 Derin B. Keskin1,2
Derin B. Keskin1,2 Bruce Reinhold1,2 Sun Young Lee3
Bruce Reinhold1,2 Sun Young Lee3 Guanglan Zhang1,2 Simon Lank4 David H. O’Connor4 Ross S. Berkowitz1,2,5
Guanglan Zhang1,2 Simon Lank4 David H. O’Connor4 Ross S. Berkowitz1,2,5 Vladimir Brusic1,2
Vladimir Brusic1,2 Seung Jo Kim3
Seung Jo Kim3 Ellis L. Reinherz1,2*
Ellis L. Reinherz1,2*- 1 Dana-Farber Cancer Institute, Boston, MA, USA
- 2 Harvard Medical School, Boston, MA, USA
- 3 Gynecologic Cancer Center, CHA University, Seongnam, Korea
- 4 University of Wisconsin, Madison, WI, USA
- 5 Brigham and Women’s Hospital, Boston, MA, USA
Persistent infection with high-risk human papilloma viruses (HPV) is the worldwide cause of many cancers, including cervical, anal, vulval, vaginal, penile, and oropharyngeal. Since T cells naturally eliminate the majority of chronic HPV infections by recognizing epitopes displayed on virally altered epithelium, we exploited Poisson detection mass spectrometry (MS3) to identify those epitopes and inform future T cell-based vaccine design. Nine cervical cancer biopsies from HPV-16 positive HLA-A*02 patients were obtained, histopathology determined, and E7 oncogene PCR-amplified from tumor DNA and sequenced. Conservation of E7 oncogene coding segments was found in all tumors. MS3 analysis of HLA-A*02 immunoprecipitates detected E711–19 peptide (YMLDLQPET) in seven of the nine tumor biopsies. The remaining two samples were E711–19 negative and lacked the HLA-A*02 binding GILT thioreductase peptide despite possessing binding-competent HLA-A*02 alleles. Thus, the conserved E711–19 peptide is a dominant HLA-A*02 binding tumor antigen in HPV-16 transformed cervical squamous and adenocarcinomas. Findings that a minority of HLA-A*02:01 tumors lack expression of both E711–19 and a peptide from a thioreductase important in processing of cysteine-rich proteins like E7 underscore the value of physical detection, define a potential additional tumor escape mechanism and have implications for therapeutic cancer vaccine development.
Introduction
Human papilloma viruses (HPV) are double-stranded DNA viruses that infect epithelial cells of the skin and mucosa (zur Hausen, 2002). Of the more than 200 known HPV types, 30–40 are transmitted through sexual contact, infecting the anogenital region and oropharynx. Among these, approximately 15 are designated “high-risk” (i.e., oncogenic) and have been linked to cervical cancer as well as anal, vulva, vaginal, penile, and oropharyngeal cancers (de Villiers et al., 2004). Worldwide, >5% of all new cancers are attributable to high-risk HPV infections (Parkin, 2006). HPV is the cause of virtually all cases of cervical cancer, the second most common female cancer globally (Alani and Munger, 1998; Walboomers et al., 1999). Furthermore, in the developing world where HPV disease burden is greatest, cervical carcinoma is the leading cause of cancer mortality among women. The HPV-16 subtype alone is responsible for 50% of cervical cancers and high-grade cervical intraepithelial lesions (Stone et al., 2002).
Human papilloma viruses is linked to malignant transformation following persistent infection with one or more of the high-risk HPV types (including -16, -18, -31, -33, -35, -39, -45, -51, -52, -56, -58, and -59; Munoz et al., 2003). The viral E6 and E7 encoded proteins have transforming activities through functional inactivation of the p53 and retinoblastoma (Rb) tumor suppressor proteins, respectively (Munger et al., 1989a,b, 1992; Scheffner et al., 1990). Not surprisingly, therefore, abrogation of activities of E6 and/or E7 experimentally terminates the malignant state of HPV-transformed cells in vitro (Desaintes et al., 1997).
A key advance in combating HPV infection and its causally related diseases has been based on virus-like particle prophylactic vaccine development (Kirnbauer et al., 1993). This vaccine consists of recombinant HPV L1 capsid protein that self-assembles to create virus-like particles against which protective, high-titered anti-L1 neutralizing antibodies are elicited in vivo (Kirnbauer et al., 1992, 1993). In both females and males, such vaccines comprising two high-risk capsids effectively prevent development of anogenital diseases (Frazer et al., 2010). However, capsid-based therapeutic HPV vaccine strategies cannot be employed once infection/disease is established for at least two reasons. First, humoral protection is associated with antibody binding to the capsid L1 protein thereby blocking virion entry into the cell. The L1 protein is exclusively expressed late in the HPV replication cycle and only in differentiated keratinocytes within the upper layers of the epithelium to create infectious virions. Persistent infection in the host is maintained by low copy numbers of unencapsulated virus in basal stem cells (Stanley et al., 2007). As humoral immune responses against capsid proteins do not affect persistently infected basal cells, they are consequently unable to clear infection (Stoler et al., 1990; Frazer et al., 2010). Second, high-risk HPV-associated cancers arise with genomic integration of E6 and E7 viral oncogenes. Although L1 and L2 genes may also be integrated and capsid proteins expressed (Bellone et al., 2009) these are intracellular and hence, antibody inaccessible.
During natural infection, HPV is cleared by the immune system in the vast majority of subjects. A few percent of persistent infections evolve into neoplasias that may progress to malignancy with high-risk HPV types (Bhat et al., 2011). Although immune suppression is associated with chronic infection, most patients with persistent HPV infections do not show immune deficiencies in response to other pathogens (Frazer, 2004). Persistent infection with malignant or premalignant lesions, is often associated with HPV-specific T cells whose significance for antitumor immunity is not always clear. For example, one study that detected HPV-responding T cells in peripheral blood of patients with high-grade cervical intraepithelial neoplasia (CIN2/3) was unable to correlate IFN-γ levels or antigen specificity of T cell response with the 26% of the patients that showed spontaneous lesion regression (Trimble et al., 2011). In another cervical cancer study, a third of the patients exhibited HPV-specific proliferative T cell responses prior to surgery, but long-term survival after surgery was not associated with T cell response (Heusinkveld et al., 2011). Whereas the presence of circulating HPV-specific CD8+ cytotoxic T lymphocytes (CTL) did not correlate with disease prognosis in early stage cervical cancer, the degree of CD8+ tumor infiltration and balance (ratio) of CD8+ T cells to regulatory T cells were correlated (Piersma et al., 2007; Jordanova et al., 2008). Consistent with a critical role of inflammatory signals in inducing T cell responses, vulval intraepithelial lesions treated with the toll-like receptor (TLR) 7 activator Imiquimod showed regression and viral clearance in association with circulating HPV-specific T cells (van Seters et al., 2008; Terlou et al., 2010). Recently, vaccination with synthetic long-peptides covering the E6 and E7 oncoproteins plus conventional adjuvant was reported to clear HPV-16 induced high-grade vulva intraepithelial neoplastic lesions in ∼50% of patients (Kenter et al., 2009).
While it is widely acknowledged that antitumor cellular immunity is influenced by regulatory mechanisms, tumor microenvironment, and tumor escape mechanisms (Finn, 2008), the nature of T cell antigen specificity is less defined despite its critical role. Ex vivo T cell assays that mark HPV-specificity identify a broad priming display by professional antigen presenting cells (pAPCs) but do not interrogate antitumor CTL activity nor do they identify the breadth of HPV-specific major histocompatibility complex (MHC) I display of the tumor per se (de Vos van Steenwijk et al., 2010). Pointedly, in the context of a narrow tumor display, antitumor vaccines that prime a broad T cell response generate a small fraction of tumor lytic CD8+ T cells, resulting in low densities of useful tumor infiltrating T cells.
In order to focus the cytotoxic T cell response on E7 and/or E6 targets with precision, it is first critical to define the peptide epitopes presented on the human leukocyte antigen (HLA) molecules of human tumor cells in vivo. These are the targets for CD8+ CTL, after all. Because of the pitfalls of indirect functional identification of presumptive tumor antigens (Reinherz and Acuto, 2011), a definitive physical identification of tumor antigens by mass spectrometry is important. This can be very challenging, given a limited number of tumor cells available from clinical biopsy samples and potential downregulation of HLA by tumor escape mechanisms (Campo et al., 2010). To this end, we have developed MS3 Poisson detection mass spectrometry and exploited it here to identify an E711–19 9-mer peptide as a dominant T cell epitope presented by HLA-A*02:01 on the majority of HPV-16 cervical squamous and adenocarcinomas before and post-adjuvant chemotherapy. Detailed analysis of viral E7 gene sequences, high resolution HLA typing and definition of peptide presentation on tumor cells permit rational assessment of a cancer’s vulnerability to immunotherapy.
Materials and Methods
Samples
Nine HPV-16+ cervical cancer biopsies were obtained from CHA Bundang Medical Center, CHA University in Korea. Patients consented to donate part of the surgical tumor biopsy material and RNA for research under IRB approved protocol.
HLA Typing
Patients’ peripheral blood mononuclear cells (PBMC) were screened for BB7.2 antibody reactivity by flow cytometry to identify patients expressing HLA-A*02 alleles. RNA was extracted from PBMC to obtain detailed molecular HLA typing data for HLA-A, HLA-B, and HLA-C alleles by 454 sequencing methods.
RNA Extraction
Total peripheral blood mononuclear cells (PBMC) were isolated from whole blood using ficoll/paque density centrifugation. RNA was isolated from PBMC using Qiagen RNAeasy kits.
454 HLA-Amplicon Sequencing
Clonal HLA-amplicon pyrosequencing was performed as previously described (Lank et al., 2010), with additional novel PCR amplicon binding sites to allow higher resolution genotyping. Briefly, patient RNA was reverse transcribed to cDNA using the Superscript III First-Strand Synthesis System with oligo(d)T primer (Invitrogen, Carlsbad, CA, USA). For each sample, universally conserved HLA class I primers were used to generate a 581-bp amplicon (Forward primer: 5′-GTGGGCTACGTGGACGAC-3′; Reverse primer: 5′-TGCCAGGTCAGTGTGATCTC-3′) and a 1021-bp amplicon (Forward primer: 5′-TGGCCCTGACCSAGACCTG-3′; Reverse primer: 5′-CAGAGCCCTGGGCACTGT-3′), which together completely cover exons 2, 3, 4, 5, and 6, as well as parts of exons 1 and 7. The amplification oligos were fusion primers containing these sequences as well as standard Roche/454 “A” and “B” sequencing adapters and unique multiplex identifier DNA barcodes (MIDs). PCR products were purified using Amplure-XP SPRI beads (Beckman Coulter Genomics, Danvers, MA, USA) and subsequently pooled to create a multiplexed sequencing library. This library was sequenced on a GS-Jr pyrosequencer with Titanium series reagents using the standard bi-directional amplicon sequencing protocol and base calling (454 Life Sciences, Branford, CT, USA). Sequence reads were parsed by MID, quality filtered, then forward, and reverse reads were aligned to the library of known HLA class I alleles (IMGT database, version 3.3) using BLAT. The pattern of perfectly matched alleles for each amplicon and direction was used to determine comprehensive, high resolution class I genotype for each class I locus.
Tumor Protein Extraction and Immunoprecipitation
Bulk tumor tissue was resuspended in 2 ml of protein lysis buffer containing 20 mM Tris (pH 8.0), 1 mM EDTA, 100 mM NaCl, 1% Triton X-100, 60 mM n-octylglucoside, phenylmethylsulfonyl fluoride (all from Sigma-Aldrich), and protease inhibitors (Complete protease inhibitor cocktail tablets, Roche). Tumor tissue was disrupted using a Tissueraptor (Qiagen). Each lysate was pre-cleared using centrifugation and the pellet containing insoluble membrane and nuclear fractions was frozen for further processing and DNA extraction. Soluble lysates were co-incubated with 20 μl of GammaBind Plus Sepharose beads (GE Lifesciences, Piscataway, NJ, USA) non-covalently linked to BB7.2 (BD biosciences, San Diego, CA, USA) antibody for 3 h. Beads were washed and prepared for MS analysis as described previously (Reinhold et al., 2010; Riemer et al., 2010).
MS3 Detection of Tumor-Derived E7 Peptides Isolated by Anti-HLA Immunoprecipitation
Bound peptides, released from peptide–HLA-A*02 complexes by acid wash of the GammaBind Plus Sepharose beads, were trapped on a C18 tip and eluted into a needle for nanospray ionization and mass analyses. Typically, the sample needle was mounted on a Sciex Elite quadrupole–TOF for high resolution mass spectra and then transferred to a QTrap 4000 quadrupole–LIT for MS3 ion fragmentation analyses. MS3 data are generated from two successive stages of ion isolation and fragmentation. The second stage fragmentation data are analyzed for molecular targets by a detection algorithm based on sampling a stochastic Poisson process. Briefly, one calculates an amplitude at which the MS3 spectrum of the target peptide can be embedded in the MS3 spectrum of the peptide mixture at a fixed probability. The detection is scored based on comparing the embedding of the target spectrum to a set of trial spectra generated by translating the target spectrum. Plotting the embedding amplitude as a function of m/z translation, a graph is created that marks high detection significance with a prominent 0-translation peak. For detecting the E711–19 peptide YMLDLQPET the primary MS3 reference pattern used for detection is generated by first dissociating the doubly changed molecular ions at m/z 555.3 and then dissociating the product ions in the window at m/z 764.4 (corresponding to the b6 ion fragment YMLDLQ-). The MS3 555.3/764.4 data of peptides from the tumor samples is Poisson measured against the dissociation spectrum of the b6 ion fragment of the YMLDLQPET peptide. The less intense b8 fragment YMLDLQPE- can also be used for detection by Poisson analysis of MS3 555.3/990.5 data. MS3 Poisson detection does not require chromatographic separation of the recovered peptides, allowing targeted analyses for samples on smaller scales (Reinhold et al., 2010; Riemer et al., 2010).
E7–PCR and Sequencing
Nuclear cell pellets remaining after protein immunoprecipitation were utilized for DNA extraction. The pellets were further solubilized and digested using Proteinase K-supplemented nuclear lysis buffer. Total tumor genomic DNA was extracted utilizing the Wizard SV genomic DNA extraction system (Promega, Madison, WI, USA). The integrated HPV-16 E7 gene was amplified from total DNA using the E7-specific primers 5′-TGTCAAAAGCCACTGTGTCC-3′ (forward) and 5′-AGTGGACTACCAAATACTTTCGTT-3′ (reverse). PCR products were purified using the Wizard PCR cleanup kit (Promega, Madison, WI, USA). The E7 gene of each sample was sequenced at the Dana-Farber Cancer Center DNA sequencing core facilities. The resulting sequences were translated to protein sequences via the ExPASy proteomics server – Translate tool1. Protein sequences were aligned using online MAFFT multiple alignment software2.
35-mer E7 HPV Peptide Synthesis, Dendritic and B Lymphocyte Loading, and Presentation Analysis
The E71–35 peptide (MHGDTPTLHEYMLDLQPETTDLYCYEQLNDSSEEE), identical to a previously published sequence (Kenter et al., 2009), was synthesized (RS Synthesis, Louisville, KY, USA) and HPLC purified to greater than 95% and stored in DMSO prior to use in presentation studies. Adherent monocytes were purified from HLA-A02 donor PBMC’s and differentiated using 50 ng/ml GM-CSF and 50 ng/ml IL-4 (Peprotech, NJ, USA) for 7 days. Dendritic cells (DCs) were induced to mature using 1 μg/ml LPS and loaded with 100 μg/ml E71–35 peptide simultaneously overnight. B cells were stained with CD19 and sterile-sorted from HLA-A02 donor PBMCs using a FACS Aria. B cells were activated using 1 μg/ml LPS and simultaneously loaded with 100 μg/ml E71–35 peptide overnight. MHC peptide complexes were immunoprecipitated from 2 to 2.5 × 106 DC or B lymphocytes and peptide presentation was analyzed by MS3.
Suppression of γ-Interferon-Inducible Lysosomal Thiol Reductase (GILT) Protein Expression Utilizing siRNA
Lentiviral siRNA silencers and siRNA oligomers targeted for GILT suppression as well as vector controls were purchased from Santa Cruz Biotechnology. GILT siRNA lentiviral particles and control lentiviral particles were used to infect CaSki cells in the presence of 5 μg/ml polybrene. Cells with stable lentiviral integration were selected in vitro using 2 μg/ml puromycin. In parallel, CaSki cells were also transfected with GILT siRNA oligomers or FITC-labeled control siRNA using Lipofectamine 2000. Transfected cells were cultured for three additional days. MHC peptide complexes were isolated from 2 × 107 cells from each treatment group by immunoprecipitation and peptide presentation was analyzed by MS3 (see Appendix for additional details).
Results
Patient Population and Overall Assessment Strategy
Biopsies of HPV-16 positive cervical cancer were obtained from HLA-A*02 positive patients either prior to or following neoadjuvant chemotherapy with cisplatin and etoposide (Bae et al., 2008), defining the type and extent of tumor pathology. The HPV-16 status of the tumors and patients was determined by HPV-specific gene array analysis (An et al., 2003). A frozen tumor biopsy was provided for DNA extraction and E7 sequencing and for MS analysis of HLA-associated peptide epitopes. In addition, PBMC were isolated and RNA extracted for high resolution HLA typing by 454 sequencing. Korea was selected as a geographical locale in view of the significant prevalence of HPV-16 positive cervical cancer and availability of modern medical practices.
The tripartite approach using genomics, proteomics (MS3) and immunomics (HLA typing and epitope binding prediction) illustrated in Figure 1, offers a general strategy for identifying CD8+ CTL target antigens on tumors or infected tissues. DNA sequencing provides viral strain-specific information, permitting protein sequence and epitope predictions in conjunction with bioinformatics. Poisson detection MS3 proteomics identifies those peptides among many possibilities that are physically displayed on the surface of cells. High resolution HLA typing defines the specific alleles born by a given patient to which peptides can be predicted to bind and then verified by MS3. Note, for example, that among 13 distinct predicted HLA-A*0201 binding E7 peptides, only one, E711–19 (YMLDLQPET), was found to be naturally processed and displayed on HPV-16 transformed HLA-A*0201 expressing cell lines in vitro (Riemer et al., 2010).
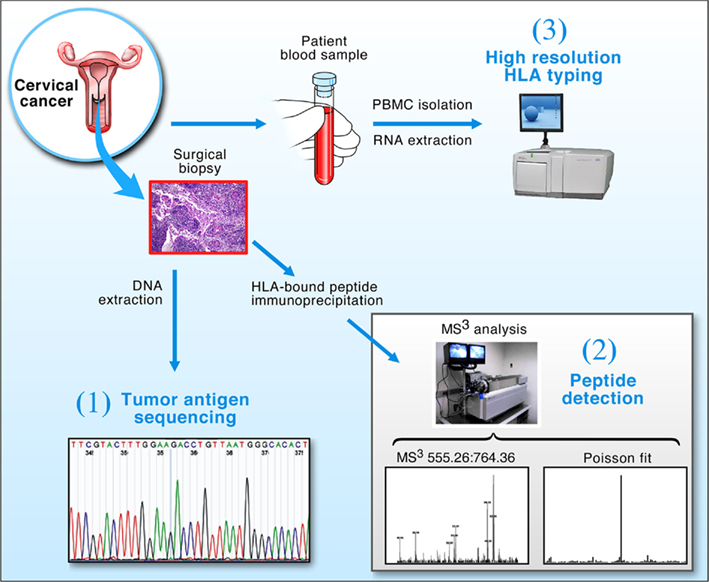
Figure 1. Strategy for the detection of tumor antigens from clinical biopsy samples. Cervical cancer biopsy material and blood were obtained from patients and processed in three ways: (1) Tumor DNA was extracted and the E7 oncogene sequenced to determine its conservation; (2) The protein fraction from the tumor was used to extract peptide–HLA complexes for MS3 analysis; and (3) RNA was isolated from patient PBMCs and used to obtain high resolution HLA typing via 454 sequencing. Collectively, the technologies allow DNA sequencing and T cell epitope predictions within conserved segments of tumor antigens bound to genotypically defined HLA molecules to be verified by MS3 analysis.
Clinical characteristics are reviewed in Table 1. All patients were pre-selected for PBMC BB7.2 antibody reactivity and hence were HLA-A*02 positive, limiting the potential complexity of tumor antigens. Notwithstanding, there are more than 280 different HLA-A*02 alleles that form the HLA-A2 supertype (Robinson et al., 2011), exhibiting overlapping yet differential peptide binding repertoires (Sudo et al., 1995). Hence, it was essential to precisely HLA type samples. Four digit molecular typing was possible for all HLA-A, -B, and -C alleles (Lank et al., 2010) aside from several ambiguities in HLA-A and -C in KTS1 and HLA-A in KTS9.
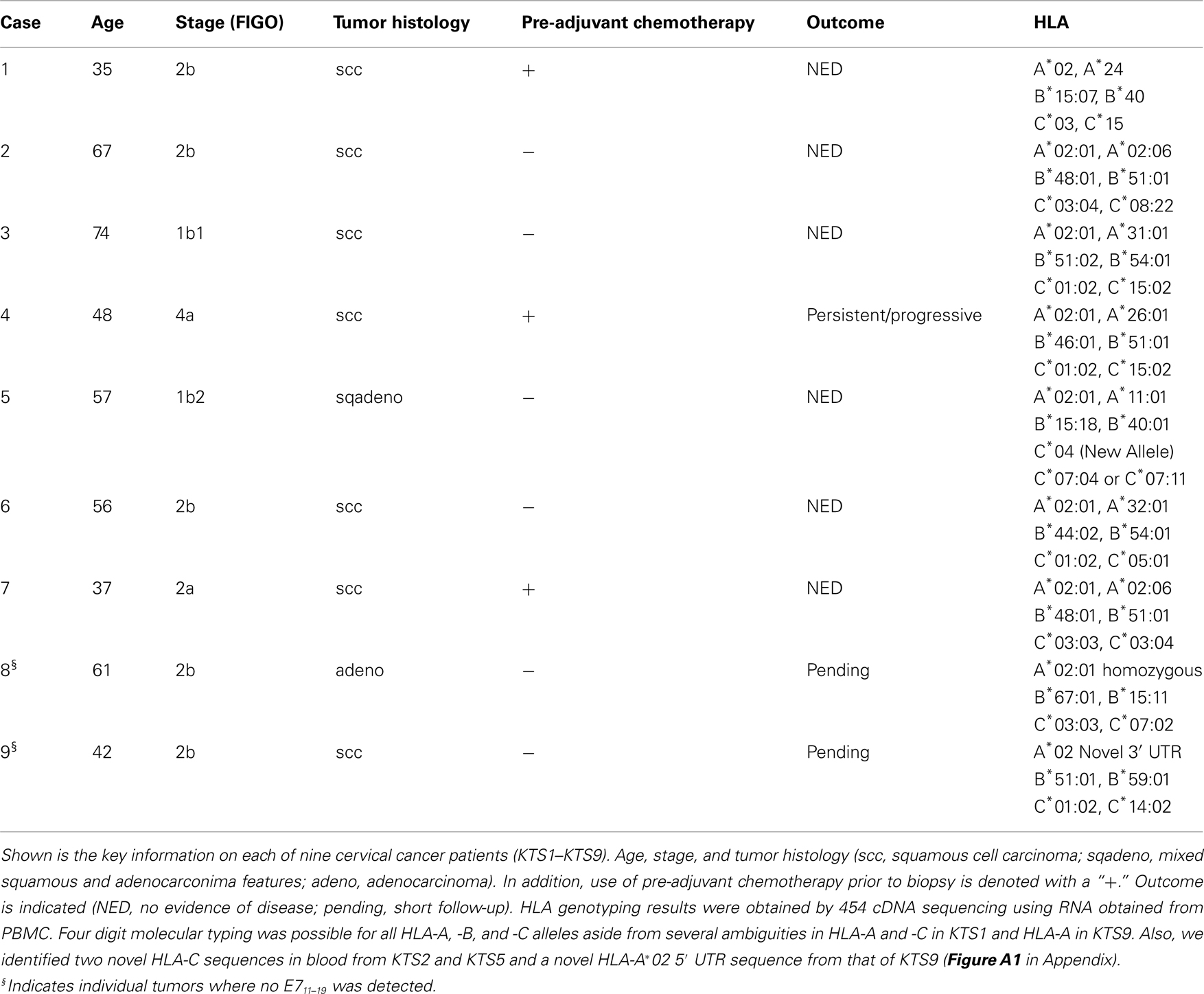
Table 1. Human papilloma viruses-16+ cervical tumor samples from HLA-A*02+ patients.
Histopathology of Cervical Tumor Samples
Nine HPV-16+ cervical cancer tumor biopsies representing a variety of pathologies were used in the present study. Tumor staging (FIGO) ranged from 1b1 to 4a. Biopsies were obtained either at the time of initial diagnosis (samples KTS2, 3, 5, 6, 8, 9) or at the time of radical operation after three cycles of neoadjuvant chemotherapy (samples KTS1, 4, and 7). H&E sections of representative tumors are presented in Figure 2. Seven of nine were defined as squamous cell carcinomas, in agreement with studies showing that HPV-16 infection is most commonly associated with squamous cell carcinomas while HPV-18 is strongly associated with adenocarcinomas (Wilczynski et al., 1988).
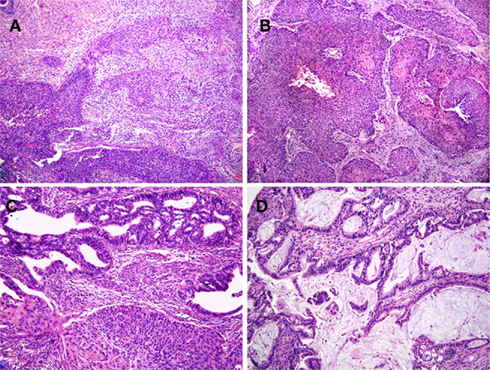
Figure 2. Human papilloma viruses-16+ cervical cancer tumor samples display a variety of pathologies. Histopathology of tumor samples stained with hematoxylin and eosin. (A,B) are representatives of squamous cell carcinoma samples. The tumor sample shown in (A) (KTS2) is an invasive, moderate to poorly differentiated squamous cell carcinoma (200×). The tumor sample in (B) (KTS9) represents an invasive, moderately differentiated squamous cell carcinoma but with focal keratinization (right; 100×). The remaining two samples shown are adenocarcinomas with or without squamous features [(C,D), respectively, 200×]. (C) Shows an adeno-squamous carcinoma (KTS5), demonstrating glandular differentiation with a squamous component and exhibiting the sheet growth characteristics of squamous epithelium. (D) Shows a mucin producing, invasive adenocarcinoma (KTS8). Results are from pathology slides prepared from nine different tumors.
HPV-16 E7 Sequencing
DNA was extracted from each tumor mass and its HPV-16 E7 oncogene was amplified by gene specific primers to analyze the sequence of the HPV-16 E7 genomic integrant. As shown in Figure 3, we determined that the sequence for the dominant E711–19 epitope YMLDLQPET was conserved in all of the tumors samples. Two HPV E7 sequence variants are observed: Asn is found at position 29 (N29) in KTS2, KTS3, and KTS6 whereas Ser (S29) is present in the remaining samples. Sequence comparison with known HPV-16 virus sequences in the NIH GenBank suggests that the N29 variant belongs to an HPV-16 strain that has spread worldwide (Seedorf et al., 1985). In contrast, the S29 variant belongs to an HPV-16 strain common in Asia and East Asia (Figure A2 in Appendix). Other than amino acid position 29, the remainder of the protein sequence was conserved and identical to our reference sequence E7 KTS7.

Figure 3. Multiple alignment of HPV-16 E7 protein sequences determined from tumor DNA sequencing. HPV-16 E7 DNA sequences were PCR-amplified from total tumor DNA and sequenced. DNA sequences were translated to and protein sequences aligned for comparison. Two HPV-16 E7 sequence variants are present in the analyzed tumor samples: N29 in KTS2, KTS3, and KTS6 marked as red, and S29 in the remaining samples. The E7 11–19 YMLDLQPET T-cell epitope highlighted as yellow was conserved in all samples. Results are representative of two independent sequencing reactions performed on nine different tumor DNA samples.
Poisson MS3 Detection of the HPV-16 E711–19 Peptide YMLDLQPET
The amount of available tumor biopsy material for the MS3 samples varied from 7.9 to 19 mg. Samples were immunoprecipitated with BB7.2 mAb to isolate peptide–HLA-A*02 complexes and peptides eluted and analyzed by MS3 for presence of the E711–19 YMLDLQPET. Figure 4 shows Poisson detection plots for the nine tumor samples (KTS1–KTS9) and control HLA-A*02:01 peptides from an HPV negative T cell hybridoma (Figure 4J). Detection is based on a probabilistic Poisson process, gaging the likelihood that the sample contains a natural target peptide whose MS3 fragmentation pattern was previously determined using a synthetic peptide (see Materials and Methods). In the Poisson plots of Figure 4, this significance corresponds to the amplitude of the 0-offset peak relative to the peaks in the m/z offset range from −50 to 50. Setting the detection threshold at 4× the average of the offset amplitudes (shaded regions, Figure 4), tumor samples KTS1–KTS7 (Figures 4A–G) were positive for E711–19 while samples KTS8, KTS9, and control (Figures 4H–J, respectively) have 0-amplitudes below this level and were negative (Figures A3–A5 in Appendix). Samples KTS1–KTS4 were also examined by MS3 555.3/990.5 and were positive for the b8 fragment as expected (Figures A3–A4 in Appendix).
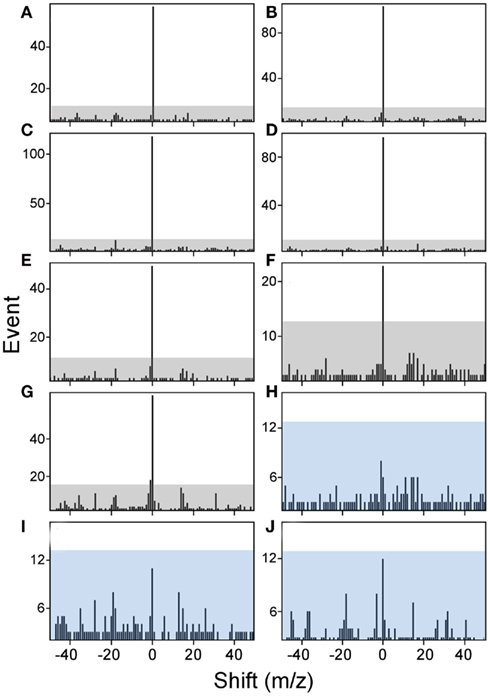
Figure 4. Poisson detection for the E711–19 ion fragment YMLDLQ- in MS3 555.3/764.4 spectra of HLA-A*02 bound peptides from biopsied tumor samples. Shaded regions mark a significance boundary set at 4 times the average amplitude excluding the 0-peak. Poisson fits to the unshifted reference spectrum (the 0-peak on the x-axis) that remain within the shaded region do not represent significantly ion events in the MS3 spectra of the samples. KTS1–KTS9 (A–I), respectively. Biopsied cervical tumors with the (A–G) representing tumors samples KTS1–7 indicating positive detection. (H,I) Represent negative detection on tumors samples KTS8 and 9. (J) Illustrates peptides from a non-HPV-transformed T cell line, as a negative control. Results are from single immunoprecipitation experiments repeated on nine different tumor samples.
Poisson MS3 Analysis does not Detect the HPV-16 E711–20 Peptide YMLDLQPETT on Patients’ Tumors
The E711–19 peptide was previously detected in all HPV-16 E7 expressing cell lines examined, whereas the E711–20 peptide was absent (Riemer et al., 2010). This difference does not reflect a technical limitation, as it was previously shown that MS3 Poisson detection for the HLA-A*02 binding E711–20 peptide YMLDLQPETT is as sensitive as that of E711–19 (Reinhold et al., 2010; Riemer et al., 2010). Tumor samples KTS1–2, KTS4, and KTS8 were also examined by MS3 Poisson detection for the longer E711–20 peptide. The E711–20 peptide was not detected in any patient tumors (Figures A6 and A7 in Appendix). In contrast, pAPCs like human monocyte-derived dendritic cells as well as B cells process a synthetic long peptide, E71–35, to cross-present both E711–20 and E711–19 peptide bound to HLA-A*02. In fact, cross-presented E711–20 is more abundant than E711–19 (Figure A8 in Appendix).
Failure to Detect γ-Interferon-Inducible Lysosomal Thiol Reductase (GILT) Signal Peptides in E711–19 Negative Tumor Samples
The negative detection of E711–19 in samples KTS8 and KTS9 (Figures 4H,I) was not associated with lower overall peptide recovery (Figures A9 and A10 in Appendix). Unexpectedly, these two samples were additionally negative for the HLA-A*02 binding peptides LLDVPTAAV and LLLDVPTAAVQA derived from the signal peptide of GILT, also termed Ifi30 or IP30. This enzyme is involved in antigen presentation and hence its downregulation may have significant consequences for immune responses against tumors (O’Donnell et al., 2004; Rausch et al., 2010; Srinivasan and Maric, 2011). Six of the seven E711–19 positive samples were checked for one or both of the two GILT-associated peptides which were each abundant and could be trivially detected by MS2 analysis (Figures A11–A13 in Appendix). The lack of GILT signal peptide was noticed initially in analyzing incidentally collected MS2 data from KTS8 (Figure A11D in Appendix) and characterized as negative, in contrast with abundant peptide amounts observed in all E711–19 positive samples. A subsequent E711–19 negative sample, KTS9. was examined by MS3 analysis for LLDVPTAAV and here the negative result extends to the very highest sensitivity (Figure 5).
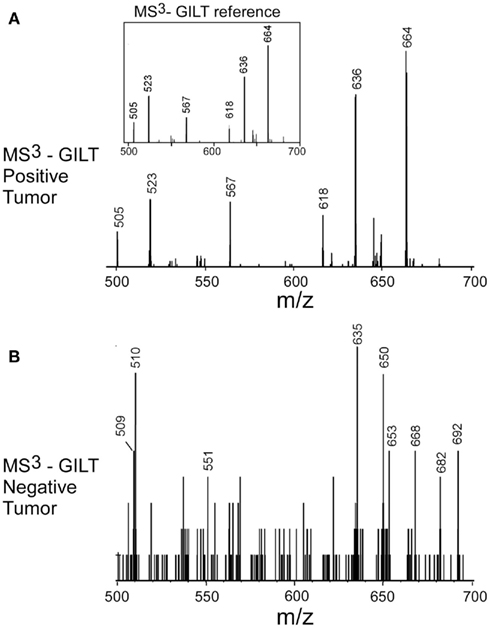
Figure 5. Identifying the HLA-A*02 bound peptide LLDVPTAAV from the signal sequence of γ-interferon-inducible lysosomal thiol reductase (GILT). (A) MS3 449.76/781.4 spectrum from a tumor sample that is positive for E711–19 identifies abundant GILT signal peptide; the inset shows the MS3 449.76/781.4 spectrum of a synthetic LLDVPTAAV. (B) MS3 449.76/781.4 spectrum from tumor sample KTS9 (negative for E711–19) does not identify the GILT signal peptide even with the high sensitivity analysis employed. Results are from single immunoprecipitation experiments repeated on two different tumor samples.
Although loss of transporter associated with antigen processing (TAP)-1 or -2 gene function is associated with very high relative levels of GILT signal peptides on HLA-A2 molecules (Henderson et al., 1992; Wei and Cresswell, 1992),TAP expression does not account for the observed differential display of GILT signal peptides in tumor samples KTS1–7 vs. KTS8–9. The pool of peptides associated with A2 molecules from TAP-deficient cells is a low complexity set dominated by a few signal peptides. On the other hand, those from TAP-sufficient cells are of very high complexity, a feature which is immediately apparent in the mass spectrum (Figure A14 in Appendix) and clearly characterizes peptide recovery from both the GILT positive and GILT negative tumor samples (Figure A10 in Appendix). Furthermore, lentiviral siRNA silencing of GILT in the HPV-16 transformed CaSki cell line (Figure 6) resulted in 80% specific suppression of GILT peptide expression and concomitant E711–19 reduction, consistent with the correlation between loss of HLA-associated GILT peptide and absent E711–19 in patient tumor samples. Since GILT expression could not be extinguished in the CaSki cell line, we cannot prove that the association is causal (that is, the link between lack of GILT and E711–19 in patient tumors). Other factors that might account for lack of E711–19 expression on KTS8–9 remain to be investigated.
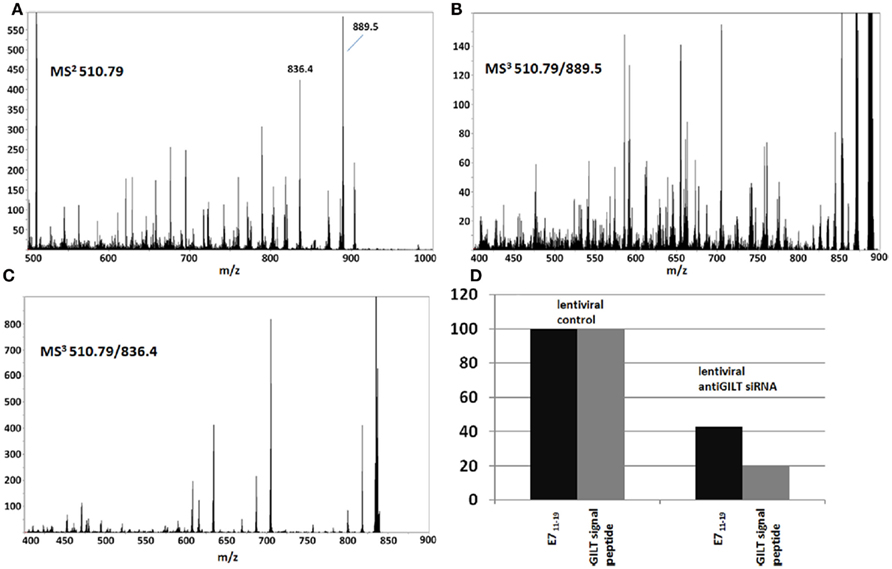
Figure 6. siRNA suppression of GILT in CaSki cells shows that the abundance of GILT correlates with the abundance of the E711–19 peptide. siRNA suppression of GILT protein is correlated with a reduction in the amount of E711–19 peptide associated with HLA-A*02 in CaSki cells. The suppression of GILT is monitored by its signal peptide LLDVPTAAV and both the GILT and E711–19 peptides are characterized relative to the overall peptides recovered. (A–C) Show the MS2 and MS3 spectra that identify the MS3 510.79/889.5 spectrum as a good ion flux calibrant (see Appendix for further details). (D) Shows the percentage reduction in the GILT and E711–19 peptides relative to the overall HLA-A*02 associated peptide signal as a function of lentiviral siRNA silencing of GILT protein expression.
Discussion
Almost all currently licensed vaccines against infectious pathogens stimulate generation of antibody production (Bambini and Rappuoli, 2009). However, because of recent advances in immunobiology, T cell-based vaccines will soon evolve (Reinherz and Acuto, 2011). Engagement of this limb of the immune response is important since CTL target virally transformed cells such as HPV-16 induced tumor cells, whereas antibodies cannot readily do so. In addition, T cells recognize internal viral proteins that are much less variable than envelope proteins, the targets of neutralizing antibodies. Influenza A is a prime example of a RNA virus where such a T cell approach has the potential to lead to a universal vaccine with coverage of seasonal variants as well as pandemic threats (Reinherz and Acuto, 2011).
T cell recognition is referred to as MHC-restricted. The T cell receptor on a given T cell interacts with a specific peptide in complex with an MHC molecule (pMHC) being expressed on the surface of infected targets, tumors, or antigen presenting cells. During viral infection, proteolytic cleavage in the cytosol leads to the generation of peptides, i.e., so-called “processing.” These peptides traffic into the endoplasmic reticulum and are loaded onto HLA molecules in the human, followed by expression of the complex on the cell surface. CTL recognition is of peptide epitopes generally 9–10 amino acids in length bound to HLA molecules (H-2 in the mouse), flagging a target for destruction. Each human expresses on his (her) nucleated cells, three to six of these HLA-A, -B, and -C molecules (number dependent on zygosity at each locus) with ∼5000 HLA class I allelic variants identified globally. Peptide binding differs for distinct alleles while subtle differences are observed for peptide repertoires binding to similar HLA alleles. An additional complexity is that as many as 100,000 distinct pMHC complexes may be arrayed on the surface of each target cells, with the T cell having the capacity to detect fewer than a dozen of the relevant peptides among a sea of related yet distinct, irrelevant pMHC complexes. As T cell recognition is exquisitely sensitive and precise it follows that detailed information must be acquired on the exact viral peptide epitope (or tumor antigen) that is expressed on infected/transformed cells being targeted for CTL destruction.
This study utilizes a new approach to directly identify tumor antigens in clinical biopsy samples of women with HPV-16 induced cervical cancers obtained either prior to or following adjuvant chemotherapy. A nanospray MS3 method is deployed on a quadrupole linear ion trap hybrid, detecting targeted peptides with high dynamic range and sensitivity from complex cellular peptide mixtures without prior separation requirements (Reinhold et al., 2010). This technique represents a major advance in translational medicine compared with prior attempts at detection of tumor antigens from bulk surgical resection or cultured tumor cells. Peptides bound to HLA-A*02 family could be recovered and identified from as little as ≤8 mg of tissue, offering an approximately 1000-fold improvement in sensitivity compared to previous studies (Kawahara et al., 2006; Hawkins et al., 2008; Weinzierl et al., 2008). In seven of nine HLA-A*02 patient tumors, the E711–19 9-mer bound to HLA-A*02 alleles was readily detected. The remaining two patients’ samples (KTS8 and KTS9) lacked E711–19 as well as the GILT HLA-A*02 binding 9-mer and 11-mer signal peptides, LLDVPTAAV and LLDVPTAAVQA, respectively.
The current findings underscore the heterogeneity of HPV-16 induced tumor antigen presentation while highlighting the value of direct detection in individual tumor samples. Furthermore, the data imply that inhibition of GILT protein expression in these tumors, as manifested by absent GILT peptide derivatives, may be a mechanism of tumor escape since this enzyme impacts antigen display (vide infra). The frequency of cysteine residues in mammalian proteins is around 2.26% of the amino acids (Miseta and Csutora, 2000). The cysteine content of HPV-16 E6 and E7 are 8.86 and 7.14% respectively. Given that E6 and E7 are extremely cysteine-rich proteins, proteolysis, and therefore presentation of E6 and E7 related peptides may be significantly impaired in the absence of GILT (Yee et al., 1985; Cresswell et al., 1999). Of note, overall peptide expression on KTS8 and KTS9 was not reduced in complexity or amount relative to that of the other samples. Currently, in the mouse GILT is known to be targeted to lysosomal and phagosomal compartments involved in impacting MHC class II molecules and cross-presentation of class I molecules (orthologs of HLA molecules; Maric et al., 2001; Singh and Cresswell, 2010). Our studies in the human suggest that GILT may also contribute to class I presentation in the setting of HPV transformation. Notwithstanding this, the loss of GILT is not the key basis of tumor escape in view of the fact that the majority of patients retain GILT and E711–19 peptide–HLA-associated display.
We sequenced the HPV-16 genomic DNA to determine the translated E7 amino acid sequence and found that the E711–19 epitope is conserved in all HPV-16 patient isolates. MS3 on tumor biopsies identified the presence of the 9-mer but not a 10-mer, E711–20. We postulate that prior failure of an E711–20 peptide vaccine to elicit a positive clinical outcome was due to the lack of display of the 10-mer on tumor samples (van Driel et al., 1999). Furthermore, T cells elicited against the 9-mer show specificity against the 9-mer but have little, if any, crossreactivity with the 10-mer. This 9-mer serves to direct cytolysis by T cell lines, whereas the related 10-mer (E711–20) was neither detected by MS3 on HPV-transformed tumor cells nor effectively recognized by 9-mer specific CTL (Riemer et al., 2010). That study also showed that the direct tumor analysis of pMHC complexes is required for antigen identification since most conserved HPV E7 and E6 epitopes predicted and experimentally shown to bind HLA-A*02 are not naturally processed and presented.
We predicted E7-derived peptide binding to 88 distinct HLA-A2 supertype alleles (Zhang et al., 2010). E711–19 was predicted to bind with high affinity to 64 HLA-A2 alleles (72.7%). Of these binders, HLA-A*02:01 and HLA-A*02:06 comprise 85% of the Korean A2 population. While failure to express a binding allele could be a basis for absent E711–19 display on 15% of patients, high resolution HLA typing excluded this possibility among our patients, instead defining a conjoint loss of E711–19 and GILT signal peptide display.
The above constellation of data can foster a personalized medicine approach for vaccine design based on genomics, proteomics, and HLA typing considerations. In addition, universal vaccine approaches are also tractable given the advancing knowledge on HLA class I supertypes. Nine HLA class I supertypes (A1, A2, A3, A24, B7, B27, B44, B58, and B62) have been identified (Sidney et al., 1996). A combination of the four major HLA class I supertypes (A2, A3, B7, and B44) affords general population coverage >90%, regardless of ethnicity (Reche and Reinherz, 2005; Zhang et al., 2010).
Our approach combines the speed and breadth of coverage afforded by computational predictions with the depth and precision of MS3. Detection of the subset of predicted peptides actually displayed on infected/tumor cells by evolving MS methods allows for precise epitope targeting, excluding those pAPC “cross-presented only” epitopes or irrelevant crossreactivities. Formulations that incorporate CTL epitopes with the universal CD4 helper stimulator and TLR or other molecules are practical. The alternative approach already showing promise in HPV-16 induced vulva intraepithelial neoplasms utilizes vaccines with synthetic long-peptides derived from E6 and E7 (Kenter et al., 2009). However, in the absence of directed epitope selectivity, it is difficult to prevent irrelevant cross-presentation and attendant inflammation directed to non-protective epitopes (Figure A8 in Appendix). Such non-tumor targeting CTL could dilute the effective immune response. Furthermore, while this long peptide approach is an important advance, only a subset of patients responds to such therapy (Kenter et al., 2009). This variability in outcome is presumably a consequence of HLA differences among patients with differential epitope presentation capacity, display heterogeneity as noted here for GILT and tumor microenvironmental differences (Welters et al., 2010). Our approach is a first step to rational tumor vaccine design. It should foster clinical success in conjunction with blockade of intrinsic and extrinsic tumor escape mechanisms.
In addition to the high-risk HPVs, human cancer viruses include Epstein–Barr virus, hepatitis B virus, hepatitis C virus, human T cell lymphotropic virus type 1, Kaposi’s sarcoma-associated herpes virus and Merkel cell polyoma virus (reviewed in Sarid and Gao, 2011). It has been suggested that viral infections contribute to upward of 15–20% of all human cancers worldwide (Sarid and Gao, 2011). As screening and molecular detection methods improve, the number of viruses associated with human tumors is likely to increase even further. Our approach represents a means to identify key tumor target antigens for therapeutic vaccine development. Rules established through this approach may also serve to guide vaccine development for tumors of non-viral etiology.
Conclusion
Characterization by Poisson detection MS3 of tumor biopsy samples from HPV-16 induced cervical cancers in HLA-A*02:01 patients studied herein in conjunction with earlier analysis of HPV-16 transformed HLA-A*02 epithelial cell lines (Riemer et al., 2010) provides considerable collective insight into HLA-associated E7 peptide display on human cancer cells. Only one epitope among 13 in silico-based bioinformatic predictions was observed using an exquisitely sensitive physical detection method. Thus, bioinformatic prediction should be viewed as offering an inclusion list of potential epitopes for physical detection, the vast majority of which are absent on tumor cell targets. These conclusions are in stark contrast to prevailing notions derived from in silico epitope landscape predictions in breast and colon cancer, suggesting that missense mutations are sufficiently common to give rise to multiple immunogenic T cell epitopes in each tumor without need for knowledge about the target antigens (Segal et al., 2008). Instead, we suggest that precise detailing of T cell epitopes and their MHC restriction elements are required to focus CTL for useful T cell vaccine development.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by NIH grant UO1 AI090043, DFCI institutional funding for the Cancer Vaccine Center, NIH grant R24RR021745 and The Wisconsin Alumni research Foundation award MSN 100252. We gratefully acknowledge the pathological consultation of Dr. Christopher Crum, Director of Women’s and Perinatal Pathology, Brigham and Women’s Hospital.
Footnotes
References
Alani, R. M., and Munger, K. (1998). Human papillomaviruses and associated malignancies. J. Clin. Oncol. 16, 330–337.
An, H. J., Cho, N. H., Lee, S. Y., Kim, I. H., Lee, C., Kim, S. J., Mun, M. S., Kim, S. H., and Jeong, J. K. (2003). Correlation of cervical carcinoma and precancerous lesions with human papillomavirus (HPV) genotypes detected with the HPV DNA chip microarray method. Cancer 97, 1672–1680.
Bae, J. H., Lee, S. J., Lee, A., Park, Y. G., Bae, S. N., Park, J. S., and Namkoong, S. E. (2008). Neoadjuvant cisplatin and etoposide followed by radical hysterectomy for stage 1B-2B cervical cancer. Gynecol. Oncol. 111, 444–448.
Bambini, S., and Rappuoli, R. (2009). The use of genomics in microbial vaccine development. Drug Discov. Today 14, 252–260.
Bellone, S., El-Sahwi, K., Cocco, E., Casagrande, F., Cargnelutti, M., Palmieri, M., Bignotti, E., Romani, C., Silasi, D. A., Azodi, M., Schwartz, P. E., Rutherford, T. J., Pecorelli, S., and Santin, A. D. (2009). Human papillomavirus type 16 (HPV-16) virus-like particle L1-specific CD8+ cytotoxic T lymphocytes (CTLs) are equally effective as E7-specific CD8+ CTLs in killing autologous HPV-16-positive tumor cells in cervical cancer patients: implications for L1 dendritic cell-based therapeutic vaccines. J. Virol. 83, 6779–6789.
Bhat, P., Mattarollo, S. R., Gosmann, C., Frazer, I. H., and Leggatt, G. R. (2011). Regulation of immune responses to HPV infection and during HPV directed immunotherapy. Immunol. Rev. 239, 85–98.
Campo, M. S., Graham, S. V., Cortese, M. S., Ashrafi, G. H., Araibi, E. H., Dornan, E. S., Miners, K., Nunes, C., and Man, S. (2010). HPV-16 E5 down-regulates expression of surface HLA class I and reduces recognition by CD8 T cells. Virology 407, 137–142.
Cresswell, P., Arunachalam, B., Bangia, N., Dick, T., Diedrich, G., Hughes, E., and Maric, M. (1999). Thiol oxidation and reduction in MHC-restricted antigen processing and presentation. Immunol. Res. 19, 191–200.
de Villiers, E. M., Fauquet, C., Broker, T. R., Bernard, H. U., and zur Hausen, H. (2004). Classification of papillomaviruses. Virology 324, 17–27.
de Vos van Steenwijk, P. J., Heusinkveld, M., Ramwadhdoebe, T. H., Lowik, M. J., van der Hulst, J. M., Goedemans, R., Piersma, S. J., Kenter, G. G., and van der Burg, S. H. (2010). An unexpectedly large polyclonal repertoire of HPV-specific T cells is poised for action in patients with cervical cancer. Cancer Res. 70, 2707–2717.
Desaintes, C., Demeret, C., Goyat, S., Yaniv, M., and Thierry, F. (1997). Expression of the papillomavirus E2 protein in HeLa cells leads to apoptosis. EMBO J. 16, 504–514.
Frazer, I. H. (2004). Prevention of cervical cancer through papillomavirus vaccination. Nat. Rev. Immunol. 4, 46–54.
Frazer, I. H., Leggatt, G. R., and Mattarollo, S. R. (2010). Prevention and treatment of papillomavirus-related cancers through immunization. Annu. Rev. Immunol. 29, 111–138.
Hawkins, O. E., Vangundy, R. S., Eckerd, A. M., Bardet, W., Buchli, R., Weidanz, J. A., and Hildebrand, W. H. (2008). Identification of breast cancer peptide epitopes presented by HLA-A*0201. J. Proteome Res. 7, 1445–1457.
Henderson, R. A., Michel, H., Sakaguchi, K., Shabanowitz, J., Appella, E., Hunt, D. F., and Engelhard, V. H. (1992). HLA-A2.1-associated peptides from a mutant cell line: a second pathway of antigen presentation. Science 255, 1264–1266.
Heusinkveld, M., Welters, M. J., van Poelgeest, M. I., van der Hulst, J. M., Melief, C. J., Fleuren, G. J., Kenter, G. G., and van der Burg, S. H. (2011). The detection of circulating human papillomavirus-specific T cells is associated with improved survival of patients with deeply infiltrating tumors. Int. J. Cancer 128, 379–389.
Jordanova, E. S., Gorter, A., Ayachi, O., Prins, F., Durrant, L. G., Kenter, G. G., van der Burg, S. H., and Fleuren, G. J. (2008). Human leukocyte antigen class I, MHC class I chain-related molecule A, and CD8+/regulatory T-cell ratio: which variable determines survival of cervical cancer patients? Clin. Cancer Res. 14, 2028–2035.
Kawahara, M., Hori, T., Matsubara, Y., Okawa, K., and Uchiyama, T. (2006). Identification of HLA class I-restricted tumor-associated antigens in adult T cell leukemia cells by mass spectrometric analysis. Exp. Hematol. 34, 1496–1504.
Kenter, G. G., Welters, M. J., Valentijn, A. R., Lowik, M. J., Berends-van der Meer, D. M., Vloon, A. P., Essahsah, F., Fathers, L. M., Offringa, R., Drijfhout, J. W., Wafelman, A. R., Oostendorp, J., Fleuren, G. J., van der Burg, S. H., and Melief, C. J. (2009). Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N. Engl. J. Med. 361, 1838–1847.
Kirnbauer, R., Booy, F., Cheng, N., Lowy, D. R., and Schiller, J. T. (1992). Papillomavirus L1 major capsid protein self-assembles into virus-like particles that are highly immunogenic. Proc. Natl. Acad. Sci. U.S.A. 89, 12180–12184.
Kirnbauer, R., Taub, J., Greenstone, H., Roden, R., Durst, M., Gissmann, L., Lowy, D. R., and Schiller, J. T. (1993). Efficient self-assembly of human papillomavirus type 16 L1 and L1-L2 into virus-like particles. J. Virol. 67, 6929–6936.
Lank, S. M., Wiseman, R. W., Dudley, D. M., and O’Connor, D. H. (2010). A novel single cDNA amplicon pyrosequencing method for high-throughput, cost-effective sequence-based HLA class I genotyping. Hum. Immunol. 71, 1011–1017.
Maric, M., Arunachalam, B., Phan, U. T., Dong, C., Garrett, W. S., Cannon, K. S., Alfonso, C., Karlsson, L., Flavell, R. A., and Cresswell, P. (2001). Defective antigen processing in GILT-free mice. Science 294, 1361–1365.
Miseta, A., and Csutora, P. (2000). Relationship between the occurrence of cysteine in proteins and the complexity of organisms. Mol. Biol. Evol. 17, 1232–1239.
Munger, K., Phelps, W. C., Bubb, V., Howley, P. M., and Schlegel, R. (1989a). The E6 and E7 genes of the human papillomavirus type 16 together are necessary and sufficient for transformation of primary human keratinocytes. J. Virol. 63, 4417–4421.
Munger, K., Werness, B. A., Dyson, N., Phelps, W. C., Harlow, E., and Howley, P. M. (1989b). Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J. 8, 4099–4105.
Munger, K., Scheffner, M., Huibregtse, J. M., and Howley, P. M. (1992). Interactions of HPV E6 and E7 oncoproteins with tumour suppressor gene products. Cancer Surv. 12, 197–217.
Munoz, N., Bosch, F. X., de Sanjose, S., Herrero, R., Castellsague, X., Shah, K. V., Snijders, P. J., and Meijer, C. J. (2003). Epidemiologic classification of human papillomavirus types associated with cervical cancer. N. Engl. J. Med. 348, 518–527.
O’Donnell, P. W., Haque, A., Klemsz, M. J., Kaplan, M. H., and Blum, J. S. (2004). Cutting edge: induction of the antigen-processing enzyme IFN-gamma-inducible lysosomal thiol reductase in melanoma cells is STAT1-dependent but CIITA-independent. J. Immunol. 173, 731–735.
Parkin, D. M. (2006). The global health burden of infection-associated cancers in the year 2002. Int. J. Cancer 118, 3030–3044.
Piersma, S. J., Jordanova, E. S., van Poelgeest, M. I., Kwappenberg, K. M., van der Hulst, J. M., Drijfhout, J. W., Melief, C. J., Kenter, G. G., Fleuren, G. J., Offringa, R., and van der Burg, S. H. (2007). High number of intraepithelial CD8+ tumor-infiltrating lymphocytes is associated with the absence of lymph node metastases in patients with large early-stage cervical cancer. Cancer Res. 67, 354–361.
Rausch, M. P., Irvine, K. R., Antony, P. A., Restifo, N. P., Cresswell, P., and Hastings, K. T. (2010). GILT accelerates autoimmunity to the melanoma antigen tyrosinase-related protein 1. J. Immunol. 185, 2828–2835.
Reche, P. A., and Reinherz, E. L. (2005). PEPVAC: a web server for multi-epitope vaccine development based on the prediction of supertypic MHC ligands. Nucleic Acids Res. 33, W138–W142.
Reinherz, E. L., and Acuto, O. (2011). Molecular T cell biology-basic and translational challenges in the 21st century. Front. Immunol. 2:3. doi:10.3389/fimmu.2011.00003
Reinhold, B., Keskin, D. B., and Reinherz, E. L. (2010). Molecular detection of targeted major histocompatibility complex I-bound peptides using a probabilistic measure and nanospray MS(3) on a hybrid quadrupole-linear ion trap. Anal. Chem. 82, 9090–9099.
Riemer, A., Keskin, D. B., Zhang, G., Handley, M., Anderson, K. S., Brusic, V., Reinhold, B., and Reinherz, E. L. (2010). A conserved E7-derived CTL epitope expressed on human papillomavirus-16 transformed HLA-A2+ human epithelial cancers. J. Biol. Chem. 285, 29608–29622.
Robinson, J., Mistry, K., McWilliam, H., Lopez, R., Parham, P., and Marsh, S. G. (2011). The IMGT/HLA database. Nucleic Acids Res. 39, D1171–D1176.
Sarid, R., and Gao, S. J. (2011). Viruses and human cancer: from detection to causality. Cancer Lett. 305, 218–227.
Scheffner, M., Werness, B. A., Huibregtse, J. M., Levine, A. J., and Howley, P. M. (1990). The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 63, 1129–1136.
Seedorf, K., Krammer, G., Durst, M., Suhai, S., and Rowekamp, W. G. (1985). Human papillomavirus type 16 DNA sequence. Virology 145, 181–185.
Segal, N. H., Parsons, D. W., Peggs, K. S., Velculescu, V., Kinzler, K. W., Vogelstein, B., and Allison, J. P. (2008). Epitope landscape in breast and colorectal cancer. Cancer Res. 68, 889–892.
Sidney, J., Grey, H. M., Kubo, R. T., and Sette, A. (1996). Practical, biochemical and evolutionary implications of the discovery of HLA class I supermotifs. Immunol. Today 17, 261–266.
Singh, R., and Cresswell, P. (2010). Defective cross-presentation of viral antigens in GILT-free mice. Science 328, 1394–1398.
Srinivasan, P., and Maric, M. (2011). Signal transducer and activator of transcription 1 negatively regulates constitutive gamma interferon-inducible lysosomal thiol reductase expression. Immunology 132, 209–216.
Stanley, M. A., Pett, M. R., and Coleman, N. (2007). HPV: from infection to cancer. Biochem. Soc. Trans. 35, 1456–1460.
Stoler, M. H., Whitbeck, A., Wolinsky, S. M., Broker, T. R., Chow, L. T., Howett, M. K., and Kreider, J. W. (1990). Infectious cycle of human papillomavirus type 11 in human foreskin xenografts in nude mice. J. Virol. 64, 3310–3318.
Stone, K. M., Karem, K. L., Sternberg, M. R., McQuillan, G. M., Poon, A. D., Unger, E. R., and Reeves, W. C. (2002). Seroprevalence of human papillomavirus type 16 infection in the United States. J. Infect. Dis. 186, 1396–1402.
Sudo, T., Kamikawaji, N., Kimura, A., Date, Y., Savoie, C. J., Nakashima, H., Furuichi, E., Kuhara, S., and Sasazuki, T. (1995). Differences in MHC class I self peptide repertoires among HLA-A2 subtypes. J. Immunol. 155, 4749–4756.
Terlou, A., Seters, M., Kleinjan, A., Heijmans-Antonissen, C., Santegoets, L. A., Beckmann, I., Beurden, M., Helmerhorst, T. J., and Blok, L. J. (2010). Imiquimod-induced clearance of HPV is associated with normalization of immune cell counts in usual type vulvar intraepithelial neoplasia. Int. J. Cancer 127, 2831–2840.
Trimble, C. L., Peng, S., Thoburn, C., Kos, F., and Wu, T. C. (2011). Naturally occurring systemic immune responses to HPV antigens do not predict regression of CIN2/3. Cancer Immunol. Immunother. 59, 799–803.
van Driel, W. J., Ressing, M. E., Kenter, G. G., Brandt, R. M., Krul, E. J., van Rossum, A. B., Schuuring, E., Offringa, R., Bauknecht, T., Tamm-Hermelink, A., van Dam, P. A., Fleuren, G. J., Kast, W. M., Melief, C. J., and Trimbos, J. B. (1999). Vaccination with HPV16 peptides of patients with advanced cervical carcinoma: clinical evaluation of a phase I-II trial. Eur. J. Cancer 35, 946–952.
van Seters, M., van Beurden, M., ten Kate, F. J., Beckmann, I., Ewing, P. C., Eijkemans, M. J., Kagie, M. J., Meijer, C. J., Aaronson, N. K., Kleinjan, A., Heijmans-Antonissen, C., Zijlstra, F. J., Burger, M. P., and Helmerhorst, T. J. (2008). Treatment of vulvar intraepithelial neoplasia with topical imiquimod. N. Engl. J. Med. 358, 1465–1473.
Walboomers, J. M., Jacobs, M. V., Manos, M. M., Bosch, F. X., Kummer, J. A., Shah, K. V., Snijders, P. J., Peto, J., Meijer, C. J., and Munoz, N. (1999). Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 189, 12–19.
Wei, M. L., and Cresswell, P. (1992). HLA-A2 molecules in an antigen-processing mutant cell contain signal sequence-derived peptides. Nature 356, 443–446.
Weinzierl, A. O., Maurer, D., Altenberend, F., Schneiderhan-Marra, N., Klingel, K., Schoor, O., Wernet, D., Joos, T., Rammensee, H. G., and Stevanovic, S. (2008). A cryptic vascular endothelial growth factor T-cell epitope: identification and characterization by mass spectrometry and T-cell assays. Cancer Res. 68, 2447–2454.
Welters, M. J., Kenter, G. G., de Vos van Steenwijk, P. J., Lowik, M. J., Berends-van der Meer, D. M., Essahsah, F., Stynenbosch, L. F., Vloon, A. P., Ramwadhdoebe, T. H., Piersma, S. J., van der Hulst, J. M., Valentijn, R. P. M., Fathers, L. M., Drijfhout, J. W., Franken, K. M. C., Oostendorp, J., Fleuren, G. J., Melief, C. J. M., and van der Burg, S. H. (2010). Success or failure of vaccination for HPV16-positive vulvar lesions correlates with kinetics and phenotype of induced T-cell responses. Proc. Natl. Acad. Sci. U.S.A. 107, 11895–11899.
Wilczynski, S. P., Bergen, S., Walker, J., Liao, S. Y., and Pearlman, L. F. (1988). Human papillomaviruses and cervical cancer: analysis of histopathologic features associated with different viral types. Hum. Pathol. 19, 697–704.
Yee, C., Krishnan-Hewlett, I., Baker, C. C., Schlegel, R., and Howley, P. M. (1985). Presence and expression of human papillomavirus sequences in human cervical carcinoma cell lines. Am. J. Pathol. 119, 361–366.
Zhang, G. L., Deluca, D. S., Keskin, D. B., Chitkushev, L., Zlateva, T., Lund, O., Reinherz, E. L., and Brusic, V. (2010). MULTIPRED2: a computational system for large-scale identification of peptides predicted to bind to HLA supertypes and alleles. J. Immunol. Methods 374, 53–61.
zur Hausen, H. (2002). Papillomaviruses and cancer: from basic studies to clinical application. Nat. Rev. Cancer 2, 342–350.
Appendix
Detection of YMLDLQPET in Cervical Tumor Samples by MS3 Poisson Detection of b8 Fragment
Collisional dissociation of the doubly charged E711–19 peptide at m/z 555.263 produces a series of b-ions at m/z 636, 764, and 990 that can be selected for MS3 Poisson detection (Figure A3). The major b6 fragment at m/z 764 is optimal but the lower intensity m/z 636 or 990 fragments can provide additional evidence. However, this evidence is available only at a substantially reduced sensitivity. The two low mass y-ions at m/z 346 and 474 are less effective as MS3 targets for Poisson detection in complex peptide mixtures since the lower m/z range has a higher ion background and the reference dissociation patterns of smaller ion fragments provide fewer peaks for pattern discrimination.
Tumor samples KTS1, KTS2, KTS4, and KTS7 were analyzed by Poisson MS3 detection for additional evidence of YMLDLQPET using MS3 555.3/990.5 spectra (Figure A4). No other tumor samples were analyzed using MS3 555.3/990.5 data acquisition. Tumor samples KTS1, KTS2, and KTS4 were positive, but detection significance in KTS7 was below threshold.
MS Analysis of Tumor Sample KTS6 was Compromised
A stubborn technical problem in the nano affinity purification with nanospray MS analyses is the occasional appearance of higher molecular weight fragments that elute from the C18 tip in the organic phase along with the MHC peptides. This ion signature has been associated with a highly variable loss of sensitivity for the lower m/z peptides. KTS6 showed a relatively low significance in MS3 Poisson detection of the E711–19 peptide compared with the other E711–19 positive samples (KTS1–5 and KTS7). However, KTS6 also distinguished itself with a substantial ion background of high molecular weight fragments (Figure A5) and this may account for the low detection significance. For contrast, Figure A10 shows more typical MS spectra of HLA-A2 peptides from other tumor samples that do not present a high molecular weight ion background.
E711–20 Peptide YMLDLQPETT was not Detected in any of the Tumor Samples
MS2 m/z 605.787 of synthetic peptide YMLDLQPETT shows the b6 ion fragment YMLDLQ- is generated in high abundance by collision activated dissociation, similar to MS2 m/z 555.263 of YMLDLQPET. The reverse phase trapping and ionization of the two peptides are also similar as measured with the synthetic standards. As m/z 605.787 is in the upper wing of the m/z distribution of peptide molecular ions recovered from HLA-A*02 complexes, there is less co-selected ion background, and one would expect detection of YMLDLQPETT in tumor extracts by MS3 Poisson analysis should be at least as sensitive as detecting YMLDLQPET, if not more so.
The tumor samples KTS1, KTS2, KTS4, and KTS8 were checked my MS3 605.8/764.4 Poisson detection for the E711–20 peptide YMLDLQPETT. Figure A7 shows the E711–20 Poisson detection plots for these tumor samples in Figures A7A–D. No tumor sample gave a positive detection signature for E711–20.
Negative Detection of E711–19 in Tumor Samples KT8 and KTS9 is not Due to Poor Peptide Recovery
A quadrupole–oTOF mass spectrum of HLA-A2 bound peptides from each tumor sample is taken after the nanospray needle is loaded with the peptide sample. The same needle is then transferred to the quadrupole–LIT for MS3 data acquisition. For the quadrupole–oTOF mass spectrum, ion peak amplitudes (y-axis) are in units of counts per second. The ion counts depend on analyte concentration but they also depend on the nanospray needle’s position, the tip’s aperture, the charged aerosol’s plume geometry and the electrodynamic flow induced by the ionization voltage. These are not generally reproducible, making the overall ion flux an unreliable metric of peptide recovery. A more consistent signature of peptide recovery is to use an internal ion metric, in particular, to compare the m/z signature of doubly charged peptide ions to the singly charged ions of the solvent background. A sample showing good peptide recovery produces an ion signature which has a pair of roughly equivalent peaks per m/z unit in the m/z range of 450–550 (inset, Figure A9) whereas the ion chemical or solvent noise produces only singly charged ions or a single peak per m/z unit. The doubled density of peaks over m/z 450–550 defines a hump that is easily observed in the mass spectrum of samples with good MHC I peptide recovery (Figures A9 and A10).
Figure A10 shows quadrupole–oTOF MS spectra of peptides recovered from four tumor samples. In each spectrum the MHC I peptide hump stands out against the chemical noise background. This indicates the peptide recoveries for the E711–19 negative tumor samples KTS8 and KTS9 (Figures A10C,D) are comparable with the E711–19 positive tumor samples KTS5 and KTS7 (Figures A10A,B). The lack of MS3 E711–19 ion events in KTS8 and KTS9 is not correlated with a lack of peptide molecular ions.
Abundant γ-Interferon-Inducible Lysosomal Thiol Reductase (GILT) Signal Peptides of GILT are Identified by MS2 Spectra Only in the E711–19 Positive Tumor Samples
The association of the GILT signal peptides LLDVPTAAV and LLLDVPTAAVQA with the Poisson detection of E711–19 was not anticipated at the outset of this study. During the collection of mass spectral data for tumor KTS8 only the lack of the GILT signal peptides was immediately noted. The calculations for Poisson detection are post-acquisition, but the MS2 449.75 spectrum of LLDVPTAAV is often monitored in optimizing needle alignment and ionization voltage since the fragmentation of LLDVPTAAV in HLA-A2+ samples is almost universally observed as a strong signal that can be immediately recognized (Figure A11).
In tumor samples that were run before recognizing that the GILT signal peptide might be absent, the MS2 449.75 spectrum was not always collected, even if it was monitored in optimizing ion signal prior to MS3 analysis on the quadrupole–LIT instrument. In a number of the tumor samples MS2 605.787 spectra were collected as part of monitoring for the E711–20 peptide YMLDLQPETT. Selecting the 605.787 m/z window also transmits the doubly charged GILT signal peptide LLLDVPTAAVQA at m/z 605.856 and since this peptide is again a dominant component, the dissociation signature of the synthetic peptide (Figure A12) can be immediately recognized against the ion background. The combination of MS2 449.75, MS2 605.787, and MS3 449.75/781.4 experiments then identifies tumor samples KTS1, KTS2, KTS4–7 as GILT positive and KTS8, KTS9 as GILT negative. The MS2 data is shown in Figure A13. Only tumor sample KTS3 was not checked for GILT either directly or incidentally as the nanospray needle tip failed early in the analysis.
Peptides Recovered from HLA-A2 Complexes Isolated from TAP-negative Cells Comprise a Set of Relatively Low Complexity Compared with TAP-Positive Cells
Loss of transporter associated with antigen processing (TAP) stops the flow of proteasome-generated peptides from the cytosol into the ER, leaving only ER-resident signal peptides to associate with HLA-A2 molecules. This results in a dramatically simplified pool of peptides recovered from BB7.2 (anti-HLA-A2) affinity purified peptide–HLA complexes when comparing TAP-deficient to TAP-sufficient cells. The difference in complexity translates into a very distinct mass spectral signature in which a few dominant signal peptides is replaced by a hump of overlapped doubly charged peptides (Figure A14). This signature is also evident in Figure A10, indicating TAP-sufficiency in these tumor samples. None of the tumor samples examined produced a signature characteristic of TAP-deficiency.
siRNA Suppression of GILT in CaSki Cells Shows the Abundance of GILT is Correlated with the Abundance of the E711–19 Peptide
To causally connect the correlation between GILT signal peptide and E711–19 observed in tumor samples, GILT protein expression in CaSki cells was suppressed by siRNA techniques (see Materials and Methods) and the impact on E711–19 presentation was examined. The suppression of GILT was monitored through the GILT signal peptide LLDVPTAAV, which is a very abundant HLA-A*02 associated peptide in CaSki cells. Both LLDVPTAAV and the E711–19 peptide were measured by MS3 and Poisson fitting against the background of all HLA-A*02 bound peptides affinity purified from CaSki. Poisson fitting returns event numbers that depend on collection period, sample recovery and ion beam luminosity. Instead of compensating for these factors in order to translate the Poisson fit events into moles, we focused on siRNA-induced changes in relative abundance, since this can be directly measured. Essentially, the ion current that can be identified as LLDVPTAAV and E711–19 is determined by Poisson fitting, and these event numbers are scaled or normalized by a measure of the overall peptide ion flux. The scaled LLDVPTAAV and E711–19 events are then assessed in GILT suppressed CaSki cells and control CaSki cells to determine if downregulation of GILT is associated with downregulation of E711–19. Although different measures of ion flux are possible, normalization using an MS3 spectrum has advantages. MS3 spectra are less impacted by background solvent ion noise and can be collected under the same ion optical conditions (long accumulation periods, ion trapping in the Q0 focusing rods) as the target MS3 spectra. Not all MS3 spectra are equivalent for monitoring ion flux. Ideally, the normalization would employ MS3 spectra that have many events and represent a large set of endogenous peptides, as this limits the impact on overall flux due to changes in a few abundant peptides that may vary with the probe and control status. Figure 6 shows the MS2 510.79 spectrum of HLA-A*02 peptides recovered from CaSki cells. The precursor ion peak at m/z 510.79 is near the center of the molecular ion “hump” that is characteristic of affinity purified HLA-A*02 peptides (see, for example, Figure A10) and is more likely to contain multiple peptides. The two most abundant fragment ion peaks in the MS2 510.79 spectrum are observed at m/z 889.5 and 836.4. The MS3 510.79/889.5 and 510.79/836.4 spectra (Figures 6B,C) show markedly different degrees of peak complexity; the ion composition of the 510.79/836.4 peak is relatively simple, while that of 510.79/889.5 is substantially more complex. As the loss of leucine or isoleucine from the carboxy terminus would generate the m/z 889.5 fragment from the doubly charged 510.79 ion peak, and these are common terminal residues, it is of little surprise to find a large set of peptides at m/z 510.79 generating a m/z 889.5 b-ion. In this study MS3 510.79/889.5 spectra are used for ion flux normalization.
Temporal variation in the ion beam luminosity is often observed during the long period over which nanospray MS3 spectra are collected. To correct for this the target spectra (here LLDVPTAAV and E711–19) are collected in series with the MS3 spectra used for flux normalization. That is, an MS3 spectrum for LLDVPTAAV is collected, then a spectrum for E711–19 and then the spectrum for ion flux. This cycle is repeated to collect adequate ion statistics.
Figure 6D shows the change in the ratio of LLDVPTAAV and E711–19 events to ion counts in the MS3 510.79/889.5 flux normalization spectrum for lentiviral siRNA suppression of GILT. If the control lentiviral infection of CaSki cells define 100% values, lentiviral siRNA suppression of GILT reduces the relative amount of GILT signal peptide to 20% and the relative amount of E711–19 to 43%. For Lipofectamine 2000 transfection, a reduction in GILT signal peptide to 29% was observed while the relative amount of E711–19 was reduced to 55% of the Lipofectamine 2000 control (scrambled siRNA). Although these results are consistent with a mechanistic correlation between GILT and the processing of E711–19, siRNA suppression was unable to reduce the GILT signal peptide to the undetectable levels observed in tumor samples KTS8 and KTS9.

Figure A1. Novel HLA sequences identified from patients KTS2 and KTS5. HLA RNA pyrosequencing identified two novel HLA-C sequences in the patient cohort. KTS2 presented a novel C*08:22-like allele with a T587C mutation, resulting in a leucine to proline substitution. KTS5 presented a putative 9 bp insertion at position 974 in a C*04:01:01-like allele, resulting in a three amino acid insertion toward the 3′ end of the class I open reading frame. Due to the positioning of this insertion within a palindromatic sequence motif we cannot presently rule out the possibility that it is a sequencing artifact of the Roche/454 pyrosequencing process.

Figure A2. Multiple alignment of Raw E7 DNA sequences. Multiple sequence alignments of HPV E7 sequences from tumor biopsies KTS1–9 are shown. The reference sequence is E7 from KTS6, identical to worldwide strain that has 100% identity to more than 50 reported sequences (representative sequence: GenBank K02718; Seedorf et al., 1985). E7 KTS1 is a novel sequence showing a single nt difference to East Asian (AF534061) and Thai (FJ610150) strains. KTS2 represents another worldwide strain (AY686582). E7 KTS3 is a novel sequence showing a single nt difference to the worldwide strain (AY686582). E7 KTS4, KTS5, and KTS8 sequences represent Asian strains (AF534061). E7 KTS7 sequence represents East Asian strain (AF486333). E7 KTS9 sequence represents East Asian strain (AF486332). Positions 31–57 encode HLA-A02 T cell epitope YMLDLQPET. Codon 85–87 encodes amino acid variability (N or S), while all other variable positions, shown as shaded, represent silent mutations. Unresolved nucleotides in KTS1 and KTS4 are represented by ambiguity code “n”. In summary, the HPV E7 sequence variability in the cohort is relatively high, representing multiple HPV-16 species. At the protein level, on the contrary, the sequence is extremely well conserved indicating high stability of the protein product.
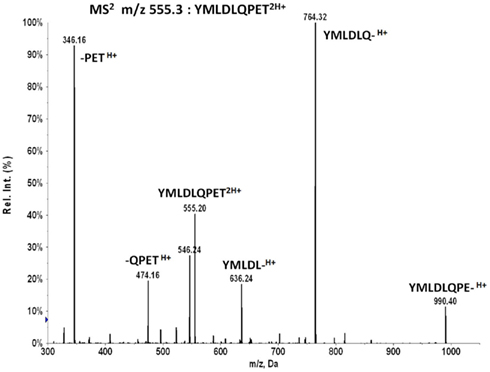
Figure A3. MS2 555.3 spectrum by collisional activation of the doubly charged molecular ion from synthetic peptide YMLDLQPET.
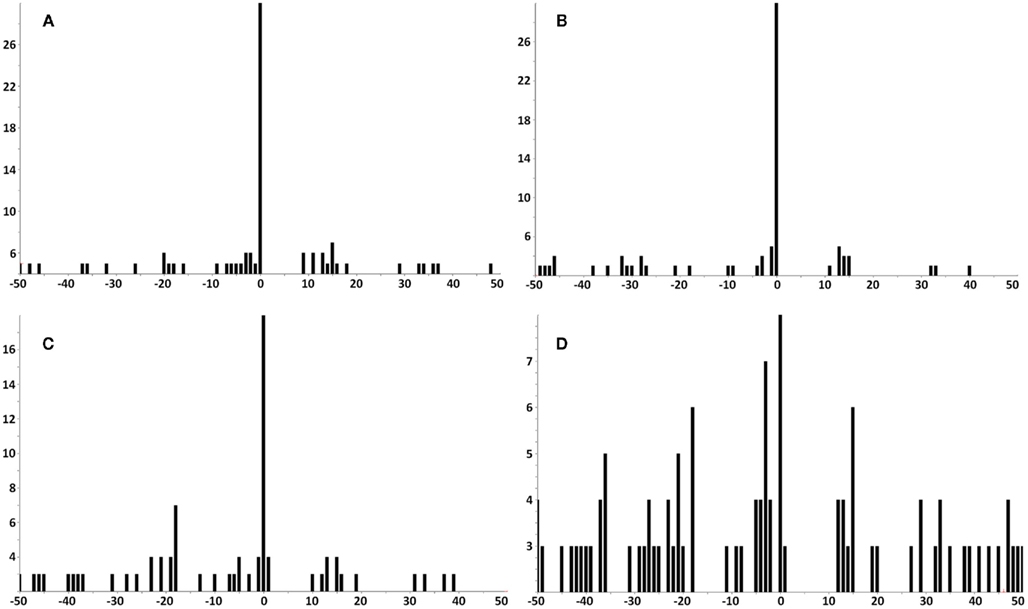
Figure A4. Lower sensitivity MS3 555.3/990.5 Poisson plots for detecting YMLDLQPET in HLA-A2 bound peptides from tumor samples. (A) Tumor sample #1; (B) sample #2; (C) sample #4; (D) sample #7. YMLDLQPET detection in samples 1, 2, and 4 are confirmed, but the detection significance in sample 7 is below threshold.
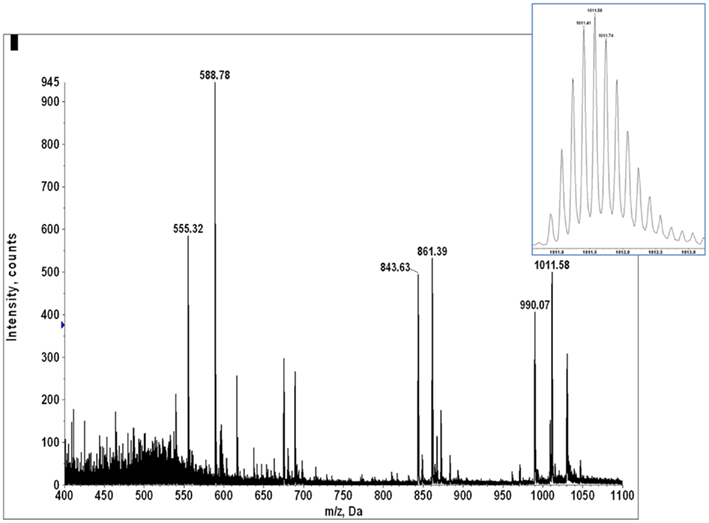
Figure A5. The nanospray MS spectrum of peptides recovered from KTS6 taken with a quadrupole–oTOF instrument shows a substantial background of ion masses around 6 kDa (inset showing isotope peaks in the sixth charge state).
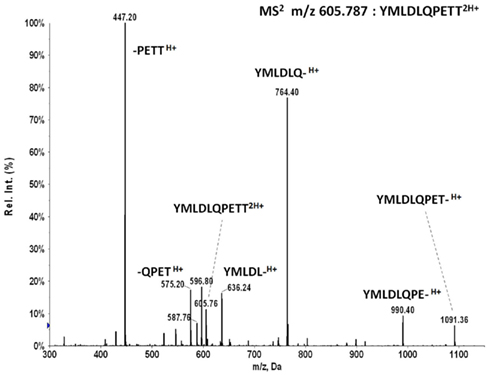
Figure A6. MS2 605.8 of synthetic peptide YMLDLQPETT generates a prominent b6 ion fragment YMLDLQ- which is used for MS3 605.8/764.4 Poisson detection.
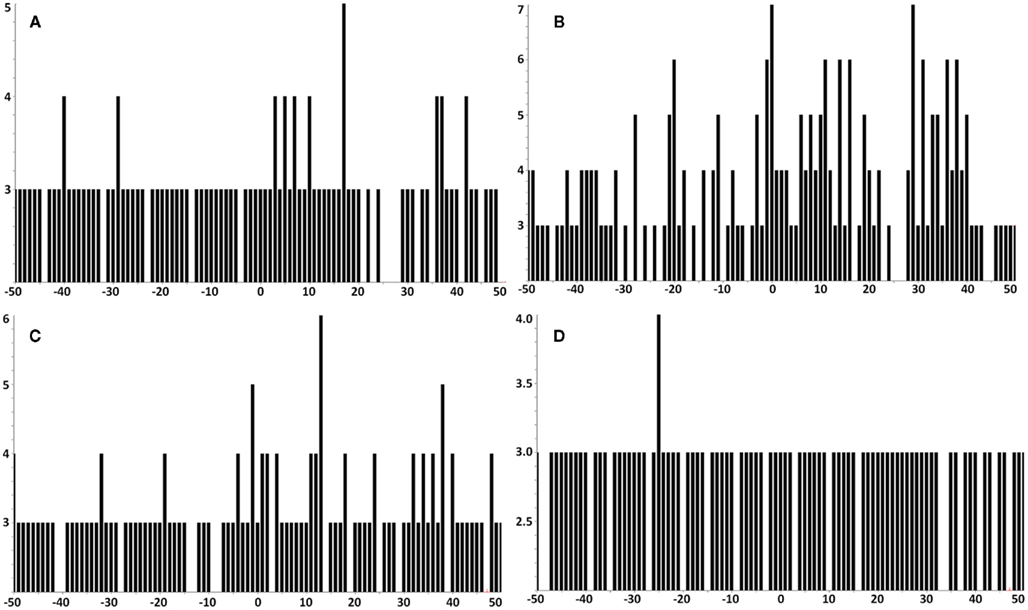
Figure A7. In all tumor samples where E711–20 was checked, it was not detected. MS3 Poisson detection plots for E711–20 are negative for both the E711–19 positive samples KTS1, KTS2, and KTS4 (A–C) and the E711–19 negative sample KTS8 (D).
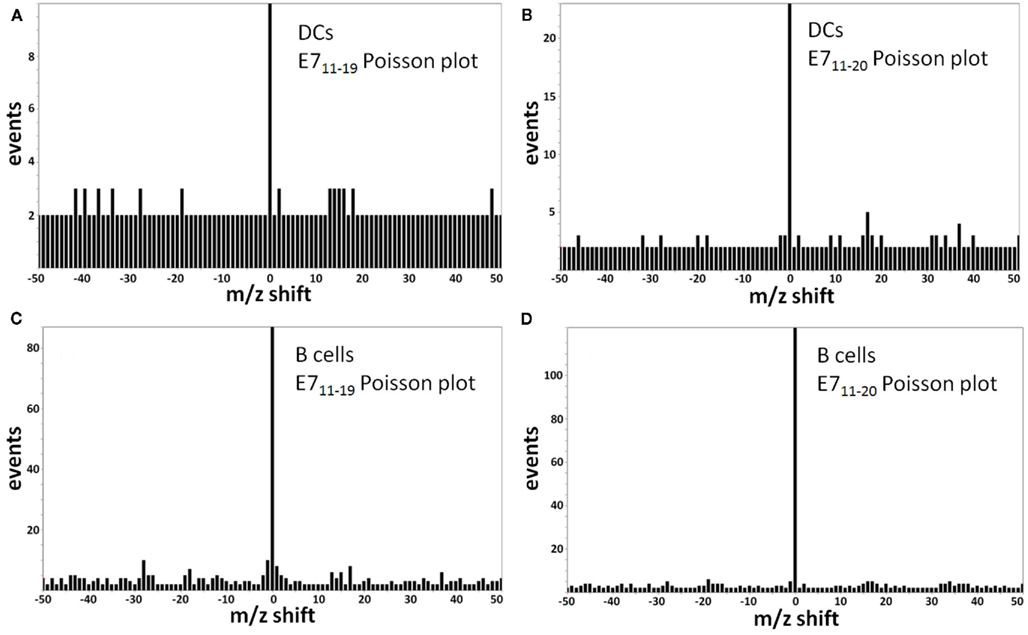
Figure A8. Monocyte-derived DCs (moDCs) and B cells from human blood take up and process a long peptide consisting of the first 35 amino acids of the E7 oncoprotein, MHGDTPTLHEYMLDLQPETTDLYCYEQLNDSSEEE. Both the E711–19 and E711–20 peptides are cross-presented as surface HLA-A*02 complexes with the E711–20 peptide more abundant. (A) Poisson detection plot of the MS3 555.3/764.4 spectrum (for E711–19) of peptides recovered from moDCs loaded with 35-mer; (B) Poisson detection plot of the MS3 605.8/764.4 spectrum (for E711–20) of peptides recovered from moDCs. The A, B pair were collected as averages over the sequence A, B, A, B,… so their intensities can be directly compared. (C) Poisson detection of the MS3 555.3/764.4 spectrum (for E711–19) of peptides recovered from B cells; (D) Poisson detection of the MS3 605.8/764.4 spectrum (for E711–20) of peptides recovered from B cells. The C, D pair can also be directly compared.
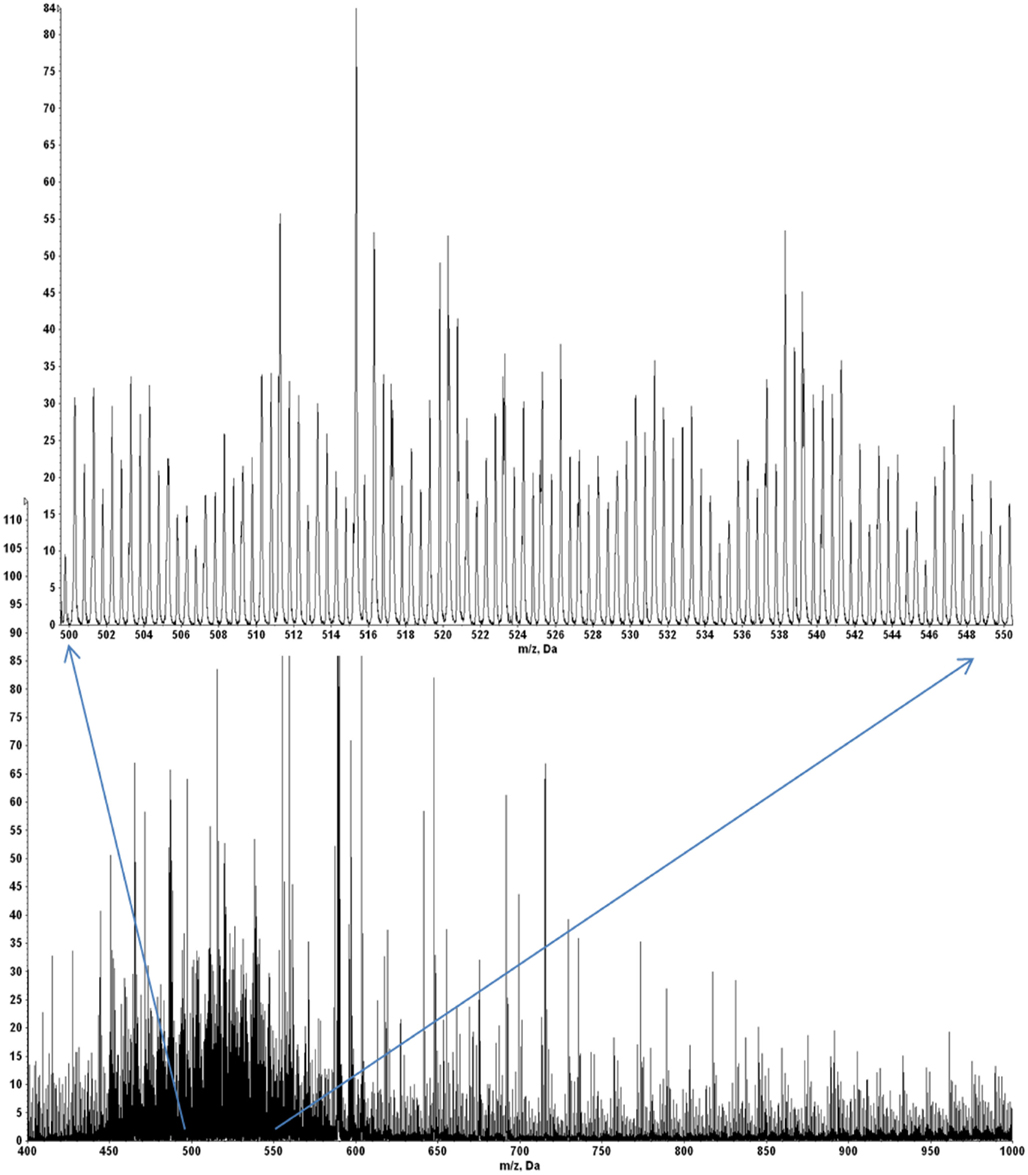
Figure A9. The mass spectrum of HLA-A2 bound peptides from tumor sample KTS5. Doubly charged peptides from m/z 450 to 600 (inset) provide a “hump” that roughly characterizes the overall recovery.
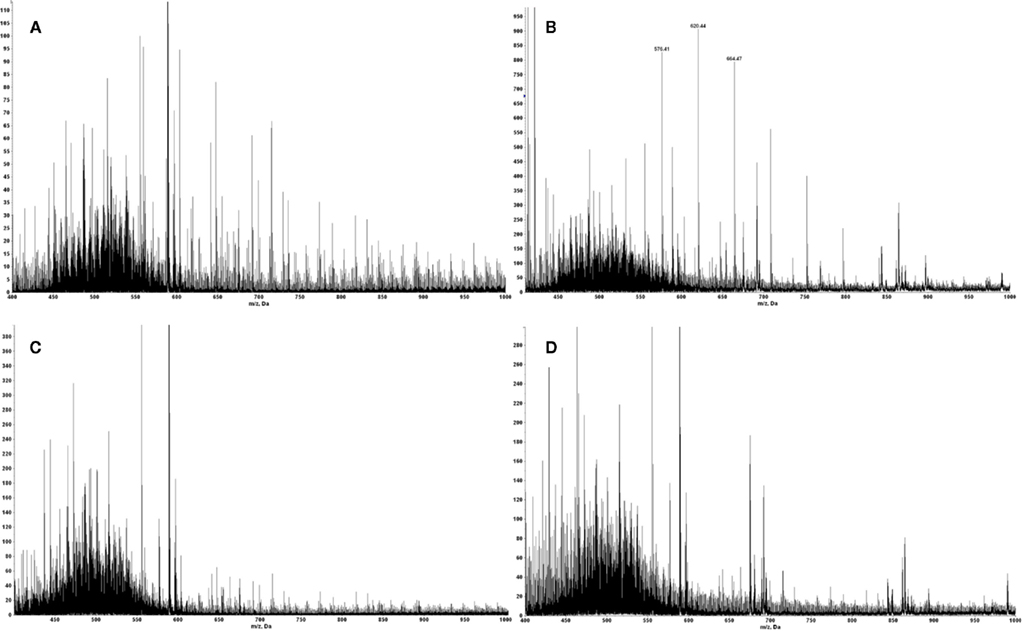
Figure A10. Overall HLA-A2 peptide recovery is marked by a “hump” in the m/z range from 450 to 550 due to doubly charged peptide molecular ions relative to singly charged background ions (see Figure A9 and related discussion). This figure illustrates good MHC I peptide recovery in all four samples shown. (A) E711–19 positive sample KTS5; (B) E711–19 positive sample KTS7; (C) E711–19 negative sample KTS8; (D) E711–19 negative sample KTS9.
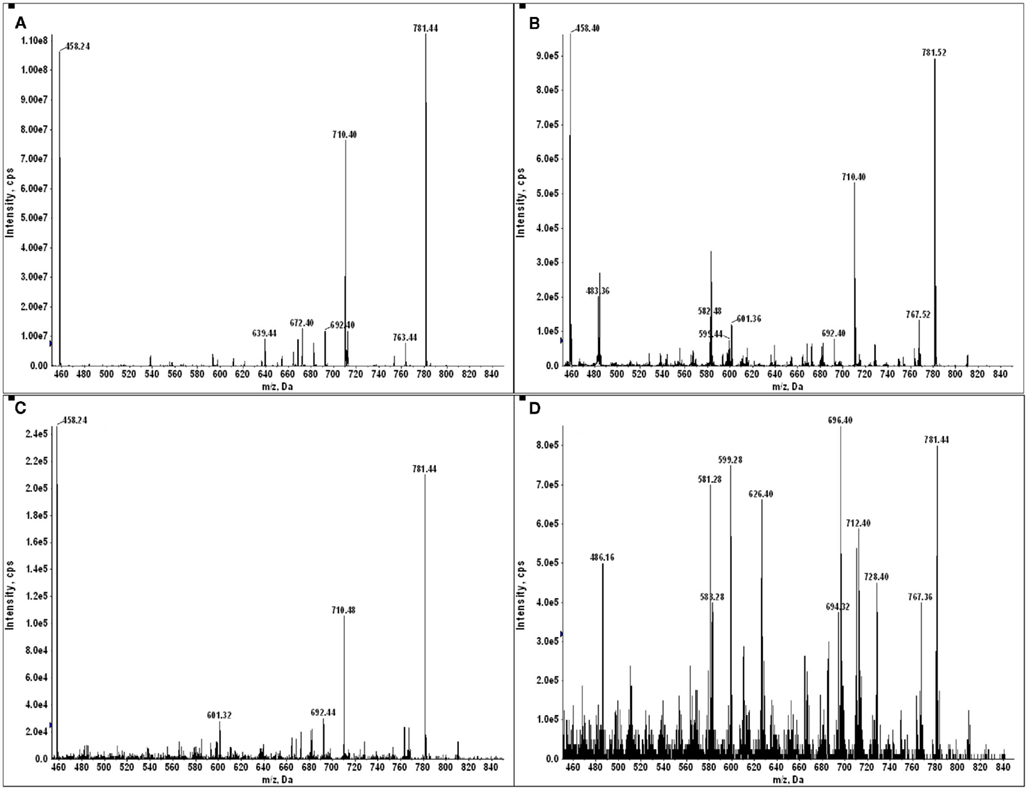
Figure A11. MS2 449.75 of HLA-A2 bound peptides is almost universally dominated by the fragmentation of LLDVPTAAV from the GILT signal peptide. The lack of this signature in tumor sample KTS8 was exceptional and immediately noted. (A) MS2 449.75 of synthetic peptide LLDVPTAAV; (B) MS2 449.75 of CaSki, an HLA-A*0201 positive HPV-16 transformed cell line; (C) MS2 449.75 of an HLA-A*0201 positive respiratory epithelial cell line BEAS; (D) MS2 449.75 of tumor sample KTS8. Note in particular the lack of the major m/z 458 fragment in (D).
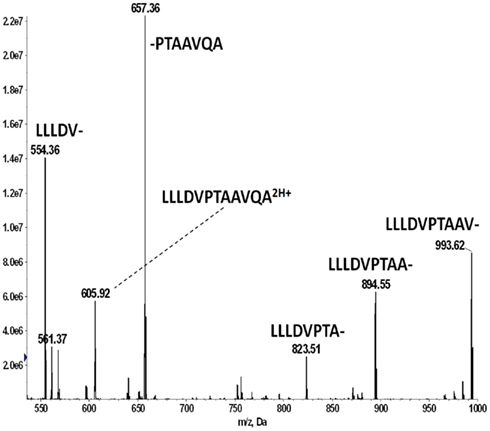
Figure A12. MS2 605.8 spectrum of synthetic peptide LLLDVPTAAVQA.
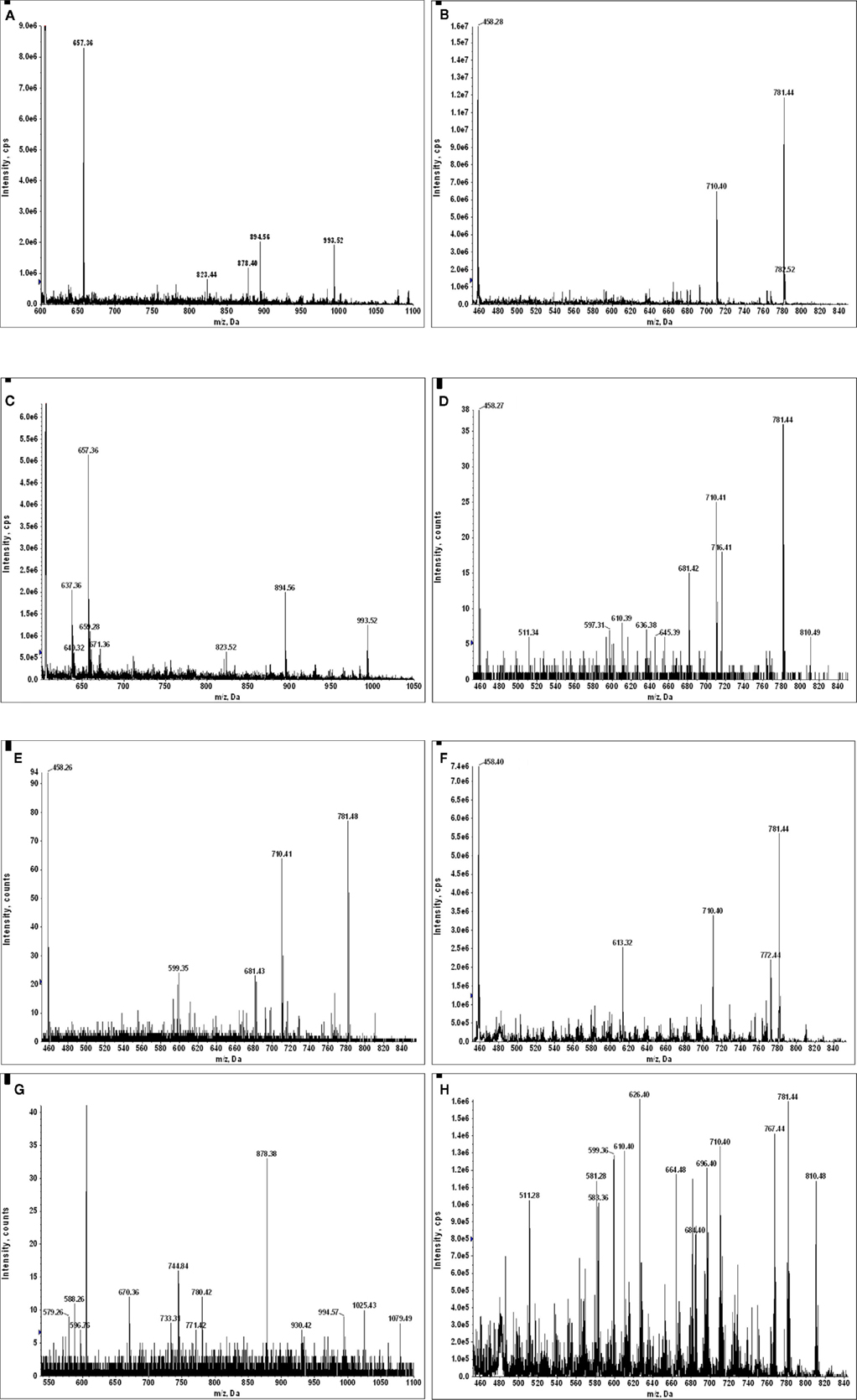
Figure A13. (A) MS2 605.8 of sample KTS1; (B) MS2 449.8 of sample KTS2; (C) MS2 605.8 of sample KTS4; (D) MS2 449.8 of sample KTS5; (E) MS2 449.8 of sample KTS6; (F) MS2 449.8 of sample KTS7; (G) MS2 605.8 of sample KTS8; (H) MS2 449.8 of sample KTS9. Only in the MS2 spectra of (G,H) can one not recognize the dissociation spectrum of a GILT signal peptide.
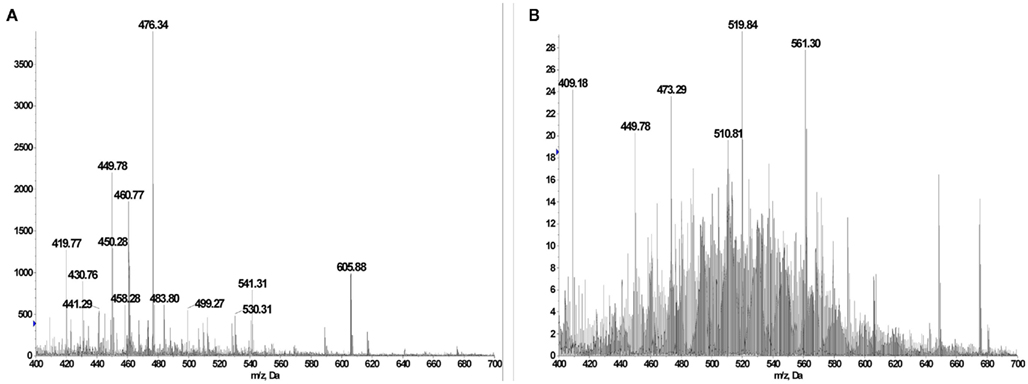
Figure A14. (A) BB7.2 pulldown and acid extracted peptide pool from TAP-deficient T2 cells showing prominent GILT signal peptides at m/z 449.78 (LLDVPTAAV2H+), m/z 460.77 (LLDVPTAAVNa+H+), and m/z 605.88 (LLLDVPTAAVQA2H+). (B) BB7.2 pulldown and acid extracted peptide pool from TAP-sufficient T1 cells. The m/z 449.78 peak is still primarily the GILT signal peptide LLDVPTAAV2H+ but a complex set of other doubly charged peptides cover the m/z range 420–620.
Keywords: T cell epitopes, mass spectrometry, tumor antigens, human papilloma virus, E7, GILT, CTL targets, T cell vaccines
Citation: Keskin DB, Reinhold B, Lee SY, Zhang G, Lank S O’Connor DH, Berkowitz RS, Brusic V, Kim SJ and Reinherz EL (2011) Direct identification of an HPV-16 tumor antigen from cervical cancer biopsy specimens. Front. Immun. 2:75. doi: 10.3389/fimmu.2011.00075
Received: 12 October 2011;
Paper pending published: 07 November 2011;
Accepted: 26 November 2011;
Published online: 13 December 2011.
Edited by:
Nick Gascoigne, Scripps Research Institute, USAReviewed by:
Edward State Collins, The University of North Carolina at Chapel Hill, USAShoba Ranganathan, Macquarie University, Australia
Salvatore Valitutti, INSERM, France
David Kranz, University of Illinois Urbana-Champaign, USA
Copyright: © 2011 Keskin, Reinhold, Lee, Zhang, Lank, O’Connor, Berkowitz, Brusic, Kim and Reinherz. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Ellis L. Reinherz, Dana-Farber Cancer Institute, 77 Avenue Louis Pasteur, HIM 418, Boston, MA 02115, USA. e-mail:ZWxsaXNfcmVpbmhlcnpAZGZjaS5oYXJ2YXJkLmVkdQ==