

 Derin B. Keskin3,4
Derin B. Keskin3,4

- 1 Cancer Vaccine Center, Dana-Farber Cancer Institute, Boston, MA, USA
- 2 Department of Systems Biology, Center for Biological Sequence Analysis, Technical University of Denmark, Lyngby, Denmark
- 3 Department of Medicine, Harvard Medical School, Boston, MA, USA
- 4 Laboratory of Immunobiology, Dana-Farber Cancer Institute, Boston, MA, USA
Broad coverage of the pathogen population is particularly important when designing CD8+ T-cell epitope vaccines against viral pathogens. Traditional approaches are based on combinations of highly conserved T-cell epitopes. Peptide block entropy analysis is a novel approach for assembling sets of broadly covering antigens. Since T-cell epitopes are recognized as peptides rather than individual residues, this method is based on calculating the information content of blocks of peptides from a multiple sequence alignment of homologous proteins rather than using the information content of individual residues. The block entropy analysis provides broad coverage of variant antigens. We applied the block entropy analysis method to the proteomes of the four serotypes of dengue virus (DENV) and found 1,551 blocks of 9-mer peptides, which cover 99% of available sequences with five or fewer unique peptides. In contrast, the benchmark study by Khan et al. (2008) resulted in 165 conserved 9-mer peptides. Many of the conserved blocks are located consecutively in the proteins. Connecting these blocks resulted in 78 conserved regions. Of the 1551 blocks of 9-mer peptides 110 comprised predicted HLA binder sets. In total, 457 subunit peptides that encompass the diversity of all sequenced DENV strains of which 333 are T-cell epitope candidates.
Introduction
T-cell mediated immunity is a key factor in host responses against human pathogens. It is important for clearance of infection and for anticancer immunity. Peptide-based vaccines offer significant potential advantages in comparison to vaccines using whole proteins or pathogens. These advantages include absence of infectious agents, minimization of negative effects (such as oncogenicity or allergenicity), minimal biological risk (such as reassortment, recombination, or genome integration), ease of production and quality control, and flexibility in inclusion of peptides from multiple molecular targets and their variants (Purcell et al., 2007). Peptides that are recognition targets of CTLs are proposed as key components of the efforts to develop the next generation of vaccines against various diseases including influenza (Brown and Kelso, 2009), HIV (Barouch et al., 2010), and cancers (Pilla et al., 2009). Despite significant research effort, the development of efficient T-cell vaccines has proven difficult (Appay, 2009). The obstacles related to antigenic targeting include diversity of antigenic targets, human leukocyte antigen (HLA) diversity, and availability of peptide targets, i.e., effectiveness of antigen processing and presentation (Brusic and August, 2004; Riemer et al., 2010). Conserved peptides have been studied as targets for epitope-based vaccines in influenza (Tan et al., 2010), variola (Sette et al., 2009), hepatitis C (Yerly et al., 2008), and HIV (Reche et al., 2006; Fischer et al., 2007; Nickle et al., 2007), among others.
Reverse vaccinology (Rappuoli, 2000) provides a conceptual framework where vaccine targets are initially defined from pathogen proteomes using bioinformatics pre-screening, followed by target selection and experimental validation (De Groot and Rappuoli, 2004; Sette and Rappuoli, 2010). The scientific community has long been aware of the need for vaccine strategies which address viral diversity (Hu et al., 1996). In highly variable pathogens, such as HIV, influenza, or dengue, polyvalent vaccines are considered as a solution to viral diversity (Fischer et al., 2007; Morrison et al., 2010).
Strategies for Dealing with Host and Viral Diversity in Vaccine Design
The design of broadly protective T-cell-based vaccines involves identification and selection of vaccine targets composed of conserved antigens containing T-cell epitopes that are both protective and broadly cross-reactive to viral subtypes. The proposed methods of consensus (CON; Gaschen et al., 2002; De Groot et al., 2005), ancestor (ANC; Gao et al., 2005), and center of tree (COT; Nickle et al., 2007) involve assembling individual amino acids into “centralized” consensus sequences of viral proteins that compress antigenic diversity into a small set of artificial immunogens representative of the virus population. Others, such as the mosaic vaccine design (Fischer et al., 2007), cover viral diversity by assembling naturally occurring peptides identified as T-cell epitopes into artificial proteins, thereby collectively achieving polyvalent coverage. Mosaic vaccines have been tested in preclinical trials with rhesus monkeys showing that these immunogens can induce responses against peptide targets (Barouch et al., 2010; Santra et al., 2010). Common to these methods is a systematic inclusion of highly conserved epitope candidates and exclusion of rare peptides, despite the fact that it has recently been demonstrated that MHC class I epitopes in the flaviviruses have exceptionally low targeting efficiency (i.e., low correlation between MHC binding affinities and conservation of the targeted proteomic regions; Hertz et al., 2011). Furthermore, some low frequency peptides have been shown to be favorable T-cell epitopes in HIV (Rolland et al., 2011). The exclusive focus on highly conserved epitope candidates, for example those maintained in 90% or more of sequences, presents a serious limitation in target selection in dengue virus (DENV) and in other virus species that have multiple serotypes or clades (such as HIV and influenza). In these viruses position many peptides may be intra-clade conserved, but not inter-clade conserved.
A large-scale systematic analysis of peptide conservation and diversity using Shannon entropy calculation and prediction of HLA specificity for conserved peptides in DENV proteins was previously performed for identification of conserved T-cell epitope candidates (Khan et al., 2008). The authors sought to find peptides of nine residues or longer in length, conserved in a minimum of 80% of the 12,404 DENV protein sequences in their dataset. They identified 44 such peptides, of which 34 were conserved in more than 95% of their dataset. They furthermore showed that a subset comprising 34 of the 44 sequences contained 9-mer peptides which were computationally predicted to bind HLA molecules thus representing candidate HLA super type-restricted T-cell epitopes.
Our study extends the analysis of conservation and variability of the DENV proteome. Its focus is the identification of pools of peptides that broadly cover both intra-serotype and inter-serotype diversity. In DENV, the vast majority of experimentally validated epitopes are not conserved across the proteome of all four serotypes. Yet, an efficient vaccine against DENV infection must be protective against all four serotypes. Therefore, the optimal assembly of vaccine targets presents a combinatorial problem where conservation of antigens, diversity of possible HLA interactions, and functional relevance of individual peptides must be assessed. Furthermore, tetravalent formulations (Guy et al., 2010; Murrell et al., 2011) represent main candidate dengue vaccines currently in development. Despite 60 years of research, effective dengue vaccine is not yet available (Murrell et al., 2011). We, therefore, developed and deployed a set of analytical tools to identify, assess, and combine peptide pools suitable for vaccine targeting. It is well-established that the complications associated with secondary infection involve humoral immunity (Halstead and O’rourke, 1977; Halstead et al., 1977), and that immunity against DENV is primarily antibody mediated. Recent research has shown that cellular immunity also plays a role in these complications (Duangchinda et al., 2010), and that cross-reactive DENV-specific T-cells may contribute to the development of dengue hemorrhagic fever (DHF). In this paper we focus on identifying T-cell epitopes, although the block entropy method can be readily used for identification of both linear and discontinuous motifs such as functional B-cell epitopes.
Human CD8+ T-cell epitopes and naturally processed HLA ligands are, with only a few exceptions, 8–11 residues long (Rammensee et al., 1999). CD4+ T-cell epitopes bind HLA molecules mainly through the 9-mer binding core while flanking residues modulate binding (Brown et al., 1993). Therefore, the variability and conservation of antigens should be examined for peptides rather than for individual residues. Block entropy calculations – i.e., calculating information content of a set of aligned sequences – have previously been applied to the analysis of motifs in DNA sequences (Lio et al., 1996). Also, the entropy of 9-mer peptides can be derived from values of individual positions determined using center of the 9-mer calculation (Khan et al., 2008). We applied similar formula for entropy calculations for 8-, 9-, 10-, and 11-mer peptides as blocks. The peptide block entropy method is based on calculating the entropy of blocks of peptides and frequency of individual peptides in a multiple sequence alignment (MSA) of homologous viral protein sequences. The block entropy analysis becomes a useful tool when searching for conserved peptides; for example, a given block may contain only two unique peptides at 50% frequency each, but despite such a modest variability these peptides will not be deemed conserved using the approach that considers conservation of individual amino acids, as used in previous efforts (Fischer et al., 2007; Khan et al., 2008). Because peptide blocks are extracted from a MSA of homologous protein sequences from DENV, a relatively high level of homology of the peptides is expected to be found within an average block. Blocks of homologous peptides are more likely to display similar binding affinity to the HLA class I than randomly assembled sets of non-homologous peptides. However, certain residue variation(s), such as T-cell epitope anchor positions (Falk et al., 1991), can significantly change binding affinity. Similarly, the regions surrounding the block are likely to display inter-sequence homology, suggesting blocks of peptides are more likely to have similar processing characteristics, including proteasomal cleavage and TAP affinity (Martinez et al., 2009), than randomly assembled peptide pools. A block consisting of several epitope candidates with the combined capacity to cover the diversity of all known DENV strains could therefore be a valuable target for prophylactic polyvalent vaccine design.
Materials and Methods
Variability and Conservation Metrics
The calculation of information content of residues in a MSA of homologous protein sequences is based on the calculation of Shannon entropy (Shannon, 1948):
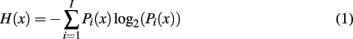
where H is the entropy, x is the position in the MSA, i represents a given individual amino acid at position x, I is the number of different amino acids on position x, and Pi(x) is the frequency of amino acid i at position x. The conservation of a given position is defined as the frequency of the consensus amino acid (most frequent at a given position).
Block Entropy and Conservation
Shannon entropy can be calculated for each peptide in a block. Each block contains a total of W unique peptides of length l in a dataset of N sequences of length L. We can thus extract L–l blocks, B, of N or fewer unique peptides. The application in conservation analysis is the identification of peptides, which together as a subset, Sw, of W represents a given fraction of W. The formula for calculation of block entropy is:
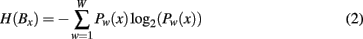
Where H(Bx) is the total entropy of a block of peptides starting at position x, w is a single unique peptide in the space of W unique peptides in block Bx. Pw(x) is the frequency of peptide w at position x.
Four variables are used to classify a block as conserved or not conserved:
1. the minimum number of unique peptides, u, required to reach a pre-defined cumulative frequency in the block in which they are found;
2. the minimum percentage, yx, of a block that must be covered by the subset of peptides, Su, for a block to be considered conserved;
3. the maximum allowed fraction, gx, of peptides containing gaps in the block;
4. the minimum percentage with which each serotype should be covered individually, sx.
In this analysis we used u = 5, yx = 0.99, gx = 0.01, and sx = 0.99. The peptides which are collectively only present in 1% of all sequences (1 − yx) are unlikely to be stable peptides and many may also be data noise originating from sequencing errors, database entry errors, etc. Assuming that the subset of peptides which collectively occur in less than 1% of known sequences represent variants of low fitness, sequencing errors, or rare variants, we consider that 99% coverage represents a practical threshold for complete conservation. The identification of conserved blocks is combined with the assessment of HLA binding potential for each peptide in each block. Blocks in which all peptides, u, in Su show similar binding affinity to the same HLA molecule, are classified as “immuno-functionally conserved.” Blocks in which not all u in Su are predicted as HLA binders with the same HLA restriction were discarded.
Prediction of Peptide Binding to MHC Class I
Human leukocyte antigen binding affinities of peptides in conserved blocks were predicted using NetMHC 3.2 (Lundegaard et al., 2008). Binding affinity to HLA class I was predicted for peptides of nine residues long for the following HLA alleles: HLA-A*0201, HLA-A*03:01, HLA-A*11:01, HLA-A*24:02, HLA-B*07:02, HLA-B*08:01, HLA-B*15:01. These HLA class I alleles were selected for the analysis because NetMHC3.2 predictions of peptide binding to these variants were shown to be highly accurate (Lin et al., 2008). The default thresholds for binding level affinity (IC50 < 500 nM for weak binders and IC50 < 50 nM for strong binders) were used for binding classification in this study. Thus a minimum binding affinity of 500 nM was required for a peptide to be considered a potential binder.
Dealing with Alignment Gaps and Ambiguous Characters in the MSA
Gap insertions in the alignment correspond to insertion or deletion (indel) variation in one or more sequences in the dataset. The DENV diversity is generally caused by substitution mutations rather than indels, but some gaps were observed. Indels of residues can lead to significant change of binding potential or, if both variants are binders, completely different T-cell recognition (Riemer et al., 2010). Therefore, in block entropy based conservation analysis we consider blocks with gaps problematic. In most cases gaps in the alignment were caused by a fraction of the sequences lower than 1% (rare sequences) which were simply removed. If gaps could not be eliminated in this way, the blocks in which more than 1% of the peptides contained gaps were considered too variable and were classified as not conserved. Similarly, peptides containing ambiguous amino acid characters (such as “X”) were omitted from the analysis.
Sequence Logos
We used sequence logos to visualize the information content (measured in bits) of each position within the blocks (Schneider and Stephens, 1990). Sequence logos are visual representations of the Shannon entropy of the positions within a given sequence. The theoretical maximum entropy of a position in a protein sequence is log220 ≈ 4.32 (corresponding to equal representation of all 20 amino acids), so each amino acid on a position can be represented by its fractional information content of the maximum. To generate sequence logos we used WebLogo (Crooks et al., 2004).
Block Logos
We designed a logo for visualizing information content of blocks by modifying the sequence logo representation. Sequence logos are very informative about the occurrence of residues on each position, but do not carry valuable information about the frequencies of peptides. Since the theoretical maximum entropy of a block of unlimited size is log2209 ≈ 39 (corresponding to an equal representation of all possible 9-mers), we use the total entropy, H(B), of a block as the maximum bit on the y axis. The information content of each unique peptide, w, in each block, Bx, can be calculated as follows:

where H(w) is the entropy of peptide w, Pw(x) is the frequency of peptide w, and H(Bx) is the total entropy of the block, B, starting at position, x, in the MSA. The peptides are displayed from most to least frequent starting from the base of the x axis.
DENV Sequences and T-cell Epitope Data
The immune epitope database (IEDB; Vita et al., 2010) was queried for known DENV MHC class I binders. For the block entropy analysis we used only complete DENV protein sequences extracted from GenPept (Benson et al., 2010). These sequences were aligned using MAFFT (Katoh and Toh, 2008). Individual protein products were annotated only in a small fraction (roughly 30%) of the polyprotein sequences retrieved from NCBI. The remaining proteomes were annotated using annotation from GenPept reference sequences within the MAFFT alignments. The sequences were deposited in an in-house, publically available database for easy access <cvc.dfci.harvard.edu/flavi> (Olsen et al., 2011). The numbers of sequences classified by protein and serotype are listed in Table 1.
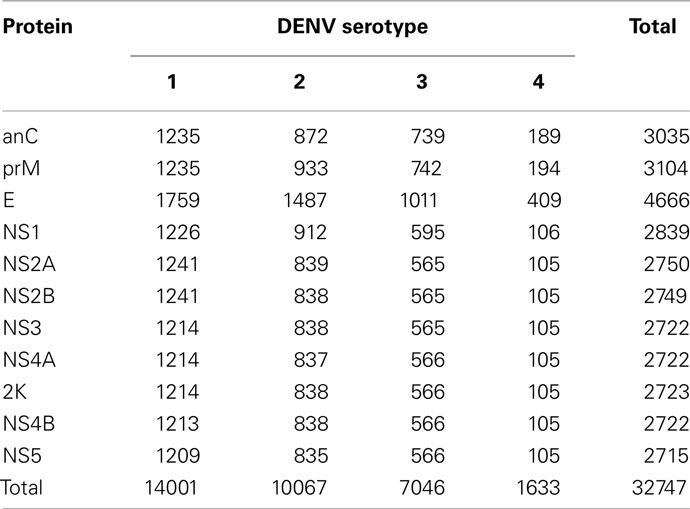
Table 1. Sequence data used in this analysis.
Due to sampling bias and natural frequency differences between the four serotypes of DENV, sequences for the serotypes were not found in similar numbers. For example, relatively small number of available DENV4 sequences meant that extra care was required to ensure that DENV4 diversity was covered properly. We therefore adjusted the size of the datasets of DENV2, DENV3, and DENV4 simply by multiplying the datasets of less frequent serotypes to match the most frequent serotype, DENV1. Upon inspection of the adjusted dataset, we concluded that no further significant strain redundancy was present.
Results and Discussion
Conservation of Known T-cell Epitopes
Querying the IEDB database (Vita et al., 2010) for experimentally determined DENV CD8+ T-cell epitopes yielded a list of 190 verified 9-mer T-cell epitopes. The average conservation of known T-cell epitopes across the DENV1–4 proteins was 37.13%. Only 18 (10%) of all known epitopes are found in >90% of the DENV1–4 strains (Figure 1). Thus only 10% of the known epitopes would be included as potential vaccine targets using a residue-based definition of conservation.
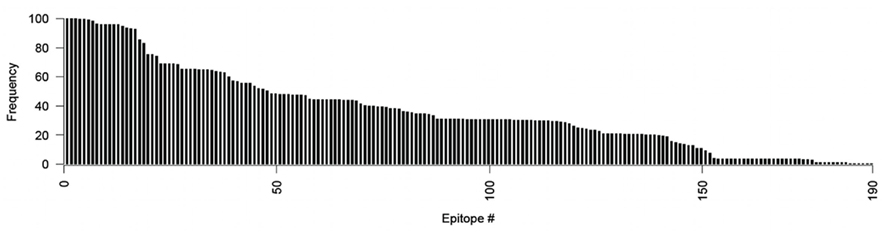
Figure 1. The frequency of the 190 known epitopes sorted from most to least frequent.
Summary of Conserved Pan-DENV Peptide Blocks
We analyzed all blocks of 8, 9, 10, and 11 residues long in the MSA of DENV polyproteins. For each block we calculated the block entropy, the minimum number of peptides needed to cover 99% of a block, the coverage of each of the four DENV serotypes, and also identified the total number of peptides in each block. The conservation of 8-, 9-, 10-, and 11-mer blocks is summarized in Table 2.
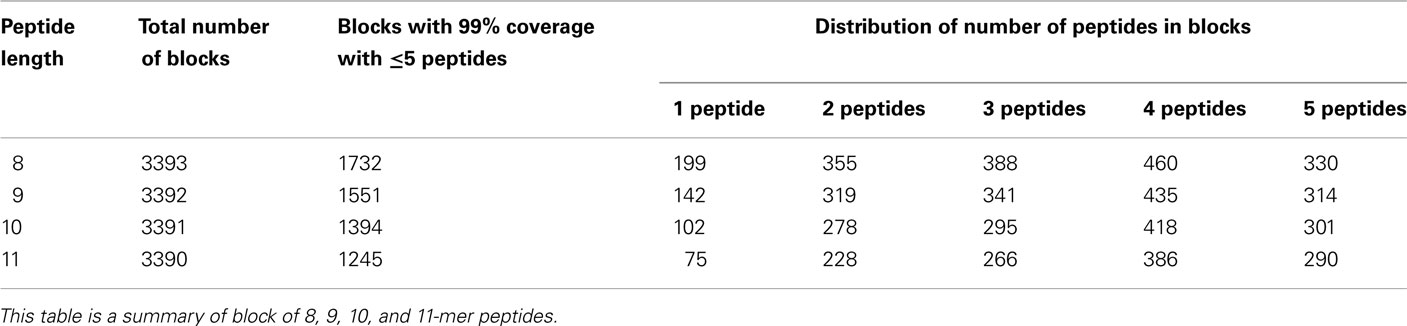
Table 2. The total number of blocks which covers 99% of the sequences with five peptides or less, as well as the relative distribution of numbers of peptides in each block.
There are 1,732, 1,551, 1,394, and 1,245 conserved blocks of 8-, 9-, 10-, and 11-mer peptides respectively. Khan et al. (2008) identified 206, 165, 118, and 88 conserved 8-, 9-, 10-, and 11-mer peptides, respectively, by their criteria for conservation (individual peptides conserved in 80% or more sequences). Using peptide block entropy approach to conservation analysis, we found an approximately 10-fold larger conserved target space, which can be examined for potential T-cell epitope candidates. We found the conserved blocks in anC, prM, NS2A, NS2B, NS4A, and 2K proteins, which have previously been considered as too variable for mapping T-cell epitope candidates for cross-protective vaccine constructs.
By using the conservation thresholds defined in the Section “Materials and Methods,” we examined each protein for conservation of blocks. The number of peptides (9-mers) required to cover 99% of the block for each position in the proteome is shown in Figure 2. Peptide block conservation relative to protein length was highest in the NS4B and lowest in NS2A proteins. In NS4B, 166 blocks of 9-mers (69.1% of blocks within this protein) were conserved whereas NS2A showed only 8.83% block conservation.
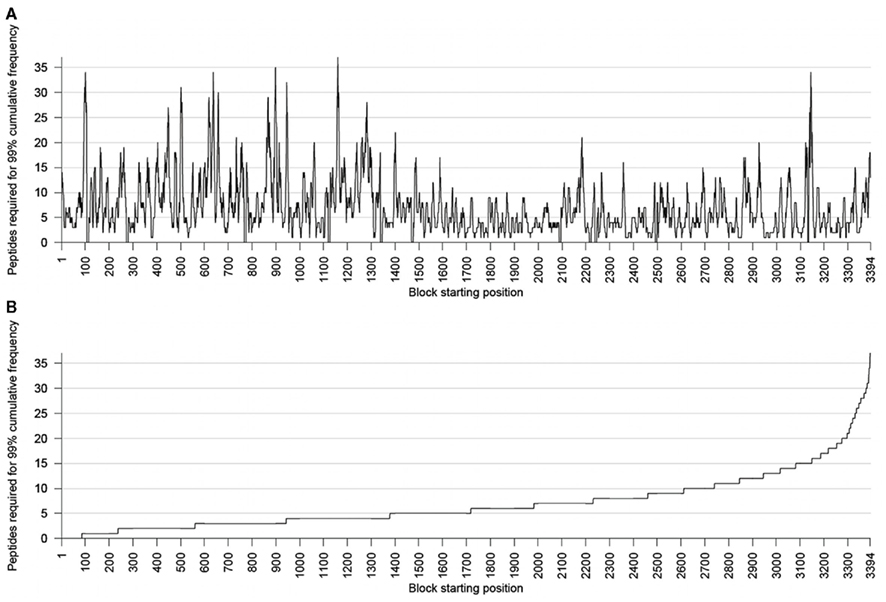
Figure 2. (A) The number of 9-mer peptides required to cover 99% of a block, for each possible start position in the proteome. (B) The number of 9-mer peptides required to cover 99% of a block, sorted in increasing order.
The average entropy for blocks of 9-mer peptides was 1.70 ± 0.71. This is almost double the entropy of individual positions where values larger than 1 indicate highly variable positions (Koo et al., 2009). The entropy of blocks, with only a very few exceptions, where five or less peptides are required to cover 99% of sequences within a block is as high as 2.4 bits (Figure 3). This indicates that the block entropy analysis is a robust method, making it suitable for identification of conserved regions of antigenic proteins, where antigenic diversity can be covered by a small number of peptides. This result shows that block entropy analysis is suitable for target selection in polyvalent vaccine formulations.
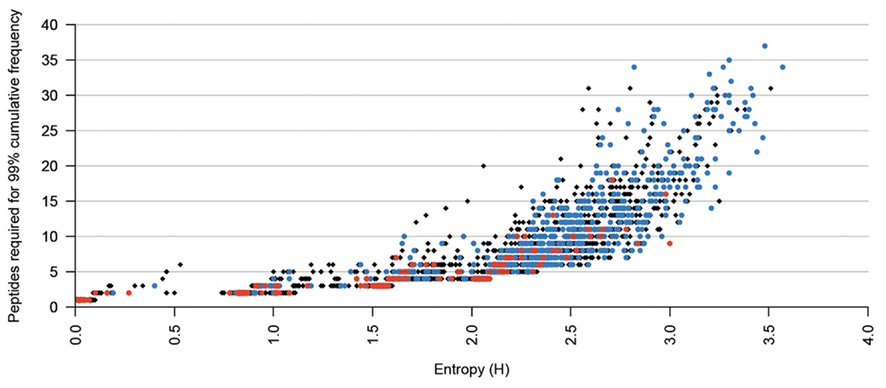
Figure 3. X, Y scatter plot of the number of peptides required for 99% coverage of a given block, against the entropy of each given block. The black diamonds correspond to blocks in which no peptides are predicted to bind to the HLA. The blue circles indicate that some, but not all, peptides within that block are predicted to be epitopes. The red squares indicate that all peptides within the block are predicted to bind the same HLA type.
Block Information Content
We examined the information content of individual blocks. Table 3 shows a representative 9-mer block (position 388 of the MSA of NS3 proteins). We calculated the frequency and information content of each peptide in the block and assessed serotype distribution of these peptides. Five peptides are needed to cover >99% of the sequences within this block, covering approximately 65, 29, 4, 1, and 0.3% of sequenced DENV strains respectively. None of these peptides would have been included in a traditional conservation analysis, in which 80–90% is a typical conservation threshold. This analysis also brings an insight into the effects of threshold selection. If a loose block conservation threshold was used (90%), the three least frequent peptides in the MSA would be excluded and this would exclude DENV4 peptides from the target set. The 99% threshold, on the other hand, would exclude only extremely rare peptides across DENV1–3 serotypes. The peptide number 5 was only found in five strains isolated in Senegal from the late 1960s and three strains from Nigeria from late 1990s. Peptide 5 therefore appears to be a geographically and historically isolated low fitness variant. Peptides 3 and 4 were, however, found in strains isolated almost every year from 1944 to present, and 1983 to present, respectively. Furthermore, peptides 3 and 4 were found distributed across Asia and Australasia and peptide 3 was also observed in Latin America and parts of South America. It is therefore highly likely that strains containing these particular peptides will resurface again given that geographic barriers to spread of DENV are diminishing in the wake of climate changes and increased travel. We, therefore, expect that these strains will continue to spread and proliferate across the world (Mackenzie et al., 2004; Franco et al., 2010). Modern vaccine development clearly requires variant inclusion beyond target selection resulting from a simple conservation analysis. Block entropy analysis enables identification and further analysis of historical strains, while it is much more difficult to identify relevant low frequency peptides using individual position analysis.
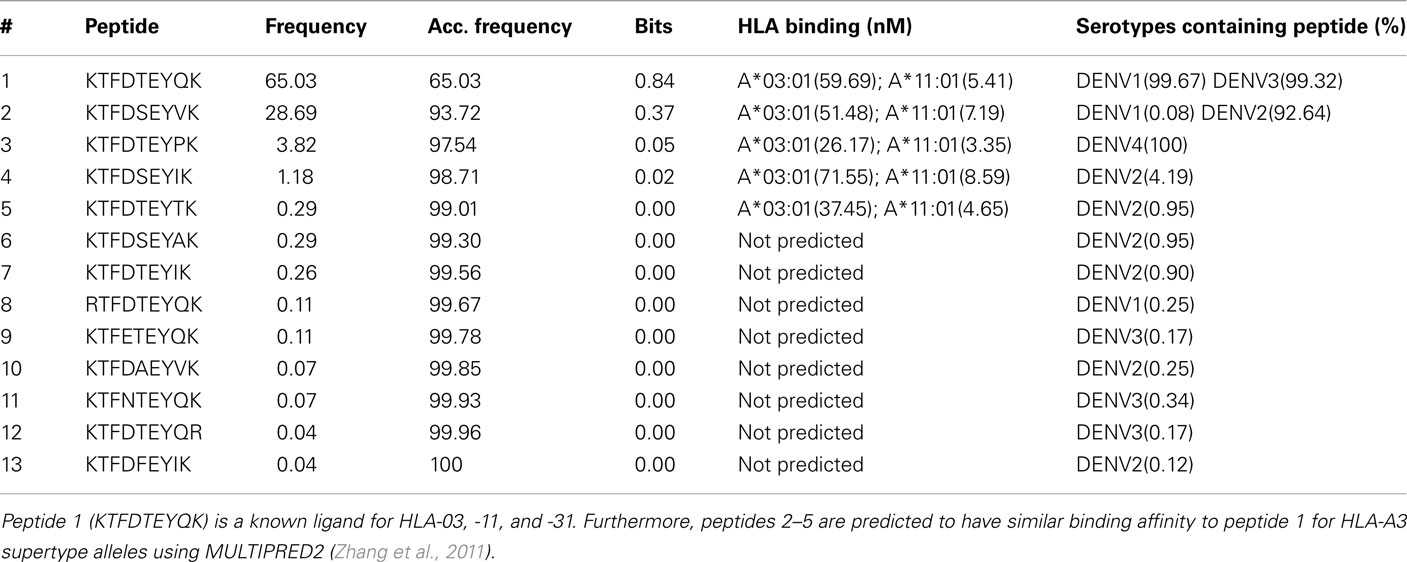
Table 3. Details of the peptides observed in block 388 of the NS3 protein.
We compared conservation analysis using block entropy with the analysis based on frequency of individual positions. This comparison can be supported by the visualization tools; the sequence logo (Crooks et al., 2004) and our new tool, the Block Logo (Figure 4). From the sequence logo one can picture a combinatorial space in which up to eight different peptides maybe present. From the block logo we can see that only two peptides cover 94% while only four peptides within the block show any notable presence.
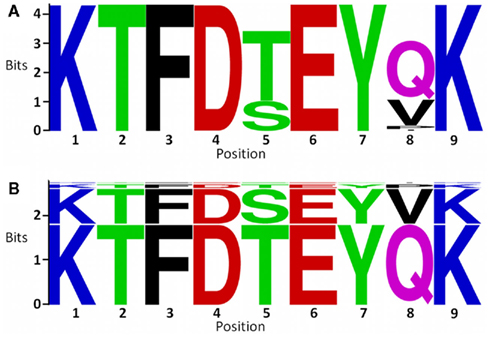
Figure 4. (A) Sequence logo plot of the residues in the block starting at position 388 of the MSA of NS3 protein sequences. The sequence logo was generated using WebLogo (http://weblogo.berkeley.edu/). (B) Shows a peptide block logo of the peptides in the same block. The block logo was generated locally using the Block Logo tool. The colors of the amino acids correspond to their chemical properties: polar amino acids (G, S, T, Y, C, Q, and N) are shown in green, basic amino acids (K, R, and H) are shown in blue, acidic amino acids (D and E) are shown in red, and hydrophobic amino acids (A, V, L, I, P, W, F, and M) are shown in black.
Prediction of HLA Binding of Conserved DENV Peptide Blocks
Binding affinities were predicted for each of the 5,113 peptides in the 1,551 blocks of conserved 9-mer peptides. If all peptides in a block were predicted to bind to the same HLA type, we consider the block “immuno-functionally conserved” (further defined in Materials and Methods). In total 110 blocks, comprising 333 peptides, were predicted to be immuno-functionally conserved. The distribution of immuno-functionally conserved peptides from different proteins with the number of peptides in each block is shown in Table 4.
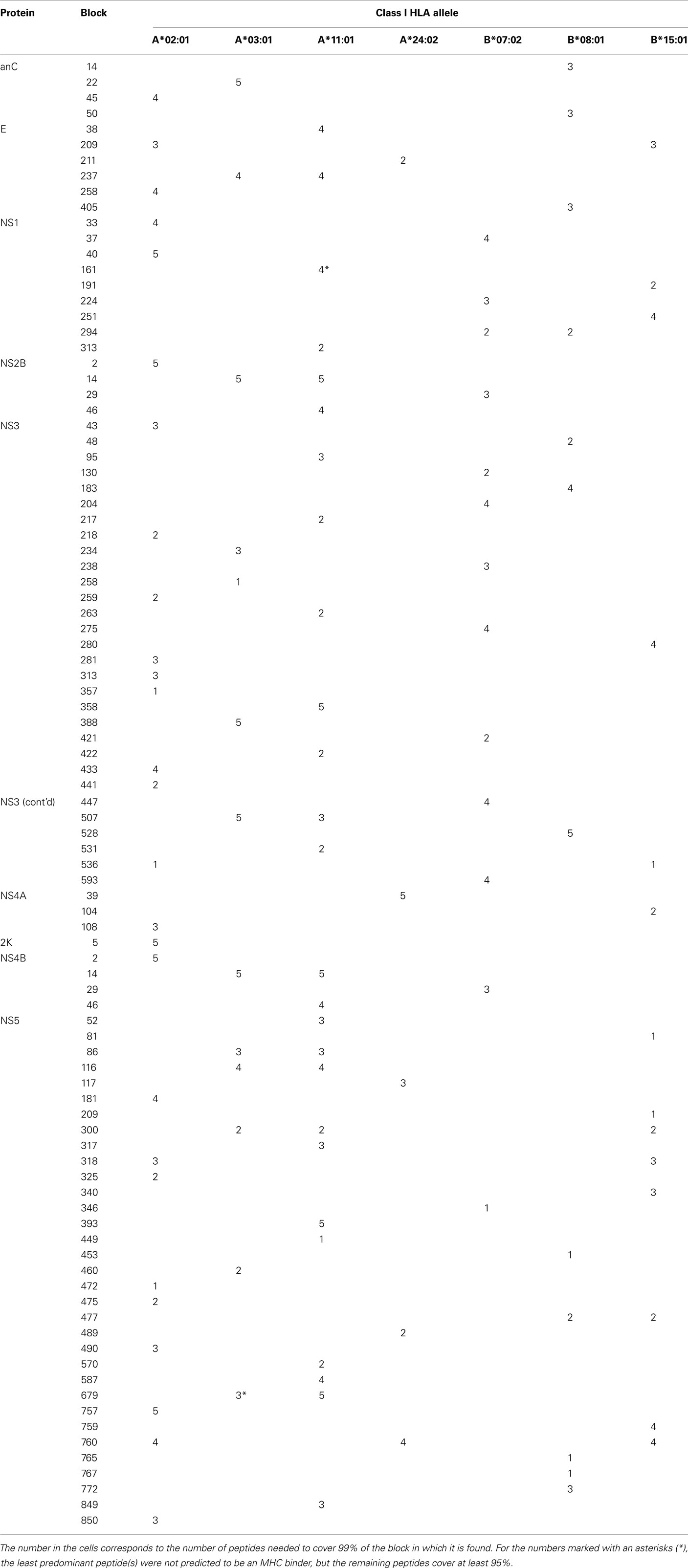
Table 4. Conserved blocks with all peptides are predicted (NetMHC) to have high HLA binding affinity.
The antigenic potential differs between individual proteins, as shown by the number of predicted epitope blocks relative to the size of the protein (Table 5). A protein that has a high conservation to size ratio is traditionally assumed to have high antigenic potential for vaccine design. Proteins NS3, NS5, NS2B, and anC have high antigenic potential while others, particularly prM and NS2A have low antigenic potential. The 2K protein is predicted to have only one immuno-functionally conserved block, but it is very small in size (23 amino acids), resulting in a high conservation to size ratio.
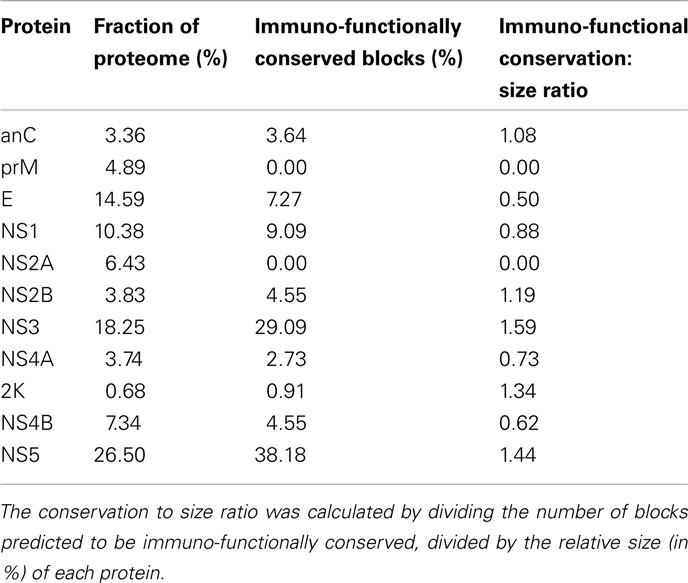
Table 5. The ratio of conservation to size of each DENV protein is shown.
As the number of peptides in immune-conserved blocks increases, the higher the entropy. However, blocks that have identical number of peptides can vary in their entropy due to the diversity in the set of peptides making up the remaining 1% of the block excluded from the conserved set. Figure 3 shows blocks in which some (but not all) peptides are predicted to be epitopes (662 of 3309 peptides), blocks in which all peptides are predicted to bind to the same HLA (151 of 3309 peptides), and blocks predicted to have no HLA binders (2496 of 3309 peptides). The highest concentration of immune-conserved blocks is found in the body of blocks conserved by two, three, and four peptides. We can hypothesize that the immunogenicity of these regions cause higher evolutionary pressure, but that in some regions DENV have only limited space for variability due to possible loss of biological function. This limited variability means that in many cases the peptide variants within a given block may have immunogenic potential with the same HLA restriction and with similar binding affinity, but may require different T-cell clones for immune recognition. Such peptides make excellent targets for polyvalent vaccines for complete coverage.
Although the accuracy of the MHC binding prediction algorithm is high, experimental validation of these epitopes should be performed to ensure first that the peptides are processed and presented to the immune system, and also that they are functional T-cell epitopes. Prediction and experimental validation of 8-, 10-, and 11-mers has yet to be performed. Likewise, prediction and experimental validation of binding can be extended to MHC class II.
Compressing Antigenic Diversity for Vaccinology Applications
A common approach to designing vaccines against DENV involves polyvalent constructs (Murrell et al., 2011). The block entropy method facilitates the design of polyvalent vaccines by identifying sequences that offer broad coverage of the diversity of all DENV. An example of a broadly neutralizing DENV vaccine design is a tetravalent chimeric live attenuated vaccine developed by Sanofi Pasteur. The vaccine covers all four DENV serotypes and offers protection after three doses are delivered over a 15-months period (Morrison et al., 2010). The long period between the first immunization and protection state presents a limitation, since the risk of DHF from natural secondary infection would be significantly increased between the first and the last dose. Given these limitations of current tetravalent vaccine design (four constructs), and the lack of effective protection with current vaccines, we considered an additional construct and analyzed blocks which require up to five peptides (w = 5) to achieve the accumulative coverage of 99%. In regions of consecutive conserved blocks, the peptides can be extended to encompass several conserved blocks. The extended blocks represent the regions in the DENV proteins which can be covered by including five or less peptides in the vaccine construct (Table 6). This analysis can be performed for u > 5, but such design will include a much larger number of constructs in the polyvalent vaccine. While the main focus of this work is on the selection of T-cell epitope targets for DENV vaccine development, it also provides a method which can be used as input for experimental design of other immunological studies, such as examining immunodominance, competitive epitope binding, and detrimental cross-reactivity.
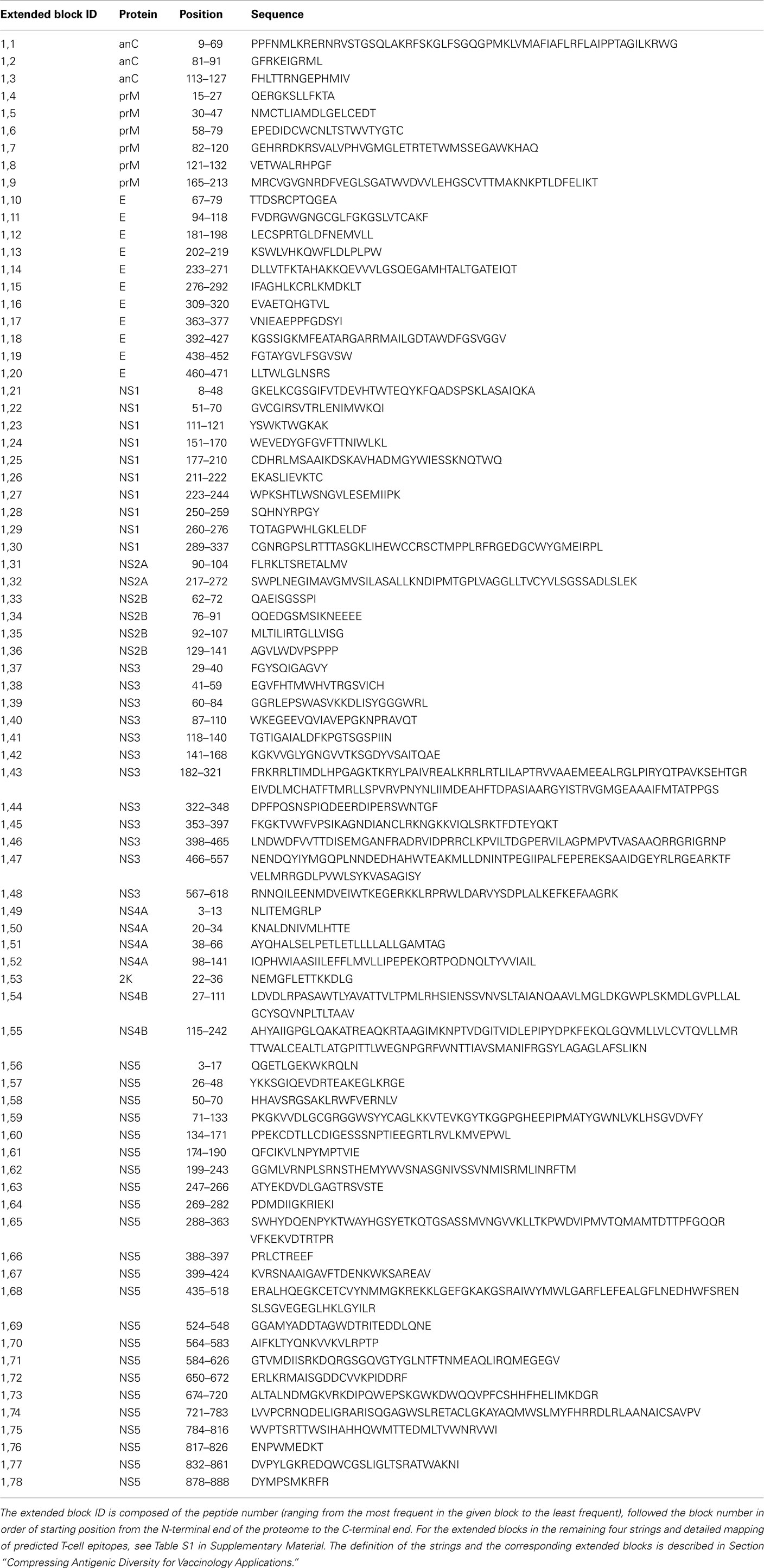
Table 6. Extended block sequences for string 1.
Experimental Support of Principle
We examined IEDB and current literature for examples of experimentally validated epitopes and compared them with our results. We found four examples where predictions correspond to experimental data (Table 7) and two examples where predictions did not match fully the experimental data (Table 7).
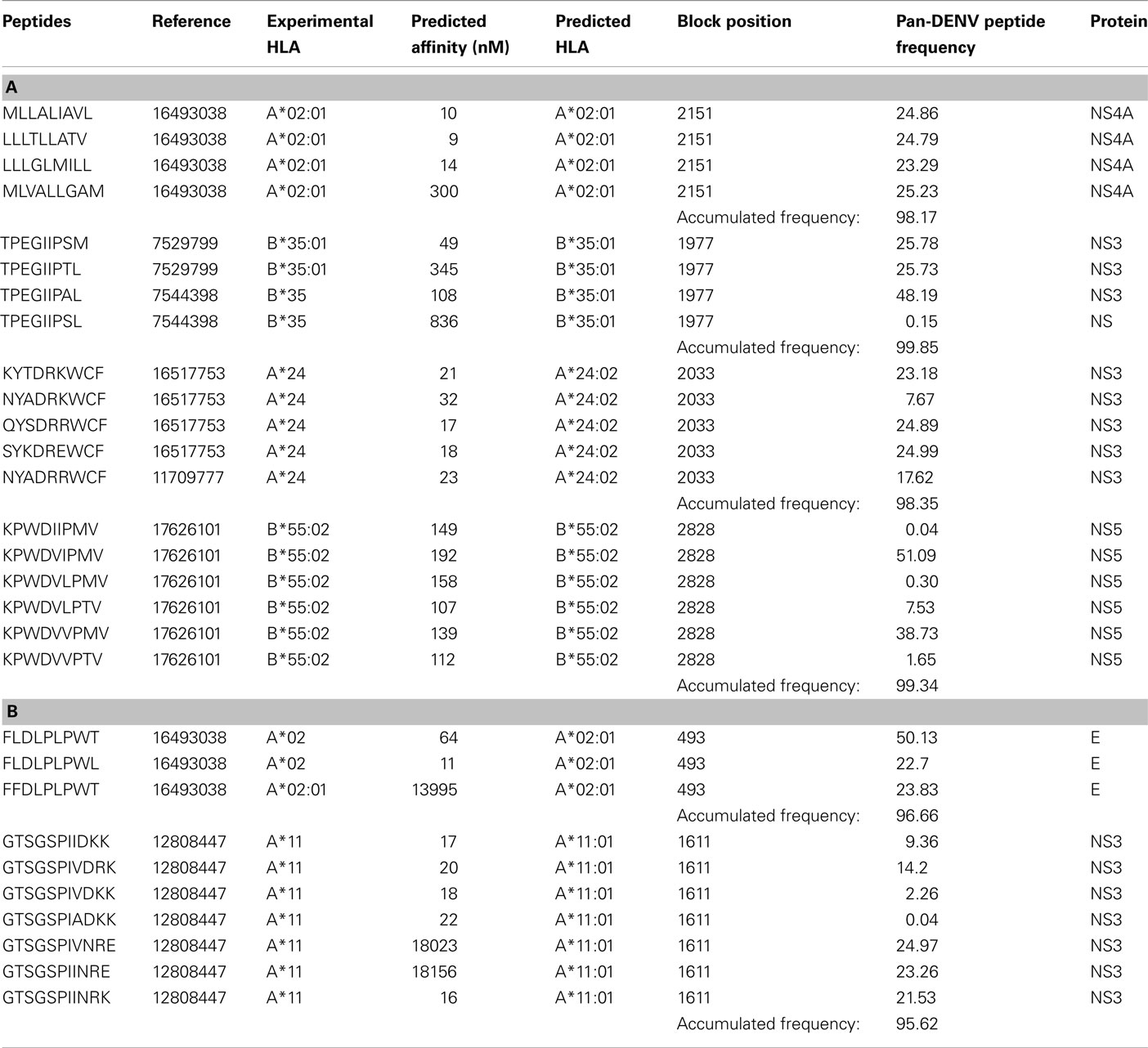
Table 7. (A) Examples of experimental evidence supporting the application of the block entropy approach to achieve broad coverage by including homologous peptide blocks in polyvalent vaccine constructs. In the four examples below, a high percentage of pan-DENV population coverage was achieved by including only blocks of peptides that have all been predicted, as well as experimentally validated, to bind HLA. Predictions for B*55:02 were done using netMHCpan 2.4 (PMID: 17726526). (B) Two examples of blocks of experimentally validated epitopes, for which the block entropy approach failed to account due to epitope predictions inconsistent with the experimental findings of the respective authors.
The blocks presented in Table 7 consist of four to six peptides with accumulated minimum frequency of 98% that were both predicted and experimentally validated HLA binders. The blocks presented in Table 7 consist of three and seven peptides respectively. Common for these two blocks is that one or more peptides experimentally shown to be HLA binders were not predicted to be binders. These two blocks, although potentially useful in a polyvalent vaccine construct, where not identified as universally binding by the prediction algorithm. The implication is that the blocks where majority or all of peptides are identified as potential binders should be experimentally validated. Conversely, the blocks where majority of peptides are identified as non-binders are less likely to be experimental T-cell epitopes.
Conclusion
The analysis of conservation of DENV antigens should consider both the pan-DENV antigenic diversity and the diversity between DENV serotypes. Furthermore, functional properties, such as HLA binding potential are essential for the assessment of immunogenic potential of antigens. DENV conservation analysis in previous studies was based on the traditional approach, in which a peptide is classified as conserved or not conserved based on analysis of each individual amino acid along with an arbitrary frequency threshold (typically 90% or higher). The premise of traditional conservation analysis of vaccine targets is that conserved epitope candidates are more likely to confer cross-protection between pathogen variants. We argue that variant inclusion is important for polyvalent coverage, since the array of factors making a peptide immunogenic is far too complex to assume that conserved predicted binders are automatically the best immunogens. Our systematic approach, that deploys analysis of conservation of blocks of peptides, has produced a 10-fold larger number of potential DENV T-cell epitope targets than the traditional approach. Similar to a previous benchmark study (Khan et al., 2008), our method also enables vaccine target discovery that considers both the conservation of antigens and the immunogenic potential of these peptides. The peptide blocks determined in our study can be used to inform and focus the design of experimental studies of polyvalent dengue vaccines. Furthermore, our approach is applicable to any variable virus such as HIV, influenza, or Hepatitis C. It is also applicable to broader vaccine approaches such as identification of shared peptide targets across major Flavivirus pathogens.
The block entropy analysis is an informed strategy for achieving broad strain coverage in vaccine design with the inclusion of significant but less frequent variants. Central to this approach is the fact that T-cell epitopes are recognized as peptides rather than single residues, and should therefore be analyzed as such. For example, the analysis of 9-mer blocks enables characterization of a set of peptides which collectively can be considered as conserved for immunological applications. In this study, we based the assessment of immunological potential using predictions of peptide binding to seven common HLA class I molecules for which prediction algorithms have been validated (Lin et al., 2008). Some of the conserved blocks contain peptides which are all predicted to bind the same HLA allele – these blocks are considered to be immuno-functionally conserved. The peptide block thus becomes a unit of analysis for building combinatorial vaccine formulations with broad coverage of both pathogen variants and diverse HLA haplotypes. This analysis provides a reasonably sized set of targets that can be experimentally validated, for example by mass spectrometry (Reinhold et al., 2010).
The premise for the concept of immuno-functional conservation is not only that all peptides in a block bind to MHC with the same affinity and HLA restriction, but also that there is enough redundancy in CTLs so that each epitope/MHC complex may elicit an equally strong T-cell response. However, immunodominance of certain epitopes and some T-cell receptors (TCRs; Nikolich-Zugich et al., 2004) can lead to an uneven response to antigens upon vaccination and thus incomplete strain coverage upon challenge. High intra-block homology could allow for a population of CTLs to recognize all epitope in a block, which may also favor the concept of including entire immuno-functionally conserved blocks in a polyvalent vaccine construct. Hence, predicting the cross-protective capacity of a peptide-based vaccine gets more difficult as the number of T-cell epitope candidates increases. Furthermore, the larger the combinatorial space needed to cover the diversity of the four serotypes; the more complex the task to combine all of the epitopes in one vaccine without compromising its efficacy. Considering these factors, we choose to include all blocks in which five peptides or less cover 99% of the block. This number maybe subject to adjustment after proper experimental validation of the epitope pools.
Applying block entropy analysis to the proteomes of DENV1–4, yielded 1,732, 1,551, 1,394, and 1,245 conserved blocks of 8-, 9-, 10-, and 11-mer peptides respectively, as opposed to the results of the benchmark study (Khan et al., 2008) which yielded 206, 165, 118, and 88 conserved 8-, 9-, 10-, and 11-mer peptides respectively, using the traditional criteria for conservation. Of the 1,551 blocks of 9-mer peptides, 110 blocks, consisting of 333 peptides, were predicted to be immuno-functionally conserved, based on their predicted binding affinity to HLA class I, which can form the basis of a T-cell-based polyvalent vaccine against DENV. The method presented here can be readily applied to other relevant viral pathogens such as influenza, HIV, or HPV, as well as extended to encompass MHC class II epitope candidates and functional B-cell epitopes.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by NIH grant U01 AI 90043 (Guang Lan Zhang, Derin B. Keskin, Ellis L. Reinherz, and Vladimir Brusic). Lars Rønn Olsen was supported by a number of Danish student grants (Otto Mønsteds Foundation; Rudolph Als Foundation; Civil Engineer Frants Alling’s Scholarship; Julie Damm’s; Rebild National Park Society, Inc.; Inge and Jørgen Larsen’s Memorial Scholarship; Mayor Niels Albrechtsen’s Scholarship; Danish Society of Engineers Scholarship; and Oticon Foundation).
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/t_cell_biology/10.3389/fimmu.2011.00069/abstract
References
Appay, V. (2009). 25 years of HIV research!…and what about a vaccine? Eur. J. Immunol. 39, 1999–2003.
Barouch, D. H., O’brien, K. L., Simmons, N. L., King, S. L., Abbink, P., Maxfield, L. F., Sun, Y. H., La Porte, A., Riggs, A. M., Lynch, D. M., Clark, S. L., Backus, K., Perry, J. R., Seaman, M. S., Carville, A., Mansfield, K. G., Szinger, J. J., Fischer, W., Muldoon, M., and Korber, B. (2010). Mosaic HIV-1 vaccines expand the breadth and depth of cellular immune responses in rhesus monkeys. Nat. Med. 16, 319–323.
Benson, D. A., Karsch-Mizrachi, I., Lipman, D. J., Ostell, J., and Sayers, E. W. (2010). GenBank. Nucleic Acids Res. 38, D46–D51.
Brown, J. H., Jardetzky, T. S., Gorga, J. C., Stern, L. J., Urban, R. G., Strominger, J. L., and Wiley, D. C. (1993). Three-dimensional structure of the human class II histocompatibility antigen HLA-DR1. Nature 364, 33–39.
Brown, L. E., and Kelso, A. (2009). Prospects for an influenza vaccine that induces cross-protective cytotoxic T lymphocytes. Immunol. Cell Biol. 87, 300–308.
Brusic, V., and August, J. T. (2004). The changing field of vaccine development in the genomics era. Pharmacogenomics 5, 597–600.
Crooks, G. E., Hon, G., Chandonia, J. M., and Brenner, S. E. (2004). WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190.
De Groot, A. S., Marcon, L., Bishop, E. A., Rivera, D., Kutzler, M., Weiner, D. B., and Martin, W. (2005). HIV vaccine development by computer assisted design: the GAIA vaccine. Vaccine 23, 2136–2148.
Duangchinda, T., Dejnirattisai, W., Vasanawathana, S., Limpitikul, W., Tangthawornchaikul, N., Malasit, P., Mongkolsapaya, J., and Screaton, G. (2010). Immunodominant T-cell responses to dengue virus NS3 are associated with DHF. Proc. Natl. Acad. Sci. U.S.A. 107, 16922–16927.
Falk, K., Rotzschke, O., Stevanovic, S., Jung, G., and Rammensee, H. G. (1991). Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature 351, 290–296.
Fischer, W., Perkins, S., Theiler, J., Bhattacharya, T., Yusim, K., Funkhouser, R., Kuiken, C., Haynes, B., Letvin, N. L., Walker, B. D., Hahn, B. H., and Korber, B. T. (2007). Polyvalent vaccines for optimal coverage of potential T-cell epitopes in global HIV-1 variants. Nat. Med. 13, 100–106.
Franco, C., Hynes, N. A., Bouri, N., and Henderson, D. A. (2010). The dengue threat to the United States. Biosecur. Bioterror. 8, 273–276.
Gao, F., Weaver, E. A., Lu, Z., Li, Y., Liao, H. X., Ma, B., Alam, S. M., Scearce, R. M., Sutherland, L. L., Yu, J. S., Decker, J. M., Shaw, G. M., Montefiori, D. C., Korber, B. T., Hahn, B. H., and Haynes, B. F. (2005). Antigenicity and immunogenicity of a synthetic human immunodeficiency virus type 1 group m consensus envelope glycoprotein. J. Virol. 79, 1154–1163.
Gaschen, B., Taylor, J., Yusim, K., Foley, B., Gao, F., Lang, D., Novitsky, V., Haynes, B., Hahn, B. H., Bhattacharya, T., and Korber, B. (2002). Diversity considerations in HIV-1 vaccine selection. Science 296, 2354–2360.
Guy, B., Saville, M., and Lang, J. (2010). Development of Sanofi Pasteur tetravalent dengue vaccine. Hum. Vaccin. 6, 696–705.
Halstead, S. B., and O’rourke, E. J. (1977). Dengue viruses and mononuclear phagocytes. I. Infection enhancement by non-neutralizing antibody. J. Exp. Med. 146, 201–217.
Halstead, S. B., O’rourke, E. J., and Allison, A. C. (1977). Dengue viruses and mononuclear phagocytes. II. Identity of blood and tissue leukocytes supporting in vitro infection. J. Exp. Med. 146, 218–229.
Hertz, T., Nolan, D., James, I., John, M., Gaudieri, S., Phillips, E., Huang, J. C., Riadi, G., Mallal, S., and Jojic, N. (2011). Mapping the landscape of host-pathogen coevolution: HLA class I binding and its relationship with evolutionary conservation in human and viral proteins. J. Virol. 85, 1310–1321.
Hu, D. J., Dondero, T. J., Rayfield, M. A., George, J. R., Schochetman, G., Jaffe, H. W., Luo, C. C., Kalish, M. L., Weniger, B. G., Pau, C. P., Schable, C. A., and Curran, J. W. (1996). The emerging genetic diversity of HIV. The importance of global surveillance for diagnostics, research, and prevention. JAMA 275, 210–216.
Katoh, K., and Toh, H. (2008). Recent developments in the MAFFT multiple sequence alignment program. Brief. Bioinformatics 9, 286–298.
Khan, A. M., Miotto, O., Nascimento, E. J., Srinivasan, K. N., Heiny, A. T., Zhang, G. L., Marques, E. T., Tan, T. W., Brusic, V., Salmon, J., and August, J. T. (2008). Conservation and variability of dengue virus proteins: implications for vaccine design. PLoS Negl. Trop. Dis. 2, e272. doi:10.1371/journal.pntd.0000272
Koo, Q. Y., Khan, A. M., Jung, K. O., Ramdas, S., Miotto, O., Tan, T. W., Brusic, V., Salmon, J., and August, J. T. (2009). Conservation and variability of West Nile virus proteins. PLoS ONE 4, e5352. doi:10.1371/journal.pone.0005352
Lin, H. H., Ray, S., Tongchusak, S., Reinherz, E. L., and Brusic, V. (2008). Evaluation of MHC class I peptide binding prediction servers: applications for vaccine research. BMC Immunol. 9, 8. doi:10.1186/1471-2172-9-8
Lio, P., Politi, A., Buiatti, M., and Ruffo, S. (1996). High statistics block entropy measures of DNA sequences. J. Theor. Biol. 180, 151–160.
Lundegaard, C., Lamberth, K., Harndahl, M., Buus, S., Lund, O., and Nielsen, M. (2008). NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8-11. Nucleic Acids Res. 36, W509–W512.
Mackenzie, J. S., Gubler, D. J., and Petersen, L. R. (2004). Emerging flaviviruses: the spread and resurgence of Japanese encephalitis, West Nile and dengue viruses. Nat. Med. 10, S98–S109.
Martinez, A. N., Tenzer, S., and Schild, H. (2009). T-cell epitope processing (the epitope flanking regions matter). Methods Mol. Biol. 524, 407–415.
Morrison, D., Legg, T. J., Billings, C. W., Forrat, R., Yoksan, S., and Lang, J. (2010). A novel tetravalent dengue vaccine is well tolerated and immunogenic against all 4 serotypes in Flavivirus-naive adults. J. Infect. Dis. 201, 370–377.
Murrell, S., Wu, S. C., and Butler, M. (2011). Review of dengue virus and the development of a vaccine. Biotechnol. Adv. 29, 239–247.
Nickle, D. C., Rolland, M., Jensen, M. A., Pond, S. L., Deng, W., Seligman, M., Heckerman, D., Mullins, J. I., and Jojic, N. (2007). Coping with viral diversity in HIV vaccine design. PLoS Comput. Biol. 3, e75. doi:10.1371/journal.pcbi.0030075
Nikolich-Zugich, J., Slifka, M. K., and Messaoudi, I. (2004). The many important facets of T-cell repertoire diversity. Nat. Rev. Immunol. 4, 123–132.
Olsen, L. R., Zhang, G. L., Reinherz, E. L., and Brusic, V. (2011). FLAVIdB: a data mining system for knowledge discovery in flaviviruses with direct applications in immunology and vaccinology. Immunome Res. 8, 1.
Pilla, L., Rivoltini, L., Patuzzo, R., Marrari, A., Valdagni, R., and Parmiani, G. (2009). Multipeptide vaccination in cancer patients. Expert Opin. Biol. Ther. 9, 1043–1055.
Purcell, A. W., Mccluskey, J., and Rossjohn, J. (2007). More than one reason to rethink the use of peptides in vaccine design. Nat. Rev. Drug Discov. 6, 404–414.
Rammensee, H., Bachmann, J., Emmerich, N. P., Bachor, O. A., and Stevanovic, S. (1999). SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics 50, 213–219.
Reche, P. A., Keskin, D. B., Hussey, R. E., Ancuta, P., Gabuzda, D., and Reinherz, E. L. (2006). Elicitation from virus-naive individuals of cytotoxic T lymphocytes directed against conserved HIV-1 epitopes. Med. Immunol. 5, 1.
Reinhold, B., Keskin, D. B., and Reinherz, E. L. (2010). Molecular detection of targeted major histocompatibility complex I-bound peptides using a probabilistic measure and nanospray MS(3) on a hybrid quadrupole-linear ion trap. Anal. Chem. 82, 9090–9099.
Riemer, A. B., Keskin, D. B., Zhang, G., Handley, M., Anderson, K. S., Brusic, V., Reinhold, B., and Reinherz, E. L. (2010). A conserved E7-derived cytotoxic T lymphocyte epitope expressed on human papillomavirus 16-transformed HLA-A2+ epithelial cancers. J. Biol. Chem. 285, 29608–29622.
Rolland, M., Frahm, N., Nickle, D. C., Jojic, N., Deng, W., Allen, T. M., Brander, C., Heckerman, D. E., and Mullins, J. I. (2011). Increased breadth and depth of cytotoxic T lymphocytes responses against HIV-1-B Nef by inclusion of epitope variant sequences. PLoS ONE 6, e17969. doi:10.1371/journal.pone.0017969
Santra, S., Liao, H. X., Zhang, R., Muldoon, M., Watson, S., Fischer, W., Theiler, J., Szinger, J., Balachandran, H., Buzby, A., Quinn, D., Parks, R. J., Tsao, C. Y., Carville, A., Mansfield, K. G., Pavlakis, G. N., Felber, B. K., Haynes, B. F., Korber, B. T., and Letvin, N. L. (2010). Mosaic vaccines elicit CD8+ T lymphocyte responses that confer enhanced immune coverage of diverse HIV strains in monkeys. Nat. Med. 16, 324–328.
Schneider, T. D., and Stephens, R. M. (1990). Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 18, 6097–6100.
Sette, A., Grey, H., Oseroff, C., Peters, B., Moutaftsi, M., Crotty, S., Assarsson, E., Greenbaum, J., Kim, Y., Kolla, R., Tscharke, D., Koelle, D., Johnson, R. P., Blum, J., Head, S., and Sidney, J. (2009). Definition of epitopes and antigens recognized by vaccinia specific immune responses: their conservation in variola virus sequences, and use as a model system to study complex pathogens. Vaccine 27(Suppl. 6), G21–G26.
Sette, A., and Rappuoli, R. (2010). Reverse vaccinology: developing vaccines in the era of genomics. Immunity 33, 530–541.
Shannon, C. E. (1948). A mathematical theory of communication. Bell Syst. Tech. J. 27, 379–423; 623–656.
Tan, P. T., Heiny, A. T., Miotto, O., Salmon, J., Marques, E. T., Lemonnier, F., and August, J. T. (2010). Conservation and diversity of influenza A H1N1 HLA-restricted T cell epitope candidates for epitope-based vaccines. PLoS ONE 5, e8754. doi:10.1371/journal.pone.0008754
Vita, R., Zarebski, L., Greenbaum, J. A., Emami, H., Hoof, I., Salimi, N., Damle, R., Sette, A., and Peters, B. (2010). The immune epitope database 2.0. Nucleic Acids Res. 38, D854–D862.
Yerly, D., Heckerman, D., Allen, T., Suscovich, T. J., Jojic, N., Kadie, C., Pichler, W. J., Cerny, A., and Brander, C. (2008). Design, expression, and processing of epitomized hepatitis C virus-encoded CTL epitopes. J. Immunol. 181, 6361–6370.
Keywords: antigenic diversity, epitope-based vaccines, immunoinformatics, polyvalent vaccines, reverse vaccinology, vaccine informatics
Citation: Olsen LR, Zhang GL, Keskin DB, Reinherz EL and Brusic V (2011) Conservation analysis of dengue virus T-cell epitope-based vaccine candidates using peptide block entropy. Front. Immun. 2:69. doi: 10.3389/fimmu.2011.00069
Received: 16 August 2011;
Accepted: 14 November 2011;
Published online: 20 December 2011.
Edited by:
Michael Dustin, NYU School of Medicine, USAReviewed by:
Christopher E. Rudd, University of Cambridge, UKBrian M. Baker, University of Notre Dame, USA
Copyright: © 2011 Olsen, Zhang, Keskin, Reinherz and Brusic. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Vladimir Brusic, Cancer Vaccine Center, Dana-Farber Cancer Institute, Harvard Institutes of Medicine 401, 77 Avenue Louis Pasteur, Boston, MA 02118, USA. e-mail:dmxhZGltaXJfYnJ1c2ljQGRmY2kuaGFydmFyZC5lZHU=