated by macrophages and red blood cells. The splenic white pulp forms a periarteriolar lymphoid sheath (PALS) populated mainly by T cells. B cell follicles, supported by FDC as in the LN, are next to the PALS. The marginal sinus and its associated layer of FRC and MOMA-1 staining metallophilic macrophages define the outer boundary of the white pulp.
LTi and LTo cells – Definition and Function
Lymph nodes develop during embryogenesis or in the first few weeks after birth through recruitment and interaction of lymphoid tissue inducer (LTi) cells and lymphoid tissue organizer (LTo) cells. The central role of the LTi cells in LN development is the expression and presentation of lymphotoxin alpha and beta (LTα1β2) to the LTβ receptor (LTβR) on LTo, and this interaction leads to organized lymphoid structures. The importance of this signal is demonstrated in mice deficient in LTβR or LTα, which lack LN and Peyer’s patches (PP; De Togni et al., 1994; Futterer et al., 1998). Although the early differentiation pathway of LTi cells from multipotent hematopoietic progenitor cells remains to be characterized, studies suggest that the early specification to the LTi lineage takes place in lineage marker-negative (Lin−) populations, and α4β7 integrin, IL-7Rα+, and CD4+CD3− are useful markers to define LTi (Mebius et al., 1996; Yoshida et al., 1999; Honda et al., 2001). A recent study shows that transcription factor Runx/Cbfb2 complexes are required for LTi cell differentiation (Tachibana et al., 2011). LTo cells, also called mesenchymal organizer cells or stromal organizer cell, express LTβR. LTo can be derived from the antimesenteric side of the intestine during development or from a hematopoietic cell lineage (Adachi et al., 1998; Nishikawa et al., 2000). The LTi–LTo interaction activates a signaling cascade resulting in the expression of adhesion molecules such as VCAM-1, intercellular cell adhesion molecule-1 (ICAM-1), and mucosal addressin cell adhesion molecule-1 and homeostatic chemokines such as CXCL13, CCL19, and CCL21 (van de Pavert and Mebius, 2010). CXCL13, the B zone chemokine, recruits circulating B cells to what becomes the B cell area; and the T zone chemokines, CCL19 and CCL21, attract T and DC to shape the T cell area (Cupedo and Mebius, 2005; Scandella et al., 2008). The human equivalent of murine LTi cells has been identified but has not been as well characterized as in mice. LTi cells in adult human are identified as IL-22 expressing NK type cells (Cupedo et al., 2009). The major phenotypic difference between mouse and human LTi cells is that mouse LTi cells are either CD4+ or CD4−, while human LTi cells are CD4− or CD4low (Kim et al., 2009).
In addition to organizing lymphoid structure during development, LTi cells are present in adult lymphoid tissues. Even after the maturation of SLO, a continuous interplay between lymphocytes and stromal cells is likely to be required for the maintenance of tissue architecture and the characteristics of adult stromal cells. Adult LTi cells express OX40L and CD30L, which are critical for memory CD4 T cell generation (Kim et al., 2005). OX40 and CD30 deficient mice have impaired CD4 T cell-dependent memory antibody responses in the spleen and gut (Lane et al., 2008; Tsuji et al., 2008). LTo give rise to various stromal cell subsets such as FDC and FRC that are present in lymphoid organs. However, it remains unknown how many different mesenchymal stromal cell types exist in SLO. The relationship between embryonic LTo cells and different types of stromal cells in adult SLO, and the postnatal fate of LTo cells are also unclear. It is possible that LTo-like cells persist in the adult and play a role in the maintenance of SLO. Katakai et al. (2008) found a layer of unique reticular cells underneath the subcapsular sinus (SCS) lining of adult LNs. These specialized mesenchymal cells, marginal reticular cells (MRS), share many characteristics with LTo cells and are observed in mucosal SLO such as PP, nasal-associated lymphoid tissues, and isolated lymphoid follicles of adult mice (Katakai et al., 2008).
Splenic LTi-like cells contribute the development of SLO and also to host defense, by producing IL-17 and IL-22 in response to pathogen or IL-23 stimulation (Takatori et al., 2009). Phenotypically, splenic LTi-like cells in adult mice are similar to LN. They are CD4+CD3−NK1.1−CD11b−Gr-1−CD11c−B220−, express RORγt, aryl hydrocarbon receptor, IL-23R and CCR6, and are located mainly in the white pulp, particularly at the junction of T- and B-zones in the follicle. Formation of splenic white pulp does not require LTi cells, but the maturation of the tissue structure depends on LTα1β2 produced by lymphocytes and LTβR signaling (Fu and Chaplin, 1999), so that LTα, LTβ, or LTβR deficient mice display disrupted splenic architecture (De Togni et al., 1994). Thus, despite some similarities, the developmental program and molecular requirements of each SLO are clearly different.
Lymphotoxin System
LT is a tumor necrosis factor (TNF)-related cytokine required for the development and organization of SLO (Cyster, 2003). Its ligands and receptors are summarized in Figure 2. There are four closely related ligands: LTα; LTβ; TNF; and “homologous to LT, inducible expression, competes with herpes simplex virus (HSV) glycoprotein D for HSV entry mediator (HVEM), a receptor expressed on T lymphocytes” (LIGHT or TNFSF14). There are four cognate receptors: TNFR1; TNFR2; LTβ; and HVEM. LTα and LTβ form three different ligands, a secreted homotrimer LTα3 and two membrane-bound heterotrimers LTα1β2 (predominant form) and LTα2β1. The ligand–receptor binding patterns overlap within the family. LTα3 binds TNFR1 and TNFR2; LTα1β2 signals through LTβR; LTα2β1 binds TNFR1 and TNFR2; and LIGHT interacts with LTβR and HVEM. LTα3 may also bind HVEM (Schneider et al., 2004). As mentioned above, LT expressing LTi cells interact with the corresponding LTβR on the LTo cells during development. LTβR signaling is also required for maintaining the structure and regulating immune responses in adult mice (McCarthy et al., 2006).
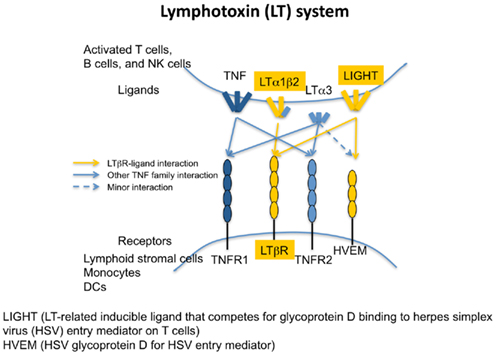
Figure 2. The lymphotoxin receptors and their ligands. The interactions of TNF cytokines with their cognate receptors are shown by arrows. Both LTα1/β2 and LIGHT bind to LTβR. HVEM binds to membrane or soluble LIGHT, soluble LTα3. Soluble LTα3 binds to TNFRI and TNFR II. Dashed line refers to weak interactions.
The development of the spleen is independent of LT, however, the microarchitecture of the splenic white pulp requires LTβR signaling for its development and maintenance in the adult mice. Treatment with LTβRIg, which blocks LT signaling, dissolves discrete B cell follicles, alters the marginal zone, prevents germinal center formation in the spleen, and impairs antibody production in response to immunization (Mackay et al., 1997).
Secondary Lymphoid Organs and Tolerance
Stromal cell function and tolerance
The appropriate structure of the SLO is integral in immune fate decisions, as following SLO entry naïve lymphocytes must decide to remain naïve, or become activated, anergic, or deleted. The presence (or absence) of antigen, co-stimulation, cell interactions, and/or chemokines/cytokines are all instructive in these decisions. Stromal cells construct intricate scaffolding within the SLO and provide architectural support. In addition to defining structure, these cells also contribute to lymphocyte trafficking, antigen presentation and cellular interactions (Mueller and Germain, 2009; Luther et al., 2011). FRC produce and secrete a fibrous antigen detected by the Erasmus University Rotterdam-thymic reticulum antibody 7 (ER-TR7; Steele et al., 2009) that forms a meshwork throughout the SLO (Van Vliet et al., 1986; Mueller and Germain, 2009). As elegantly demonstrated by Katakai et al. (2004b), the assembly of the ER-TR7 protein into this meshwork frame involves both FRC and lymphocyte collaboration and changes in response to antigen challenge, suggesting a structural plasticity that molds to the immune challenge at hand. To generate a fully formed meshwork, signaling through TNFR and LTβR via the inflammatory cytokines TNFα, LTα, and LTβ is required.
In the LN, lymphocytes leave the blood and enter via the HEV before flowing to the cortical ridge and cortical sinuses (Grigorova et al., 2010). As the HEV are the entry points for lymphocytes, the signals they present may be important factors in the genesis and shape of the immune response. Following inflammatory challenge, HEV express peripheral node addressin (PNAd), CCL21, ICAM-1, and CXCL9, attracting CD62L+, CCR7+, LFA-1+, and/or CXCR3+ T cells (Springer, 1994; Guarda et al., 2007). To attract B cells, HEV express CXCL13 resulting in cell arrest and LN entry (Kanemitsu et al., 2005). Even at this early phase of lymphocyte:LN interaction, lymphocytes encounter fate determining signals at the HEV. Activated T cells and antigen-presenting DC home to HEV following infection (Bajenoff et al., 2003), and we have shown both antigen and Treg seeding along HEV in tolerant mice following transplantation (Ochando et al., 2006). These findings suggest that these responding cells and/or antigen-presenting cells (APC) shape the migratory path of naïve lymphocytes into or through the LN and the subsequent immune response.
Following LN entry via HEV, or splenic entry via central arterioles in the marginal zone (Steiniger et al., 2001), lymphocytes encounter the FRC network, which dictates their path through the SLO (Mueller and Germain, 2009). The FRC are layered on top of and entwining the ER-TR7 scaffolding, forming a conduit system that transports molecules sampled from the lymph which has entered into the capsule and through the cortex (Katakai et al., 2004b). Lymphocytes (Gretz et al., 1997), soluble factors (Gretz et al., 1997), low molecular mass molecules (Gretz et al., 2000), and antigen (Pape et al., 2007) pass through these conduits, traveling through the reticular network from the SCS to the HEV. Conduit-transported antigen travels to varying locations within the LN. If antigen is transported to follicles, B cells take it up and become activated (Pape et al., 2007). If the antigen is acquired from the lymph, it can be presented by lymphatic vessel endothelial hyaluronan receptor 1 (LYVE-1)+, conduit-associated, DC (Gretz et al., 1997). These resident DC are associated with reticular fibers within the LN (Sixt et al., 2005), and can take up antigen within minutes following exposure (Itano and Jenkins, 2003). Interestingly, immature DC (such as resident DC) adhere much more readily than mature DC (such as recently migrated inflammatory DC) to the extracellular matrix (Sixt et al., 2005), and may be of exceptional significance in terms of tolerance, as immature DC would fail to provide the requisite co-stimulation signals needed for T cell activation and may be tolerogenic (Itano and Jenkins, 2003). Hence, if potentially graft-reactive T cells encounter these immature as opposed to mature DC, the T cells may become anergic rather than primed.
Fibroblastic reticular cells not only form the physical structure of conduits to allow the flow of antigen and cells in the LN (Link et al., 2011) and spleen (Nolte et al., 2003), they also affect chemotaxis of lymphocytes and DC by producing chemokines and expressing adhesion markers. Stromal cells lining the HEV express CCL19 (Forster et al., 2008) and CCL21 (Luther et al., 2000; Forster et al., 2008), which are indispensable for attracting T cells and DC via surface CCR7 engagement, both to the LN and then to T cell area (Forster et al., 2008). Further, stromal cells express the adhesion molecules ICAM-1 (Boscacci et al., 2010), ICAM-2 (Boscacci et al., 2010), VCAM-1 (Katakai et al., 2004a; Boscacci et al., 2010), and sialic acid (Kraal et al., 1994); adhesion molecules that direct lymphocytes expressing LFA-1 or α4β1 (Lo et al., 2003) to enter the HEV in the LN (Boscacci et al., 2010) or white pulp in the spleen (Kraal et al., 1994; Lo et al., 2003), and home to distinct microdomains.
The chemokines CXCL12 (Wright et al., 2002; Katakai et al., 2004a) and CX3CL1 (Katakai et al., 2004a) further direct lymphocyte homing to and within SLO (Boscacci et al., 2010). CXCL12, displayed on HEV (Okada et al., 2002), interacts with CXCR4 on central memory CD8+ T cells (Scimone et al., 2004) and B cells (Okada et al., 2002), attracting these cells to the LN independent of CCR7 expression (Scimone et al., 2004). CXCL12 expression may be of special importance in terms of transplant tolerance as it results in plasmacytoid DC (pDC) LN recruitment (Vanbervliet et al., 2003), and pDC presentation of donor antigen is integral to inducing tolerance to vascularized grafts (Ochando et al., 2006). Further, stromal cells guide activated follicular B cells to T cell zones and sites of T cell help following their upregulation of CCR7 (Reif et al., 2002).
Fibroblastic reticular cells may also deliver inhibitory or negative signals to lymphocytes, preventing their homing to and within SLO. FRC produce CCL2 (Katakai et al., 2004b), resulting in decreased lymphocyte homing and effector function by preventing activation of LFA-1, inhibiting ICAM-1 attachment, and subsequent adhesion steps and travel to SLO (Flaishon et al., 2008). FRC also express class I MHC and the inhibitory molecule programmed cell death ligand 1 (PD-L1), and are thus capable of delivering signals to lymphocytes moving across their surfaces, influencing tolerance versus immune activation by tuning the responsiveness or activation state of the lymphocytes (Mueller and Ernst, 2007). Thus, FRC not only help attract lymphocytes to the LN and to specific areas within the LN, they also help dictate the interactions of the cells inside the LN. Interactions in the LN can also dictate where the activated lymphocytes travel following LN activation. By expressing retinoic acid in the LN, stromal cells (Hammerschmidt et al., 2008), and resident DC (Iwata et al., 2004) induce α4β7 and CCR9 upregulation of activated T cells, resulting in their travel to the gut.
Fibroblastic reticular cells provide both cues and scaffolding, dictating the movements, and interactions of the diverse cell populations residing and migrating through the SLO. These observations suggest a model in which a transplant recipient encounters donor antigen, and as the recipient responds to these antigens lymphoid organ structure is remodeled. SLO structure affects both primary and subsequent immune responses to alloantigens, altering where antigen is presented, which cell types encounter antigen, the activation signals detected by these cell types, and the interactions of the various cell populations. Further levels of complexity include that SLO structure is influenced by previous inflammatory and antigen challenges, so the structure within the SLO of the transplant recipient may be as unique as a fingerprint. Hence, detection of these distinctive structural and environmental pressures may provide novel and unique targets for designing therapeutic protocols.
Spleen
The spleen’s role in graft rejection is different between models such as vascularized or non-vascularized organ transplantation models. In some vascularized models, it contributes to graft rejection; however, in non-vascularized transplantation models, LN but not the spleen are essential for the rejection of skin allografts. Rather, the spleen appears to enhance graft prolongation (Souther et al., 1974; Streilein and Wiesner, 1977; Coons and Goldberg, 1978; Lakkis et al., 2000). The rejection of skin allografts depends on the presence of LN, whereas the rejection of vascularized heart allografts occurs in the presence of either the spleen or LN (Lakkis et al., 2000). On the contrary, Chosa et al. (2007) showed that the spleen plays an important role in maintaining tolerance after removal of the vascularized heart graft. The APC migration route might explain the difference between models. In vascularized transplantation models, passenger leukocytes, including DC, are thought to migrate from the vascularized graft into the recipient spleen, where they activate T cells and cause rejection (Larsen et al., 1990a,b; Saiki et al., 2001). In the case of tolerance maintenance, naïve DC migrate to a tolerant graft and become regulatory or immature DC, these DC may then migrate to the spleen where they generate Treg (Chosa et al., 2007). CD4+ T cells from spleen of unresponsive cardiac allograft transplant recipients showed increased IL-10 and decreased IL-4 and IFNγ (DePaz et al., 2003).
In non-vascularized grafts, DC migrate from the graft to the regional draining LN. Activated DC are trapped in the LN where they generate effector T cells from naïve T cells. Regulatory or immature DC may pass through the LN, reach the spleen, and generate Treg. Several studies indicate that the expression pattern of chemokines and cytokine receptors between LN and spleen is different, explaining the different roles in transplant models (Tang and Cyster, 1999; Alferink et al., 2003).
Suppression and regulatory mechanisms of the spleen are shown in several studies. A subset of splenic red pulp F4/80hiMac-1low macrophages, whose differentiation is regulated by CSF-1, regulates CD4+ T cell responses using TGFβ and IL-10 and inducing differentiation of Foxp3+ Treg (Kurotaki et al., 2011). In a vascularized transplant model, Li et al. (2010) showed that CD8+ Treg and pDC in the allograft and spleen induce tolerance. In mice, a rare cell type located in splenic red pulp with phenotypic attributes of DC (CD11c, CD8, CD80/86, MHCII) are uniquely competent to mediate T cell suppression via indoleamine oxidase after in vivo treatment with TLR9 ligands (Mellor et al., 2005; Johnson et al., 2010). NK cells have been shown to contribute to both graft rejection and tolerance both in cardiac and skin transplant models (Heidecke et al., 1985; Rabinovich et al., 2000; Beilke et al., 2005; Kroemer et al., 2008; Zecher et al., 2010). Thus, activated, regulatory, or immature DC and NK cells trapped in the spleen interact with other innate and adaptive immune cells, generate effector T cells and Treg, which contribute either to rejection or acceptance depending on the model (Murphy et al., 2011). The possibility that the spleen has the two functions of graft rejection and graft prolongation has been demonstrated in spleen transplantation models (Dor et al., 2003).
Lymph node
In addition to dictating movement of lymphocytes within the LN by supplying both structure and directional cues, stromal cells may also dictate lymphocyte survival. Stromal cells affect T cell viability, and correct positioning within the LN is integral to T cell survival (Table 1). Highlighting this importance is the effect of HIV pathogenesis on CD4+ T cell survival. Following HIV infection, the LN T cell zones become areas of significant pathology as viral replication occurs almost exclusively in these areas (Schacker et al., 2001). Innate immune cells attack these areas in attempts at viral eradication, resulting in scarring, and collagen deposition within LN niches (Estes et al., 2008), along with structural dissolution within the LN (Biberfeld et al., 1985). This scaring inhibits T cell access to FRC-produced IL-7, resulting in the depletion of T cells via apoptosis (Zeng et al., 2011). As T cells are depleted, the source of LTβ production is decreased, leading to further disruption of the FRC network (Zeng et al., 2011). The interdependence between FRC and T cells for the maintenance of LN structure demonstrates the balance among activation, ignorance, or tolerance that is dependent upon the microdomains within the LN structure.
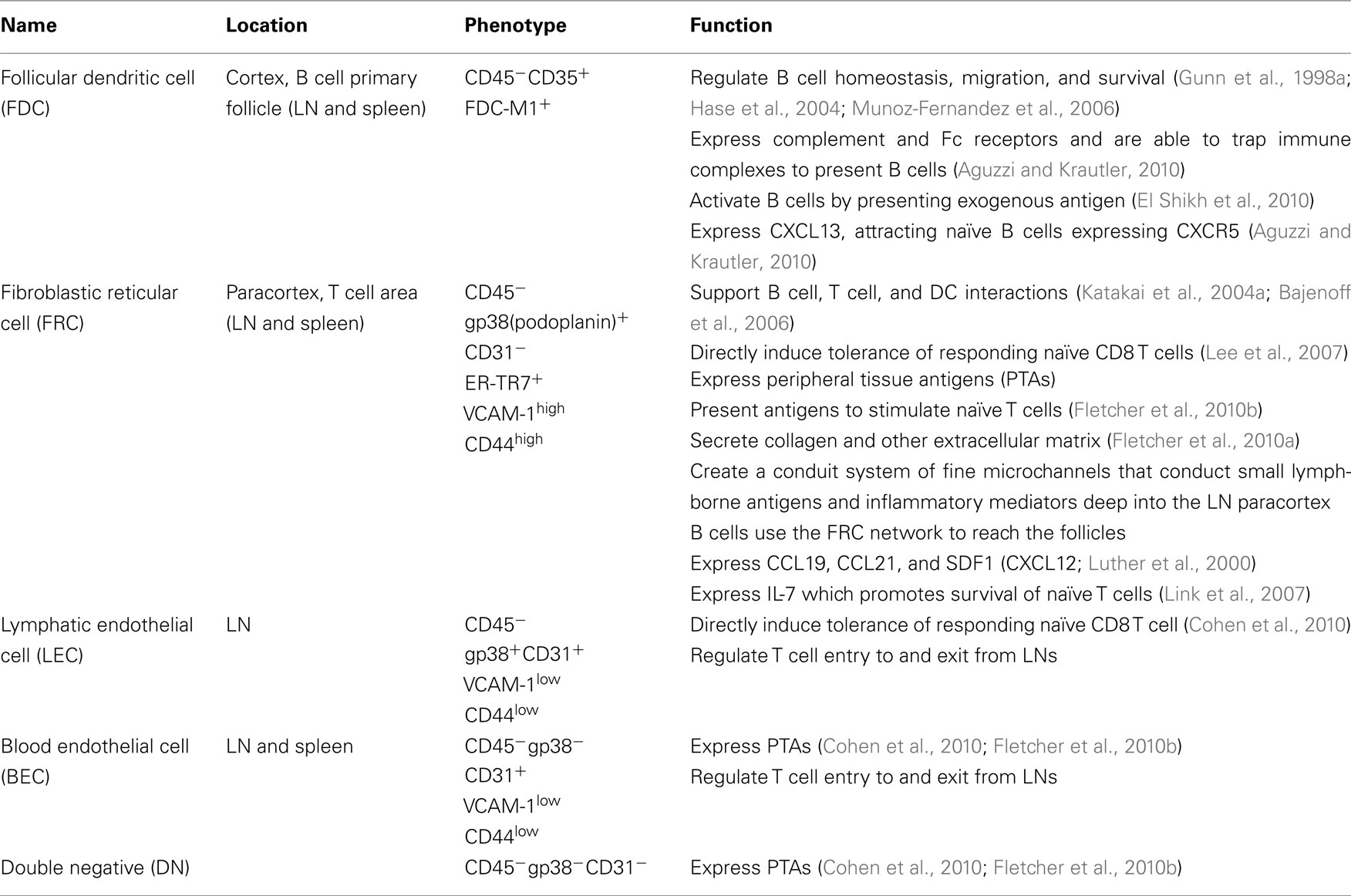
Table 1. Stromal cells in SLO.
Stromal cells also participate in peripheral tolerance by expressing autoimmune regulator gene (Aire). Aire expression is most commonly associated with thymic expression of tissue-specific antigens; antigens that represent autologous proteins not typically present within the thymus. By presenting these antigens during thymic selection, potentially autoimmune T cells are deleted (Anderson et al., 2002). More recently, a role for Aire in peripheral tolerance has been demonstrated (Metzger and Anderson, 2011). Under steady-state conditions, tissue resident DC in the periphery pick up peripheral tissue-restricted antigen (PTA), and migrate to the LN (Waithman et al., 2007). There, Aire-expressing gp38+CD31+ lymphatic endothelial (Cohen et al., 2010) and gp38+CD31− stromal cells (Gardner et al., 2008) express these antigens. These stromal cells express a variety of antigen processing and presentation genes, including MHC II, having phenotypic similarities to DC (Gardner et al., 2008). Hence, it is possible that allograft antigens, entering the LN either via lymphatic conduits or tissue DC, may be picked up by endothelial and/or stromal cells and presented to potentially graft-reactive lymphocytes, resulting in tolerance instead of activation. Indeed, the Aire-expressing stromal cells are preferentially located along boundaries of T and B cell interactions within the LN (Gardner et al., 2008), and are sufficient to drive both proliferation and deletion of self-reactive CD8+ T cells (Lee et al., 2007). Significantly, these stromal cells also express PD-L1, suggestive of a potential mechanism for peripheral cell deletion (Gardner et al., 2008). This CD8+ T cell deletion strategy is mirrored in chronic viral infection; LCMV upregulates PD-L1 in FRC, deleting virus-specific potential effector cells and allowing for viral persistence (Mueller et al., 2007).
Different subsets of stromal cells express Aire and PTA to varying degrees (Cohen et al., 2010). FRC express PTA under steady-state conditions, and down-modulate their antigen expression following an inflammatory stimulus (Fletcher et al., 2010a). In contrast, other LN stromal cell subsets increase their PTA expression in response to bystander inflammation (Fletcher et al., 2010a). Thus, specific subsets of stromal cells may be responsible for specific antigen tolerance. These findings may be exploited in transplant tolerance, as presentation of donor antigen in a non-inflammatory environment may engage several different tolerance mechanisms. Donor antigen could potentially be targeted to stromal cells for presentation, or therapeutic interventions may be able to exploit naturally occurring peripheral tolerance mechanisms.
Blood Vascular Endothelium
Dendritic cell and lymphocytes enter the LN through HEV and afferent lymphatics (von Andrian and Mempel, 2003). During this process, lymphatics and HEV not only play a role as a route for cell trafficking but also interact with the migrating immune cells to influence their immunologic properties and thereby immune responses. HEV are specialized postcapillary venules found in LN, where the lymphocytes circulating in the blood enter the LN by diapedesis (Girard and Springer, 1995). In order to facilitate the migration of lymphocytes, endothelial cells in HEV secrete CCL21, the CCR7 ligand, and express adhesion molecules such as PNAd (ligand for CD62L) and ICAM-1 (ligand for LFA-1; Campbell et al., 1998; Gunn et al., 1998b; Forster et al., 2008). An additional chemokine CX3CL1, expressed on inflamed endothelial cells, interacts with its receptor CX3CR1 on activated cytotoxic lymphocytes, functioning as a cell adhesion molecule (Umehara et al., 2004). Another CCR7 ligand, CCL19, is produced by FRC in the T cell area and can be transported to the luminal surface of HEV to mediate T cell recruitment (Baekkevold et al., 2001; Link et al., 2007). CCR7 signaling is reported to induce cell cycle arrest in T cell receptor (TCR)-stimulated T cells and inhibit T cell proliferation (Ziegler et al., 2007). CCR7 signaling acts differently on DC. CCL19 and CCL21 induce DC to become mature; produce proinflammatory cytokines such as IL-1β, IL-12, and TNFα; and drive Th1 responses (Marsland et al., 2005). Thus, by engaging CCR7 and directing the migration of T cells and DC from blood into the LN, HEV induce significant changes in the maturation, differentiation, and responses of the migrating leukocytes.
Whether HEV enhance immune regulatory capacity of Treg is not certain. In an islet allograft model, adoptively transferred Treg that enter LN through HEV do not acquire an activated phenotype to suppress the alloimmune response (Zhang et al., 2009). On the other hand, activated endothelial cells from other sources such as thoracic aorta and lung induce and activate Treg using the PD-1/PD-L1 interaction and IL-10 (Krupnick et al., 2005; Bedke et al., 2010). Under inflammatory conditions, human endothelial cells amplify Treg in a contact-ICAM-1-dependent mechanism (Taflin et al., 2011). Under tolerogenic conditions, alloantigen-presenting pDC also migrate through HEV when homing to LN, to mediate Treg development and tolerance to vascularized cardiac allografts (Ochando et al., 2006).
Lymphatic Endothelium
Lymphatics are lined with LEC expressing LYVE-1. Lymphatics are found throughout the LN and have different functions and phenotypes in different regions. DC and T cells in peripheral tissues enter LN through afferent lymphatics, which typically end in the SCS, a hollow space below the fibrous capsule of the LN (Randolph et al., 2005). Thereafter, DC and T cells use different routes for entry into the LN parenchyma. By injecting cells directly into afferent lymph vessels, Braun et al. (2011) showed that DC transmigrate through the floor of the SCS on the afferent side in CCR7-dependent fashion to enter the LN parenchyma. On the other hand, T cells enter the LN parenchyma mainly from peripheral medullary sinuses and this is not CCR7-dependent. However, CCR7 signals are absolutely required for the directional migration of both DC and T cells into the T cell zone. Interestingly, when DC are injected before T cells, T cells are able to transmigrate through the SCS floor. This suggests that DC transmigrating through the SCS induce changes in SCS morphology. In the steady state, Treg also enter the LN via afferent lymphatics, which is increased in inflammatory conditions (Tomura et al., 2010).
When leaving the LN, lymphocytes in the parenchyma enter medullary networks of lymphatic sinuses and from there the efferent lymphatics (von Andrian and Mempel, 2003). Lymphatics associated with the egress of lymphocytes are also found adjacent to HEV and B cell follicles in cortex (Sinha et al., 2009; Grigorova et al., 2010). These cortical sinuses extend to medullary sinuses. Emigration of lymphocytes into the efferent lymphatics for egress from the LN requires sphingosine-1-phosphate (S1P) interaction with the S1P receptor 1 (S1P1) on lymphocytes, and LEC are the main producers of S1P required for LN egress (Matloubian et al., 2004; Pham et al., 2010). In some immune responses and inflammatory conditions, lymphocytes down-regulate S1P1 expression, which blocks the egress of lymphocytes from the LN (Matloubian et al., 2004). Down-regulation of S1P1 by internalization is one of the major mechanisms of immune modulation by the drug FTY720 (Brinkmann et al., 2004).
Lymphatic vessels play an important role in immune tolerance. In an islet allograft model, Treg that are adoptively transferred and migrate from the graft to LN via afferent lymphatics prevent graft rejection. However, Treg which enter LN via HEV do not (Zhang et al., 2009). During a cutaneous immune response, Treg that move from skin to LN via afferent lymphatics are more potent in immune suppressive capacity than LN-resident Treg (Tomura et al., 2010). These findings suggest that migration through lymphatics may enhance the immunosuppressive capacity of Treg. Alternatively, it is possible that Treg are primed and activated in the peripheral tissues before migrating to LN. The chemokines CCL19 and CCL21, which are secreted by LEC, inhibit T cell proliferation (Ziegler et al., 2007). LN-resident LEC mediate peripheral tolerance by expressing and presenting multiple peripheral tissue antigens to CD8+ T cells and inducing their deletion (Cohen et al., 2010). Inflamed, TNFα-stimulated LEC reduce the expression of the co-stimulatory molecule CD86 by DC, and suppress the ability of DC to induce T cell proliferation via a Mac-1 (CD11b/CD18)/ICAM-1 (CD54)-dependent mechanism (Podgrabinska et al., 2009). Thus, LEC can engage multiple mechanisms to negatively regulate immune and inflammatory responses.
Vascular endothelial growth factors (VEGF), especially VEGF-A and VEGF-C, are involved in LEC proliferation and lymphangiogenesis (Shibuya and Claesson-Welsh, 2006; Tammela and Alitalo, 2010). Among VEGF receptors (VEGFR), VEGFR-2 is expressed both on blood vascular endothelium and lymphatic endothelium, but VEGFR-3 is specific for lymphatic endothelium. VEGF-A promotes lymphatic vessel formation via signaling through VEGFR-2 and the effect of VEGF-C is mediated by VEGFR-3. Chronic inflammatory conditions are associated with lymphangiogenesis in the draining LN, which enhances the migration of DC to LN and DC–T cell interactions. VEGF-C is induced in response to proinflammatory cytokines such as TNFα and IL-1β in several cells, including macrophages and granulocytes (Tammela and Alitalo, 2010). In inflammatory conditions, entry of these immune cells into the LN is markedly increased, which may be associated with inflammatory lymphangiogenesis in LN. B cells in inflamed LN produce VEGF-A and promote lymphangiogenesis (Angeli et al., 2006). Chronically inflamed tissue also induces lymphangiogenesis in draining LN by producing VEGF-A, and this is independent of B cells (Halin et al., 2007). On the other hand, T cells negatively regulate LN lymphatic vessel formation through IFNγ (Kataru et al., 2011). TGFβ also inhibits the proliferation and migration of cultured human LEC as well as lymphangiogenesis (Oka et al., 2008). Together, these reports suggest a highly complex network of tissue and LN interactions that regulate lymphangiogenesis.
In an islet allograft model, anti-VEGFR-3 mAb inhibits lymphangiogenesis and prolongs allograft survival, suggesting that inhibition of lymphangiogenesis may prevent immunity and inflammation (Yin et al., 2011). However, in an arthritis model of TNF-transgenic mice and a chronic cutaneous inflammation model, anti-VEGFR-3 mAb increases inflammation in joints and skin despite suppressing lymphangiogenesis (Guo et al., 2009; Huggenberger et al., 2010). In contrast to anti-VEGFR-3 mAb, anti-VEGFR-2 mAb suppresses both lymphangiogenesis and inflammation in these two models. It is suggested that VEGFR-2 and VEGFR-3 signaling pathways have different mechanisms for reducing lymphangiogenesis, and that increased inflammation after specific blockade of VEGFR-3 is a result of improper lymphatic drainage with failure to resolve inflammatory infiltrates.
Lymph node lymphatics and HEV seem to be in synchrony. B cell-derived VEGF-A promotes HEV expansion as well as lymphangiogenesis in LN (Shrestha et al., 2010). Both LN lymphatics and HEV express LTβR, and lymphangiogenesis is accompanied by upregulation of PNAd and LTβR by HEV, and is inhibited by LTβRIg treatment (Liao and Ruddle, 2006). These findings suggest that the two vascular systems engage in cross-talk through B cells and LTβR (Table 1).
T Cells
When T cells enter LN through HEV or lymphatics, they are under the influence of chemokines secreted by LEC or vascular endothelial cells such as CCL19 and CCL21, which can inhibit the proliferation of T cells (Ziegler et al., 2007). Contact with endothelial cells also can affect the immunologic properties of migrating T cells. On the other hand, T cells can influence lymphatics and negatively regulate LN lymphatic vessel formation through IFNγ (Kataru et al., 2011).
T cell homing to LN plays a critical role in tolerance to alloantigen because Treg develop and are required within the LN during tolerance induction. If T cell homing to LN is inhibited by anti-L-selectin (CD62L) mAb, cardiac allograft survival is prevented despite a tolerogenic regimen of anti-CD40L mAb plus donor-specific transfusion (Ochando et al., 2005). Only the CD62L+ subpopulation of Treg, which have the LN-homing adhesion molecule, are reported to protect from lethal GVHD in allogeneic bone marrow transplantation model and delay adoptive transfer of diabetes (Szanya et al., 2002; Ermann et al., 2005). Follicular Treg are present in LN, limit T follicular helper cell and germinal center B cell numbers, and suppress germinal center reactions (Chung et al., 2011; Linterman et al., 2011). In LN, Treg directly interact with antigen-bearing DC by forming long-lasting conjugates to inhibit T cell priming and activation by DC (Tang et al., 2006). In the peripheral tissue, Treg inhibit DC migration to LN in a TGFβ and IL-10 dependent fashion, which prevents priming of effector T cells by DC in the LN (Zhang et al., 2009). These findings suggest that LN are a major place where Treg exert their immune modulatory actions in various ways. Treg limit the access of immune cells to LN and inhibit DC–T cell interactions in the LN for the regulation of immune reactivity (Table 2).

Table 2. Treg in lymphoid organs.
Primary Lymphoid Organs
Thymus
The thymus is a lymphoid organ that is critical for tolerance. It is not only the site for eliminating self-reactive T cells through negative selection, but also for controlling self-reactive T cells through CD4+Foxp3+ regulatory T cell (Treg) development. Delayed generation of Treg by thymectomy at day 3, but not day 7, results in autoimmune disease development (Fontenot et al., 2005). Treg development is observed only when the antigen is expressed in the thymus (Jordan et al., 2001). Foxp3 is induced during very early thymic developmental events before TCR rearrangement (Pennington et al., 2006), suggesting a major role in determining T cell fate prior to or coincident with TCR engagement. These reports all demonstrate the critical role of the thymus for maintaining a protective suppressor cell population.
A number of observations show that Treg develop in the medulla: expression of Aire within medullary thymic epithelial cells (Aschenbrenner et al., 2007); the overwhelming majority of Foxp3+ cells are found within the medulla (Fontenot et al., 2005); and thymic stromal-derived lymphopoietin (TSLP) produced in the medullary region of the human thymus is critical for Treg development (Jiang et al., 2006). However, thymic DC also make TSLP and these cells may be found in the cortex (Watanabe et al., 2005); mice expressing MHC II only in the cortex are able to produce natural Treg (nTreg; Bensinger et al., 2001); and when thymic migration from the cortex to the medulla was blocked by administering pertussis toxin (Liston et al., 2008), or through CCR7 deficiency (Kurobe et al., 2006), CD4+Foxp3+ Treg accumulate within the cortex. Thus, both the thymic medulla and cortex participate in nTreg development. In a study using TCR transgenic models naturally devoid of Foxp3+ cells, it was observed that de novo generation of Treg occurred intrathymically under non-inflammatory conditions of antigen encounter and was essential for robust tolerance induction (Zelenay et al., 2010).
Foxp3 expression does not commence until day 3 in neonates, suggesting that only organized thymic architecture provides proper co-stimulatory signals in the early thymus. CD28, IL-2R, TSLP receptor, CD154, glucocorticoid-induced TNF receptor, and Stat5 signals are all implicated in the development and lineage commitment of thymus-derived nTreg (Bettini and Vignali, 2010). Although mice deficient in TGFβ or TGFβ receptor II (TGFβRII) have normal numbers of Treg in adult thymus, the numbers of CD4+CD25+Foxp3+ thymocytes are greatly reduced in young mice (Liu et al., 2008). Further, CD4+CD25+Foxp3+ thymocytes are completely lost in mice lacking both TGFβRI and IL-2, indicating TGFβR signaling is involved in development and maintenance of nTreg.
Various experimental models demonstrate that the thymus is required for Treg development and consequent tolerance induction to alloantigens. In a rat model using immature myeloid DC (imDC) primed with immune-dominant allopeptide in vitro, DC re-circulate through the recipient thymus and result in acquired graft-as-self-tolerance (Gopinathan et al., 2001; Oluwole et al., 2001). CD4+CD25+Foxp3+ anti-donor Treg development is dependent on the presence of the thymus for at least 2–3 weeks after imDC injection, and early thymectomy abolishes tolerance (Oluwole et al., 2003). Trafficking of donor alloantigen to the thymus or inoculating the thymus directly with allopeptides induces tolerance in large animal models, and thymectomy in recipients prevents tolerance induction (Griesemer et al., 2010). Transplanting porcine thymus tissue to mice leads to the development of donor-specific tolerance in vitro (Lee et al., 1994), and indefinite donor-matched porcine skin graft survival (Zhao et al., 1996). Transplanting vascularized porcine thymus tissue to baboons is associated with donor-specific T cell unresponsiveness (Barth et al., 2003), and the presence of thymic chimerism and donor bone marrow engraftment correlate precisely with tolerance to subsequent solid organ transplants (Horner et al., 2006). Overall, these reports demonstrate that both T cell development in allogeneic or xenogeneic thymus can induce tolerance, and that DC can traffic to the thymus to present or cross-present alloantigens for tolerance induction (Table 2).
Bone Marrow
Bone marrow is an essential part of the immature and mature lymphocyte recirculation network, and it harbors mature CD4+CD25+ Treg and serves as a Treg reservoir. Studies show that CXCR4/CXCL12 signals play an important role in regulating Treg trafficking from bone marrow and in maintaining homeostatic levels of Treg in the periphery. G-CSF treatment decreases bone marrow CXCL12 expression, and results in Treg mobilization from bone marrow into the periphery, and is consistent with the low prevalence of acute GVHD and the improvement in autoimmune diseases following G-CSF treatment (Zou et al., 2004). In a ret transgenic mouse spontaneous melanoma model, significantly higher numbers of Treg are found in skin tumors and metastatic LN at early stages of melanoma progression, compared with more advanced stages, and inversely correlate with Treg numbers in the bone marrow, suggesting trafficking from bone marrow to melanoma lesions. Elevated CCR4 expression is observed on Treg, while higher production of its ligand CCL2 is observed in tumor lesions, indicating CCR4/CCL2 signals play a role in Treg mobilization (Kimpfler et al., 2009). Recent studies show that the hematopoietic stem/progenitor cell (HSPC) niche is immune-privileged in the bone marrow. Persistence and survival of both allogeneic and syngeneic HSPCs are observed after 30 days in non-irradiated recipient mice without immunosuppression. High-resolution in vivo imaging shows that HSPC are colocalized with Treg and accumulate on the endosteal surface in the calvarial and trabecular bone marrow, Treg actively participate in protection of allo-HSPC, and depleting FoxP3 regulatory Treg cells results in allogeneic HSPC destruction (Fujisaki et al., 2011).
The bone marrow is the primary site for B cell maturation. Naive B cells then migrate to SLO, become plasmablasts upon antigenic stimulation in antigen-activated T cell areas, secrete low-affinity antibody and eventually undergo apoptosis. Some activated B cells enter into the long-lived memory compartment as either memory B cells or long-lived plasma cells (PC). Long-lived PC remain in either the LN or spleen, but most home to and reside in the bone marrow. Long-lived PC in the marrow are a major source of persistent donor-specific alloantibody (Stegall et al., 2010). Long-lived PC are sensitive to proteasome inhibition including bortezomib, but are not affected by rituximab, IVIG, or thymoglobulin (Perry et al., 2009). Although PC also may exist in SLO, the contribution of PC from areas other than the bone marrow to alloantibody production is not clear. These documentations all point to the bone marrow as a major site of immune regulation for responses to alloantigens, and that real therapeutic strategies must account for both Treg and PC in the bone marrow (Table 3).
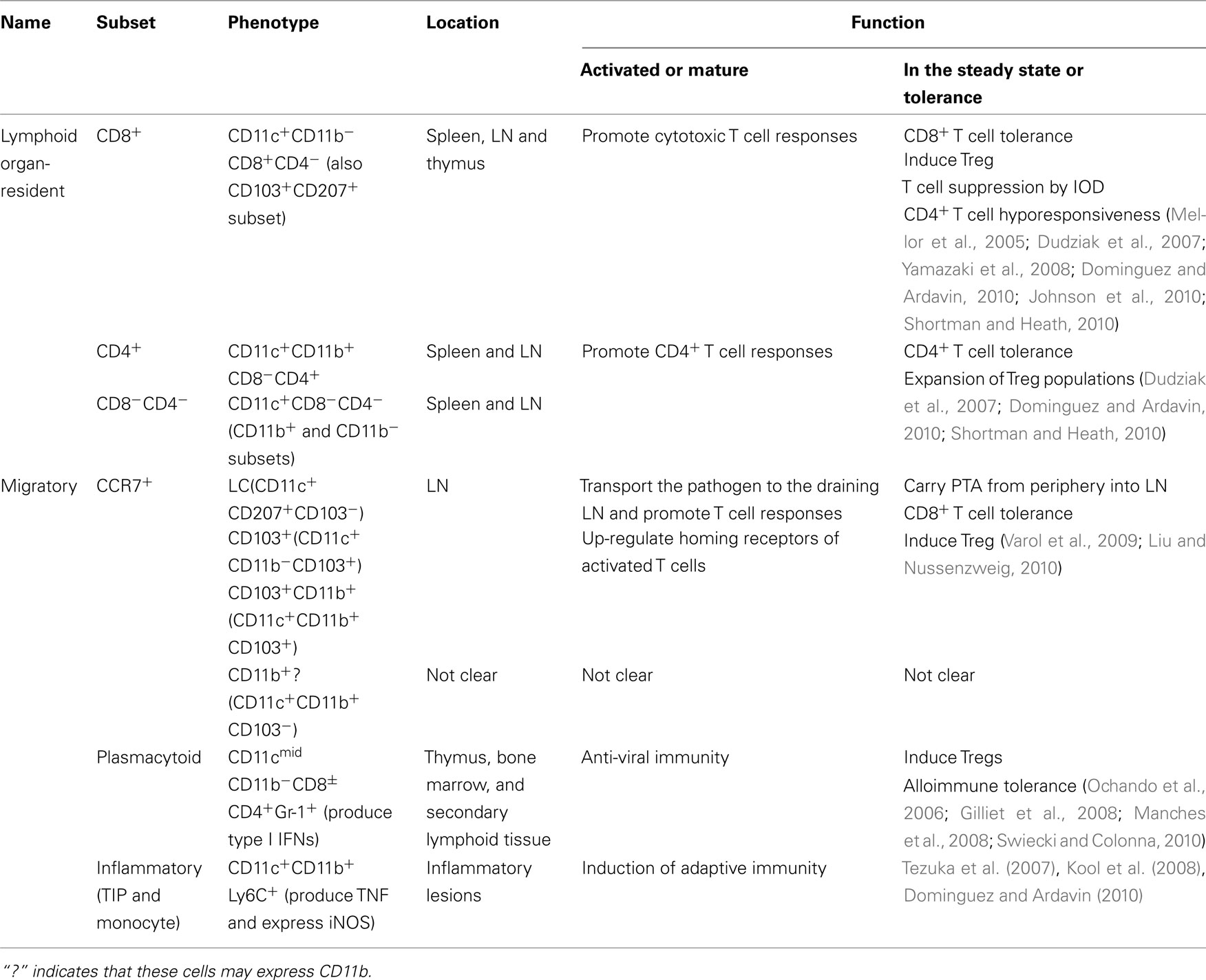
Table 3. Dendritic cell function in lymphoid organs.
Tertiary Lymphoid Organs
Ectopic or tertiary lymphoid organs (TLO) are often induced at sites of chronic infection or inflammation in peripheral non-lymphoid organs. These tissues are architecturally similar to SLO, with separate B and T cell areas, specialized populations of DC, well-differentiated stromal cells, and HEV (Carragher et al., 2008). It seems likely that similar signaling mechanisms that are responsible for SLO ontogeny are involved in TLO formation (Motallebzadeh et al., 2008). Like SLO, TLO are formed in a highly regulated manner via production of homeostatic chemokines (CXCL13 and CCL19/CCL21), and in response to signaling from the heterotrimer lymphotoxin (LT) α1β2 acting on the LTβR on stromal organizer cells (Drayton et al., 2006; van de Pavert and Mebius, 2010). The expression of LTα, LTβ, and LIGHT all contribute to the formation of TLO via signaling through TNFR1 and LTβR (Lee et al., 2006). Although the organogenesis of LN clearly requires LTi cells, whether LTi cells are required for formation of TLO follicles is less clear (Carragher et al., 2008). Even with deletion of the inhibitor of differentiation two gene, essential for generation of CD3−CD4+ LTi cells and development of SLO, TLO still form in the thyroid (Marinkovic et al., 2006).
Tertiary lymphoid organs have been described in a variety of autoimmune diseases including gastritis, thyroiditis, and systemic lupus erythematosus (Carragher et al., 2008). The formation of TLO correlates with the development of diabetes, whereas blocking their formation prevents the development of diabetes (Wu et al., 2001; Lee et al., 2006; Penaranda et al., 2010). The chronic phase of the immune response to an allograft is in many ways similar to the responses that typify relapsing autoimmunity. Autoimmune responses are characterized by immune environments in which target antigens persist, and the inability to eradicate alloantigens may provide a similar stimulus for formation of TLO within the graft. After organ transplantation, there are continuous low levels of dynamic interactions between donor alloantigens and the recipient immune system, a situation similar to chronic inflammation. The positioning of lymphoid tissue within an inflamed allograft may thus result in more aggressive effector lymphocyte responses that focuses on locally presented alloantigens (Motallebzadeh et al., 2008). For example, TLO or PNAd+ HEV without organized lymphoid accumulation are observed in murine heart transplants and in association with chronic allograft rejection, but to a much lesser extent with acute rejection (Baddoura et al., 2005). Germinal center-like structures are present in chronically rejected human allografts (Motallebzadeh et al., 2008). Some reports have shown that chronically rejected allografts are simultaneously the target and the site of production of alloantibodies (Smith et al., 2002; Kerjaschki et al., 2004; Thaunat et al., 2005; Thaunat and Nicoletti, 2008). TLO occur in rat donor allografts latently infected with cytomegalovirus (CMV), and in part mediate ganciclovir-insensitive rejection by providing a scaffold for immune activation (Orloff et al., 2011). Similarly, pre-transplant human CMV infection is associated with the acceleration of renal transplant vascular sclerosis and chronic allograft rejection (Fitzgerald et al., 2004). TLO within skin allografts are able to support effective alloresponses, leading to rejection, and development of a memory response (Nasr et al., 2007). Thus, it appears that the generation of TLO is associated with the chronic phase of destructive auto- or alloimmune responses.
The ectopic accumulation of lymphoid cells has been considered to signify destructive inflammation that is accompanied by tissue damage (Drayton et al., 2006). However, there are also examples in which TLO appear to contribute to local protective immune responses. A high proportion of Treg are detected within the TLO in gastric mucosa of mice with autoimmune gastritis and atherosclerotic aortic media of apoE−/− mice (Katakai et al., 2006; Grabner et al., 2009). T2-6AB transgenic mice, in which T cells express a T cell receptor (TCR) specific for the H+/K+-ATPase α subunit, spontaneously develop TLO, although autoimmune responses are clearly limited. There is locally restricted T cell activation and Th2 skewing of the self-reactive T cells, as well as the accumulation of Treg in the target organ (Katakai et al., 2006). Many tumors manipulate the immune environment to escape immune surveillance. By secretion of CCL21, melanoma tumor cells attract LTi cells, which are needed for the formation of TLO-like structures within the tumor. Knockdown of endogenous CCL21 expression in tumor cells induces antigen-specific immunity and inhibits tumor growth. Hence, in melanoma tumors, the TLO provide a tolerogenic environment by recruitment of regulatory leukocyte populations (Shields et al., 2010). Thus, depending on the type of immune responses, generation of TLO at sites of inflammation can lead to detrimental or pathological immune responses, or may help resolve local inflammation or infection (Motallebzadeh et al., 2008).
Conclusion and Perspectives
While great progress has been made in understanding molecules, signaling pathways, and cells important for alloimmunity, and how to manipulate, interfere with, and suppress immune cell activation, migration, and effectors functions to benefit transplantation, evidence has emerged that various lymphoid organs, their anatomic structures, and particular microenvironments that lymphocytes encounter or reside in during the course of the alloresponse are also critical in determining the final outcome of graft acceptance and transplant tolerance. The appropriate tissue architecture may decide whether immune cells remain naïve, or become activated, anergic, or deleted by affecting antigen presentation, adhesion molecule expression, co-stimulatory signal activation, cytokine and chemokine production, thus affect regulatory and effector cell differentiation, trafficking and effector activities. In order to further overcome barriers to transplant tolerance, precise models and investigations on the arrangement of cells and molecules in lymphoid structures and anatomic pathways are required. For example, how LTi/LTo interactions, LT signal cascades, stromal cells, and specific microdomains affect immune responses and transplant tolerance induction and maintenance will all be productive areas for investigation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
Aire, autoimmune regulator; APC, antigen-presenting cell; DC, dendritic cell; ER-TR7, Erasmus University Rotterdam- thymic reticulum antibody 7; FDC, follicular dendritic cell; FRC, fibroblastic reticular cell; FTreg, follicular regulatory T cells; GITR, glucocorticoid-induced tumor necrosis factor receptor; HEV, high endothelial venules; HSPC, hematopoietic stem/progenitor cells; HVEM, herpes simplex virus (HSV) glycoprotein D for HSV entry mediator; imDC, immature myeloid DC; LEC, lymphatic endothelial cells; LN, lymph node; LT, lymphotoxin; LTβR, Lymphotoxin β receptor; LTi, lymphoid tissue inducer; LTo, lymphoid tissue organizer; LYVE-1, lymphatic vessel endothelial hyaluronan receptor 1; nTreg, natural Treg; PALS, periarteiolar lymphoid sheath; pDC, plasmacytoid DC; PD-L1, programmed death ligand 1; PNAd, peripheral lymph node addressin; PP, Peyer’s patches; PTA, peripheral tissue-restricted antigen; SCS, subcapsular sinus; SIV, Simian immunodeficiency virus; SLO, secondary lymphoid organs; TCR, T cell receptor; TFH, follicular helper T cells; TGFβR, TGFβ receptor; TLO, tertiary lymphoid organs; TNF, tumor necrosis factor; Treg, regulatory T cells; TSLP, thymic stromal lymphopoietin; VEGF, vascular endothelial growth factor.
References
Adachi, S., Yoshida, H., Honda, K., Maki, K., Saijo, K., Ikuta, K., Saito, T., and Nishikawa, S. I. (1998). Essential role of IL-7 receptor alpha in the formation of Peyer’s patch anlage. Int. Immunol. 10, 1–6.
Aguzzi, A., and Krautler, N. J. (2010). Characterizing follicular dendritic cells: a progress report. Eur. J. Immunol. 40, 2134–2138.
Alferink, J., Lieberam, I., Reindl, W., Behrens, A., Weiss, S., Huser, N., Gerauer, K., Ross, R., Reske-Kunz, A. B., Ahmad-Nejad, P., Wagner, H., and Forster, I. (2003). Compartmentalized production of CCL17 in vivo: strong inducibility in peripheral dendritic cells contrasts selective absence from the spleen. J. Exp. Med. 197, 585–599.
Anderson, M. S., Venanzi, E. S., Klein, L., Chen, Z., Berzins, S. P., Turley, S. J., Von Boehmer, H., Bronson, R., Dierich, A., Benoist, C., and Mathis, D. (2002). Projection of an immunological self shadow within the thymus by the aire protein. Science 298, 1395–1401.
Angeli, V., Ginhoux, F., Llodra, J., Quemeneur, L., Frenette, P. S., Skobe, M., Jessberger, R., Merad, M., and Randolph, G. J. (2006). B cell-driven lymphangiogenesis in inflamed lymph nodes enhances dendritic cell mobilization. Immunity 24, 203–215.
Aschenbrenner, K., D’cruz, L. M., Vollmann, E. H., Hinterberger, M., Emmerich, J., Swee, L. K., Rolink, A., and Klein, L. (2007). Selection of Foxp3+ regulatory T cells specific for self antigen expressed and presented by Aire+ medullary thymic epithelial cells. Nat. Immunol. 8, 351–358.
Baddoura, F. K., Nasr, I. W., Wrobel, B., Li, Q., Ruddle, N. H., and Lakkis, F. G. (2005). Lymphoid neogenesis in murine cardiac allografts undergoing chronic rejection. Am. J. Transplant. 5, 510–516.
Baekkevold, E. S., Yamanaka, T., Palframan, R. T., Carlsen, H. S., Reinholt, F. P., Von Andrian, U. H., Brandtzaeg, P., and Haraldsen, G. (2001). The CCR7 ligand elc (CCL19) is transcytosed in high endothelial venules and mediates T cell recruitment. J. Exp. Med. 193, 1105–1112.
Bajenoff, M., Egen, J. G., Koo, L. Y., Laugier, J. P., Brau, F., Glaichenhaus, N., and Germain, R. N. (2006). Stromal cell networks regulate lymphocyte entry, migration, and territoriality in lymph nodes. Immunity 25, 989–1001.
Bajenoff, M., Granjeaud, S., and Guerder, S. (2003). The strategy of T cell antigen-presenting cell encounter in antigen-draining lymph nodes revealed by imaging of initial T cell activation. J. Exp. Med. 198, 715–724.
Barth, R. N., Yamamoto, S., Lamattina, J. C., Kumagai, N., Kitamura, H., Vagefi, P. A., Awwad, M., Colvin, R. B., Cooper, D. K., Sykes, M., Sachs, D. H., and Yamada, K. (2003). Xenogeneic thymokidney and thymic tissue transplantation in a pig-to-baboon model: I. Evidence for pig-specific T-cell unresponsiveness. Transplantation 75, 1615–1624.
Bedke, T., Pretsch, L., Karakhanova, S., Enk, A. H., and Mahnke, K. (2010). Endothelial cells augment the suppressive function of CD4+ CD25+ Foxp3+ regulatory T cells: involvement of programmed death-1 and IL-10. J. Immunol. 184, 5562–5570.
Beilke, J. N., Kuhl, N. R., Van Kaer, L., and Gill, R. G. (2005). NK cells promote islet allograft tolerance via a perforin-dependent mechanism. Nat. Med. 11, 1059–1065.
Bensinger, S. J., Bandeira, A., Jordan, M. S., Caton, A. J., and Laufer, T. M. (2001). Major histocompatibility complex class II-positive cortical epithelium mediates the selection of CD4(+)25(+) immunoregulatory T cells. J. Exp. Med. 194, 427–438.
Bettini, M. L., and Vignali, D. A. (2010). Development of thymically derived natural regulatory T cells. Ann. N. Y. Acad. Sci. 1183, 1–12.
Biberfeld, P., Porwit-Ksiazek, A., Bottiger, B., Morfeldt-Mansson, L., and Biberfeld, G. (1985). Immunohistopathology of lymph nodes in HTLV-III infected homosexuals with persistent adenopathy or AIDS. Cancer Res. 45, 4665s–4670s.
Boscacci, R. T., Pfeiffer, F., Gollmer, K., Sevilla, A. I., Martin, A. M., Soriano, S. F., Natale, D., Henrickson, S., Von Andrian, U. H., Fukui, Y., Mellado, M., Deutsch, U., Engelhardt, B., and Stein, J. V. (2010). Comprehensive analysis of lymph node stroma-expressed Ig superfamily members reveals redundant and nonredundant roles for ICAM-1, ICAM-2, and VCAM-1 in lymphocyte homing. Blood 116, 915–925.
Braun, A., Worbs, T., Moschovakis, G. L., Halle, S., Hoffmann, K., Bolter, J., Munk, A., and Forster, R. (2011). Afferent lymph-derived T cells and DCs use different chemokine receptor CCR7-dependent routes for entry into the lymph node and intranodal migration. Nat. Immunol. 9, 879–887.
Brinkmann, V., Cyster, J. G., and Hla, T. (2004). FTY720: sphingosine 1-phosphate receptor-1 in the control of lymphocyte egress and endothelial barrier function. Am. J. Transplant. 4, 1019–1025.
Campbell, J. J., Bowman, E. P., Murphy, K., Youngman, K. R., Siani, M. A., Thompson, D. A., Wu, L., Zlotnik, A., and Butcher, E. C. (1998). 6-C-kine (SLC), a lymphocyte adhesion-triggering chemokine expressed by high endothelium, is an agonist for the MIP-3beta receptor CCR7. J. Cell Biol. 141, 1053–1059.
Carragher, D. M., Rangel-Moreno, J., and Randall, T. D. (2008). Ectopic lymphoid tissues and local immunity. Semin. Immunol. 20, 26–42.
Chosa, E., Hara, M., Watanabe, A., Matsuzaki, Y., Nakamura, K., Hamano, K., Wood, K. J., and Onitsuka, T. (2007). Spleen plays an important role in maintaining tolerance after removal of the vascularized heart graft. Transplantation 83, 1226–1233.
Chung, Y., Tanaka, S., Chu, F., Nurieva, R. I., Martinez, G. J., Rawal, S., Wang, Y. H., Lim, H., Reynolds, J. M., Zhou, X. H., Fan, H. M., Liu, Z. M., Neelapu, S. S., and Dong, C. (2011). Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat. Med. 17, 983–988.
Cohen, J. N., Guidi, C. J., Tewalt, E. F., Qiao, H., Rouhani, S. J., Ruddell, A., Farr, A. G., Tung, K. S., and Engelhard, V. H. (2010). Lymph node-resident lymphatic endothelial cells mediate peripheral tolerance via Aire-independent direct antigen presentation. J. Exp. Med. 207, 681–688.
Coons, T. A., and Goldberg, E. H. (1978). Rejection of male skin grafts by splenectomized female mice. Science 200, 320–321.
Cupedo, T., Crellin, N. K., Papazian, N., Rombouts, E. J., Weijer, K., Grogan, J. L., Fibbe, W. E., Cornelissen, J. J., and Spits, H. (2009). Human fetal lymphoid tissue-inducer cells are interleukin 17-producing precursors to RORC+ CD127+ natural killer-like cells. Nat. Immunol. 10, 66–74.
Cupedo, T., and Mebius, R. E. (2005). Cellular interactions in lymph node development. J. Immunol. 174, 21–25.
De Togni, P., Goellner, J., Ruddle, N. H., Streeter, P. R., Fick, A., Mariathasan, S., Smith, S. C., Carlson, R., Shornick, L. P., Strauss-Schoenberger, J., Russell, J. H., Karr, R., Chaplin, and David, D. (1994). Abnormal development of peripheral lymphoid organs in mice deficient in lymphotoxin. Science 264, 703–707.
DePaz, H. A., Oluwole, O. O., Adeyeri, A. O., Witkowski, P., Jin, M. X., Hardy, M. A., and Oluwole, S. F. (2003). Immature rat myeloid dendritic cells generated in low-dose granulocyte macrophage-colony stimulating factor prolong donor-specific rat cardiac allograft survival. Transplantation 75, 521–528.
Dominguez, P. M., and Ardavin, C. (2010). Differentiation and function of mouse monocyte-derived dendritic cells in steady state and inflammation. Immunol. Rev. 234, 90–104.
Dor, F. J., Gollackner, B., and Cooper, D. K. (2003). Can spleen transplantation induce tolerance? A review of the literature. Transpl. Int. 16, 451–460.
Drayton, D. L., Liao, S., Mounzer, R. H., and Ruddle, N. H. (2006). Lymphoid organ development: from ontogeny to neogenesis. Nat. Immunol. 7, 344–353.
Dudziak, D., Kamphorst, A. O., Heidkamp, G. F., Buchholz, V. R., Trumpfheller, C., Yamazaki, S., Cheong, C., Liu, K., Lee, H. W., Park, C. G., Steinman, R. M., and Nussenzweig, M. C. (2007). Differential antigen processing by dendritic cell subsets in vivo. Science 315, 107–111.
El Shikh, M. E., El Sayed, R. M., Sukumar, S., Szakal, A. K., and Tew, J. G. (2010). Activation of B cells by antigens on follicular dendritic cells. Trends Immunol. 31, 205–211.
Ermann, J., Hoffmann, P., Edinger, M., Dutt, S., Blankenberg, F. G., Higgins, J. P., Negrin, R. S., Fathman, C. G., and Strober, S. (2005). Only the CD62L+ subpopulation of CD4+CD25+ regulatory T cells protects from lethal acute GVHD. Blood 105, 2220–2226.
Estes, J. D., Haase, A. T., and Schacker, T. W. (2008). The role of collagen deposition in depleting CD4+ T cells and limiting reconstitution in HIV-1 and SIV infections through damage to the secondary lymphoid organ niche. Semin. Immunol. 20, 181–186.
Fitzgerald, J. T., Gallay, B., Taranto, S. E., Mcvicar, J. P., Troppmann, C., Chen, X., Mcintosh, M. J., and Perez, R. V. (2004). Pretransplant recipient cytomegalovirus seropositivity and hemodialysis are associated with decreased renal allograft and patient survival. Transplantation 77, 1405–1411.
Flaishon, L., Hart, G., Zelman, E., Moussion, C., Grabovsky, V., Lapidot Tal, G., Feigelson, S., Margalit, R., Harmelin, A., Avin-Wittenberg, T., Shoseyov, D., Alon, R., Girard, J. P., and Shachar, I. (2008). Anti-inflammatory effects of an inflammatory chemokine: CCL2 inhibits lymphocyte homing by modulation of CCL21-triggered integrin-mediated adhesions. Blood 112, 5016–5025.
Fletcher, A. L., Lukacs-Kornek, V., Reynoso, E. D., Pinner, S. E., Bellemare-Pelletier, A., Curry, M. S., Collier, A. R., Boyd, R. L., and Turley, S. J. (2010a). Lymph node fibroblastic reticular cells directly present peripheral tissue antigen under steady-state and inflammatory conditions. J. Exp. Med. 207, 689–697.
Fletcher, A. L., Malhotra, D., and Turley, S. J. (2010b). Lymph node stroma broaden the peripheral tolerance paradigm. Trends Immunol. 32, 12–18.
Fontenot, J. D., Dooley, J. L., Farr, A. G., and Rudensky, A. Y. (2005). Developmental regulation of Foxp3 expression during ontogeny. J. Exp. Med. 202, 901–906.
Forster, R., Davalos-Misslitz, A. C., and Rot, A. (2008). CCR7 and its ligands: balancing immunity and tolerance. Nat. Rev. Immunol. 8, 362–371.
Fu, Y. X., and Chaplin, D. D. (1999). Development and maturation of secondary lymphoid tissues. Annu. Rev. Immunol. 17, 399–433.
Fujisaki, J., Wu, J., Carlson, A. L., Silberstein, L., Putheti, P., Larocca, R., Gao, W., Saito, T. I., Lo Celso, C., Tsuyuzaki, H., Sato, T., Cote, D., Sykes, M., Strom, T. B., Scadden, D. T., and Lin, C. P. (2011). In vivo imaging of Treg cells providing immune privilege to the haematopoietic stem-cell niche. Nature 474, 216–219.
Futterer, A., Mink, K., Luz, A., Kosco-Vilbois, M. H., and Pfeffer, K. (1998). The lymphotoxin beta receptor controls organogenesis and affinity maturation in peripheral lymphoid tissues. Immunity 9, 59–70.
Gardner, J. M., Devoss, J. J., Friedman, R. S., Wong, D. J., Tan, Y. X., Zhou, X., Johannes, K. P., Su, M. A., Chang, H. Y., Krummel, M. F., and Anderson, M. S. (2008). Deletional tolerance mediated by extrathymic Aire-expressing cells. Science 321, 843–847.
Gilliet, M., Cao, W., and Liu, Y. J. (2008). Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat. Rev. Immunol. 8, 594–606.
Girard, J. P., and Springer, T. A. (1995). High endothelial venules (HEVs): specialized endothelium for lymphocyte migration. Immunol. Today 16, 449–457.
Gopinathan, R., Depaz, H. A., Oluwole, O. O., Ali, A. O., Garrovillo, M., Engelstad, K., Hardy, M. A., and Oluwole, S. F. (2001). Role of reentry of in vivo alloMHC peptide-activated T cells into the adult thymus in acquired systemic tolerance. Transplantation 72, 1533–1541.
Grabner, R., Lotzer, K., Dopping, S., Hildner, M., Radke, D., Beer, M., Spanbroek, R., Lippert, B., Reardon, C. A., Getz, G. S., Fu, Y. X., Hehlgans, T., Mebius, R. E., Van Der Wall, M., Kruspe, D., Englert, C., Lovas, A., Hu, D., Randolph, G. J., Weih, F., and Habenicht, A. J. (2009). Lymphotoxin beta receptor signaling promotes tertiary lymphoid organogenesis in the aorta adventitia of aged ApoE-/- mice. J. Exp. Med. 206, 233–248.
Gretz, J. E., Anderson, A. O., and Shaw, S. (1997). Cords, channels, corridors and conduits: critical architectural elements facilitating cell interactions in the lymph node cortex. Immunol. Rev. 156, 11–24.
Gretz, J. E., Norbury, C. C., Anderson, A. O., Proudfoot, A. E., and Shaw, S. (2000). Lymph-borne chemokines and other low molecular weight molecules reach high endothelial venules via specialized conduits while a functional barrier limits access to the lymphocyte microenvironments in lymph node cortex. J. Exp. Med. 192, 1425–1440.
Griesemer, A. D., Sorenson, E. C., and Hardy, M. A. (2010). The role of the thymus in tolerance. Transplantation 90, 465–474.
Grigorova, I. L., Panteleev, M., and Cyster, J. G. (2010). Lymph node cortical sinus organization and relationship to lymphocyte egress dynamics and antigen exposure. Proc. Natl. Acad. Sci. U.S.A. 107, 20447–20452.
Guarda, G., Hons, M., Soriano, S. F., Huang, A. Y., Polley, R., Martin-Fontecha, A., Stein, J. V., Germain, R. N., Lanzavecchia, A., and Sallusto, F. (2007). L-selectin-negative CCR7- effector and memory CD8+ T cells enter reactive lymph nodes and kill dendritic cells. Nat. Immunol. 8, 743–752.
Gunn, M. D., Ngo, V. N., Ansel, K. M., Ekland, E. H., Cyster, J. G., and Williams, L. T. (1998a). A B-cell-homing chemokine made in lymphoid follicles activates Burkitt’s lymphoma receptor-1. Nature 391, 799–803.
Gunn, M. D., Tangemann, K., Tam, C., Cyster, J. G., Rosen, S. D., and Williams, L. T. (1998b). A chemokine expressed in lymphoid high endothelial venules promotes the adhesion and chemotaxis of naive T lymphocytes. Proc. Natl. Acad. Sci. U.S.A. 95, 258–263.
Guo, R., Zhou, Q., Proulx, S. T., Wood, R., Ji, R. C., Ritchlin, C. T., Pytowski, B., Zhu, Z., Wang, Y. J., Schwarz, E. M., and Xing, L. (2009). Inhibition of lymphangiogenesis and lymphatic drainage via vascular endothelial growth factor receptor 3 blockade increases the severity of inflammation in a mouse model of chronic inflammatory arthritis. Arthritis Rheum. 60, 2666–2676.
Halin, C., Tobler, N. E., Vigl, B., Brown, L. F., and Detmar, M. (2007). VEGF-A produced by chronically inflamed tissue induces lymphangiogenesis in draining lymph nodes. Blood 110, 3158–3167.
Hammerschmidt, S. I., Ahrendt, M., Bode, U., Wahl, B., Kremmer, E., Forster, R., and Pabst, O. (2008). Stromal mesenteric lymph node cells are essential for the generation of gut-homing T cells in vivo. J. Exp. Med. 205, 2483–2490.
Hase, H., Kanno, Y., Kojima, M., Hasegawa, K., Sakurai, D., Kojima, H., Tsuchiya, N., Tokunaga, K., Masawa, N., Azuma, M., Okumura, K., and Kobata, T. (2004). BAFF/BLyS can potentiate B-cell selection with the B-cell coreceptor complex. Blood 103, 2257–2265.
Heidecke, C. D., Araujo, J. L., Kupiec-Weglinski, J. W., Abbud-Filho, M., Araneda, D., Stadler, J., Siewert, J., Strom, T. B., and Tilney, N. L. (1985). Lack of evidence for an active role for natural killer cells in acute rejection of organ allografts. Transplantation 40, 441–444.
Honda, K., Nakano, H., Yoshida, H., Nishikawa, S., Rennert, P., Ikuta, K., Tamechika, M., Yamaguchi, K., Fukumoto, T., Chiba, T., and Nishikawa, S. I. (2001). Molecular basis for hematopoietic/mesenchymal interaction during initiation of Peyer’s patch organogenesis. J. Exp. Med. 193, 621–630.
Horner, B. M., Cina, R. A., Wikiel, K. J., Lima, B., Ghazi, A., Lo, D. P., Yamada, K., Sachs, D. H., and Huang, C. A. (2006). Predictors of organ allograft tolerance following hematopoietic cell transplantation. Am. J. Transplant. 6, 2894–2902.
Huggenberger, R., Ullmann, S., Proulx, S. T., Pytowski, B., Alitalo, K., and Detmar, M. (2010). Stimulation of lymphangiogenesis via VEGFR-3 inhibits chronic skin inflammation. J. Exp. Med. 207, 2255–2269.
Itano, A. A., and Jenkins, M. K. (2003). Antigen presentation to naive CD4 T cells in the lymph node. Nat. Immunol. 4, 733–739.
Iwata, M., Hirakiyama, A., Eshima, Y., Kagechika, H., Kato, C., and Song, S. Y. (2004). Retinoic acid imprints gut-homing specificity on T cells. Immunity 21, 527–538.
Jiang, Q., Su, H., Knudsen, G., Helms, W., and Su, L. (2006). Delayed functional maturation of natural regulatory T cells in the medulla of postnatal thymus: role of TSLP. BMC Immunol. 7, 6. doi: 10.1186/1471-2172-7-6
Johnson, B. A. III, Kahler, D. J., Baban, B., Chandler, P. R., Kang, B., Shimoda, M., Koni, P. A., Pihkala, J., Vilagos, B., Busslinger, M., Munn, D. H., and Mellor, A. L. (2010). B-lymphoid cells with attributes of dendritic cells regulate T cells via indoleamine 2,3-dioxygenase. Proc. Natl. Acad. Sci. U.S.A. 107, 10644–10648.
Jordan, M. S., Boesteanu, A., Reed, A. J., Petrone, A. L., Holenbeck, A. E., Lerman, M. A., Naji, A., and Caton, A. J. (2001). Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat. Immunol. 2, 301–306.
Kanemitsu, N., Ebisuno, Y., Tanaka, T., Otani, K., Hayasaka, H., Kaisho, T., Akira, S., Katagiri, K., Kinashi, T., Fujita, N., Tsuruo, T., and Miyasaka, M. (2005). CXCL13 is an arrest chemokine for B cells in high endothelial venules. Blood 106, 2613–2618.
Katakai, T., Hara, T., Lee, J. H., Gonda, H., Sugai, M., and Shimizu, A. (2004a). A novel reticular stromal structure in lymph node cortex: an immuno-platform for interactions among dendritic cells, T cells and B cells. Int. Immunol. 16, 1133–1142.
Katakai, T., Hara, T., Sugai, M., Gonda, H., and Shimizu, A. (2004b). Lymph node fibroblastic reticular cells construct the stromal reticulum via contact with lymphocytes. J. Exp. Med. 200, 783–795.
Katakai, T., Nomura, T., Gonda, H., Sugai, M., Agata, Y., Nishio, A., Masuda, T., Sakaguchi, S., and Shimizu, A. (2006). Spontaneous large-scale lymphoid neogenesis and balanced autoimmunity versus tolerance in the stomach of H+/K+-ATPase-reactive TCR transgenic mouse. J. Immunol. 177, 7858–7867.
Katakai, T., Suto, H., Sugai, M., Gonda, H., Togawa, A., Suematsu, S., Ebisuno, Y., Katagiri, K., Kinashi, T., and Shimizu, A. (2008). Organizer-like reticular stromal cell layer common to adult secondary lymphoid organs. J. Immunol. 181, 6189–6200.
Kataru, R. P., Kim, H., Jang, C., Choi, D. K., Koh, B. I., Kim, M., Gollamudi, S., Kim, Y. K., Lee, S. H., and Koh, G. Y. (2011). T lymphocytes negatively regulate lymph node lymphatic vessel formation. Immunity 34, 96–107.
Kerjaschki, D., Regele, H. M., Moosberger, I., Nagy-Bojarski, K., Watschinger, B., Soleiman, A., Birner, P., Krieger, S., Hovorka, A., Silberhumer, G., Laakkonen, P., Petrova, T., Langer, B., and Raab, I. (2004). Lymphatic neoangiogenesis in human kidney transplants is associated with immunologically active lymphocytic infiltrates. J. Am. Soc. Nephrol. 15, 603–612.
Kim, M. Y., Anderson, G., White, A., Jenkinson, E., Arlt, W., Martensson, I. L., Erlandsson, L., and Lane, P. J. (2005). OX40 ligand and CD30 ligand are expressed on adult but not neonatal CD4+CD3- inducer cells: evidence that IL-7 signals regulate CD30 ligand but not OX40 ligand expression. J. Immunol. 174, 6686–6691.
Kim, M. Y., Kim, K. S., Mcconnell, F., and Lane, P. (2009). Lymphoid tissue inducer cells: architects of CD4 immune responses in mice and men. Clin. Exp. Immunol. 157, 20–26.
Kimpfler, S., Sevko, A., Ring, S., Falk, C., Osen, W., Frank, K., Kato, M., Mahnke, K., Schadendorf, D., and Umansky, V. (2009). Skin melanoma development in ret transgenic mice despite the depletion of CD25+Foxp3+ regulatory T cells in lymphoid organs. J. Immunol. 183, 6330–6337.
Kool, M., Soullie, T., Van Nimwegen, M., Willart, M. A., Muskens, F., Jung, S., Hoogsteden, H. C., Hammad, H., and Lambrecht, B. N. (2008). Alum adjuvant boosts adaptive immunity by inducing uric acid and activating inflammatory dendritic cells. J. Exp. Med. 205, 869–882.
Kraal, G., Hoeben, K., Breve, J., and Van Den Berg, T. K. (1994). The role of sialic acid in the localization of lymphocytes in the spleen. Immunobiology 190, 138–149.
Kroemer, A., Edtinger, K., and Li, X. C. (2008). The innate natural killer cells in transplant rejection and tolerance induction. Curr. Opin. Organ Transplant. 13, 339–343.
Krupnick, A. S., Gelman, A. E., Barchet, W., Richardson, S., Kreisel, F. H., Turka, L. A., Colonna, M., Patterson, G. A., and Kreisel, D. (2005). Murine vascular endothelium activates and induces the generation of allogeneic CD4+25+Foxp3+ regulatory T cells. J. Immunol. 175, 6265–6270.
Kurobe, H., Liu, C., Ueno, T., Saito, F., Ohigashi, I., Seach, N., Arakaki, R., Hayashi, Y., Kitagawa, T., Lipp, M., Boyd, R. L., and Takahama, Y. (2006). CCR7-dependent cortex-to-medulla migration of positively selected thymocytes is essential for establishing central tolerance. Immunity 24, 165–177.
Kurotaki, D., Kon, S., Bae, K., Ito, K., Matsui, Y., Nakayama, Y., Kanayama, M., Kimura, C., Narita, Y., Nishimura, T., Iwabuchi, K., Mack, M., Van Rooijen, N., Sakaguchi, S., Uede, T., and Morimoto, J. (2011). CSF-1-dependent red pulp macrophages regulate CD4 T cell responses. J. Immunol. 186, 2229–2237.
Lakkis, F. G., Arakelov, A., Konieczny, B. T., and Inoue, Y. (2000). Immunologic ‘ignorance’ of vascularized organ transplants in the absence of secondary lymphoid tissue. Nat. Med. 6, 686–688.
Lane, P., Kim, M. Y., Withers, D., Gaspal, F., Bekiaris, V., Desanti, G., Khan, M., Mcconnell, F., and Anderson, G. (2008). Lymphoid tissue inducer cells in adaptive CD4 T cell dependent responses. Semin. Immunol. 20, 159–163.
Larsen, C. P., Austyn, J. M., and Morris, P. J. (1990a). The role of graft-derived dendritic leukocytes in the rejection of vascularized organ allografts. Recent findings on the migration and function of dendritic leukocytes after transplantation. Ann. Surg. 212, 308–315; discussion 316–307.
Larsen, C. P., Morris, P. J., and Austyn, J. M. (1990b). Migration of dendritic leukocytes from cardiac allografts into host spleens. A novel pathway for initiation of rejection. J. Exp. Med. 171, 307–314.
Lee, J. W., Epardaud, M., Sun, J., Becker, J. E., Cheng, A. C., Yonekura, A. R., Heath, J. K., and Turley, S. J. (2007). Peripheral antigen display by lymph node stroma promotes T cell tolerance to intestinal self. Nat. Immunol. 8, 181–190.
Lee, L. A., Gritsch, H. A., Sergio, J. J., Arn, J. S., Glaser, R. M., Sablinski, T., Sachs, D. H., and Sykes, M. (1994). Specific tolerance across a discordant xenogeneic transplantation barrier. Proc. Natl. Acad. Sci. U.S.A. 91, 10864–10867.
Lee, Y., Chin, R. K., Christiansen, P., Sun, Y., Tumanov, A. V., Wang, J., Chervonsky, A. V., and Fu, Y. X. (2006). Recruitment and activation of naive T cells in the islets by lymphotoxin beta receptor-dependent tertiary lymphoid structure. Immunity 25, 499–509.
Li, X. L., Menoret, S., Bezie, S., Caron, L., Chabannes, D., Hill, M., Halary, F., Angin, M., Heslan, M., Usal, C., Liang, L., Guillonneau, C., Le Mauff, B., Cuturi, M. C., Josien, R., and Anegon, I. (2010). Mechanism and localization of CD8 regulatory T cells in a heart transplant model of tolerance. J. Immunol. 185, 823–833.
Liao, S., and Ruddle, N. H. (2006). Synchrony of high endothelial venules and lymphatic vessels revealed by immunization. J. Immunol. 177, 3369–3379.
Link, A., Hardie, D. L., Favre, S., Britschgi, M. R., Adams, D. H., Sixt, M., Cyster, J. G., Buckley, C. D., and Luther, S. A. (2011). Association of T-zone reticular networks and conduits with ectopic lymphoid tissues in mice and humans. Am. J. Pathol. 178, 1662–1675.
Link, A., Vogt, T. K., Favre, S., Britschgi, M. R., Acha-Orbea, H., Hinz, B., Cyster, J. G., and Luther, S. A. (2007). Fibroblastic reticular cells in lymph nodes regulate the homeostasis of naive T cells. Nat. Immunol. 8, 1255–1265.
Linterman, M. A., Pierson, W., Lee, S. K., Kallies, A., Kawamoto, S., Rayner, T. F., Srivastava, M., Divekar, D. P., Beaton, L., Hogan, J. J., Fagarasan, S., Liston, A., Smith, K. G., and Vinuesa, C. G. (2011). Foxp3(+) follicular regulatory T cells control the germinal center response. Nat. Med. 17, 975–982.
Liston, A., Nutsch, K. M., Farr, A. G., Lund, J. M., Rasmussen, J. P., Koni, P. A., and Rudensky, A. Y. (2008). Differentiation of regulatory Foxp3+ T cells in the thymic cortex. Proc. Natl. Acad. Sci. U.S.A. 105, 11903–11908.
Liu, K., and Nussenzweig, M. C. (2010). Origin and development of dendritic cells. Immunol. Rev. 234, 45–54.
Liu, Y., Zhang, P., Li, J., Kulkarni, A. B., Perruche, S., and Chen, W. (2008). A critical function for TGF-beta signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nat. Immunol. 9, 632–640.
Lo, C. G., Lu, T. T., and Cyster, J. G. (2003). Integrin-dependence of lymphocyte entry into the splenic white pulp. J. Exp. Med. 197, 353–361.
Luther, S. A., Tang, H. L., Hyman, P. L., Farr, A. G., and Cyster, J. G. (2000). Coexpression of the chemokines ELC and SLC by T zone stromal cells and deletion of the ELC gene in the plt/plt mouse. Proc. Natl. Acad. Sci. U.S.A. 97, 12694–12699.
Luther, S. A., Vogt, T. K., and Siegert, S. (2011). Guiding blind T cells and dendritic cells: a closer look at fibroblastic reticular cells found within lymph node T zones. Immunol. Lett. 138, 9–11.
Mackay, F., Majeau, G. R., Lawton, P., Hochman, P. S., and Browning, J. L. (1997). Lymphotoxin but not tumor necrosis factor functions to maintain splenic architecture and humoral responsiveness in adult mice. Eur. J. Immunol. 27, 2033–2042.
Manches, O., Munn, D., Fallahi, A., Lifson, J., Chaperot, L., Plumas, J., and Bhardwaj, N. (2008). HIV-activated human plasmacytoid DCs induce Tregs through an indoleamine 2,3-dioxygenase-dependent mechanism. J. Clin. Invest. 118, 3431–3439.
Marinkovic, T., Garin, A., Yokota, Y., Fu, Y. X., Ruddle, N. H., Furtado, G. C., and Lira, S. A. (2006). Interaction of mature CD3+CD4+ T cells with dendritic cells triggers the development of tertiary lymphoid structures in the thyroid. J. Clin. Invest. 116, 2622–2632.
Marsland, B. J., Battig, P., Bauer, M., Ruedl, C., Lassing, U., Beerli, R. R., Dietmeier, K., Ivanova, L., Pfister, T., Vogt, L., Nakano, H., Nembrini, C., Saudan, P., Kopf, M., and Bachmann, M. F. (2005). CCL19 and CCL21 induce a potent proinflammatory differentiation program in licensed dendritic cells. Immunity 22, 493–505.
Matloubian, M., Lo, C. G., Cinamon, G., Lesneski, M. J., Xu, Y., Brinkmann, V., Allende, M. L., Proia, R. L., and Cyster, J. G. (2004). Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 427, 355–360.
McCarthy, D. D., Summers-Deluca, L., Vu, F., Chiu, S., Gao, Y., and Gommerman, J. L. (2006). The lymphotoxin pathway: beyond lymph node development. Immunol. Res. 35, 41–54.
Mebius, R. E., Streeter, P. R., Michie, S., Butcher, E. C., and Weissman, I. L. (1996). A developmental switch in lymphocyte homing receptor and endothelial vascular addressin expression regulates lymphocyte homing and permits CD4+ CD3- cells to colonize lymph nodes. Proc. Natl. Acad. Sci. U.S.A. 93, 11019–11024.
Mellor, A. L., Baban, B., Chandler, P. R., Manlapat, A., Kahler, D. J., and Munn, D. H. (2005). Cutting edge: CpG oligonucleotides induce splenic CD19+ dendritic cells to acquire potent indoleamine 2,3-dioxygenase-dependent T cell regulatory functions via IFN Type 1 signaling. J. Immunol. 175, 5601–5605.
Metzger, T. C., and Anderson, M. S. (2011). Control of central and peripheral tolerance by Aire. Immunol. Rev. 241, 89–103.
Motallebzadeh, R., Bolton, E. M., and Pettigrew, G. J. (2008). Lymphoid tissue formation in allografts: innocent until proven guilty. Transplantation 85, 309–311.
Mueller, E. W., and Ernst, N. E. (2007). Antibiotic therapy and immunosuppression: choosing an edge on a familiar double-edged sword. Crit. Care Med. 35, 1430–1431.
Mueller, S. N., and Germain, R. N. (2009). Stromal cell contributions to the homeostasis and functionality of the immune system. Nat. Rev. Immunol. 9, 618–629.
Mueller, S. N., Matloubian, M., Clemens, D. M., Sharpe, A. H., Freeman, G. J., Gangappa, S., Larsen, C. P., and Ahmed, R. (2007). Viral targeting of fibroblastic reticular cells contributes to immunosuppression and persistence during chronic infection. Proc. Natl. Acad. Sci. U.S.A. 104, 15430–15435.
Munoz-Fernandez, R., Blanco, F. J., Frecha, C., Martin, F., Kimatrai, M., Abadia-Molina, A. C., Garcia-Pacheco, J. M., and Olivares, E. G. (2006). Follicular dendritic cells are related to bone marrow stromal cell progenitors and to myofibroblasts. J. Immunol. 177, 280–289.
Murphy, S. P., Porrett, P. M., and Turka, L. A. (2011). Innate immunity in transplant tolerance and rejection. Immunol. Rev. 241, 39–48.
Nasr, I. W., Reel, M., Oberbarnscheidt, M. H., Mounzer, R. H., Baddoura, F. K., Ruddle, N. H., and Lakkis, F. G. (2007). Tertiary lymphoid tissues generate effector and memory T cells that lead to allograft rejection. Am. J. Transplant. 7, 1071–1079.
Nishikawa, S. I., Hashi, H., Honda, K., Fraser, S., and Yoshida, H. (2000). Inflammation, a prototype for organogenesis of the lymphopoietic/hematopoietic system. Curr. Opin. Immunol. 12, 342–345.
Nolte, M. A., Belien, J. A., Schadee-Eestermans, I., Jansen, W., Unger, W. W., Van Rooijen, N., Kraal, G., and Mebius, R. E. (2003). A conduit system distributes chemokines and small blood-borne molecules through the splenic white pulp. J. Exp. Med. 198, 505–512.
Ochando, J. C., Homma, C., Yang, Y., Hidalgo, A., Garin, A., Tacke, F., Angeli, V., Li, Y., Boros, P., Ding, Y., Jessberger, R., Trinchieri, G., Lira, S. A., Randolph, G. J., and Bromberg, J. S. (2006). Alloantigen-presenting plasmacytoid dendritic cells mediate tolerance to vascularized grafts. Nat. Immunol. 7, 652–662.
Ochando, J. C., Yopp, A. C., Yang, Y., Garin, A., Li, Y., Boros, P., Llodra, J., Ding, Y., Lira, S. A., Krieger, N. R., and Bromberg, J. S. (2005). Lymph node occupancy is required for the peripheral development of alloantigen-specific Foxp3+ regulatory T cells. J. Immunol. 174, 6993–7005.
Oka, M., Iwata, C., Suzuki, H. I., Kiyono, K., Morishita, Y., Watabe, T., Komuro, A., Kano, M. R., and Miyazono, K. (2008). Inhibition of endogenous TGF-beta signaling enhances lymphangiogenesis. Blood 111, 4571–4579.
Okada, T., Ngo, V. N., Ekland, E. H., Forster, R., Lipp, M., Littman, D. R., and Cyster, J. G. (2002). Chemokine requirements for B cell entry to lymph nodes and Peyer’s patches. J. Exp. Med. 196, 65–75.
Oluwole, O. O., Depaz, H. A., Adeyeri, A., Jin, M. X., Hardy, M. A., and Oluwole, S. F. (2003). Role of CD4+CD25+ regulatory T cells from naive host thymus in the induction of acquired transplant tolerance by immunization with allo-major histocompatibility complex peptide. Transplantation 75, 1136–1142.
Oluwole, O. O., Depaz, H. A., Gopinathan, R., Ali, A., Garrovillo, M., Jin, M. X., Hardy, M. A., and Oluwole, S. F. (2001). Indirect allorecognition in acquired thymic tolerance: induction of donor-specific permanent acceptance of rat islets by adoptive transfer of allopeptide-pulsed host myeloid and thymic dendritic cells. Diabetes 50, 1546–1552.
Orloff, S. L., Hwee, Y. K., Kreklywich, C., Andoh, T. F., Hart, E., Smith, P. A., Messaoudi, I., and Streblow, D. N. (2011). Cytomegalovirus latency promotes cardiac lymphoid neogenesis and accelerated allograft rejection in CMV naive recipients. Am. J. Transplant. 11, 45–55.
Pape, K. A., Catron, D. M., Itano, A. A., and Jenkins, M. K. (2007). The humoral immune response is initiated in lymph nodes by B cells that acquire soluble antigen directly in the follicles. Immunity 26, 491–502.
Penaranda, C., Tang, Q., Ruddle, N. H., and Bluestone, J. A. (2010). Prevention of diabetes by FTY720-mediated stabilization of peri-islet tertiary lymphoid organs. Diabetes 59, 1461–1468.
Pennington, D. J., Silva-Santos, B., Silberzahn, T., Escorcio-Correia, M., Woodward, M. J., Roberts, S. J., Smith, A. L., Dyson, P. J., and Hayday, A. C. (2006). Early events in the thymus affect the balance of effector and regulatory T cells. Nature 444, 1073–1077.
Perry, D. K., Burns, J. M., Pollinger, H. S., Amiot, B. P., Gloor, J. M., Gores, G. J., and Stegall, M. D. (2009). Proteasome inhibition causes apoptosis of normal human plasma cells preventing alloantibody production. Am. J. Transplant. 9, 201–209.
Pham, T. H., Baluk, P., Xu, Y., Grigorova, I., Bankovich, A. J., Pappu, R., Coughlin, S. R., Mcdonald, D. M., Schwab, S. R., and Cyster, J. G. (2010). Lymphatic endothelial cell sphingosine kinase activity is required for lymphocyte egress and lymphatic patterning. J. Exp. Med. 207, 17–27.
Podgrabinska, S., Kamalu, O., Mayer, L., Shimaoka, M., Snoeck, H., Randolph, G. J., and Skobe, M. (2009). Inflamed lymphatic endothelium suppresses dendritic cell maturation and function via Mac-1/ICAM-1-dependent mechanism. J. Immunol. 183, 1767–1779.
Rabinovich, B. A., Shannon, J., Su, R. C., and Miller, R. G. (2000). Stress renders T cell blasts sensitive to killing by activated syngeneic NK cells. J. Immunol. 165, 2390–2397.
Randolph, G. J., Angeli, V., and Swartz, M. A. (2005). Dendritic-cell trafficking to lymph nodes through lymphatic vessels. Nat. Rev. Immunol. 5, 617–628.
Reif, K., Ekland, E. H., Ohl, L., Nakano, H., Lipp, M., Forster, R., and Cyster, J. G. (2002). Balanced responsiveness to chemoattractants from adjacent zones determines B-cell position. Nature 416, 94–99.
Saiki, T., Ezaki, T., Ogawa, M., and Matsuno, K. (2001). Trafficking of host- and donor-derived dendritic cells in rat cardiac transplantation: allosensitization in the spleen and hepatic nodes. Transplantation 71, 1806–1815.
Scandella, E., Bolinger, B., Lattmann, E., Miller, S., Favre, S., Littman, D. R., Finke, D., Luther, S. A., Junt, T., and Ludewig, B. (2008). Restoration of lymphoid organ integrity through the interaction of lymphoid tissue-inducer cells with stroma of the T cell zone. Nat. Immunol. 9, 667–675.
Schacker, T., Little, S., Connick, E., Gebhard, K., Zhang, Z. Q., Krieger, J., Pryor, J., Havlir, D., Wong, J. K., Schooley, R. T., Richman, D., Corey, L., and Haase, A. T. (2001). Productive infection of T cells in lymphoid tissues during primary and early human immunodeficiency virus infection. J. Infect. Dis. 183, 555–562.
Schneider, K., Potter, K. G., and Ware, C. F. (2004). Lymphotoxin and LIGHT signaling pathways and target genes. Immunol. Rev. 202, 49–66.
Scimone, M. L., Felbinger, T. W., Mazo, I. B., Stein, J. V., Von Andrian, U. H., and Weninger, W. (2004). CXCL12 mediates CCR7-independent homing of central memory cells, but not naive T cells, in peripheral lymph nodes. J. Exp. Med. 199, 1113–1120.
Shibuya, M., and Claesson-Welsh, L. (2006). Signal transduction by VEGF receptors in regulation of angiogenesis and lymphangiogenesis. Exp. Cell Res. 312, 549–560.
Shields, J. D., Kourtis, I. C., Tomei, A. A., Roberts, J. M., and Swartz, M. A. (2010). Induction of lymphoidlike stroma and immune escape by tumors that express the chemokine CCL21. Science 328, 749–752.
Shrestha, B., Hashiguchi, T., Ito, T., Miura, N., Takenouchi, K., Oyama, Y., Kawahara, K., Tancharoen, S., Ki, I. Y., Arimura, N., Yoshinaga, N., Noma, S., Shrestha, C., Nitanda, T., Kitajima, S., Arimura, K., Sato, M., Sakamoto, T., and Maruyama, I. (2010). B cell-derived vascular endothelial growth factor A promotes lymphangiogenesis and high endothelial venule expansion in lymph nodes. J. Immunol. 184, 4819–4826.
Sinha, R. K., Park, C., Hwang, I. Y., Davis, M. D., and Kehrl, J. H. (2009). B lymphocytes exit lymph nodes through cortical lymphatic sinusoids by a mechanism independent of sphingosine-1-phosphate-mediated chemotaxis. Immunity 30, 434–446.
Sixt, M., Kanazawa, N., Selg, M., Samson, T., Roos, G., Reinhardt, D. P., Pabst, R., Lutz, M. B., and Sorokin, L. (2005). The conduit system transports soluble antigens from the afferent lymph to resident dendritic cells in the T cell area of the lymph node. Immunity 22, 19–29.
Smith, R. N., Chang, Y., Houser, S., Dec, G. W., and Grazette, L. (2002). Higher frequency of high-grade rejections in cardiac allograft patients after Quilty B lesions or grade 2/4 rejections. Transplantation 73, 1928–1932.
Souther, S. G., Morris, R. E., and Vistnes, L. M. (1974). Prolongation of rat cardiac allograft survival by splenectomy following transplantation. Transplantation 17, 317–319.
Springer, T. A. (1994). Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell 76, 301–314.
Steele, K. E., Anderson, A. O., and Mohamadzadeh, M. (2009). Fibroblastic reticular cells and their role in viral hemorrhagic fevers. Expert Rev. Anti. Infect. Ther. 7, 423–435.
Stegall, M. D., Raghavaiah, S., and Gloor, J. M. (2010). The (re)emergence of B cells in organ transplantation. Curr. Opin. Organ Transplant. 15, 451–455.
Steiniger, B., Barth, P., and Hellinger, A. (2001). The perifollicular and marginal zones of the human splenic white pulp: do fibroblasts guide lymphocyte immigration? Am. J. Pathol. 159, 501–512.
Streilein, J. W., and Wiesner, J. (1977). Influence of splenectomy on first set rejection reactions of C57BL/6 females to male skin isografts. J. Exp. Med. 146, 809–816.
Swiecki, M., and Colonna, M. (2010). Unraveling the functions of plasmacytoid dendritic cells during viral infections, autoimmunity, and tolerance. Immunol. Rev. 234, 142–162.
Szanya, V., Ermann, J., Taylor, C., Holness, C., and Fathman, C. G. (2002). The subpopulation of CD4+CD25+ splenocytes that delays adoptive transfer of diabetes expresses L-selectin and high levels of CCR7. J. Immunol. 169, 2461–2465.
Tachibana, M., Tenno, M., Tezuka, C., Sugiyama, M., Yoshida, H., and Taniuchi, I. (2011). Runx1/Cbfbeta2 complexes are required for lymphoid tissue inducer cell differentiation at two developmental stages. J. Immunol. 186, 1450–1457.
Taflin, C., Favier, B., Baudhuin, J., Savenay, A., Hemon, P., Bensussan, A., Charron, D., Glotz, D., and Mooney, N. (2011). Human endothelial cells generate Th17 and regulatory T cells under inflammatory conditions. Proc. Natl. Acad. Sci. U.S.A. 108, 2891–2896.
Takatori, H., Kanno, Y., Watford, W. T., Tato, C. M., Weiss, G., Ivanov, II Littman, D. R., and O’shea, J. J. (2009). Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. J. Exp. Med. 206, 35–41.
Tammela, T., and Alitalo, K. (2010). Lymphangiogenesis: molecular mechanisms and future promise. Cell 140, 460–476.
Tang, H. L., and Cyster, J. G. (1999). Chemokine Up-regulation and activated T cell attraction by maturing dendritic cells. Science 284, 819–822.
Tang, Q., Adams, J. Y., Tooley, A. J., Bi, M., Fife, B. T., Serra, P., Santamaria, P., Locksley, R. M., Krummel, M. F., and Bluestone, J. A. (2006). Visualizing regulatory T cell control of autoimmune responses in nonobese diabetic mice. Nat. Immunol. 7, 83–92.
Tezuka, H., Abe, Y., Iwata, M., Takeuchi, H., Ishikawa, H., Matsushita, M., Shiohara, T., Akira, S., and Ohteki, T. (2007). Regulation of IgA production by naturally occurring TNF/iNOS-producing dendritic cells. Nature 448, 929–933.
Thaunat, O., Field, A. C., Dai, J., Louedec, L., Patey, N., Bloch, M. F., Mandet, C., Belair, M. F., Bruneval, P., Meilhac, O., Bellon, B., Joly, E., Michel, J. B., and Nicoletti, A. (2005). Lymphoid neogenesis in chronic rejection: evidence for a local humoral alloimmune response. Proc. Natl. Acad. Sci. U.S.A. 102, 14723–14728.
Thaunat, O., and Nicoletti, A. (2008). Lymphoid neogenesis in chronic rejection. Curr. Opin. Organ Transplant. 13, 16–19.
Tomura, M., Honda, T., Tanizaki, H., Otsuka, A., Egawa, G., Tokura, Y., Waldmann, H., Hori, S., Cyster, J. G., Watanabe, T., Miyachi, Y., Kanagawa, O., and Kabashima, K. (2010). Activated regulatory T cells are the major T cell type emigrating from the skin during a cutaneous immune response in mice. J. Clin. Invest. 120, 883–893.
Tsuji, M., Suzuki, K., Kitamura, H., Maruya, M., Kinoshita, K., Ivanov, Ii, Itoh, K., Littman, D. R., and Fagarasan, S. (2008). Requirement for lymphoid tissue-inducer cells in isolated follicle formation and T cell-independent immunoglobulin A generation in the gut. Immunity 29, 261–271.
Umehara, H., Bloom, E. T., Okazaki, T., Nagano, Y., Yoshie, O., and Imai, T. (2004). Fractalkine in vascular biology: from basic research to clinical disease. Arterioscler. Thromb. Vasc. Biol. 24, 34–40.
van de Pavert, S. A., and Mebius, R. E. (2010). New insights into the development of lymphoid tissues. Nat. Rev. Immunol. 10, 664–674.
Van Vliet, E., Melis, M., Foidart, J. M., and Van Ewijk, W. (1986). Reticular fibroblasts in peripheral lymphoid organs identified by a monoclonal antibody. J. Histochem. Cytochem. 34, 883–890.
Vanbervliet, B., Bendriss-Vermare, N., Massacrier, C., Homey, B., De Bouteiller, O., Briere, F., Trinchieri, G., and Caux, C. (2003). The inducible CXCR3 ligands control plasmacytoid dendritic cell responsiveness to the constitutive chemokine stromal cell-derived factor 1 (SDF-1)/CXCL12. J. Exp. Med. 198, 823–830.
Varol, C., Vallon-Eberhard, A., Elinav, E., Aychek, T., Shapira, Y., Luche, H., Fehling, H. J., Hardt, W. D., Shakhar, G., and Jung, S. (2009). Intestinal lamina propria dendritic cell subsets have different origin and functions. Immunity 31, 502–512.
von Andrian, U. H., and Mempel, T. R. (2003). Homing and cellular traffic in lymph nodes. Nat. Rev. Immunol. 3, 867–878.
Waithman, J., Allan, R. S., Kosaka, H., Azukizawa, H., Shortman, K., Lutz, M. B., Heath, W. R., Carbone, F. R., and Belz, G. T. (2007). Skin-derived dendritic cells can mediate deletional tolerance of class I-restricted self-reactive T cells. J. Immunol. 179, 4535–4541.
Watanabe, N., Wang, Y. H., Lee, H. K., Ito, T., Cao, W., and Liu, Y. J. (2005). Hassall’s corpuscles instruct dendritic cells to induce CD4+CD25+ regulatory T cells in human thymus. Nature 436, 1181–1185.
Wright, N., Hidalgo, A., Rodriguez-Frade, J. M., Soriano, S. F., Mellado, M., Parmo-Cabanas, M., Briskin, M. J., and Teixido, J. (2002). The chemokine stromal cell-derived factor-1 alpha modulates alpha 4 beta 7 integrin-mediated lymphocyte adhesion to mucosal addressin cell adhesion molecule-1 and fibronectin. J. Immunol. 168, 5268–5277.
Wu, Q., Salomon, B., Chen, M., Wang, Y., Hoffman, L. M., Bluestone, J. A., and Fu, Y. X. (2001). Reversal of spontaneous autoimmune insulitis in nonobese diabetic mice by soluble lymphotoxin receptor. J. Exp. Med. 193, 1327–1332.
Yamazaki, S., Dudziak, D., Heidkamp, G. F., Fiorese, C., Bonito, A. J., Inaba, K., Nussenzweig, M. C., and Steinman, R. M. (2008). CD8+ CD205+ splenic dendritic cells are specialized to induce Foxp3+ regulatory T cells. J. Immunol. 181, 6923–6933.
Yin, N., Zhang, N., Xu, J., Shi, Q., Ding, Y., and Bromberg, J. S. (2011). Targeting lymphangiogenesis after islet transplantation prolongs islet allograft survival. Transplantation 92, 25–30.
Yoshida, H., Honda, K., Shinkura, R., Adachi, S., Nishikawa, S., Maki, K., Ikuta, K., and Nishikawa, S. I. (1999). IL-7 receptor alpha+ CD3(-) cells in the embryonic intestine induces the organizing center of Peyer’s patches. Int. Immunol. 11, 643–655.
Zecher, D., Li, Q., Oberbarnscheidt, M. H., Demetris, A. J., Shlomchik, W. D., Rothstein, D. M., and Lakkis, F. G. (2010). NK cells delay allograft rejection in lymphopenic hosts by downregulating the homeostatic proliferation of CD8+ T cells. J. Immunol. 184, 6649–6657.
Zelenay, S., Bergman, M. L., Paiva, R. S., Lino, A. C., Martins, A. C., Duarte, J. H., Moraes-Fontes, M. F., Bilate, A. M., Lafaille, J. J., and Demengeot, J. (2010). Cutting edge: intrathymic differentiation of adaptive Foxp3+ regulatory T cells upon peripheral proinflammatory immunization. J. Immunol. 185, 3829–3833.
Zeng, M., Smith, A. J., Wietgrefe, S. W., Southern, P. J., Schacker, T. W., Reilly, C. S., Estes, J. D., Burton, G. F., Silvestri, G., Lifson, J. D., Carlis, J. V., and Haase, A. T. (2011). Cumulative mechanisms of lymphoid tissue fibrosis and T cell depletion in HIV-1 and SIV infections. J. Clin. Invest. 121, 998–1008.
Zhang, N., Schroppel, B., Lal, G., Jakubzick, C., Mao, X., Chen, D., Yin, N., Jessberger, R., Ochando, J. C., Ding, Y., and Bromberg, J. S. (2009). Regulatory T cells sequentially migrate from inflamed tissues to draining lymph nodes to suppress the alloimmune response. Immunity 30, 458–469.
Zhao, Y., Swenson, K., Sergio, J. J., Arn, J. S., Sachs, D. H., and Sykes, M. (1996). Skin graft tolerance across a discordant xenogeneic barrier. Nat. Med. 2, 1211–1216.
Ziegler, E., Oberbarnscheidt, M., Bulfone-Paus, S., Forster, R., Kunzendorf, U., and Krautwald, S. (2007). CCR7 signaling inhibits T cell proliferation. J. Immunol. 179, 6485–6493.
Keywords: tolerance, lymph node, structure
Citation: Burrell BE, Ding Y, Nakayama Y, Park K-S, Xu J, Yin N and Bromberg JS (2011) Tolerance and lymphoid organ structure and function. Front. Immun. 2:64. doi: 10.3389/fimmu.2011.00064
Received: 30 September 2011; Accepted: 07 November 2011;
Published online: 07 December 2011.
Edited by:
Xian Chang Li, Brigham and Women’s Hospital, USAReviewed by:
Julian Dyson, Imperial College London, UKGraham Anderson, University of Birmingham, UK
Copyright: © 2011 Burrell, Ding, Nakayama, Park, Xu, Yin and Bromberg. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Jonathan S. Bromberg, University of Maryland, 29 Greene Street, Suite 9200, Baltimore, MD 21201, USA. e-mail:amJyb21iZXJnQHNtYWlsLnVtYXJ5bGFuZC5lZHU=