 Susanne Herold*
Susanne Herold* Konstantin Mayer and Juergen Lohmeyer
Konstantin Mayer and Juergen Lohmeyer
- Department of Internal Medicine II, University of Giessen Lung Center, Giessen, Germany
Lung macrophages are long living cells with broad differentiation potential, which reside in the lung interstitium and alveoli or are organ-recruited upon inflammatory stimuli. A role of resident and recruited macrophages in initiating and maintaining pulmonary inflammation in lung infection or injury has been convincingly demonstrated. More recent reports suggest that lung macrophages are main orchestrators of termination and resolution of inflammation. They are also initiators of parenchymal repair processes that are essential for return to homeostasis with normal gas exchange. In this review we will discuss cellular cross-talk mechanisms and molecular pathways of macrophage plasticity which define their role in inflammation resolution and in initiation of lung barrier repair following lung injury.
Introduction
Alveolar macrophages are tissue-resident or recruited cells with key functions in recognition of pathogens, initiation of host defense via protective inflammation, and in clearance of pathogens from the airways. Forming the first line of defense toward foreign invaders, alveolar macrophages scavenge and phagocytose pathogens and sense microbial patterns via toll-like receptors (TLRs), NOD-like receptors (NODs), and intracellular helicases like retinoic acid inducible gene I (RIG-I) and other pattern recognition receptors. Upon activation they release early response cytokines such as type I IFN, TNF-α, and IL-1β in an IRF- or NF-κB-dependent way. These cytokines stimulate neighboring alveolar epithelial cells and tissue-resident macrophages in an auto- and paracrine manner to produce a variety of chemokines which in turn mediate the recruitment of neutrophils, and later on, exudate macrophages and lymphocytes to the site of infection, ultimately resulting in clearance of pathogens.
Lung inflammation is not merely terminated when the pathogen is cleared and pro-inflammatory signaling events, previously initiated by recognition of foreign antigen or host-derived alarmins, decline. In fact, resolution of lung inflammation and return to tissue homeostasis is an active, tightly coordinated process which reverses all of the steps involved in initiation of the inflammatory response and induces counter-regulatory mechanisms which terminate these. This process includes cessation of granulocyte emigration from blood vessels, restoration of normal vascular permeability and removal of extravasated fluids, termination of monocyte emigration and induction of their maturation into resident alveolar macrophages, removal of apoptotic neutrophils, and finally, repair of “bystander” injury to restore functional endothelial and epithelial monolayers. Apart from their well-known role in phagocytosis and recognition of foreign antigens it is increasingly recognized that alveolar macrophages are endowed with high functional plasticity allowing them to acquire different pro- or anti-inflammatory as well as tissue-reparative phenotypes during the course of inflammation, dependent on the signals they receive from surrounding cells or from the pathogen itself. The ability to integrate these various signals in the course of inflammation and to mount a differential response empowers the mononuclear phagocyte, either lung resident or recruited, to terminate and resolve alveolar inflammation in the later phases of acute lung injury and to tightly coordinate parenchymal repair processes that are essential for return to homeostasis (Figure 1).
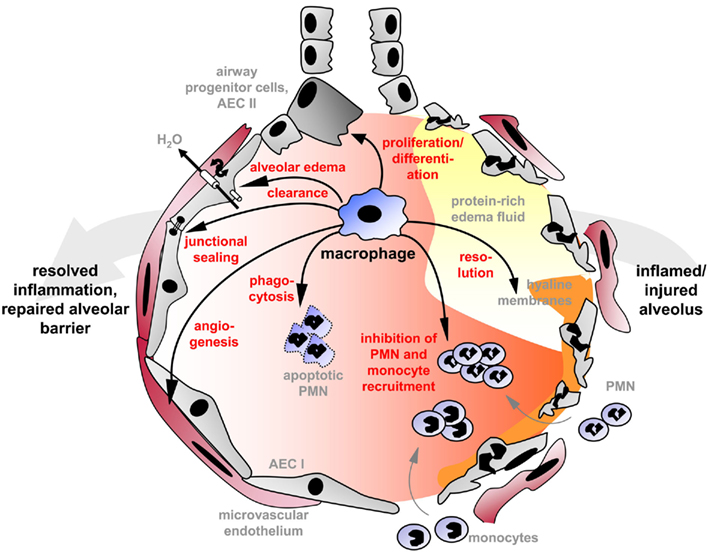
Figure 1. Macrophages terminate and resolve alveolar inflammation after acute inflammatory lung injury and coordinate structural and functional parenchymal repair processes that are essential for return to homeostasis. Inflammation resolution and tissue repair after injury involve a variety of timely coordinated, active processes in which lung macrophages are directly or indirectly involved: Inhibition of granulocyte (PMN) and monocyte recruitment from the circulation, phagocytosis of apoptotic neutrophils or parenchymal cells, removal of fibrin, clearance of alveolar edema fluid, and repair of the endo- and epithelial barrier by junctional sealing and induction of angiogenesis and proliferation/differentiation of epithelial progenitor cells including type II alveolar epithelial cells (AEC).
Change in Local Lipid and Mediator Profile Initiates Macrophage-Mediated Resolution of Inflammation
Lipoxins
Lipid mediators are key players in termination of pulmonary inflammation and initiation of resolution (Serhan et al., 2008), characterized by an active switch of the lipid mediator profile found at the inflamed site (Levy et al., 2001). During the initial inflammatory response, prostaglandins and leukotrienes, generated from arachidonic acid, an omega-6 polyunsaturated fatty acid (PUFA) by endothelial cells, neutrophils, and tissue-recruited and resident macrophages, amplify inflammation (Funk, 2001). Later on, the prostaglandins PGE2 and PGD2, generated in a cyclooxygenase-dependent way, gradually promote the synthesis of lipid mediators with anti-inflammatory and pro-resolving activity, such as the lipoxins. Lipoxins are lipoxygenase-derived double oxygenated eicosanoids which were shown to inhibit neutrophil recruitment to inflamed sites and suppress their pro-inflammatory actions, but promote recruitment of macrophage precursors (Maddox et al., 1997; Chiang et al., 2006). Lipoxin A4 rapidly stimulates macrophages to phagocytose apoptotic neutrophils (Godson et al., 2000), induces RhoA- and Rac-dependent cytoskeleton re-organization of macrophages (Maderna et al., 2002), and inhibits macrophage CXCL8 release (Jozsef et al., 2002), supporting macrophage-mediated resolution of inflammation. In turn, as a result of engulfment of apoptotic neutrophils, macrophages themselves become a primary source of lipoxins (Freire-de-Lima et al., 2006). With regard to acute lung injury, it was recently demonstrated that Lipoxin A4 acts as a potent pro-apoptotic signal for alveolar neutrophils, thereby increasing their engulfment by macrophages (El Kebir et al., 2009) and triggering further release of anti-inflammatory agents.
Resolvins and Protectins
Resolvins and protectins represent another class of pro-resolving lipid mediators derived from omega-3 PUFA, eicosapentaenoic acid (EPA), and docosahexaenoic acid (DHA; Serhan et al., 2002, 2008; Ariel and Serhan, 2007). Resolvin (Rv)E1 binds to the receptor ChemR23 expressed on macrophages and their precursors and attenuate TNF-mediated NF-κB activation, thus activating an anti-inflammatory signaling pathway (Arita et al., 2007). Ligation of the pro-inflammatory leukotriene B4 receptor BLT1 on leukocytes by RvE1 has antagonizing, anti-inflammatory effects (Arita et al., 2005, 2007). Similar to the lipoxins, RvD1 and the related DHA-derived lipid mediator protectin D1 stimulate clearance of inflammatory infiltrates by macrophage phagocytosis (Schwab et al., 2007). Recently, another anti-inflammatory lipid mediator termed macrophage mediator in resolving inflammation 1 (maresin 1) was identified (Serhan et al., 2009) which is synthesized by conversion of DHA by resident tissue macrophages involving 12/15-lipoxygenase. Similarly to resolvins, maresin 1 was found to decrease neutrophil accumulation while enhancing the recruitment of macrophage precursors to sites of inflammation in a murine peritonitis model. Furthermore, maresin 1 induces the uptake of zymosan particles by macrophages and might therefore promote macrophage uptake of apoptotic neutrophils (Serhan et al., 2009). Recent evidence highlights a crucial role of resolvins in mediating the emergence of a “pro-resolution” CD11blow tissue macrophage subset, which was characterized by a distinct protein expression profile, enhanced apoptotic leukocyte engulfment, unresponsiveness to TLR ligands, and increased emigration to draining lymph nodes (Schif-Zuck et al., 2011). Resolvins were demonstrated to be important players in resolution of chronic (Uddin and Levy, 2011) and acute lung injury, as demonstrated in mouse models of aspiration and intratracheal LPS challenge, by decreasing the pro-inflammatory potential of macrophages via cross-talk with the lipoxin A4 pathway (Seki et al., 2009; Wang et al., 2011). Increasing the Rv precursor omega-3 PUFA in the transgenic fat-1 mouse model likewise attenuated LPS-induced lung injury (Mayer et al., 2009). Acute administration of these PUFA seems to exert beneficial effects on alveolar macrophages and monocytes by decreasing the adhesion and release of pro-inflammatory cytokines like TNF-α. These effects were mediated in part by platelet activating factor (PAF), another important lipid mediator (Mayer et al., 2002; Schaefer et al., 2007).However, other pathways seem to be operative under chronic exposure as in fat-1 mice (Mayer et al., 2009) the TNF-α generation remained unchanged.
Chemerin
A similar role was recently ascribed to chemerin-derived peptides. Chemerin is a chemoattractant present in diverse inflammatory exudates. It was identified as a natural ligand for the G protein-coupled receptor ChemR23 expressed by epithelial cells (Campbell et al., 2007), dendritic cells (Vermi et al., 2005), and macrophages (Luangsay et al., 2009). ChemR23 binds RvE1 and shares phylogenetic homology with other chemoattractant receptors, including those for lipoxin A4 and the neutrophil chemotaxins C5a and C3a. Recently, another chemerin receptor, GPR1, was identified (Cash et al., 2008, 2010). In a murine model of LPS-induced acute lung injury, chemerin binding to ChemR23 decreased both neutrophil invasion into the lung and pro-inflammatory cytokine generation while increasing recruitment of macrophages (Luangsay et al., 2009).
Macrophages Actively Terminate and Resolve Neutrophil Infiltrates
Termination of Neutrophil Influx
Apart from initiating neutrophil influx after recognition of pathogens or intrinsic danger signals, macrophages acquire functional profiles which actively terminate neutrophil recruitment. As recently outlined by our group, GR-1highCCR2high exudate macrophages express IL-1ra upon recruitment into the lung parenchyma in LPS- and Klebsiella pneumoniae-induced lung injury. Upon blockade of IL-1β actions at the receptor IL-1R1 expressed on alveolar epithelium, macrophage-derived IL-1ra downregulates alveolar release of the neutrophil chemokine MIP-2 and of the epithelial adhesion molecule ICAM-1, attenuating alveolar neutrophil recruitment (Herold et al., 2011). In a model of LPS-induced lung inflammation (Dean et al., 2008), MMP12 that is mainly macrophage-derived cleaves CXC-chemokines within the ELR motif, which is crucial for receptor binding resulting in loss of neutrophil-recruiting activity. MMP-dependent chemokine cleavage also affects CC-chemokines such as CCL7, which may result in dampened inflammation. Similar findings were reported for CCL2, CCL8, and CCL13 following cleavage by MMP1 and MMP3 (McQuibban et al., 2000, 2002).
Induction of Neutrophil Apoptosis
Neutrophils are rather short-lived cells, but once they have reached inflammatory sites they might initially be exposed to survival signals such as G-CSF or IL-1β (Kantari et al., 2008) to prolong their anti-bacterial actions. By providing IL-1β antagonism at the receptor level, it is likely that exudate macrophage-derived IL-1ra might force neutrophil apoptosis (Herold et al., 2011) as a first step to clear the inflammatory infiltrate in the lung. Furthermore, alveolar macrophages are a primary source of TNF-α in different models of pulmonary inflammation (Herold et al., 2006; Cabanski et al., 2008; Cakarova et al., 2009) which, at higher concentrations such as found during human ARDS (Maus et al., 1998; Park et al., 2001), promotes apoptosis of neutrophils (van den Berg et al., 2001). Similarly, resident and GR-1highCCR2high exudate alveolar macrophages were found to highly express the death ligand TRAIL in murine and human influenza and RSV infection (Zhou et al., 2006; Herold et al., 2008; Bem et al., 2010) and TRAIL significantly contributed to neutrophil apoptosis in LPS-induced lung injury (McGrath et al., 2011).
Phagocytosis of Apoptotic Neutrophils – “Find Me” and “Eat Me”
Coordinated removal of apoptotic cells by alveolar macrophages prevents the release of their toxic, tissue-damaging intracellular contents. In contrast to necrosis, apoptosis of neutrophils provides signals to alveolar macrophages to initiate clearance to limit tissue injury and to promote resolution, rather than persistence, of inflammation. First, apoptotic neutrophils advertise their own presence at the earliest stages of death and attract their scavengers via specific “find me” signals. Apart from the well-described lysophosphatidylcholine, recognized by the G-protein-coupled macrophage chemotaxis receptor G2A (Peter et al., 2008), these include fractalkine (CX3CL1), the nucleotides ATP and uridine 5′ triphosphate (UTP), S19 ribosomal protein dimer, split tyrosyl-tRNA synthetase, thrombospondin 1, and sphingosine-1-phosphate (S1P; Savill and Fadok, 2000; Ravichandran, 2010; Soehnlein and Lindbom, 2010). Just recently, Pannexin 1 channels were identified as mediators to release nucleotides as “find me” signals (Elliott et al., 2009). All of these are capable of attracting macrophages or their precursors, although only fractalkine and nucleotides have been shown to act as “find me” signals in vivo (Truman et al., 2008; Elliott et al., 2009). Whereas ATP and UTP are recognized by the G-protein-coupled macrophage receptor P2Y2, the receptor for CX3CL1, CX3CR1, defines a GR-1lowCCR2low circulating lung macrophage precursor (Landsman et al., 2007) which has been attributed a wound healing and tissue-reparative phenotype similar to the one ascribed to “alternatively activated” macrophages (Geissmann et al., 2010).
Surfaces of dying cells express or allow the access to a number of “eat me” signals that replace the native “don’t eat me” signals such as CD31 or CD47/SIRP-α present on living cells (Janssen et al., 2008). These signals may be membrane-associated (e.g., phosphatidylserine) or are released from intracellular compartments at later stages of programmed cell death (Savill and Fadok, 2000). Macrophages express a variety of receptors that bind either directly to the exposed “eat me” flags or indirectly through bridging molecules. These receptors include a phosphatidylserine receptor, the tyrosine kinase receptor MeR, integrins, scavenger receptors, and complement receptors (Mevorach et al., 1998; Grimsley and Ravichandran, 2003; Li et al., 2003; Greenberg et al., 2006; Miyanishi et al., 2007; Kennedy and DeLeo, 2009). Soluble innate immune pattern recognition proteins identifying non-self or altered-self molecular patterns are found in the immune-privileged surfaces of the lung and serve as bridging molecules. These include ficolins, pentraxins, thrombospondin, sCD14, MFG-E8, natural IgM, collections, C1q, and annexin A1 (Janssen et al., 2008; Kennedy and DeLeo, 2009; Litvack and Palaniyar, 2010). Annexin A1, released from neutrophil granules upon activation, inhibits the recruitment of leukocytes including inflammatory macrophage precursors, promotes neutrophil apoptosis, and acts on macrophages to enhance removal of dead neutrophils (Perretti and D’acquisto, 2009). In a mouse model of LPS-induced acute lung injury alveolar recruited, exudate rather than resident macrophages were shown to clear apoptotic granulocytes from the airways (Janssen et al., 2008).
Ingestion of Apoptotic Neutrophils Changes the Macrophage Phenotype
Following ingestion of apoptotic neutrophils, macrophages are stimulated to release anti-inflammatory and pro-repair mediators. One of the first studies in this field showed that co-culture of LPS-activated monocytes with apoptotic lymphocytes inhibited monocyte expression of the pro-inflammatory TNF-α and increased the release of the immunosuppressive cytokines TGF-β and IL-10 (Voll et al., 1997). In following studies, ingestion of apoptotic neutrophils by macrophages, more recently termed “efferocytosis,” had a similar effect on human monocyte-derived and murine alveolar macrophages, inducing the anti-inflammatory mediators TGF-β, PGE2, and PAF (Fadok et al., 1998; Medeiros et al., 2009). Phagocytosis of apoptotic – but not of necrotic – cells not only prevented these macrophages from killing tissue-resident cells but also triggered the release of growth factors such as vascular endothelial growth factor (VEGF; Golpon et al., 2004) or hepatocyte growth factor (HGF; Amano et al., 2004) being crucial for tissue repair after injury. Impairment of efficient phagocytosis of apoptotic airway cells may therefore contribute to the pathogenesis of chronic airways diseases like COPD, asthma, and cystic fibrosis (Krysko et al., 2010; Mukaro and Hodge, 2011).
The signaling pathways activated during the phagocytosis-dependent induction of an anti-inflammatory macrophage program in the resolution phase of tissue injury were studied in detail (Patel et al., 2007). The anti-inflammatory activity of apoptotic cells lead to an inhibition of the release of pro-inflammatory mediators from phagocytosing macrophages (Voll et al., 1997; Fadok et al., 1998). In contrast, necrotic cells, which are recognized by another distinct mechanism, rather enhance a pro-inflammatory macrophage program (Cocco and Ucker, 2001). Acquisition of anti-inflammatory activity consists in the loss of the pro-inflammatory response to inflammatory stimuli and a shift to an anti-inflammatory profile that is induced by the apoptotic neutrophil. This anti-inflammatory potential is maintained at all stages of neutrophil apoptotic cell death, irrespective of cell membrane integrity (Cocco and Ucker, 2001; Cvetanovic and Ucker, 2004; Patel et al., 2006). Recognition of apoptotic cells targets the pro-inflammatory transcriptional machinery of interacting macrophages, without apparent effect on proximal steps of TLR signaling. This modulatory activity is exerted directly upon binding to the macrophage and decreases IL-6, IL-8, and TNF-α expression in an NF-κB-dependent way. These effects were dependent on apoptotic cell recognition and independent of engulfment (Cocco and Ucker, 2001; Cvetanovic and Ucker, 2004). Apart from the counter-inflammatory response, phagocytosis (but not mere recognition of apoptotic cells) provides a PI3K/AKT-dependent survival signal to prolong the macrophage life-span to facilitate clearance of neutrophil corpses (Reddy et al., 2002). In contrast, the effects of apoptotic versus necrotic targets on the MAPK pathway depended on recognition. Exposure to apoptotic cells strongly inhibited phosphorylation of ERK1/2 but induced activation of JNK1/2 and p38, a process which did not require phagocytosis. Exposure to necrotic cells stimulated proliferation and activated ERK1/2 (Reddy et al., 2002; Patel et al., 2006).
Mechanisms and Effects of Alternative Macrophage Programming
Macrophage Subsets and Polarization
Pathogen elimination and restoration of homeostasis following infection and tissue damage requires resident tissue macrophages and a coordinated mobilization of two circulating precursor subsets defined according to lineage marker and chemokine receptor expression in mice, namely the GR-1lowCCR2lowCX3CR1high and the GR-1highCCR2highCX3CR1low peripheral blood monocytes. GR-1lowCCR2lowCX3CR1high monocytes patrol the resting vasculature, populate normal or inflammatory sites CX3CR1-dependently, and participate in resolution of inflammation and tissue repair (Auffray et al., 2007; Geissmann et al., 2010). GR-1highCCR2highCX3CR1low monocytes are predominantly inflammatory and migrate to injured and infected sites. In humans, most monocytes are CD14hiCD16− and are referred to as “classical” monocytes, whereas CD14+CD16+ monocytes are referred to as “non-classical” monocytes. CCR2 and its major ligand, CCL2, are evidently important in both emigration of these cells from the bone marrow into the blood stream and their immigration into inflamed tissues, where they undergo differentiation into macrophages that are categorized as either classically activated (CAM, M1) or alternatively activated (AAM, M2; Benoit et al., 2008; Martinez et al., 2008; Gordon and Martinez, 2010). Several genes define CAM and AAM, e.g., inos, tnf, il-12, and arg1, ym1, ym2, fizz1, mrc1, ccl22, respectively, although a clear-cut association of those genes with the functional profile of the respective subset is lacking, except for Fizz1, also known as RELM-α (Nair et al., 2009). The M1 program is associated with release of pro-inflammatory mediators such as iNOS-derived NO, TNF-α, IFN-γ, and IL-12 and critically contributes to pathogen elimination (Benoit et al., 2008; Serbina et al., 2008). In contrast, AAM, which secrete anti-inflammatory cytokines like IL-1ra, IL-10, and TGF-β, downregulate IL-12, upregulate scavenger receptors, promote angiogenesis, and support wound healing and tissue remodeling (Mosser and Edwards, 2008). They are renowned for their heterogeneity and plasticity, which is reflected by their further subdivision into M2a, M2b, and M2c subsets (Mosser and Edwards, 2008; Ricardo et al., 2008; Gordon and Martinez, 2010).
Signal Integration in the Shaping of Pulmonary Macrophage Phenotypes
As a key component of the inflammatory response that determines lung tissue destruction or recovery, increasing evidence suggests that pulmonary macrophages do not remain committed to a single activation profile. They may regress to a resting state and can subsequently be reactivated with a different polarization. Functionally distinct subsets of macrophages may exist in the same tissue and play critical roles in both initiation and recovery of inflammation. Therefore, the origin and activation state of the macrophages and the microenvironment, in which they reside, are critical determinants of their response to lung injury. The heterogeneity of macrophages, their diverse role in lung inflammation and tissue remodeling, and the coordinated activation and programming by other inflammatory and parenchymal cells are not fully understood. However, it becomes increasingly evident that cross-talk of various signals at different levels impinges on the generation of functional macrophage programs, with a variety of signals being integrated to shape a distinct phenotype at a defined stage of inflammation.
With respect to lung inflammation and injury, several of those signal steps have been defined. First, growth factors such as granulocyte macrophage colony-stimulating factor (GM-CSF) or M-CSF drive differentiation and activation of macrophage progenitors or lineage precursors but also of well-differentiated alveolar macrophages (Berclaz et al., 2002, 2007; Baleeiro et al., 2006; Ballinger et al., 2006). In addition, GM-CSF was shown to induce an M1 phenotype (Krausgruber et al., 2011). Second, at the stage of transendo/epithelial recruitment to the airspace, macrophages obtain signals from chemokines or CAMs (Srivastava et al., 2005). Then, at the site of inflammation, macrophages are primed by cytokines like IFN-γ (M1) or IL-4 and IL-13 (M2), or via Th2 cell-expressed IL-25 and IL-33 (M2; Gordon and Martinez, 2010). Next, PAMPs or DAMPs deliver signals via TLR, NLR, or other pattern recognition receptors. Exposure to LPS promotes the differentiation toward M1-like cells, whereas addition of further cytokines differentiates them toward M2-like macrophages (Martinez et al., 2008; Cabanski et al., 2009; Arora et al., 2011). Recently, a critical role for type I IFN/IFNAR signaling in differentiation of peripheral blood monocytes toward defined lung macrophage phenotypes, either classical or alternative, with different functions in control of alveolar inflammation, was demonstrated in influenza virus-induced lung injury (Seo et al., 2011). Later on, phagocytosis of apoptotic neutrophils by macrophages may add on these signals and support an anti-inflammatory, resolving and tissue-reparative phenotype with release of IL-10, TGF-β, VEGF, and HGF, as outlined above. IL-10 and TGF-β, for example, were shown to be protective in P. aeruginosa- (Buff et al., 2010) or LPS-induced lung injury (D’alessio et al., 2009) by abrogating alveolar neutrophil recruitment and by mediating counter-inflammatory effects of CD4+CD25+FoxP3+ regulatory T cells, respectively (Figure 2).
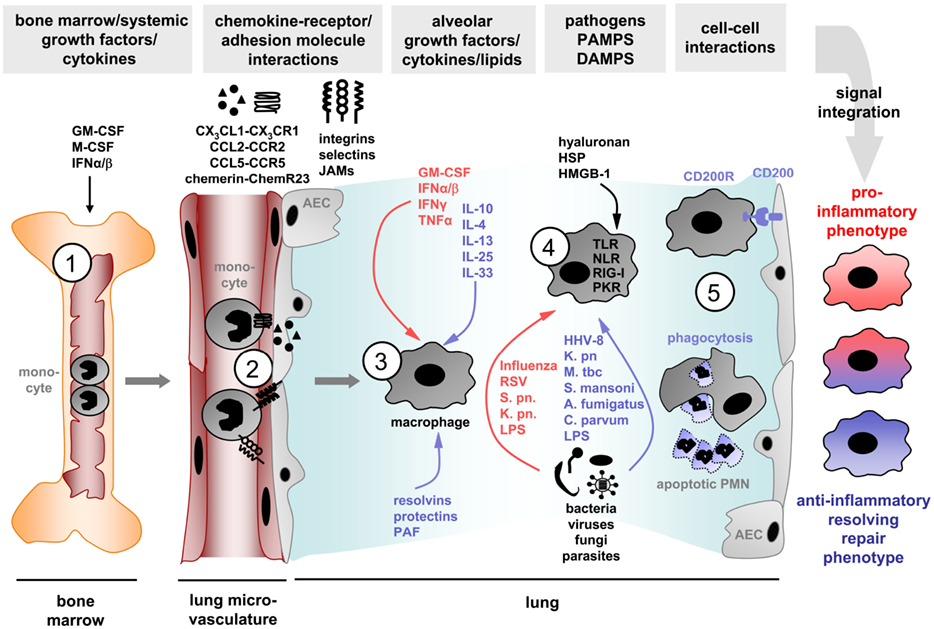
Figure 2. Different extracellular signals are integrated to shape pulmonary macrophage phenotypes during lung inflammation. First, growth factors such as GM-CSF, M-CSF, or type I interferons (IFN) drive differentiation and activation of macrophage progenitors or lineage precursors (1), second, macrophages obtain signals from chemokines or cellular adhesion molecules upon transendo/epithelial recruitment to the alveoli (2), third, macrophages receive signals from cytokines like GM-CSF and interferons (M1) or IL-4, IL-13, IL-25, or IL-33 (M2) (3); fourth, pathogens, PAMPs, or DAMPs deliver signals via TLR, NLR, or other pattern recognition receptors (4). Later on, cell–cell communications during phagocytosis of apoptotic neutrophils (PMN) or via CD200–CD200R interaction with AEC add on these signals and may support an anti-inflammatory macrophage phenotype (5). JAMs, junctional adhesion molecules; HSP, heat shock proteins; HMGB-1, high mobility group box-1; S. pn., Streptococcus pneumoniae; K. pn., Klebsiella pneumoniae; AEC, alveolar epithelial cells.
With respect to the role of macrophage phenotypes in acute lung injury, the M1 program clearly correlates with pathogen clearance, but as well with inflammation and mortality, e.g., after S. pneumoniae infection (Smith et al., 2007). A mouse model of Sendai virus infection demonstrated that NKT cells produce IL-13 through a CD1d-glycolipid-dependent mechanism, initiating pulmonary M2 amplification in late stages of infection, when virus had already been cleared from the lungs (Kim et al., 2008). Similarly, macrophage-derived IL-13 induced an M2 phenotype via STAT6 in an autocrine way upon RSV-induced lung injury (Shirey et al., 2010). GR-1highCCR2high exudate macrophages, which are tissue-recruited in LPS- or K. pneumoniae-induced lung injury, express high levels of IL-1ra, a classical M2 marker (Benoit et al., 2008) which directly antagonizes IL-1β derived from (M1 activated) resident alveolar macrophages, thereby exerting anti-inflammatory and tissue-protective effects. Adoptive transfer studies using IL-1ra−/− monocytes revealed that IL-1ra-expressing, M2 polarized exudate macrophages reduced alveolar epithelial cell damage and increased pulmonary barrier function (Herold et al., 2011).
These data suggest that, in the inflamed lung, different macrophage phenotypes are induced by distinct signals at defined time points to fulfill discriminative tasks during infection, resolution, and repair. Moreover, these differentially programmed macrophage populations cross-talk during the time course of acute pulmonary inflammation. While resident versus recruited macrophages were found to be differentially polarized (Herold et al., 2011), the question arises to what extent a functional macrophage program might be lineage-confined in monocyte/macrophage subsets (Nahrendorf et al., 2007).
Transcriptional and Epigenetic Regulation of Macrophage Polarization
The signal transduction pathways and transcription factors involved in macrophage polarization, especially with respect to lung inflammation, are still incompletely understood. Activation of the transcription factor NF-κB subunit p50 has been associated with the inhibition of M1-polarizing genes including IFN-β in vivo (Porta et al., 2009), whereas induction of the basic region-leucine zipper transcription factors CREB and C/EBPβ has been shown to upregulate M2 genes in macrophages, which promoted tissue repair after injury (Ruffell et al., 2009). Subsequent data suggested that in mice, an IRF4-dependent pathway initiates an M2 program by stimulating the expression of M2-specific markers (Satoh et al., 2010). In contrast, M1 macrophages were characterized by increased expression of IRF5, which was induced by GM-CSF during their differentiation. Forced expression of IRF5 in M2 macrophages drove M1-specific cytokines, chemokines, and costimulatory molecules and led to a potent Th1–Th17 response, whereas induction of M1-markers was impaired in irf5−/− macrophages (Krausgruber et al., 2011). Together with the data of Satoh et al. (2010) these findings establish a new paradigm of IRF5–IRF4 balance mediating M1–M2 polarization. Liao et al. (2011) identified Krüppel-like factor 4 (KLF4) as a critical regulator of macrophage polarization. KLFs represent a large family of transcription factors involved in development, differentiation, and activation of leukocytes. Macrophage KLF4 expression was robustly induced in M2 macrophages and strongly reduced in M1 macrophages, and was found to cooperate with Stat6 to induce an M2 genetic program and inhibit M1 targets via sequestration of coactivators required for NF-κB activation. KLF4-deficient macrophages demonstrated enhanced pro-inflammatory gene expression and increased bactericidal activity. Whether these transcriptional programs are operative during processes of lung macrophage polarization in lung infection, injury, and repair, however, remains to be established.
With respect to epigenetic control of macrophage polarization, reports showed that induced M2 signature genes of IL-4-treated mouse macrophages like arg1, ym1, fizz1, and mrc1, revealed reciprocal changes in histone H3K4 and H3K27 methylation (Ishii et al., 2009). These modifications depended on STAT6 activation, which bound to the demethylase Jmjd3 promoter, contributing to decreased H3K27 methylation, as well as to transcriptional activation of M2 marker genes. Moreover, the kinase AKT regulated LPS-induced microRNA in macrophages and was implicated in LPS tolerance. AKT1 and AKT2 isoforms thereby had differential effects on TLR4 and SOCS1 signaling in macrophages, depending on the microRNAs let7e and miR155 (Androulidaki et al., 2009). A possible effect of IL-4 and IL-13 on AKT isoforms has not been reported. Other studies have linked microRNA-dependent regulation with macrophage activation programs (Taganov et al., 2006; Tili et al., 2007).
Airway Epithelial–Macrophage Cross-Talk Controls Macrophage Responses
Recently, a new concept of the pulmonary “innate immune rheostat” arose from findings by Snelgrove et al. (2008) demonstrating that the phenotype of airway macrophages depends on the fine-tuned balance between negative regulatory pathways and those that amplify immunity (Snelgrove et al., 2008; Wissinger et al., 2009). As innate immunity at lung surfaces requires restraint to prevent inflammation to innocuous antigens or commensals to guarantee gas exchange, the threshold above which airway macrophages become activated must be increased by local factors. Furthermore, excessive and prolonged pathogen-induced inflammation has to be controlled to resolve infiltrates after pathogen clearance and to prevent collateral lung tissue damage. Data from an influenza virus pneumonia model demonstrated that one such key regulator is CD200R, transmitting a suppressive signal and critically regulating activation of airway macrophages on which it is expressed at high levels. CD200R levels are maintained by epithelial expression of IL-10 and TGF-β. Its ligand, CD200, is exposed on the apical side of the airway epithelium and limits alveolar macrophage-mediated inflammation. Cd200−/− mice displayed increased pro-inflammatory macrophage activity and enhanced sensitivity to influenza infection, with delayed resolution of inflammation and increased mortality. These data suggest that macrophage pro- versus anti-inflammatory phenotypes are under tight control of nearby airway epithelial cells during the course of infection, which on one side represent primary targets for infection (that has to be effectively cleared by a mounted immune response) but on the other side have to maintain lung barrier integrity and organ function. Epithelial–macrophage cross-talk by soluble and surface-expressed factors therefore seems to be an important mechanism to keep the balance between efficient host defense and excessive inflammation and injury during pneumonia.
Lung Macrophages in Tissue Repair and Remodeling
Repair of the Airway Epithelium
Acute lung inflammatory diseases or infection and the following innate and adaptive host responses leave a damaged alveolar endo/epithelial barrier. Alveolar epithelial cell apoptosis was found as major underlying cause of severe lung parenchymal damage in sterile or infectious lung injury (Albertine et al., 2002; Herold et al., 2008; Ma et al., 2010; Budinger et al., 2011). Re-epithelialization (given an intact basement membrane) and endothelial re-sealing following bronchoalveolar injury is considered as critical step to re-establish normal gas exchange conditions in the lung. Alveolar epithelium is comprised of two morphologically and functionally distinct cell types, alveolar epithelial cells type I (AEC I) and type II (AEC II). The flattened AEC I, covering a high percentage of the alveolar surface, are presumed to be terminally differentiated and exhibit a very limited potential to divide, features that make them particularly susceptible to irreparable damage (Fehrenbach, 2001; Tesfaigzi, 2003). The smaller, cuboidal AEC II retain progenitor cell properties and therefore, together with the CCSP+ Clara cells, represent a distal transit-amplifying cell pool (Stripp, 2008; Rock and Hogan, 2011). AEC II are assumed to play a central role in alveolar repair processes after injury by trans-differentiation into AEC I. This involves tightly regulated alveolar epithelial cell proliferation, migration, and polar differentiation with restoration of junctional structures (Fehrenbach, 2001). More recently, studies in mice revealed that endogenous airway epithelial progenitor cells are located within the adult lung in the basal layer of the upper airways, or within bronchoalveolar junctions. These cells, termed bronchoalveolar stem cells (BASCs) and expressing both AEC II and Clara cell properties, are defined as EpCamhigh CD104+ integrin α6β4+, are resistant to damage, proliferate after injury in vivo, are multipotent in clonal assays in vitro and give rise to different ciliated and non-ciliated epithelial cell populations of the distal lung (McQualter et al., 2010; Chapman et al., 2011). In humans, recent data define lung stem cells as positive for c-kit (Kajstura et al., 2011), whereas others found them included in the p63+ck5+ basal cell pool (Whitsett and Kalinichenko, 2011). Following acute lung injury, in accordance with data obtained from a rat model (Berthiaume et al., 2006), we demonstrated that alveolar repair processes in terms of AEC II proliferation were initiated 4 days after LPS instillation, when alveolar inflammation decreased virtually to baseline levels. However as a first step, trans-differentiation of existing AEC II into AEC I might occur fast and precedes AEC II proliferation peaking at 48–96 h post injury. We were able to delineate this feature from the notion that alveolar leakage was associated with AEC I apoptosis and declined upon recovery of the AEC I pool (Cakarova et al., 2009).
Since the earliest reports on alternatively activated M2 macrophages, it has been assumed that these cells promote repair of host tissues after inflammation, e.g., by expression of fibronectin 1 (FN-1), the TGF-β-induced matrix associated proteins BIG-H3, and IGF-1, which provide signals for tissue repair and proliferation (Gordon, 2003). However, although involvement of resident or tissue-recruited macrophages in these processes has been demonstrated for several organ systems like liver, skin, heart, kidney, and gut mucosa (Duffield, 2003; Takaba et al., 2010; Harel-Adar et al., 2011; Lee et al., 2011; Lu et al., 2011; Mahdavian Delavary et al., 2011), studies demonstrating a direct contribution of macrophages in lung epithelial regeneration after injury, e.g., by using macrophage depletion strategies, are lacking.
Several studies at least indirectly suggest that these cells are similarly involved in repair of the injured lung. In this regard, the cytokines keratinocyte growth factor (KGF, FGF7), VEGF, epidermal growth factor (EGF), heparin-binding EGF-like growth factor, platelet-derived growth factor (PDGF), GM-CSF, fibroblast growth factors 2 and 10 (FGF2, FGF10) were shown to act as potent lung epithelial mitogens (Panos et al., 1993; Melloni et al., 1996; Huffman Reed et al., 1997; Van Winkle et al., 1997; Li et al., 2001; Ray, 2005; Mura et al., 2006; Pogach et al., 2007; Gupte et al., 2009; Crosby and Waters, 2010). Anti-inflammatory or regenerative alveolar macrophages were noted to directly release the epithelial growth factors PDGF, FGFs, HGF, TGF-β, and VEGF following inflammation or lung injury (Melloni et al., 1996; Leslie et al., 1997; Morimoto et al., 2001; Miyake et al., 2007; Medeiros et al., 2009; Granata et al., 2010). Notably, our own studies demonstrate that epithelial repair processes were primed already in the pro-inflammatory phase of acute lung injury and elucidate a key role of alveolar macrophage TNF-α inducing AEC repair via induction of autocrine epithelial GM-CSF signaling (Cakarova et al., 2009). In support of these findings, GM-CSF has been recognized as potent growth factor for AEC in vitro and in lung injury models in vivo (Huffman Reed et al., 1997; Paine et al., 2003). Furthermore, we demonstrated proliferative effects of the macrophage cytokine MIF (macrophage migration inhibitory factor) which were mediated by the MIF receptor CD74 expressed on AEC II (Marsh et al., 2009). The M2 phenotype-associated cytokines IL-4 and IL-13 stimulated proliferation and migration of both murine and human bronchial epithelial cells (Booth et al., 2001; White et al., 2009). As opposed to data derived from our group, demonstrating a detrimental, tissue-damaging role of pro-apoptotic, highly inflammatory GR-1highCCR2+ exudate macrophages in murine influenza virus pneumonia (Herold et al., 2008), Narasaraju et al. (2010) argued that HGF produced by this macrophage population may contribute to the resolution of inflammation and regeneration of alveolar epithelium.
Restoration of Structural and Functional Lung Barrier Integrity
Successful lung barrier repair after injury is critically linked to the survival of the patient (Ware and Matthay, 2001). Epithelial junction formation during alveolar repair represents a crucial event for restoration of alveolar barrier function. Tight junctions are important to maintain discrete compartments in the lung and tightly regulate the flow of molecules between apical and basolateral compartments, whereas gap junctions permit direct transmission of small signaling molecules between neighboring cells. Transmembrane proteins of the occludin and claudin families are the major transmembrane structural elements of tight junctions. It has previously been shown that alveolar epithelial cells express occludins and zona occludens 1 protein (ZO-1) as part of the tight junctional complex. Tight junctions are highly dynamic structures, whose degree of sealing varies in response to external stimuli (e.g., cytokines) via MAPK, PI3K, and PKC-mediated re-organization of their sub-structures (Gonzalez-Mariscal et al., 2008). Ganter et al. (2008) previously demonstrated that IL-1β causes alveolar endothelial and epithelial permeability increase via integrin-mediated epithelial TGF-β release which induced phosphorylation of endothelial VE-cadherin and stress fiber formation. Although reports on the role of macrophages herein are limited, there is evidence that IL-1ra-expressing exudate macrophages prevent disruption and disassembly of the tight junctional protein ZO-1 in alveolar epithelial cells by IL-1β antagonism (Ganter et al., 2008; Herold et al., 2011). Macrophage-released growth factors might in turn increase tightness of junctions in airway epithelial cells (Terakado et al., 2011).
To restore normal gas exchange in the alveoli, edema fluid accumulating in the airspaces during lung injury is cleared by active sodium transport via apical membrane epithelial Na+ channels (ENaC). The electrochemical gradient for Na+ influx is maintained by the basolateral Na, K-ATPase. Transport of sodium promotes a transepithelial osmotic gradient, causing water to move passively from the airspaces to the interstitium thereby removing excess alveolar fluid (Morty et al., 2007; Eaton et al., 2009). Infection of the lung epithelium and release of pro-inflammatory mediators such as IL-1β and TNF-α, but as well TGF-β, were shown to inhibit ENaC function (Dickie et al., 2000; Kunzelmann et al., 2000; Chen et al., 2004; Roux et al., 2005; Hickman-Davis et al., 2006; Wolk et al., 2008). Similarly, LPS-stimulated, pro-inflammatory alveolar macrophages were found to decrease ENaC expression and activity (Dickie et al., 2000). In contrast, the epithelial growth factors KGF and EGF upregulated transepithelial sodium transport and increased alveolar fluid clearance in animal models of acute lung injury by affecting the Na, K-ATPase (Morty et al., 2007), and at least EGF was shown to be expressed by lung tissue macrophages in vivo (Temelkovski et al., 1997). Moreover, recombinant IL-1ra increased ENaC α and β subunit expression in primary murine and human AEC by antagonizing IL-1β in vitro, and IL-1ra-expressing M2-programmed GR-1highCCR2+ exudate macrophages similarly reverted IL-1β-induced downregulation of ENaC expression in lung parenchyma in an LPS injury mouse model in vivo (Herold et al., 2011).
Macrophages or subsets thereof are involved in neoangiogenesis either by secreting cytokines/growth factors or by providing a physical scaffold fostering endothelial cell fusion. However, signaling pathways activating an angiogenic program in macrophages, especially in the lung, are still poorly defined, and most data derive from in vitro studies in the field of tumor angiogenesis. Hence, it was shown that apoptotic cells release the lipid mediator sphingosine-1-phosphate (S1P), which activates S1P1/3 on macrophages to upregulate cyclooxygenase-2. The liberation of PGE2 then stimulates migration of endothelial cells in vitro (Brecht et al., 2011). Other findings demonstrated a role for M2 macrophages in angiogenesis which was linked to release of IL-8 (Medina et al., 2011). In the lung microvasculature, the angiopoietin (Ang)-Tie ligand-receptor system has a key regulatory role in endothelial integrity and quiescence. Whereas Ang-1-mediated Tie2 activation is required to maintain the quiescent state of the resting endothelium, Ang-2 destabilizes the quiescent endothelium and primes it to respond to exogenous stimuli, thereby facilitating the activities of inflammatory mediators, but as well of the angiogenic cytokine VEGF to promote endothelial barrier repair (Fiedler and Augustin, 2006). As outlined above, the ingestion of apoptotic cells results in release of VEFG from macrophages, one of the most important growth factors for endothelial cells (Golpon et al., 2004; Granata et al., 2010). However, although recent data suggest a link between decreased pulmonary VEGF and impaired endothelial barrier function and angiogenesis (Jesmin et al., 2011), a clear role of macrophage-derived VEGF and pulmonary microvascular angiogenesis after injury has not been defined.
Lung Tissue Remodeling and Fibrosis
Tightly controlled remodeling processes are important to restore tissue homeostasis after injury and involve transient fibroblast proliferation and production and degradation of matrix components. Excessive scarring and tissue fibrosis may result from an imbalanced action of M1 and M2 polarized macrophages after prolonged lung inflammation. Whereas M1 macrophages play a role in resolution of scarring and matrix degradation by release of a variety of anti-fibrotic cytokines such as CXCL10 (Tighe et al., 2011) or of matrix metalloproteinases (Strieter, 2008), M2 macrophages were found to support fibroproliferative tissue remodeling (Meneghin and Hogaboam, 2007; Strieter, 2008) by increased expression of TGF-β, fibronectin, proline, arginase, and tissue inhibitors of metalloproteinase (TIMPs). Prolonged IL-13 effects on alveolar macrophages, as found in several lung infectious diseases (Meneghin and Hogaboam, 2007), promotes the presence of M2-programmed macrophages and, ultimately, excessive fibrogenesis. They also express the pro-fibrotic cytokines IL-10 (Sun et al., 2011), and CCL17 which binds CCR4, and the interaction between these two drives fibrogenesis in several mouse models of lung disease (Meneghin and Hogaboam, 2007). Arginase-1-mediated metabolism of L-arginine in M2 macrophages may result in the formation of L-proline, which is used by myofibroblasts to produce collagen (Strieter, 2008). M2 macrophage numbers were increased in the lungs of patients with idiopathic pulmonary fibrosis (IPF; Prasse et al., 2006; Pechkovsky et al., 2010). Analyses of cellular cross-talk within the lung mononuclear phagocyte system revealed that Gr-1+ circulating macrophage precursors directed M2-programmed, pro-fibrotic, tissue-resident macrophages to enhance lung fibrosis in a mouse model of bleomycin injury (Gibbons et al., 2011). Apart from macrophage-mediated fibrogenic tissue remodeling, as often observed after acute or chronic infectious lung disease (Meneghin and Hogaboam, 2007), a role for M2 polarized macrophages was described in similar processes resulting in COPD or pulmonary hypertension (Benoit and Holtzman, 2010; Vergadi et al., 2011). However, although there is increasing interest in the field of macrophage polarization in tissue remodeling, the definite role of the M1/M2 balance in the process of (scarless) alveolar regeneration after acute lung injury/pneumonia is still poorly defined.
Conclusion
Although numerous studies clearly demonstrate a crucial role of resident and recruited lung macrophages with an anti-inflammatory, regenerative potential in resolution of pulmonary inflammation and in initiation of tissue repair processes, much remains to be learned about the underlying signaling pathways and the mechanisms of cell–cell communications herein. Important questions to be answered include the putative cross-talk between these macrophages and other anti-inflammatory, injury-resolving immune cells such as regulatory T cells, or their interaction with local endothelial or airway epithelial progenitors during the process of alveolar regeneration. Further issues to be addressed in future studies concern organ-related plasticity of macrophages in vivo, the question whether or to what extent macrophage phenotypes are lineage-confined or induced by an organ-specific inflammatory milieu, and the definition of robust markers for ′beneficial′ macrophage phenotypes in the context of acute lung disease, to ultimately decipher how their polarization can be manipulated to improve the outcome of acute lung disease.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the German Research Council [SFB/TR84 “Innate Immunity of the Lung,” grant LO271/4-1, and Excellence Cluster Cardio-Pulmonary System (ECCPS)], and by the German Federal Ministry of Research and Education (Clinical Research Group “Infectious Diseases” grant 01 KI 0770).
References
Albertine, K. H., Soulier, M. F., Wang, Z., Ishizaka, A., Hashimoto, S., Zimmerman, G. A., Matthay, M. A., and Ware, L. B. (2002). Fas and fas ligand are up-regulated in pulmonary edema fluid and lung tissue of patients with acute lung injury and the acute respiratory distress syndrome. Am. J. Pathol. 161, 1783–1796.
Amano, H., Morimoto, K., Senba, M., Wang, H., Ishida, Y., Kumatori, A., Yoshimine, H., Oishi, K., Mukaida, N., and Nagatake, T. (2004). Essential contribution of monocyte chemoattractant protein-1/C-C chemokine ligand-2 to resolution and repair processes in acute bacterial pneumonia. J. Immunol. 172, 398–409.
Androulidaki, A., Iliopoulos, D., Arranz, A., Doxaki, C., Schworer, S., Zacharioudaki, V., Margioris, A. N., Tsichlis, P. N., and Tsatsanis, C. (2009). The kinase Akt1 controls macrophage response to lipopolysaccharide by regulating microRNAs. Immunity 31, 220–231.
Ariel, A., and Serhan, C. N. (2007). Resolvins and protectins in the termination program of acute inflammation. Trends Immunol. 28, 176–183.
Arita, M., Bianchini, F., Aliberti, J., Sher, A., Chiang, N., Hong, S., Yang, R., Petasis, N. A., and Serhan, C. N. (2005). Stereochemical assignment, antiinflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. J. Exp. Med. 201, 713–722.
Arita, M., Ohira, T., Sun, Y. P., Elangovan, S., Chiang, N., and Serhan, C. N. (2007). Resolvin E1 selectively interacts with leukotriene B4 receptor BLT1 and ChemR23 to regulate inflammation. J. Immunol. 178, 3912–3917.
Arora, S., Olszewski, M. A., Tsang, T. M., Mcdonald, R. A., Toews, G. B., and Huffnagle, G. B. (2011). Effect of cytokine interplay on macrophage polarization during chronic pulmonary infection with Cryptococcus neoformans. Infect. Immun. 79, 1915–1926.
Auffray, C., Fogg, D., Garfa, M., Elain, G., Join-Lambert, O., Kayal, S., Sarnacki, S., Cumano, A., Lauvau, G., and Geissmann, F. (2007). Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science 317, 666–670.
Baleeiro, C. E., Christensen, P. J., Morris, S. B., Mendez, M. P., Wilcoxen, S. E., and Paine, R. III. (2006). GM-CSF and the impaired pulmonary innate immune response following hyperoxic stress. Am. J. Physiol. Lung Cell Mol. Physiol. 291, L1246–L1255.
Ballinger, M. N., Paine, R. III, Serezani, C. H., Aronoff, D. M., Choi, E. S., Standiford, T. J., Toews, G. B., and Moore, B. B. (2006). Role of granulocyte macrophage colony-stimulating factor during gram-negative lung infection with Pseudomonas aeruginosa. Am. J. Respir. Cell Mol. Biol. 34, 766–774.
Bem, R. A., Bos, A. P., Wosten-Van Asperen, R. M., Bruijn, M., Lutter, R., Sprick, M. R., and Van Woensel, J. B. (2010). Potential role of soluble TRAIL in epithelial injury in children with severe RSV infection. Am. J. Respir. Cell Mol. Biol. 42, 697–705.
Benoit, L. A., and Holtzman, M. J. (2010). New immune pathways from chronic post-viral lung disease. Ann. N. Y. Acad. Sci. 1183, 195–210.
Benoit, M., Desnues, B., and Mege, J. L. (2008). Macrophage polarization in bacterial infections. J. Immunol. 181, 3733–3739.
Berclaz, P. Y., Carey, B., Fillipi, M. D., Wernke-Dollries, K., Geraci, N., Cush, S., Richardson, T., Kitzmiller, J., O’connor, M., Hermoyian, C., Korfhagen, T., Whitsett, J. A., and Trapnell, B. C. (2007). GM-CSF regulates a PU.1-dependent transcriptional program determining the pulmonary response to LPS. Am. J. Respir. Cell Mol. Biol. 36, 114–121.
Berclaz, P. Y., Shibata, Y., Whitsett, J. A., and Trapnell, B. C. (2002). GM-CSF, via PU.1, regulates alveolar macrophage Fcgamma R-mediated phagocytosis and the IL-18/IFN-gamma -mediated molecular connection between innate and adaptive immunity in the lung. Blood 100, 4193–4200.
Berthiaume, Y., Voisin, G., and Dagenais, A. (2006). The alveolar type I cells: the new knight of the alveolus? J. Physiol. (Lond.) 572, 609–610.
Booth, B. W., Adler, K. B., Bonner, J. C., Tournier, F., and Martin, L. D. (2001). Interleukin-13 induces proliferation of human airway epithelial cells in vitro via a mechanism mediated by transforming growth factor-alpha. Am. J. Respir. Cell Mol. Biol. 25, 739–743.
Brecht, K., Weigert, A., Hu, J., Popp, R., Fisslthaler, B., Korff, T., Fleming, I., Geisslinger, G., and Brune, B. (2011). Macrophages programmed by apoptotic cells promote angiogenesis via prostaglandin E2. FASEB J. 25, 2408–2417.
Budinger, G. R., Mutlu, G. M., Urich, D., Soberanes, S., Buccellato, L. J., Hawkins, K., Chiarella, S. E., Radigan, K. A., Eisenbart, J., Agrawal, H., Berkelhamer, S., Hekimi, S., Zhang, J., Perlman, H., Schumacker, P. T., Jain, M., and Chandel, N. S. (2011). Epithelial cell death is an important contributor to oxidant-mediated acute lung injury. Am. J. Respir. Crit. Care Med. 183, 1043–1054.
Buff, S. M., Yu, H., Mccall, J. N., Caldwell, S. M., Ferkol, T. W., Flotte, T. R., and Virella-Lowell, I. L. (2010). IL-10 delivery by AAV5 vector attenuates inflammation in mice with Pseudomonas pneumonia. Gene Ther. 17, 567–576.
Cabanski, M., Steinmuller, M., Marsh, L. M., Surdziel, E., Seeger, W., and Lohmeyer, J. (2008). PKR regulates TLR2/TLR4-dependent signaling in murine alveolar macrophages. Am. J. Respir. Cell Mol. Biol. 38, 26–31.
Cabanski, M., Wilhelm, J., Zaslona, Z., Steinmuller, M., Fink, L., Seeger, W., and Lohmeyer, J. (2009). Genome-wide transcriptional profiling of mononuclear phagocytes recruited to mouse lungs in response to alveolar challenge with the TLR2 agonist Pam3CSK4. Am. J. Physiol. Lung Cell Mol. Physiol. 297, L608–L618.
Cakarova, L., Marsh, L. M., Wilhelm, J., Mayer, K., Grimminger, F., Seeger, W., Lohmeyer, J., and Herold, S. (2009). Macrophage tumor necrosis factor-alpha induces epithelial expression of granulocyte-macrophage colony-stimulating factor: impact on alveolar epithelial repair. Am. J. Respir. Crit. Care Med. 180, 521–532.
Campbell, E. L., Louis, N. A., Tomassetti, S. E., Canny, G. O., Arita, M., Serhan, C. N., and Colgan, S. P. (2007). Resolvin E1 promotes mucosal surface clearance of neutrophils: a new paradigm for inflammatory resolution. FASEB J. 21, 3162–3170.
Cash, J. L., Christian, A. R., and Greaves, D. R. (2010). Chemerin peptides promote phagocytosis in a ChemR23- and Syk-dependent manner. J. Immunol. 184, 5315–5324.
Cash, J. L., Hart, R., Russ, A., Dixon, J. P., Colledge, W. H., Doran, J., Hendrick, A. G., Carlton, M. B., and Greaves, D. R. (2008). Synthetic chemerin-derived peptides suppress inflammation through ChemR23. J. Exp. Med. 205, 767–775.
Chapman, H. A., Li, X., Alexander, J. P., Brumwell, A., Lorizio, W., Tan, K., Sonnenberg, A., Wei, Y., and Vu, T. H. (2011). Integrin alpha6beta4 identifies an adult distal lung epithelial population with regenerative potential in mice. J. Clin. Invest. 121, 2855–2862.
Chen, X. J., Seth, S., Yue, G., Kamat, P., Compans, R. W., Guidot, D., Brown, L. A., Eaton, D. C., and Jain, L. (2004). Influenza virus inhibits ENaC and lung fluid clearance. Am. J. Physiol. Lung Cell Mol. Physiol. 287, L366–L373.
Chiang, N., Serhan, C. N., Dahlen, S. E., Drazen, J. M., Hay, D. W., Rovati, G. E., Shimizu, T., Yokomizo, T., and Brink, C. (2006). The lipoxin receptor ALX: potent ligand-specific and stereoselective actions in vivo. Pharmacol. Rev. 58, 463–487.
Cocco, R. E., and Ucker, D. S. (2001). Distinct modes of macrophage recognition for apoptotic and necrotic cells are not specified exclusively by phosphatidylserine exposure. Mol. Biol. Cell 12, 919–930.
Crosby, L. M., and Waters, C. M. (2010). Epithelial repair mechanisms in the lung. Am. J. Physiol. Lung Cell Mol. Physiol. 298, L715–L731.
Cvetanovic, M., and Ucker, D. S. (2004). Innate immune discrimination of apoptotic cells: repression of proinflammatory macrophage transcription is coupled directly to specific recognition. J. Immunol. 172, 880–889.
D’alessio, F. R., Tsushima, K., Aggarwal, N. R., West, E. E., Willett, M. H., Britos, M. F., Pipeling, M. R., Brower, R. G., Tuder, R. M., Mcdyer, J. F., and King, L. S. (2009). CD4+CD25+Foxp3+ Tregs resolve experimental lung injury in mice and are present in humans with acute lung injury. J. Clin. Invest. 119, 2898–2913.
Dean, R. A., Cox, J. H., Bellac, C. L., Doucet, A., Starr, A. E., and Overall, C. M. (2008). Macrophage-specific metalloelastase (MMP-12) truncates and inactivates ELR+ CXC chemokines and generates CCL2, -7, -8, and -13 antagonists: potential role of the macrophage in terminating polymorphonuclear leukocyte influx. Blood 112, 3455–3464.
Dickie, A. J., Rafii, B., Piovesan, J., Davreux, C., Ding, J., Tanswell, A. K., Rotstein, O., and O’brodovich, H. (2000). Preventing endotoxin-stimulated alveolar macrophages from decreasing epithelium Na+ channel (ENaC) mRNA levels and activity. Pediatr. Res. 48, 304–310.
Duffield, J. S. (2003). The inflammatory macrophage: a story of Jekyll and Hyde. Clin. Sci. 104, 27–38.
Eaton, D. C., Helms, M. N., Koval, M., Bao, H. F., and Jain, L. (2009). The contribution of epithelial sodium channels to alveolar function in health and disease. Annu. Rev. Physiol. 71, 403–423.
El Kebir, D., Jozsef, L., Pan, W., Wang, L., Petasis, N. A., Serhan, C. N., and Filep, J. G. (2009). 15-epi-lipoxin A4 inhibits myeloperoxidase signaling and enhances resolution of acute lung injury. Am. J. Respir. Crit. Care Med. 180, 311–319.
Elliott, M. R., Chekeni, F. B., Trampont, P. C., Lazarowski, E. R., Kadl, A., Walk, S. F., Park, D., Woodson, R. I., Ostankovich, M., Sharma, P., Lysiak, J. J., Harden, T. K., Leitinger, N., and Ravichandran, K. S. (2009). Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 461, 282–286.
Fadok, V. A., Bratton, D. L., Konowal, A., Freed, P. W., Westcott, J. Y., and Henson, P. M. (1998). Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J. Clin. Invest. 101, 890–898.
Fehrenbach, H. (2001). Alveolar epithelial type II cell: defender of the alveolus revisited. Respir. Res. 2, 33–46.
Fiedler, U., and Augustin, H. G. (2006). Angiopoietins: a link between angiogenesis and inflammation. Trends Immunol. 27, 552–558.
Freire-de-Lima, C. G., Xiao, Y. Q., Gardai, S. J., Bratton, D. L., Schiemann, W. P., and Henson, P. M. (2006). Apoptotic cells, through transforming growth factor-beta, coordinately induce anti-inflammatory and suppress pro-inflammatory eicosanoid and NO synthesis in murine macrophages. J. Biol. Chem. 281, 38376–38384.
Funk, C. D. (2001). Prostaglandins and leukotrienes: advances in eicosanoid biology. Science 294, 1871–1875.
Ganter, M. T., Roux, J., Miyazawa, B., Howard, M., Frank, J. A., Su, G., Sheppard, D., Violette, S. M., Weinreb, P. H., Horan, G. S., Matthay, M. A., and Pittet, J. F. (2008). Interleukin-1beta causes acute lung injury via alphavbeta5 and alphavbeta6 integrin-dependent mechanisms. Circ. Res. 102, 804–812.
Geissmann, F., Manz, M. G., Jung, S., Sieweke, M. H., Merad, M., and Ley, K. (2010). Development of monocytes, macrophages, and dendritic cells. Science 327, 656–661.
Gibbons, M. A., Mackinnon, A. C., Ramachandran, P., Dhaliwal, K., Duffin, R., Phythian-Adams, A. T., Van Rooijen, N., Haslett, C., Howie, S. E., Simpson, A. J., Hirani, N., Gauldie, J., Iredale, J. P., Sethi, T., and Forbes, S. J. (2011). Ly6Chi monocytes direct alternatively activated pro-fibrotic macrophage regulation of lung fibrosis. Am. J. Respir. Crit. Care Med. 184, 569–581.
Godson, C., Mitchell, S., Harvey, K., Petasis, N. A., Hogg, N., and Brady, H. R. (2000). Cutting edge: lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J. Immunol. 164, 1663–1667.
Golpon, H. A., Fadok, V. A., Taraseviciene-Stewart, L., Scerbavicius, R., Sauer, C., Welte, T., Henson, P. M., and Voelkel, N. F. (2004). Life after corpse engulfment: phagocytosis of apoptotic cells leads to VEGF secretion and cell growth. FASEB J. 18, 1716–1718.
Gonzalez-Mariscal, L., Tapia, R., and Chamorro, D. (2008). Crosstalk of tight junction components with signaling pathways. Biochim. Biophys. Acta 1778, 729–756.
Gordon, S., and Martinez, F. O. (2010). Alternative activation of macrophages: mechanism and functions. Immunity 32, 593–604.
Granata, F., Frattini, A., Loffredo, S., Staiano, R. I., Petraroli, A., Ribatti, D., Oslund, R., Gelb, M. H., Lambeau, G., Marone, G., and Triggiani, M. (2010). Production of vascular endothelial growth factors from human lung macrophages induced by group IIA and group X secreted phospholipases A2. J. Immunol. 184, 5232–5241.
Greenberg, M. E., Sun, M., Zhang, R., Febbraio, M., Silverstein, R., and Hazen, S. L. (2006). Oxidized phosphatidylserine-CD36 interactions play an essential role in macrophage-dependent phagocytosis of apoptotic cells. J. Exp. Med. 203, 2613–2625.
Grimsley, C., and Ravichandran, K. S. (2003). Cues for apoptotic cell engulfment: eat-me, don’t eat-me and come-get-me signals. Trends Cell Biol. 13, 648–656.
Gupte, V. V., Ramasamy, S. K., Reddy, R., Lee, J., Weinreb, P. H., Violette, S. M., Guenther, A., Warburton, D., Driscoll, B., Minoo, P., and Bellusci, S. (2009). Overexpression of fibroblast growth factor-10 during both inflammatory and fibrotic phases attenuates bleomycin-induced pulmonary fibrosis in mice. Am. J. Respir. Crit. Care Med. 180, 424–436.
Harel-Adar, T., Ben Mordechai, T., Amsalem, Y., Feinberg, M. S., Leor, J., and Cohen, S. (2011). Modulation of cardiac macrophages by phosphatidylserine-presenting liposomes improves infarct repair. Proc. Natl. Acad. Sci. U.S.A. 108, 1827–1832.
Herold, S., Steinmueller, M., Von Wulffen, W., Cakarova, L., Pinto, R., Pleschka, S., Mack, M., Kuziel, W. A., Corazza, N., Brunner, T., Seeger, W., and Lohmeyer, J. (2008). Lung epithelial apoptosis in influenza virus pneumonia: the role of macrophage-expressed TNF-related apoptosis-inducing ligand. J. Exp. Med. 205, 3065–3077.
Herold, S., Tabar, T. S., Janssen, H., Hoegner, K., Cabanski, M., Lewe-Schlosser, P., Albrecht, J., Driever, F., Vadasz, I., Seeger, W., Steinmueller, M., and Lohmeyer, J. (2011). Exudate macrophages attenuate lung injury by the release of IL-1 receptor antagonist in gram-negative pneumonia. Am. J. Respir. Crit. Care Med. 183, 1380–1390.
Herold, S., Von Wulffen, W., Steinmueller, M., Pleschka, S., Kuziel, W. A., Mack, M., Srivastava, M., Seeger, W., Maus, U. A., and Lohmeyer, J. (2006). Alveolar epithelial cells direct monocyte transepithelial migration upon influenza virus infection: impact of chemokines and adhesion molecules. J. Immunol. 177, 1817–1824.
Hickman-Davis, J. M., Mcnicholas-Bevensee, C., Davis, I. C., Ma, H. P., Davis, G. C., Bosworth, C. A., and Matalon, S. (2006). Reactive species mediate inhibition of alveolar type II sodium transport during mycoplasma infection. Am. J. Respir. Crit. Care Med. 173, 334–344.
Huffman Reed, J. A., Rice, W. R., Zsengeller, Z. K., Wert, S. E., Dranoff, G., and Whitsett, J. A. (1997). GM-CSF enhances lung growth and causes alveolar type II epithelial cell hyperplasia in transgenic mice. Am. J. Physiol. 273, L715–L725.
Ishii, M., Wen, H., Corsa, C. A., Liu, T., Coelho, A. L., Allen, R. M., Carson, W. F. T., Cavassani, K. A., Li, X., Lukacs, N. W., Hogaboam, C. M., Dou, Y., and Kunkel, S. L. (2009). Epigenetic regulation of the alternatively activated macrophage phenotype. Blood 114, 3244–3254.
Janssen, W. J., Mcphillips, K. A., Dickinson, M. G., Linderman, D. J., Morimoto, K., Xiao, Y. Q., Oldham, K. M., Vandivier, R. W., Henson, P. M., and Gardai, S. J. (2008). Surfactant proteins A and D suppress alveolar macrophage phagocytosis via interaction with SIRP alpha. Am. J. Respir. Crit. Care Med. 178, 158–167.
Jesmin, S., Zaedi, S., Islam, A. M., Sultana, S. N., Iwashima, Y., Wada, T., Yamaguchi, N., Hiroe, M., and Gando, S. (2011). Time-dependent alterations of VEGF and its signaling molecules in acute lung injury in a rat model of sepsis. Inflammation. doi: 10.1007/s10753-011-9337-1. [Epub ahead of print].
Jozsef, L., Zouki, C., Petasis, N. A., Serhan, C. N., and Filep, J. G. (2002). Lipoxin A4 and aspirin-triggered 15-epi-lipoxin A4 inhibit peroxynitrite formation, NF-kappa B and AP-1 activation, and IL-8 gene expression in human leukocytes. Proc. Natl. Acad. Sci. U.S.A. 99, 13266–13271.
Kajstura, J., Rota, M., Hall, S. R., Hosoda, T., D’amario, D., Sanada, F., Zheng, H., Ogorek, B., Rondon-Clavo, C., Ferreira-Martins, J., Matsuda, A., Arranto, C., Goichberg, P., Giordano, G., Haley, K. J., Bardelli, S., Rayatzadeh, H., Liu, X., Quaini, F., Liao, R., Leri, A., Perrella, M. A., Loscalzo, J., and Anversa, P. (2011). Evidence for human lung stem cells. N. Engl. J. Med. 364, 1795–1806.
Kantari, C., Pederzoli-Ribeil, M., and Witko-Sarsat, V. (2008). The role of neutrophils and monocytes in innate immunity. Contrib. Microbiol. 15, 118–146.
Kennedy, A. D., and DeLeo, F. R. (2009). Neutrophil apoptosis and the resolution of infection. Immunol. Res. 43, 25–61.
Kim, E. Y., Battaile, J. T., Patel, A. C., You, Y., Agapov, E., Grayson, M. H., Benoit, L. A., Byers, D. E., Alevy, Y., Tucker, J., Swanson, S., Tidwell, R., Tyner, J. W., Morton, J. D., Castro, M., Polineni, D., Patterson, G. A., Schwendener, R. A., Allard, J. D., Peltz, G., and Holtzman, M. J. (2008). Persistent activation of an innate immune response translates respiratory viral infection into chronic lung disease. Nat. Med. 14, 633–640.
Krausgruber, T., Blazek, K., Smallie, T., Alzabin, S., Lockstone, H., Sahgal, N., Hussell, T., Feldmann, M., and Udalova, I. A. (2011). IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat. Immunol. 12, 231–238.
Krysko, O., Vandenabeele, P., Krysko, D. V., and Bachert, C. (2010). Impairment of phagocytosis of apoptotic cells and its role in chronic airway diseases. Apoptosis 15, 1137–1146.
Kunzelmann, K., Beesley, A. H., King, N. J., Karupiah, G., Young, J. A., and Cook, D. I. (2000). Influenza virus inhibits amiloride-sensitive Na+ channels in respiratory epithelia. Proc. Natl. Acad. Sci. U.S.A. 97, 10282–10287.
Landsman, L., Varol, C., and Jung, S. (2007). Distinct differentiation potential of blood monocyte subsets in the lung. J. Immunol. 178, 2000–2007.
Lee, S., Huen, S., Nishio, H., Nishio, S., Lee, H. K., Choi, B. S., Ruhrberg, C., and Cantley, L. G. (2011). Distinct macrophage phenotypes contribute to kidney injury and repair. J. Am. Soc. Nephrol. 22, 317–326.
Leslie, C. C., Mccormick-Shannon, K., Shannon, J. M., Garrick, B., Damm, D., Abraham, J. A., and Mason, R. J. (1997). Heparin-binding EGF-like growth factor is a mitogen for rat alveolar type II cells. Am. J. Respir. Cell Mol. Biol. 16, 379–387.
Levy, B. D., Clish, C. B., Schmidt, B., Gronert, K., and Serhan, C. N. (2001). Lipid mediator class switching during acute inflammation: signals in resolution. Nat. Immunol. 2, 612–619.
Li, C. M., Khosla, J., Hoyle, P., and Sannes, P. L. (2001). Transforming growth factor-beta(1) modifies fibroblast growth factor-2 production in type II cells. Chest 120, 60S–61S.
Li, M. O., Sarkisian, M. R., Mehal, W. Z., Rakic, P., and Flavell, R. A. (2003). Phosphatidylserine receptor is required for clearance of apoptotic cells. Science 302, 1560–1563.
Liao, X., Sharma, N., Kapadia, F., Zhou, G., Lu, Y., Hong, H., Paruchuri, K., Mahabeleshwar, G. H., Dalmas, E., Venteclef, N., Flask, C. A., Kim, J., Doreian, B. W., Lu, K. Q., Kaestner, K. H., Hamik, A., Clement, K., and Jain, M. K. (2011). Kruppel-like factor 4 regulates macrophage polarization. J. Clin. Invest. 121, 2736–2749.
Litvack, M. L., and Palaniyar, N. (2010). Review: soluble innate immune pattern-recognition proteins for clearing dying cells and cellular components: implications on exacerbating or resolving inflammation. Innate Immun. 16, 191–200.
Lu, H., Huang, D., Ransohoff, R. M., and Zhou, L. (2011). Acute skeletal muscle injury: CCL2 expression by both monocytes and injured muscle is required for repair. FASEB J. 25, 3344–3355.
Luangsay, S., Wittamer, V., Bondue, B., De Henau, O., Rouger, L., Brait, M., Franssen, J. D., De Nadai, P., Huaux, F., and Parmentier, M. (2009). Mouse ChemR23 is expressed in dendritic cell subsets and macrophages, and mediates an anti-inflammatory activity of chemerin in a lung disease model. J. Immunol. 183, 6489–6499.
Ma, X., Xu, D., Ai, Y., Ming, G., and Zhao, S. (2010). Fas inhibition attenuates lipopolysaccharide-induced apoptosis and cytokine release of rat type II alveolar epithelial cells. Mol. Biol. Rep. 37, 3051–3056.
Maddox, J. F., Hachicha, M., Takano, T., Petasis, N. A., Fokin, V. V., and Serhan, C. N. (1997). Lipoxin A4 stable analogs are potent mimetics that stimulate human monocytes and THP-1 cells via a G-protein-linked lipoxin A4 receptor. J. Biol. Chem. 272, 6972–6978.
Maderna, P., Cottell, D. C., Berlasconi, G., Petasis, N. A., Brady, H. R., and Godson, C. (2002). Lipoxins induce actin reorganization in monocytes and macrophages but not in neutrophils: differential involvement of rho GTPases. Am. J. Pathol. 160, 2275–2283.
Mahdavian Delavary, B., Van Der Veer, W. M., Van Egmond, M., Niessen, F. B., and Beelen, R. H. (2011). Macrophages in skin injury and repair. Immunobiology 216, 753–762.
Marsh, L. M., Cakarova, L., Kwapiszewska, G., Von Wulffen, W., Herold, S., Seeger, W., and Lohmeyer, J. (2009). Surface expression of CD74 by type II alveolar epithelial cells: a potential mechanism for macrophage migration inhibitory factor-induced epithelial repair. Am. J. Physiol. Lung Cell Mol. Physiol. 296, L442–L452.
Martinez, F. O., Sica, A., Mantovani, A., and Locati, M. (2008). Macrophage activation and polarization. Front. Biosci. 13, 453–461.
Maus, U., Rosseau, S., Knies, U., Seeger, W., and Lohmeyer, J. (1998). Expression of pro-inflammatory cytokines by flow-sorted alveolar macrophages in severe pneumonia. Eur. Respir. J. 11, 534–541.
Mayer, K., Kiessling, A., Ott, J., Schaefer, M. B., Hecker, M., Henneke, I., Schulz, R., Gunther, A., Wang, J., Wu, L., Roth, J., Seeger, W., and Kang, J. X. (2009). Acute lung injury is reduced in fat-1 mice endogenously synthesizing n-3 fatty acids. Am. J. Respir. Crit. Care Med. 179, 474–483.
Mayer, K., Merfels, M., Muhly-Reinholz, M., Gokorsch, S., Rosseau, S., Lohmeyer, J., Schwarzer, N., Krull, M., Suttorp, N., Grimminger, F., and Seeger, W. (2002). Omega-3 fatty acids suppress monocyte adhesion to human endothelial cells: role of endothelial PAF generation. Am. J. Physiol. Heart Circ. Physiol. 283, H811–H818.
McGrath, E. E., Marriott, H. M., Lawrie, A., Francis, S. E., Sabroe, I., Renshaw, S. A., Dockrell, D. H., and Whyte, M. K. (2011). TNF-related apoptosis-inducing ligand (TRAIL) regulates inflammatory neutrophil apoptosis and enhances resolution of inflammation. J. Leukoc. Biol. 90, 855–865.
McQualter, J. L., Yuen, K., Williams, B., and Bertoncello, I. (2010). Evidence of an epithelial stem/progenitor cell hierarchy in the adult mouse lung. Proc. Natl. Acad. Sci. U.S.A. 107, 1414–1419.
McQuibban, G. A., Gong, J. H., Tam, E. M., Mcculloch, C. A., Clark-Lewis, I., and Overall, C. M. (2000). Inflammation dampened by gelatinase A cleavage of monocyte chemoattractant protein-3. Science 289, 1202–1206.
McQuibban, G. A., Gong, J. H., Wong, J. P., Wallace, J. L., Clark-Lewis, I., and Overall, C. M. (2002). Matrix metalloproteinase processing of monocyte chemoattractant proteins generates CC chemokine receptor antagonists with anti-inflammatory properties in vivo. Blood 100, 1160–1167.
Medeiros, A. I., Serezani, C. H., Lee, S. P., and Peters-Golden, M. (2009). Efferocytosis impairs pulmonary macrophage and lung antibacterial function via PGE2/EP2 signaling. J. Exp. Med. 206, 61–68.
Medina, R. J., O’neill C, L., O’doherty T, M., Knott, H., Guduric-Fuchs, J., Gardiner, T. A., and Stitt, A. W. (2011). Myeloid angiogenic cells act as alternative M2 macrophages and modulate angiogenesis through interleukin-8. Mol. Med. 17, 1045–1055.
Melloni, B., Lesur, O., Bouhadiba, T., Cantin, A., Martel, M., and Begin, R. (1996). Effect of exposure to silica on human alveolar macrophages in supporting growth activity in type II epithelial cells. Thorax 51, 781–786.
Meneghin, A., and Hogaboam, C. M. (2007). Infectious disease, the innate immune response, and fibrosis. J. Clin. Invest. 117, 530–538.
Mevorach, D., Mascarenhas, J. O., Gershov, D., and Elkon, K. B. (1998). Complement-dependent clearance of apoptotic cells by human macrophages. J. Exp. Med. 188, 2313–2320.
Miyake, Y., Kaise, H., Isono, K., Koseki, H., Kohno, K., and Tanaka, M. (2007). Protective role of macrophages in noninflammatory lung injury caused by selective ablation of alveolar epithelial type II Cells. J. Immunol. 178, 5001–5009.
Miyanishi, M., Tada, K., Koike, M., Uchiyama, Y., Kitamura, T., and Nagata, S. (2007). Identification of Tim4 as a phosphatidylserine receptor. Nature 450, 435–439.
Morimoto, K., Amano, H., Sonoda, F., Baba, M., Senba, M., Yoshimine, H., Yamamoto, H., Ii, T., Oishi, K., and Nagatake, T. (2001). Alveolar macrophages that phagocytose apoptotic neutrophils produce hepatocyte growth factor during bacterial pneumonia in mice. Am. J. Respir. Cell Mol. Biol. 24, 608–615.
Morty, R. E., Eickelberg, O., and Seeger, W. (2007). Alveolar fluid clearance in acute lung injury: what have we learned from animal models and clinical studies? Intensive Care Med. 33, 1229–1240.
Mosser, D. M., and Edwards, J. P. (2008). Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8, 958–969.
Mukaro, V. R., and Hodge, S. (2011). Airway clearance of apoptotic cells in COPD. Curr. Drug Targets 12, 460–468.
Mura, M., Han, B., Andrade, C. F., Seth, R., Hwang, D., Waddell, T. K., Keshavjee, S., and Liu, M. (2006). The early responses of VEGF and its receptors during acute lung injury: implication of VEGF in alveolar epithelial cell survival. Crit. Care 10, R130.
Nahrendorf, M., Swirski, F. K., Aikawa, E., Stangenberg, L., Wurdinger, T., Figueiredo, J. L., Libby, P., Weissleder, R., and Pittet, M. J. (2007). The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 204, 3037–3047.
Nair, M. G., Du, Y., Perrigoue, J. G., Zaph, C., Taylor, J. J., Goldschmidt, M., Swain, G. P., Yancopoulos, G. D., Valenzuela, D. M., Murphy, A., Karow, M., Stevens, S., Pearce, E. J., and Artis, D. (2009). Alternatively activated macrophage-derived RELM-{alpha} is a negative regulator of type 2 inflammation in the lung. J. Exp. Med. 206, 937–952.
Narasaraju, T., Ng, H. H., Phoon, M. C., and Chow, V. T. (2010). MCP-1 antibody treatment enhances damage and impedes repair of the alveolar epithelium in influenza pneumonitis. Am. J. Respir. Cell Mol. Biol. 42, 732–743.
Paine, R. III, Wilcoxen, S. E., Morris, S. B., Sartori, C., Baleeiro, C. E., Matthay, M. A., and Christensen, P. J. (2003). Transgenic overexpression of granulocyte macrophage-colony stimulating factor in the lung prevents hyperoxic lung injury. Am. J. Pathol. 163, 2397–2406.
Panos, R. J., Rubin, J. S., Csaky, K. G., Aaronson, S. A., and Mason, R. J. (1993). Keratinocyte growth factor and hepatocyte growth factor/scatter factor are heparin-binding growth factors for alveolar type II cells in fibroblast-conditioned medium. J. Clin. Invest. 92, 969–977.
Park, W. Y., Goodman, R. B., Steinberg, K. P., Ruzinski, J. T., Radella, F. II, Park, D. R., Pugin, J., Skerrett, S. J., Hudson, L. D., and Martin, T. R. (2001). Cytokine balance in the lungs of patients with acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 164, 1896–1903.
Patel, V. A., Longacre, A., Hsiao, K., Fan, H., Meng, F., Mitchell, J. E., Rauch, J., Ucker, D. S., and Levine, J. S. (2006). Apoptotic cells, at all stages of the death process, trigger characteristic signaling events that are divergent from and dominant over those triggered by necrotic cells: implications for the delayed clearance model of autoimmunity. J. Biol. Chem. 281, 4663–4670.
Patel, V. A., Longacre-Antoni, A., Cvetanovic, M., Lee, D. J., Feng, L., Fan, H., Rauch, J., Ucker, D. S., and Levine, J. S. (2007). The affirmative response of the innate immune system to apoptotic cells. Autoimmunity 40, 274–280.
Pechkovsky, D. V., Prasse, A., Kollert, F., Engel, K. M., Dentler, J., Luttmann, W., Friedrich, K., Muller-Quernheim, J., and Zissel, G. (2010). Alternatively activated alveolar macrophages in pulmonary fibrosis-mediator production and intracellular signal transduction. Clin. Immunol. 137, 89–101.
Perretti, M., and D’acquisto, F. (2009). Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat. Rev. Immunol. 9, 62–70.
Peter, C., Waibel, M., Radu, C. G., Yang, L. V., Witte, O. N., Schulze-Osthoff, K., Wesselborg, S., and Lauber, K. (2008). Migration to apoptotic “find-me” signals is mediated via the phagocyte receptor G2A. J. Biol. Chem. 283, 5296–5305.
Pogach, M. S., Cao, Y., Millien, G., Ramirez, M. I., and Williams, M. C. (2007). Key developmental regulators change during hyperoxia-induced injury and recovery in adult mouse lung. J. Cell. Biochem. 100, 1415–1429.
Porta, C., Rimoldi, M., Raes, G., Brys, L., Ghezzi, P., Di Liberto, D., Dieli, F., Ghisletti, S., Natoli, G., De Baetselier, P., Mantovani, A., and Sica, A. (2009). Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor kappaB. Proc. Natl. Acad. Sci. U.S.A. 106, 14978–14983.
Prasse, A., Pechkovsky, D. V., Toews, G. B., Jungraithmayr, W., Kollert, F., Goldmann, T., Vollmer, E., Muller-Quernheim, J., and Zissel, G. (2006). A vicious circle of alveolar macrophages and fibroblasts perpetuates pulmonary fibrosis via CCL18. Am. J. Respir. Crit. Care Med. 173, 781–792.
Ravichandran, K. S. (2010). Find-me and eat-me signals in apoptotic cell clearance: progress and conundrums. J. Exp. Med. 207, 1807–1817.
Ray, P. (2005). Protection of epithelial cells by keratinocyte growth factor signaling. Proc. Am. Thorac. Soc. 2, 221–225.
Reddy, S. M., Hsiao, K. H., Abernethy, V. E., Fan, H., Longacre, A., Lieberthal, W., Rauch, J., Koh, J. S., and Levine, J. S. (2002). Phagocytosis of apoptotic cells by macrophages induces novel signaling events leading to cytokine-independent survival and inhibition of proliferation: activation of Akt and inhibition of extracellular signal-regulated kinases 1 and 2. J. Immunol. 169, 702–713.
Ricardo, S. D., Van Goor, H., and Eddy, A. A. (2008). Macrophage diversity in renal injury and repair. J. Clin. Invest. 118, 3522–3530.
Rock, J. R., and Hogan, B. L. (2011). Epithelial progenitor cells in lung development, maintenance, repair, and disease. Annu. Rev. Cell Dev. Biol. 10, 493–512.
Roux, J., Kawakatsu, H., Gartland, B., Pespeni, M., Sheppard, D., Matthay, M. A., Canessa, C. M., and Pittet, J. F. (2005). Interleukin-1beta decreases expression of the epithelial sodium channel alpha-subunit in alveolar epithelial cells via a p38 MAPK-dependent signaling pathway. J. Biol. Chem. 280, 18579–18589.
Ruffell, D., Mourkioti, F., Gambardella, A., Kirstetter, P., Lopez, R. G., Rosenthal, N., and Nerlov, C. (2009). A CREB-C/EBPbeta cascade induces M2 macrophage-specific gene expression and promotes muscle injury repair. Proc. Natl. Acad. Sci. U.S.A. 106, 17475–17480.
Satoh, T., Takeuchi, O., Vandenbon, A., Yasuda, K., Tanaka, Y., Kumagai, Y., Miyake, T., Matsushita, K., Okazaki, T., Saitoh, T., Honma, K., Matsuyama, T., Yui, K., Tsujimura, T., Standley, D. M., Nakanishi, K., Nakai, K., and Akira, S. (2010). The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat. Immunol. 11, 936–944.
Savill, J., and Fadok, V. (2000). Corpse clearance defines the meaning of cell death. Nature 407, 784–788.
Schaefer, M. B., Ott, J., Mohr, A., Bi, M. H., Grosz, A., Weissmann, N., Ishii, S., Grimminger, F., Seeger, W., and Mayer, K. (2007). Immunomodulation by n-3- versus n-6-rich lipid emulsions in murine acute lung injury – role of platelet-activating factor receptor. Crit. Care Med. 35, 544–554.
Schif-Zuck, S., Gross, N., Assi, S., Rostoker, R., Serhan, C. N., and Ariel, A. (2011). Saturated-efferocytosis generates pro-resolving CD11b low macrophages: modulation by resolvins and glucocorticoids. Eur. J. Immunol. 41, 366–379.
Schwab, J. M., Chiang, N., Arita, M., and Serhan, C. N. (2007). Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature 447, 869–874.
Seki, H., Fukunaga, K., Arita, M., Arai, H., Nakanishi, H., Taguchi, R., Miyasho, T., Takamiya, R., Asano, K., Ishizaka, A., Takeda, J., and Levy, B. D. (2009). The anti-inflammatory and proresolving mediator resolvin E1 protects mice from bacterial pneumonia and acute lung injury. J. Immunol. 184, 836–843.
Seo, S. U., Kwon, H. J., Ko, H. J., Byun, Y. H., Seong, B. L., Uematsu, S., Akira, S., and Kweon, M. N. (2011). Type I interferon signaling regulates Ly6C(hi) monocytes and neutrophils during acute viral pneumonia in mice. PLoS Pathog. 7, e1001304. doi:10.1371/journal.ppat.1001304
Serbina, N. V., Jia, T., Hohl, T. M., and Pamer, E. G. (2008). Monocyte-mediated defense against microbial pathogens. Annu. Rev. Immunol. 26, 421–452.
Serhan, C. N., Chiang, N., and Van Dyke, T. E. (2008). Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 8, 349–361.
Serhan, C. N., Hong, S., Gronert, K., Colgan, S. P., Devchand, P. R., Mirick, G., and Moussignac, R. L. (2002). Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J. Exp. Med. 196, 1025–1037.
Serhan, C. N., Yang, R., Martinod, K., Kasuga, K., Pillai, P. S., Porter, T. F., Oh, S. F., and Spite, M. (2009). Maresins: novel macrophage mediators with potent antiinflammatory and proresolving actions. J. Exp. Med. 206, 15–23.
Shirey, K. A., Pletneva, L. M., Puche, A. C., Keegan, A. D., Prince, G. A., Blanco, J. C., and Vogel, S. N. (2010). Control of RSV-induced lung injury by alternatively activated macrophages is IL-4R alpha-, TLR4-, and IFN-beta-dependent. Mucosal Immunol. 3, 291–300.
Smith, M. W., Schmidt, J. E., Rehg, J. E., Orihuela, C. J., and Mccullers, J. A. (2007). Induction of pro- and anti-inflammatory molecules in a mouse model of pneumococcal pneumonia after influenza. Comp. Med. 57, 82–89.
Snelgrove, R. J., Goulding, J., Didierlaurent, A. M., Lyonga, D., Vekaria, S., Edwards, L., Gwyer, E., Sedgwick, J. D., Barclay, A. N., and Hussell, T. (2008). A critical function for CD200 in lung immune homeostasis and the severity of influenza infection. Nat. Immunol. 9, 1074–1083.
Soehnlein, O., and Lindbom, L. (2010). Phagocyte partnership during the onset and resolution of inflammation. Nat. Rev. Immunol. 10, 427–439.
Srivastava, M., Jung, S., Wilhelm, J., Fink, L., Buhling, F., Welte, T., Bohle, R. M., Seeger, W., Lohmeyer, J., and Maus, U. A. (2005). The inflammatory versus constitutive trafficking of mononuclear phagocytes into the alveolar space of mice is associated with drastic changes in their gene expression profiles. J. Immunol. 175, 1884–1893.
Strieter, R. M. (2008). What differentiates normal lung repair and fibrosis? Inflammation, resolution of repair, and fibrosis. Proc. Am. Thorac. Soc. 5, 305–310.
Stripp, B. R. (2008). Hierarchical organization of lung progenitor cells: is there an adult lung tissue stem cell? Proc. Am. Thorac. Soc. 5, 695–698.
Sun, L., Louie, M. C., Vannella, K. M., Wilke, C. A., Levine, A. M., Moore, B. B., and Shanley, T. P. (2011). New concepts of IL-10-induced lung fibrosis: fibrocyte recruitment and M2 activation in a CCL2/CCR2 axis. Am. J. Physiol. Lung Cell Mol. Physiol. 300, L341–L353.
Taganov, K. D., Boldin, M. P., Chang, K. J., and Baltimore, D. (2006). NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. U.S.A. 103, 12481–12486.
Takaba, J., Mishima, Y., Hatake, K., and Kasahara, T. (2010). Role of bone marrow-derived monocytes/macrophages in the repair of mucosal damage caused by irradiation and/or anticancer drugs in colitis model. Mediators Inflamm. 2010, 634145.
Temelkovski, J., Kumar, R. K., and Maronese, S. E. (1997). Enhanced production of an EGF-like growth factor by parenchymal macrophages following bleomycin-induced pulmonary injury. Exp. Lung Res. 23, 377–391.
Terakado, M., Gon, Y., Sekiyama, A., Takeshita, I., Kozu, Y., Matsumoto, K., Takahashi, N., and Hashimoto, S. (2011). The Rac1/JNK pathway is critical for EGFR-dependent barrier formation in human airway epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 300, L56–L63.
Tesfaigzi, Y. (2003). Processes involved in the repair of injured airway epithelia. Arch. Immunol. Ther. Exp. (Warsz.) 51, 283–288.
Tighe, R. M., Liang, J., Liu, N., Jung, Y., Jiang, D., Gunn, M. D., and Noble, P. W. (2011). Recruited exudative macrophages selectively produce CXCL10 after noninfectious lung injury. Am. J. Respir. Cell Mol. Biol. 45, 781–788.
Tili, E., Michaille, J. J., Cimino, A., Costinean, S., Dumitru, C. D., Adair, B., Fabbri, M., Alder, H., Liu, C. G., Calin, G. A., and Croce, C. M. (2007). Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J. Immunol. 179, 5082–5089.
Truman, L. A., Ford, C. A., Pasikowska, M., Pound, J. D., Wilkinson, S. J., Dumitriu, I. E., Melville, L., Melrose, L. A., Ogden, C. A., Nibbs, R., Graham, G., Combadiere, C., and Gregory, C. D. (2008). CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood 112, 5026–5036.
Uddin, M., and Levy, B. D. (2011). Resolvins: natural agonists for resolution of pulmonary inflammation. Prog. Lipid Res. 50, 75–88.
van den Berg, J. M., Weyer, S., Weening, J. J., Roos, D., and Kuijpers, T. W. (2001). Divergent effects of tumor necrosis factor alpha on apoptosis of human neutrophils. J. Leukoc. Biol. 69, 467–473.
Van Winkle, L. S., Isaac, J. M., and Plopper, C. G. (1997). Distribution of epidermal growth factor receptor and ligands during bronchiolar epithelial repair from naphthalene-induced Clara cell injury in the mouse. Am. J. Pathol. 151, 443–459.
Vergadi, E., Chang, M. S., Lee, C., Liang, O. D., Liu, X., Fernandez-Gonzalez, A., Mitsialis, S. A., and Kourembanas, S. (2011). Early macrophage recruitment and alternative activation are critical for the later development of hypoxia-induced pulmonary hypertension. Circulation 123, 1986–1995.
Vermi, W., Riboldi, E., Wittamer, V., Gentili, F., Luini, W., Marrelli, S., Vecchi, A., Franssen, J. D., Communi, D., Massardi, L., Sironi, M., Mantovani, A., Parmentier, M., Facchetti, F., and Sozzani, S. (2005). Role of ChemR23 in directing the migration of myeloid and plasmacytoid dendritic cells to lymphoid organs and inflamed skin. J. Exp. Med. 201, 509–515.
Voll, R. E., Herrmann, M., Roth, E. A., Stach, C., Kalden, J. R., and Girkontaite, I. (1997). Immunosuppressive effects of apoptotic cells. Nature 390, 350–351.
Wang, B., Gong, X., Wan, J. Y., Zhang, L., Zhang, Z., Li, H. Z., and Min, S. (2011). Resolvin D1 protects mice from LPS-induced acute lung injury. Pulm. Pharmacol. Ther. 24, 434–441.
Ware, L. B., and Matthay, M. A. (2001). Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 163, 1376–1383.
White, S. R., Martin, L. D., Abe, M. K., Marroquin, B. A., Stern, R., and Fu, X. (2009). Insulin receptor substrate-1/2 mediates IL-4-induced migration of human airway epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 297, L164–L173.
Whitsett, J. A., and Kalinichenko, V. V. (2011). Notch and basal cells take center stage during airway epithelial regeneration. Cell Stem Cell 8, 597–598.
Wissinger, E., Goulding, J., and Hussell, T. (2009). Immune homeostasis in the respiratory tract and its impact on heterologous infection. Semin. Immunol. 21, 147–155.
Wolk, K. E., Lazarowski, E. R., Traylor, Z. P., Yu, E. N., Jewell, N. A., Durbin, R. K., Durbin, J. E., and Davis, I. C. (2008). Influenza A virus inhibits alveolar fluid clearance in BALB/c mice. Am. J. Respir. Crit. Care Med. 178, 969–976.
Keywords: macrophage, lung, inflammation, resolution, repair
Citation: Herold S, Mayer K and Lohmeyer J (2011) Acute lung injury: how macrophages orchestrate resolution of inflammation and tissue repair. Front. Immun. 2:65. doi: 10.3389/fimmu.2011.00065
Received: 03 August 2011; Accepted: 08 November 2011;
Published online: 24 November 2011.
Edited by:
Isabelle Maridonneau-Parini, Centre National de la Recherche Scientifique, FranceReviewed by:
Marco Emilio Bianchi, Universita’ Vita Salute San Raffaele, ItalyCarolyn Louise Geczy, University of New South Wales, Australia
Copyright: © 2011 Herold, Mayer and Lohmeyer. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Susanne Herold, Department of Internal Medicine II, University of Giessen Lung Center, Klinikstr. 36, D-35392 Giessen, Germany. e-mail:c3VzYW5uZS5oZXJvbGRAaW5uZXJlLm1lZC51bmktZ2llc3Nlbi5kZQ==