 Brian Jones1
Brian Jones1

- 1 Department of Pharmacology, College of Pharmacy and Pharmaceutical Sciences, The University of Toledo, Toledo, OH, USA
- 2 Veteran’s Administration, Ann Arbor, MI, USA
- 3 Department of Internal Medicine, Division of Rheumatology, University of Michigan Medical School, Ann Arbor, MI, USA
Understanding rheumatic diseases from the perspective of chemokine biology has shaped and will continue to shape our approach for targeted drug design. Among different kinds of chemokines, fractalkine/CX3CL1 has been found to play an important role in inflammation, portraying unique functional, and structural characteristics. This review summarizes the emerging role of fractalkine/CX3CL1 from a functional and clinical perspective and provides evidence to validate it as a potential therapeutic target in rheumatic diseases such as rheumatoid arthritis, Sjögren’s syndrome, systemic lupus erythematosus, scleroderma, as well as diseases related to vascular inflammation. From this, recent studies investigating potential therapeutic agents against fractalkine/CX3CL1’s role in pathology have shown promise.
Inflammation and Chemokines
Chemokines are small molecular weight heparin-binding proteins that play important roles in the recruitment and retention of inflammatory cells at the site of inflammation or injury by their chemotactic activity. Since their initial discovery, chemokines have been found to play a crucial role in directing mononuclear cells, adaptive immunity, and disease pathogenesis (Charo and Ransohoff, 2006). Chemokines elicit their impact through the chemokine receptors, the G protein-coupled class of receptors, which upon stimulation transduce an intracellular signal, and may cause additional changes within the cell and promote inflammation (Charo and Ransohoff, 2006). Fundamentally, mature chemokines are secreted and processed at the NH2 terminus, rarely at the COOH-terminus or at internal sites, to yield truncated forms with various levels of biological activity (Baggiolini et al., 1994). This cleavage at the NH2-terminal sequence preceding the first cysteine amplifies their biological activity, with little to no reported effect of the processing at the COOH-terminal amino acids (Baggiolini et al., 1994).
Fractalkine/CX3CL1
A study by Bazan et al. (1997) reported the discovery of a unique chemokine subclass, CX3C, which was different in terms of structure, biochemical features, tissue distribution, and chromosomal localization from known chemokines and represented by only one member, referred to as fractalkine or CX3CL1. Synthesized as a transmembrane molecule, fractalkine/CX3CL1, consists of an extracellular NH2-terminal domain, a mucin-like stalk, a transmembrane α helix, and a short cytoplasmic tail (Bazan et al., 1997). It was found to exist as a soluble form (consisting only of the extracellular domain and mucin-like stalk) and a membrane-bound form, which gave it an advantage to function both as a chemoattractant and an adhesion molecule as well. Soluble fractalkine/CX3CL1 is generated by the limited proteolysis of a disintegrin and metallopeptidase 10 (ADAM10) and ADAM17/tumor necrosis factor-α-converting enzyme (ADAM17/TACE) and acts as a chemoattractant for monocytes, natural killer (NK) cells, and T cells. Further mechanistic studies on the proteolytic shedding of fractalkine/CX3CL1 suggest the role of ADAM10 in the constitutive shedding and of ADAM17 in response to cell activation. Interestingly, a recent study found that ADAM17 is less responsible for constitutive shedding of fractalkine/CX3CL1 in HepG2 cells and suggested that ADAM10 may play a greater role in this process (Hundhausen et al., 2003; Turner et al., 2010). Membrane-bound fractalkine/CX3CL1, which supports integrin-independent leukocyte adhesion, can be induced on primary endothelial cells by inflammatory cytokines (Garcia et al., 2000).
Cellular Distribution and Expression
CD4+ helper cells (Th) and CD8+ cytotoxic T cells (Tc) are divided into two distinct groups based on the cytokines they produce. Th1 and Tc1 cells secrete IFN-γ, tumor necrosis factor-α (TNF-α), and IL-2, which mediate immune responses against intracellular pathogens and are associated with pathological processes such as organ-specific autoimmune diseases. Th2 and Tc2 cells produce IL-4, IL-5, and IL-13, which mediate immune responses against extracellular pathogens and are associated with allergic immune responses (Umehara et al., 2004). Recent studies have shown that various lymphocyte subsets with different tissue tropisms and functions express specific chemokine receptors. The majority of CX3CR1-expressing CD4+ and CD8+ T cells also express CCR5, but not CXCR3, which suggests that CX3CR1-expressing T cells partially overlap Th1 and Tc1 cells, respectively (Nishimura et al., 2002). CX3CR1 has also been found to have preferential expression in Th1 cells compared to Th2 cells, with only Th1 cells responding to fractalkine/CX3CL1 (Fraticelli et al., 2001). CX3CR1-expressing cells include CD4+, CD8+ T cells, γδ cells, and NK cells, and they also express CD57 and CD11b, which are good markers for cytotoxic lymphocytes, but rarely express CD27, CD28, and CD62L. Most CX3CR1-expressing cells have cytoplasmic granules which contain perforin and granzyme B (Nishimura et al., 2002).
Apart from expression in the endothelia, fractalkine/CX3CL1 and CX3CR1 are strongly expressed in the central nervous system. Fractalkine/CX3CL1 is one of the most expressed chemokines in the brain, which may help to explain its role in neuropathic pain. In the CNS, neurons express fractalkine/CX3CL1 while astrocytes express CX3CR1 (Marchesi et al., 2010). From this, fractalkine/CX3CL1 plays a role in tumor development in the CNS, where it may be induced by TNF-α or TGF-β in neurons and aid in the aggregation of glioblastomas (Sciume et al., 2010).
The expression of fractalkine/CX3CL1 in monocytes from rheumatoid arthritis (RA) peripheral blood and synovial fluid (SF) validates an important role of this chemokine in immune-related inflammatory diseases. Fractalkine/CX3CL1 is also expressed in macrophages, fibroblasts, endothelial, and dendritic cells in RA synovial tissue (ST; Ruth et al., 2001). CCL26 has recently been found to be an agonist for CX3CR1, from which it may play a dual role in allergic diseases such as atopic dermatitis and psoriasis (Nakayama et al., 2010).
Unique Mechanism of Chemotaxis by Fractalkine/CX3CL1
Before fractalkine/CX3CL1 was discovered, the mechanism of chemotaxis was thought to begin with the association of soluble chemokines with cell surface proteoglycans and tissue matrix components, followed by the interaction between chemokines and their specific receptors on leukocytes, eventually resulting in the activation of the members of the integrin family of adhesion molecules through a G-coupled protein receptor mechanism (Tanaka et al., 1993). However, fractalkine/CX3CL1 mediates the mechanism for leukocyte adhesion in a unique fashion. The chemokine domain is presented at the top of the membrane-bound, mucin-like stalk, where it acts as an adhesion molecule making the association with proteoglycans and other adhesion molecules unnecessary. Interaction between fractalkine/CX3CL1 and CX3CR1 can markedly enhance integrin affinity, which further facilitates extravasation of leukocytes during the initial tethering and transmigration steps (Jones et al., 2010). Fractalkine/CX3CL1 expression on inflamed endothelium along with the coexpression of intercellular adhesion molecule (ICAM)-1 and vascular cell adhesion molecule (VCAM)-1, greatly enhances cell adhesion function by capturing CX3CR1-expressing cells from the blood and promoting migration into tissue (Umehara et al., 2004). Studies have suggested that fractalkine/CX3CL1 expression at the site of inflammation can attract and activate NK cells, resulting in the consequent lysis of neighboring endothelial cells (Yoneda et al., 2000; Umehara et al., 2001). Increased fractalkine/CX3CL1, ICAM-1, and VCAM-1 expression on endothelial cell membranes can result from certain conditions such as viruses/bacteria, ischemia, or cytokine induction (IL-1, TNF-α, IL-6; Lanier et al., 1997). The resulting expression of adhesion molecules and fractalkine/CX3CL1 increases the adhesion and transmigration of NK cells between endothelial cells, causing inflammation.
Inducers of Fractalkine/CX3CL1
There are several known inducers of fractalkine/CX3CL1 that promote pathology in a variety of diseases. These inducers include proinflammatory cytokines such as TNF-α, IFN-γ, IL-1β, but also lipopolysaccharide (LPS; Garcia et al., 2000; Yoshida et al., 2001; Schroder et al., 2004; Matsumiya et al., 2010), providing a possible link between fractalkine/CX3CL1 and rheumatic diseases.
Tumor Necrosis Factor-α
Tumor necrosis factor-α is a pleiotropic cytokine that exerts potent proinflammatory effects and is implicated in atherosclerosis and other metabolic and inflammatory disorders such as obesity and RA. TNF-α was originally found to upregulate fractalkine/CX3CL1 in brain inflammation and subsequently was termed “neurotactin” until later being designated fractalkine (Okamoto et al., 1997). Recent studies have shown that TNF-α significantly increases fractalkine/CX3CL1 expression (Garcia et al., 2000; Moon et al., 2006). A study by Moon et al. (2006) showed that fractalkine/CX3CL1 mRNA expression was significantly increased by TNF-α stimulation in human umbilical vein endothelial cells (HUVECs). Evaluation of signaling pathways suggested that TNF-α induced fractalkine/CX3CL1 expression is mediated primarily through the expression of NF-κB, a highly regulated transcription factor involved in inflammatory responses (Brasier, 2006; Moon et al., 2006). This notion was further supported by another study in which fractalkine/CX3CL1 induction by TNF-α in rat aortic endothelial cells was suppressed through the blockade of NF-κB (Garcia et al., 2000).
Interferon-γ
Interferon-γ mediates a wide variety of immunological functions, including upregulation of pathogen recognition, antigen processing and presentation, viral defense, inhibition of cellular proliferation and effects on apoptosis, activation of microbicidal effector functions, immunomodulation, and leukocyte trafficking (Schroder et al., 2004). TNF-α and IFN-γ both serve as agonists for the induction of a variety of cytokines, while synergism of TNF-α and IFN-γ leads to superinduction in certain types of cytokines (Paludan, 2000). One study reports that treatment of HUVECs with a combination of IFN-γ or TNF-α synergistically induced fractalkine/CX3CL1 expression (Matsumiya et al., 2010). Costimulation with TNF-α and IFN-γ is also known to induce fractalkine/CX3CL1 expression by more than 10-fold in astrocytes (Yoshida et al., 2001). In RA synovial fibroblasts, synergistic induction with TNF-α and IFN-γ enhances fractalkine/CX3CL1 expression more than either cytokine alone (Volin et al., 2007). These studies propose fractalkine/CX3CL1 regulation through the post-transcriptional mechanism mediated by the p38 mitogen activated protein kinase (MAPK) mechanism, then HuR (RNA binding protein) shuttling from the nucleus to the cytoplasm, where it stabilizes mRNA and helps regulate gene expression (Hasegawa et al., 2009; Matsumiya et al., 2010).
Interleukin-1β
Interleukin-1β is secreted systemically from blood monocytes, tissue macrophages, dendritic cells, B lymphocytes, and NK cells. IL-1β has been shown to induce fractalkine/CX3CL1 expression in some studies. Garcia et al. (2000) reported NF-κB dependent fractalkine/CX3CL1 induction by IL-1β in rat aortic endothelial cells. In a parallel study by Denes et al. (2008), the lack of CX3CR1 significantly reduced IL-1β and TNF-α expression, which resulted in decreased leukocyte infiltration in CX3CR1−/− animals and consequent ischemic damage and inflammation in the cerebrum in mice. Inhibiting IL-1β expression may cause a decrease in inflammatory chemokine expression, which provides one possible way to control fractalkine/CX3CL1 synthesis.
Pathological Role of Fractalkine/CX3CL1 in Rheumatic Diseases
Rheumatoid Arthritis
Rheumatoid arthritis is the most common form of inflammatory arthritis in which the recruitment and retention of monocytes and T lymphocytes into joints is an integral pathological phenomenon (Pope, 2002). Edema, synovial hyperplasia, and destruction of cartilage and bone in RA is caused by the infiltration of inflammatory cells such as macrophages, B cells, T cells, and dendritic cells into the joint (Iwamoto et al., 2008). Leukocyte infiltration into the synovium is regulated through a multi-step process that involves interactions between leukocytes and endothelial cells, cellular adhesion molecules, and chemokines and their receptors (Iwamoto et al., 2008). Proinflammatory cytokines such as 1L-1β, TNF-α, and IFN-γ not only start a vicious cycle of synovial inflammation, but also produce chemokines that further stimulate different cell types in the affected joint including synovial fibroblasts and chondrocytes to release inflammatory mediators and matrix metalloproteinases (MMPs), to cause pathological remodeling, leading to joint destruction (Sawai et al., 2007).
Playing a unique role in RA, fractalkine/CX3CL1 acts as a chemotactic agent for monocytes and lymphocytes and as a cellular adhesion molecule (Koch, 2005). By attracting macrophages that express the CX3CR1 receptor within RA synovium, fractalkine/CX3CL1 plays an important role in the development of RA as shown in Figure 1. The study by Ruth et al. (2001) found high levels of soluble fractalkine/CX3CL1 in RA SF. Analysis of the cell-specific expression in peripheral blood and SF in this study show that primarily monocytes expressed fractalkine/CX3CL1, while macrophages, fibroblasts, endothelial, and dendritic cells expressed this chemokine in ST. In terms of pathological relevance, these levels in RA SF were significantly elevated compared with SF from osteoarthritis (OA) patients suggesting a more aggressive role of fractalkine/CX3CL1 in RA pathogenesis (Ruth et al., 2001). Some parallel studies also showed that the surface expression of CX3CR1 on CD8+ T cells from RA patients was roughly three fold when compared to the cells derived from the healthy subjects (Nanki et al., 2002). In addition to RA, fractalkine/CX3CL1 can induce chemotaxis in osteoarthritis (OA) fibroblasts, which may indicate a novel role in regulating OA fibroblast migration to bone fragments, contributing to synovitis (Klosowska et al., 2009). Administration of anti-fractalkine/CX3CL1 monoclonal antibody to mice significantly reduced the incidence of arthritis and tissue destructive trafficking of monocytes to the synovium (Nanki et al., 2002, 2004).
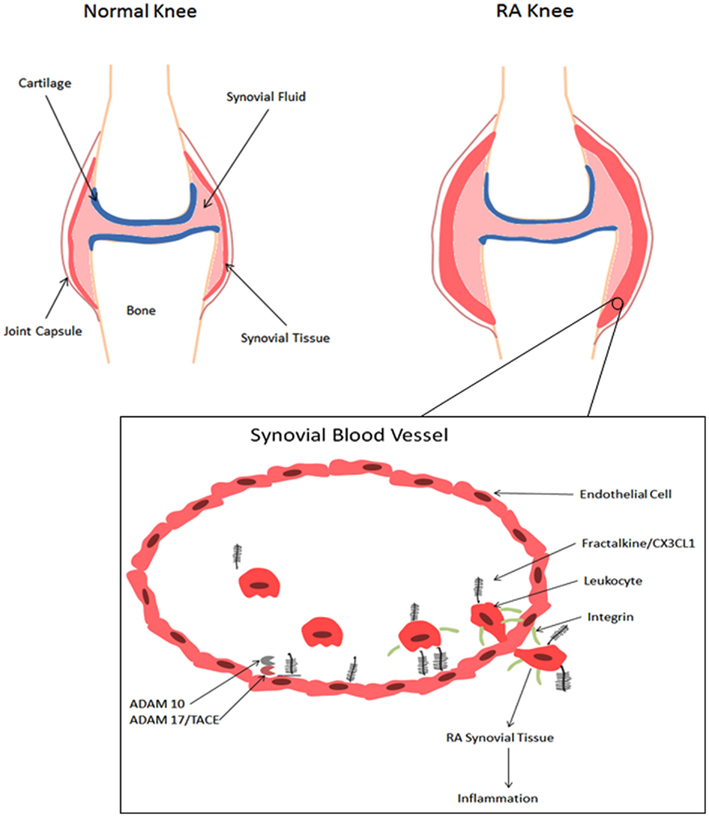
Figure 1. Role of fractalkine/CX3CL1 in the development of RA. A schematic representation of a normal knee and an RA knee. Infiltration of leukocytes results in edema and synovial hyperplasia and consequent destruction of cartilage and bone (Iwamoto et al., 2008). When fractalkine/CX3CL1 is present, the chemokine domain is presented at the top of the membrane-bound mucin-like stalk, where it acts as an adhesion molecule (Umehara et al., 2004). Immobilized fractalkine/CX3CL1 exhibits rapid and high affinity binding to CX3CR1, which consequently increases integrin affinity and leukocyte adhesion (Jones et al., 2010). Soluble fractalkine/CX3C1 binds to CX3CR1 aiding leukocyte extravasation through the vascular wall and into the tissue, further initiating the inflammatory response (Umehara et al., 2004). Membrane-bound fractalkine/CX3CL1 is cleaved by ADAM10 and ADAM17/TACE to generate soluble fractalkine/CX3CL1, which consists of the chemokine domain and the mucin-like stalk (Bazan et al., 1997;Garton et al., 2001;Hundhausen et al., 2003, 2007).
Detrimental tissue remodeling in RA is supported by neovascularization and an angiogenic microenvironment that plays an important role in chronic inflammation in RA. Some proinflammatory cytokines may directly stimulate angiogenesis or act indirectly by enhancing the production of angiogenic chemokines. To investigate the mediatory role of fractalkine/CX3CL1 in this process, one study showed that RA SF depleted of fractalkine/CX3CL1 exhibits a lower angiogenic effect as compared to control SF (Volin et al., 2001). Because of significant expression in RA, there is subsequently an increased cardiovascular risk that may contribute to the mortality of RA patients through enhanced systemic inflammation (Ruth et al., 2001; Volin et al., 2001).
Fractalkine/CX3CL1 plays a role in an uncommon but severe complication of RA, rheumatoid vasculitis (RV) which causes skin and eye disorders, neuropathy, and systemic inflammation (Genta et al., 2006). In patients suffering from RV, soluble fractalkine/CX3CL1 is significantly expressed and its levels correlated with the severity of vasculitis (Matsunawa et al., 2006). Patients receiving treatment for RV with glucocorticoids, methotrexate, or infliximab expressed significantly diminished soluble fractalkine/CX3CL1 levels in serum samples (Matsunawa et al., 2006). These therapies may target fractalkine/CX3CL1 production in various ways to help regulate the progression of RV in patients with RA.
Sjögren’s Syndrome
Sjögren’s syndrome (SS) is a systemic autoimmune disease in which immune cells attack and destroy the exocrine glands that produce tears and saliva (Delaleu and Jonsson, 2008). SS symptoms include dry eyes (xerophthalmia) and dry mouth (xerostomia; Kruszka and O’Brian, 2009). SS occurs in two basic forms: (1) primary SS, which is a solitary process and is not associated with any other illness, and (2) secondary SS, in which the disease develops in the presence of another autoimmune disease such as RA (Kruszka and O’Brian, 2009; Ramos-Casals et al., 2010). SS represents a complex, multifaceted activation of the immune system in which lymphocyte dysregulation and hyperactivity play a major role in the disease (Kruszka and O’Brian, 2009).
A study by Wildenberg et al. (2008) provides evidence that fractalkine/CX3CL1 cleavage is enhanced in SS, which may contribute to the increased chemotaxis. Interestingly, this study found an organ-specific fractalkine/CX3CL1 cleavage, with a 17-kDa fragment in the salivary glands of non-obese diabetic (NOD) mice. This phenomenon was observed as early as 10 weeks of age, where the animals were probably in a pre-disease or subclinical stage of SS, and existed until 20 weeks of age, where the disease may have advanced to overt stage. Surprisingly, the protease responsible for this cleavage has not been identified and appears to not involve ADAM10 or ADAM17/TACE. The authors propose that fractalkine/CX3CL1 is potentially a new autoantigen in SS (Wildenberg et al., 2008). In another study by Tsubota et al., fractalkine/CX3CL1 expression was upregulated in SS affected mice. Furthermore, lacrimal and salivary gland-specific fractalkine/CX3CL1 transgenic mice showed the expression of fragmented fractalkine/CX3CL1 and lymphocytic infiltration in their lacrimal glands (Tsubota et al., 2009). In SS patients, serum samples exhibit elevated fractalkine/CX3CL1, ∼1.8-fold higher than RA patients and ∼3.6-fold higher than healthy patients (Sato et al., 2006). These results suggest that fractalkine/CX3CL1 not only plays an important role in the progression of SS, but also may be processed differently in various organs in different disease states, which further warrants additional studies to validate the role and mechanism of fractalkine/CX3CL1 mediated pathogenesis in SS.
Systemic Lupus Erythematosus
Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by multiorgan damage with infiltration and sequestration of various immune cells (Umehara et al., 2006). Yajima et al. (2005) reported that serum fractalkine/CX3CL1 levels were significantly more elevated in patients with SLE than in the healthy controls. Both fractalkine/CX3CL1 expression and CD16 accumulation are markedly elevated in patients with proliferative lupus nephritis (Nakatani et al., 2010). In another study, a fractalkine/CX3CL1 antagonist was administered to MRL/lpr mice before the onset or during the early stages of lupus nephritis. Results from the study showed that the mice exhibited significantly reduced glomerular hypercellularity, glomerulosclerosis, crescent formation, and vasculitis when compared with control mice (Inoue et al., 2005). SLE patients have a predisposition to develop cardiovascular diseases. In SLE, the incidence of myocardial infarction is five times higher than that of the general population and as much as 50 times higher in young women (Cypiene et al., 2010). In a clinical study of patients with SLE, soluble fractalkine/CX3CL1 from serum samples were measured and compared to patients with other autoimmune diseases. The serum levels of soluble fractalkine in SLE patients were higher than patients with RA and SS (Sato et al., 2006). Recently, Li et al. (2010b) has proposed CX3CR1 as an indicator in clinical surveillance of SLE based on mRNA analysis from RT-PCR on peripheral blood mononuclear cells (PBMCs) of SLE patients. These studies suggest that a fractalkine/CX3CL1 antagonist may delay the progression of human SLE.
Scleroderma
Scleroderma (systemic sclerosis) is a chronic systemic autoimmune disease characterized by fibrosis and vascular alterations primarily of the skin. Scleroderma has two major forms: limited scleroderma, involving cutaneous manifestations mainly of the hands, arm, and face, and diffuse systemic scleroderma, affecting large areas of the skin as well as internal one or more internal organs such as the kidneys, esophagus, heart, and lungs. Patients with scleroderma may develop other rheumatic diseases such as SLE, RA, and SS as well as pulmonary arterial hypertension (Gabrielli et al., 2009). Enhanced expression of fractalkine/CX3CL1 has been shown in the affected skin and lung tissues of patients with cutaneous scleroderma (Hasegawa et al., 2005). This increased expression augments the recruitment of mononuclear cells into the affected tissue leading to inflammation (Hasegawa et al., 2005). Polymorphisms of CX3CR1 promote pathogenesis; where the 429I and 480M alleles are significantly increased in scleroderma patients with pulmonary arterial hypertension. Increased susceptibility to scleroderma is associated with the 249II CX3CR1 polymorphism (Marasini et al., 2005). Intravenous administration of prostaglandin E1 was reported to down-regulate serum fractalkine/CX3CL1 levels as well as CD11a and CD49d integrin expression on peripheral blood lymphocytes, adding another potential mechanism to the efficacy of prostaglandin E1 in scleroderma associated pulmonary hypertension (Sicinska et al., 2008).
Systemic Inflammatory Conditions
Fractalkine/CX3CL1 has been shown to play an important role in systemic inflammation, which also contributes as a secondary complication of rheumatic diseases (Table 1). Possible mechanisms for the role of fractalkine/CX3CL1 in coronary plaque rupture include an increase in CX3CR1-expressing monocytes or macrophage activation by CX3CR1-expressing T lymphocytes and NK cells at the site of inflammation (Ikejima et al., 2010). Studies are finding evidence that fractalkine/CX3CL1 plays an important role in atherosclerosis. Fractalkine/CX3CL1 expression is upregulated in atherosclerotic lesions of apolipoprotein E (apoE)-deficient mice, and crossing CX3CR1−/− with apoE−/− decreases atherosclerotic lesion formation with reduced macrophage accumulation (Combadiere et al., 2003; Lesnik et al., 2003; Umehara et al., 2004). Hence, addressing a therapeutic need of regulating CX3CL1–CX3CR1 duo mediated atherosclerosis may have a significant impact on the treatment of RA or SLE in terms of minimizing the detrimental vascular remodeling as one of these disease co-morbidities. In a study by Liuzzo et al. (2001) showed that IFN-γ derived from stimulated T lymphocytes activated monocytes obtained from the patients with unstable angina pectoris. These findings suggest that increased CX3CR1-expressing monocytes, T lymphocytes, and NK cells may result in a burst of macrophage activation, which could lead to plaque rupture (Ikejima et al., 2010). Fractalkine/CX3CL1 also plays a role in inflammatory cardiomyopathy, in which its upregulation affects the functional potential of PBMCs in inducing a cardiodepressive effect on heart tissue (Escher et al., 2011). Through in vitro and in vivo models, fractalkine/CX3CL1 has also been shown to induce angiogenesis in HepG2 cells (Li et al., 2010a). This suggests that fractalkine/CX3CL1 may influence hepatic biology during the inflammatory phase of rheumatic conditions such as RA or SLE and contribute to amplifying systemic inflammation in the established phase of the disease through the enhanced synthesis or expression of acute phase reactive proteins such as C-reactive protein, fibrinogen, or serum amyloid A. Further studies are awaited to pinpoint the exact role of fractalkine/CX3CL1 in chronic vascular diseases, which may shed some light on its mechanism of action and possible contribution in cardiovascular co-morbidities in the rheumatic population.
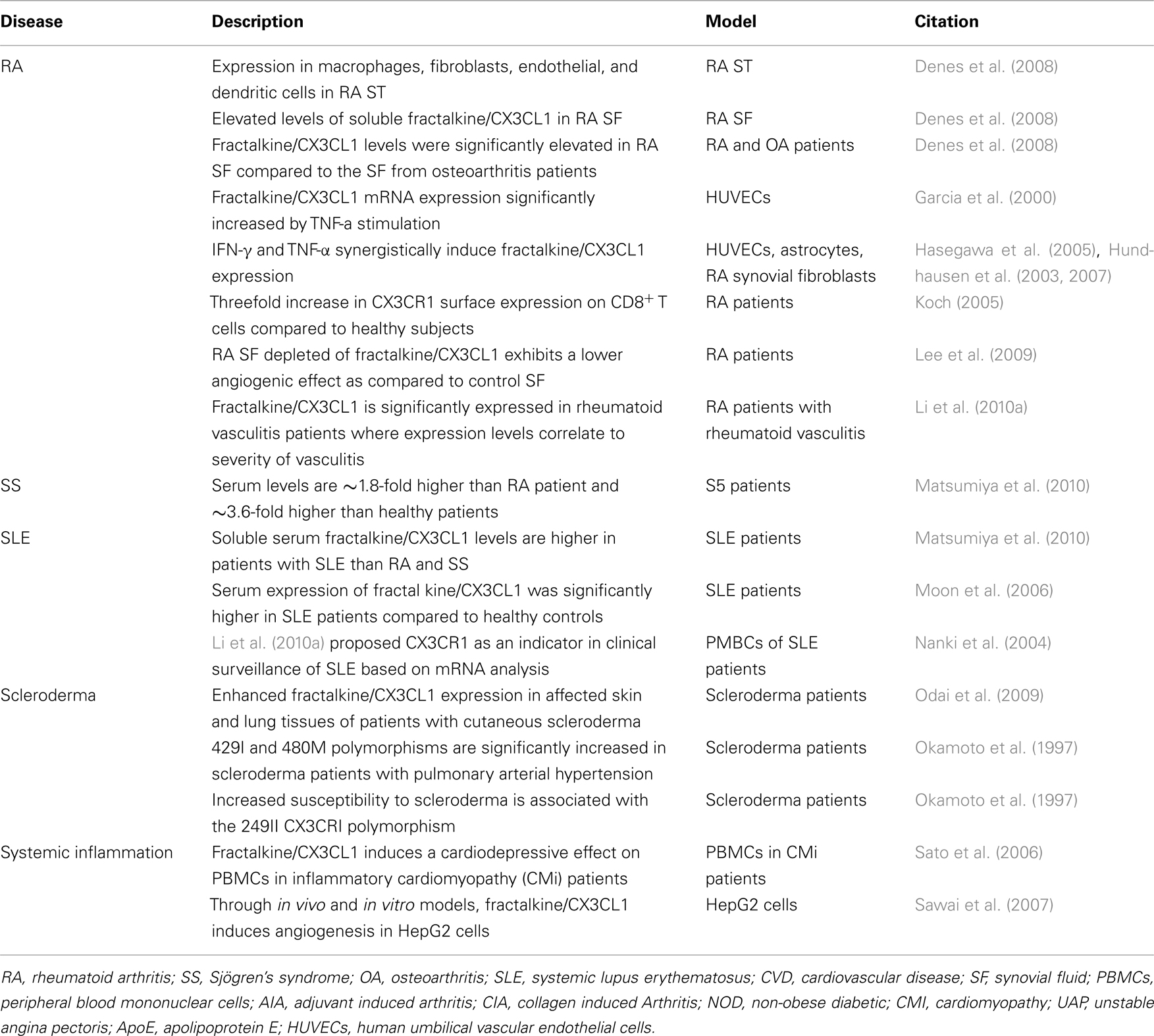
Table 1. Summary of some basic and clinical human studies on the role of fractalkine/CX3CL1 in rheumatic diseases.
Fractalkine/CX3CL1 as a Therapeutic Target
Limited understanding of the exact mechanism of fractalkine/CX3CL1 synthesis and proteolytic shedding and the signaling pathways that mediate the downstream events initiated by the fractalkine/CX3CL1–CX3CR1 duo has been a major hurdle in the path of designing therapeutic molecules targeting this chemokine selectively. However, this has not dampened the enthusiasm of pharmacologists and medicinal chemists as some of the alternative therapeutic strategies such as the inhibition of enzymes that process soluble fractalkine/CX3CL1 or CX3CR1 antagonists have been tried and tested with some promising results (Wiener et al., 2010). Furthermore, elucidation of the structure of fractalkine/CX3CL1 has made it possible to develop analogs to inhibit CX3CR1. In this regard, Inoue et al. (2005) developed fractalkine/CX3CL1 analogs by truncating four N-terminal amino acids, inhibiting the expression in rodent glomerular endothelial cells. Another therapeutic study has proposed using fractalkine/CX3CL1 antagonists with IL-4 antagonists in treating HIV-1 patients to limit the pathogenic potential of the virus (Becker, 2007).
Testing of the existing therapies used for inflammatory diseases in the suppression of fractalkine/CX3CL1 expression and release has gained some momentum (see Table 2). For example, in vitro testing of aspirin showed that inhibition of TNF-α induced fractalkine/CX3CL1 expression in HUVECs through the regulation of the NF-κB pathway (Jiang et al., 2009). Along those lines, several other studies have shown that TNF-α inhibitors decrease fractalkine/CX3CL1 expression (Feldmann and Maini, 2001; Scott and Kingsley, 2006; Odai et al., 2009). However, TNF antagonists such as infliximab, etanercept, and adalimumab have only limited efficacy against conditions related to systemic vascular pathology, which warrants further correlative studies (Aries et al., 2007; Lin et al., 2008). On a positive note, a recent study showed that the mimetic peptide Apo-A1 regulates TNF-α-induced monocyte chemotaxis partly by inhibiting fractalkine/CX3CL1 synthesis in human coronary artery endothelial cells (Di Bartolo et al., 2011).
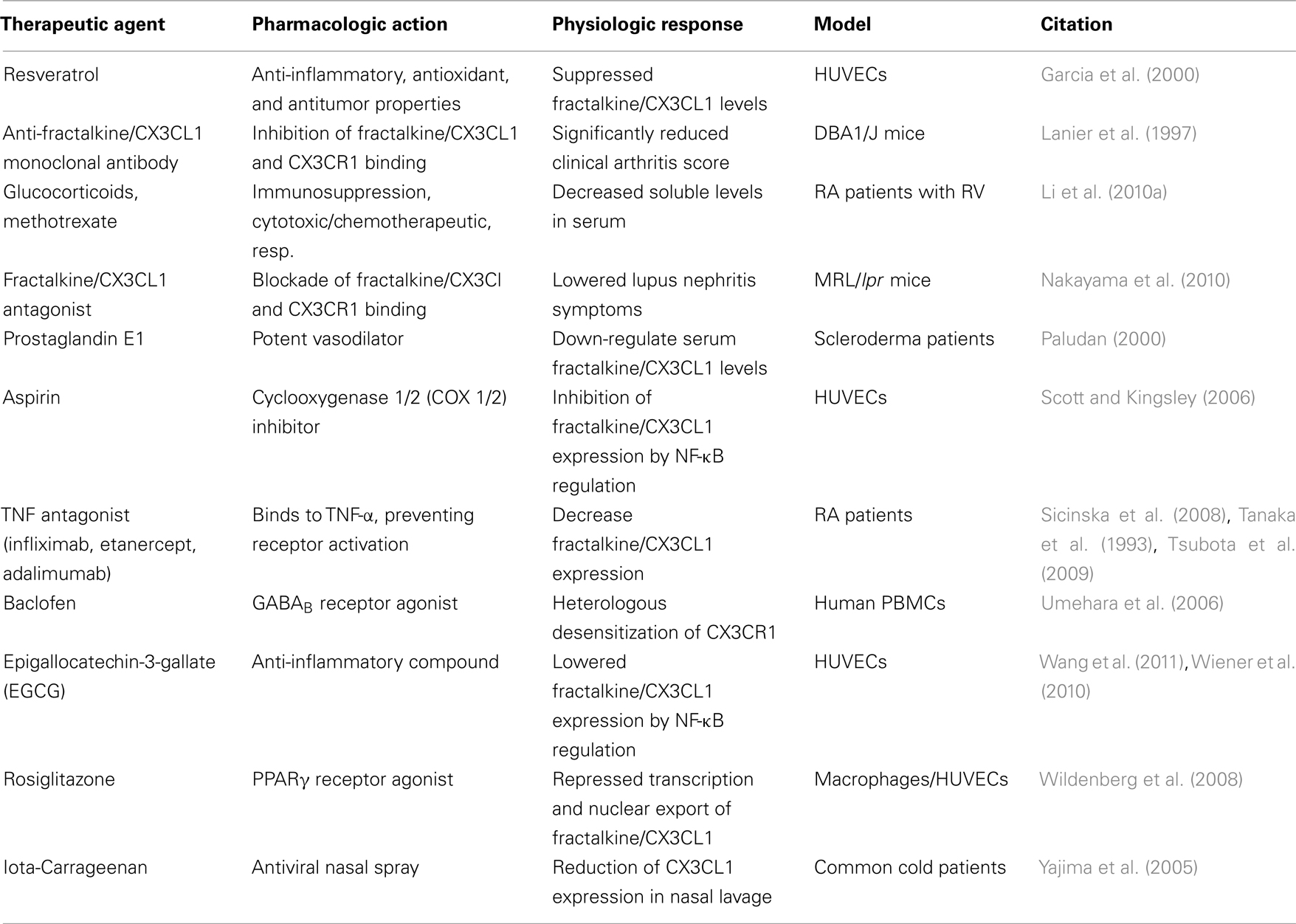
Table 2. Potential therapeutic strategies to regulate fractalkine/CX3CL1 in rheumatic diseases.
Similarly, baclofen (a GABAB receptor agonist) was shown to induce heterologous desensitization of CX3CR1 as well as other chemokine receptors (Duthey et al., 2010). Complementary and alternative medicine (CAM) approaches that have shown benefit in rheumatic diseases have been tested for their efficacy in regulating fractalkine/CX3CL1 production (Chen et al., 2011; Pirvulescu et al., 2011; Wang et al., 2011). A recent example of this includes epigallocatechin-3-gallate (EGCG), a potent anti-inflammatory compound found in green tea known to inhibit experimental arthritis, which has been found to decrease TNF-α induced fractalkine/CX3CL1 expression in HUVECs through NF-κB pathway regulation (Ahmed et al., 2008; Lee et al., 2009). Another study by Moon et al. (2006) also found suppression of TNF-α induced fractalkine/CX3CL1 expression in HUVECs by resveratrol, a polyphenolic compound present in grapes and red wine with anti-inflammatory, antioxidant, and antitumor properties. Wan and Evans (2010) investigated the effects of rosiglitazone (PPARγ agonist) on fractalkine/CX3CL1 expression. The activation of PPARγ by rosiglitazone in macrophages and HUVECs repressed the transcription of the fractalkine/CX3CL1 and nuclear export respectively, suggesting that PPARγ activation may suppress fractalkine/CX3CL1 signaling (Wan and Evans, 2010). Eccles et al. (2010) has tested an antiviral nasal spray called Iota-Carrageenan, which is derived from seaweed and is characterized by the US Food and Drug Administration as “generally regarded as safe” (GRAS). In patients with symptoms of the common cold treatment with Iota-Carrageenan reduced fractalkine/CX3CL1 expression by roughly 45–50% in nasal lavage.
Conclusion
Fractalkine/CX3CL1 is a unique chemokine with chemotactic activity for monocytes, NK cells, and T cells. It also acts as an adhesion molecule for leukocytes. Emerging evidence of its active involvement in many rheumatic diseases such as RA, SS, SLE, and scleroderma as well as other diseases related to vascular inflammation has made fractalkine/CX3CL1 an attractive therapeutic target. In the last few years, therapeutic approaches, both direct (CX3CR1 antagonist or CX3CL1 analogs) and indirect (signaling inhibitors, TACE activity inhibitors, or immunosuppressive drugs) have been tried with some success. However, a better understanding of the cell-specific activation, synthesis, and shedding of fractalkine/CX3CL1 and regulated manipulation of such steps will further assist pharmacologists to develop highly targeted therapeutic approaches to limit its pathological role in rheumatic diseases and other cardiovascular diseases.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported in part by the NIH grants AT-003633 and AR-055741 (Salahuddin Ahmed), start-up funds from The University of Toledo (Salahuddin Ahmed), the Veteran’s Administration (Alisa E. Koch), and the Frederick G. L. Huetwell and William D. Robinson, M.D. Professorship (Alisa E. Koch). Authors thank Ms. Charisse N. Montgomery for critical reading of the review.
References
Ahmed, S., Marotte, H., Kwan, K., Ruth, J. H., Campbell, P. L., Rabquer, B. J., Pakozdi, A., and Koch, A. E. (2008). Epigallocatechin-3-gallate inhibits IL-6 synthesis and suppresses transsignaling by enhancing soluble gp130 production. Proc. Natl. Acad. Sci. U.S.A. 105, 14692–14697.
Aries, P. M., Lamprecht, P., and Gross, W. L. (2007). Biological therapies: new treatment options for ANCA-associated vasculitis? Expert Opin. Biol. Ther. 7, 521–533.
Baggiolini, M., Dewald, B., and Moser, B. (1994). Interleukin-8 and related chemotactic cytokines – CXC and CC chemokines. Adv. Immunol. 55, 97–179.
Bazan, J. F., Bacon, K. B., Hardiman, G., Wang, W., Soo, K., Rossi, D., Greaves, D. R., Zlotnik, A., and Schall, T. J. (1997). A new class of membrane-bound chemokine with a CX3C motif. Nature 385, 640–644.
Becker, Y. (2007). The spreading of HIV-1 infection in the human organism is caused by fractalkine trafficking of the infected lymphocytes – a review, hypothesis and implications for treatment. Virus Genes 34, 93–109.
Charo, I. F., and Ransohoff, R. M. (2006). The many roles of chemokines and chemokine receptors in inflammation. N. Engl. J. Med. 354, 610–621.
Chen, T., Guo, Z. P., Jiao, X. Y., Jia, R. Z., Zhang, Y. H., Li, J. Y., Huang, X. L., and Liu, H. J. (2011). Peoniflorin suppresses tumor necrosis factor-alpha induced chemokine production in human dermal microvascular endothelial cells by blocking nuclear factor-kappaB and ERK pathway. Arch. Dermatol. Res. 303, 351–360.
Combadiere, C., Potteaux, S., Gao, J. L., Esposito, B., Casanova, S., Lee, E. J., Debre, P., Tedgui, A., Murphy, P. M., and Mallat, Z. (2003). Decreased atherosclerotic lesion formation in CX3CR1/apolipoprotein E double knockout mice. Circulation 107, 1009–1016.
Cypiene, A., Dadoniene, J., Rugiene, R., Ryliskyte, L., Kovaite, M., Petrulioniene, Z., Venalis, A., and Laucevicius, A. (2010). The influence of mean blood pressure on arterial stiffening and endothelial dysfunction in women with rheumatoid arthritis and systemic lupus erythematosus. Medicina (Kaunas) 46, 522–530.
Delaleu, N., and Jonsson, R. (2008). Altered fractalkine cleavage results in an organ-specific 17 kDa fractalkine fragment in salivary glands of NOD mice. Arthritis Res. Ther. 10, 114.
Denes, A., Ferenczi, S., Halasz, J., Kornyei, Z., and Kovacs, K. J. (2008). Role of CX3CR1 (fractalkine receptor) in brain damage and inflammation induced by focal cerebral ischemia in mouse. J. Cereb. Blood Flow Metab. 28, 1707–1721.
Di Bartolo, B. A., Nicholls, S. J., Bao, S., Rye, K. A., Heather, A. K., Barter, P. J., and Bursill, C. (2011). The apolipoprotein A-I mimetic peptide ETC-642 exhibits anti-inflammatory properties that are comparable to high density lipoproteins. Atherosclerosis 217, 395–400.
Duthey, B., Hubner, A., Diehl, S., Boehncke, S., Pfeffer, J., and Boehncke, W. H. (2010). Anti-inflammatory effects of the GABA(BM) receptor agonist baclofen in allergic contact dermatitis. Exp. Dermatol. 19, 661–666.
Eccles, R., Meier, C., Jawad, M., Weinmullner, R., Grassauer, A., and Prieschl-Grassauer, E. (2010). Efficacy and safety of an antiviral Iota-Carrageenan nasal spray: a randomized, double-blind, placebo-controlled exploratory study in volunteers with early symptoms of the common cold. Respir. Res. 11, 108.
Escher, F., Vetter, R., Kuhl, U., Westermann, D., Schultheiss, H. P., and Tschope, C. (2011). Fractalkine in human inflammatory cardiomyopathy. Heart 97, 733–739.
Feldmann, M., and Maini, R. N. (2001). Anti-TNF alpha therapy of rheumatoid arthritis: what have we learned? Annu. Rev. Immunol. 19, 163–196.
Fraticelli, P., Sironi, M., Bianchi, G., D’ambrosio, D., Albanesi, C., Stoppacciaro, A., Chieppa, M., Allavena, P., Ruco, L., Girolomoni, G., Sinigaglia, F., Vecchi, A., and Mantovani, A. (2001). Fractalkine (CX3CL1) as an amplification circuit of polarized Th1 responses. J. Clin. Invest. 107, 1173–1181.
Gabrielli, A., Avvedimento, E. V., and Krieg, T. (2009). Scleroderma. N. Engl. J. Med. 360, 1989–2003.
Garcia, G. E., Xia, Y., Chen, S., Wang, Y., Ye, R. D., Harrison, J. K., Bacon, K. B., Zerwes, H. G., and Feng, L. (2000). NF-kappaB-dependent fractalkine induction in rat aortic endothelial cells stimulated by IL-1beta, TNF-alpha, and LPS. J. Leukoc. Biol. 67, 577–584.
Garton, K. J., Gough, P. J., Blobel, C. P., Murphy, G., Greaves, D. R., Dempsey, P. J., and Raines, E. W. (2001). Tumor necrosis factor-alpha-converting enzyme (ADAM17) mediates the cleavage and shedding of fractalkine (CX3CL1). J. Biol. Chem. 276, 37993–38001.
Genta, M. S., Genta, R. M., and Gabay, C. (2006). Systemic rheumatoid vasculitis: a review. Semin. Arthritis Rheum. 36, 88–98.
Hasegawa, H., Kakuguchi, W., Kuroshima, T., Kitamura, T., Tanaka, S., Kitagawa, Y., Totsuka, Y., Shindoh, M., and Higashino, F. (2009). HuR is exported to the cytoplasm in oral cancer cells in a different manner from that of normal cells. Br. J. Cancer 100, 1943–1948.
Hasegawa, M., Sato, S., Echigo, T., Hamaguchi, Y., Yasui, M., and Takehara, K. (2005). Up regulated expression of fractalkine/CX3CL1 and CX3CR1 in patients with systemic sclerosis. Ann. Rheum. Dis. 64, 21–28.
Hundhausen, C., Misztela, D., Berkhout, T. A., Broadway, N., Saftig, P., Reiss, K., Hartmann, D., Fahrenholz, F., Postina, R., Matthews, V., Kallen, K. J., Rose-John, S., and Ludwig, A. (2003). The disintegrin-like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1-mediated cell-cell adhesion. Blood 102, 1186–1195.
Hundhausen, C., Schulte, A., Schulz, B., Andrzejewski, M. G., Schwarz, N., Von Hundelshausen, P., Winter, U., Paliga, K., Reiss, K., Saftig, P., Weber, C., and Ludwig, A. (2007). Regulated shedding of transmembrane chemokines by the disintegrin and metalloproteinase 10 facilitates detachment of adherent leukocytes. J. Immunol. 178, 8064–8072.
Ikejima, H., Imanishi, T., Tsujioka, H., Kashiwagi, M., Kuroi, A., Tanimoto, T., Kitabata, H., Ishibashi, K., Komukai, K., Takeshita, T., and Akasaka, T. (2010). Upregulation of fractalkine and its receptor, CX3CR1, is associated with coronary plaque rupture in patients with unstable angina pectoris. Circ. J. 74, 337–345.
Inoue, A., Hasegawa, H., Kohno, M., Ito, M. R., Terada, M., Imai, T., Yoshie, O., Nose, M., and Fujita, S. (2005). Antagonist of fractalkine (CX3CL1) delays the initiation and ameliorates the progression of lupus nephritis in MRL/lpr mice. Arthritis Rheum. 52, 1522–1533.
Iwamoto, T., Okamoto, H., Toyama, Y., and Momohara, S. (2008). Molecular aspects of rheumatoid arthritis: chemokines in the joints of patients. FEBS J. 275, 4448–4455.
Jiang, D. Q., Liu, H., Zhang, S. B., and Zhang, X. L. (2009). Aspirin inhibits tumor necrosis factor-alpha-stimulated fractalkine expression in human umbilical vein endothelial cells. Chin. Med. J. 122, 1147–1153.
Jones, B. A., Beamer, M., and Ahmed, S. (2010). Fractalkine/CX3CL1: a potential new target for inflammatory diseases. Mol. Interv. 10, 263–270.
Klosowska, K., Volin, M. V., Huynh, N., Chong, K. K., Halloran, M. M., and Woods, J. M. (2009). Fractalkine functions as a chemoattractant for osteoarthritis synovial fibroblasts and stimulates phosphorylation of mitogen-activated protein kinases and Akt. Clin. Exp. Immunol. 156, 312–319.
Koch, A. E. (2005). Chemokines and their receptors in rheumatoid arthritis: future targets? Arthritis Rheum. 52, 710–721.
Kruszka, P., and O’Brian, R. J. (2009). Diagnosis and management of Sjogren syndrome. Am. Fam. Physician 79, 465–470.
Lanier, L. L., Corliss, B., and Phillips, J. H. (1997). Arousal and inhibition of human NK cells. Immunol. Rev. 155, 145–154.
Lee, A. S., Jung, Y. J., Kim, D. H., Lee, T. H., Kang, K. P., Lee, S., Lee, N. H., Sung, M. J., Kwon, D. Y., Park, S. K., and Kim, W. (2009). Epigallocatechin-3-O-gallate decreases tumor necrosis factor-alpha-induced fractalkine expression in endothelial cells by suppressing NF-kappaB. Cell. Physiol. Biochem. 24, 503–510.
Lesnik, P., Haskell, C. A., and Charo, I. F. (2003). Decreased atherosclerosis in CX3CR1-/- mice reveals a role for fractalkine in atherogenesis. J. Clin. Invest. 111, 333–340.
Li, F., Wang, Z., Liu, Y., and Li, J. (2010a). Down-regulation of fractalkine inhibits the in vitro and in vivo angiogenesis of the hepatocellular carcinoma HepG2 cells. Oncol. Rep. 24, 669–675.
Li, Y. M., Chen, Z. Q., Yao, X., Yang, A. Z., Li, A. S., Liu, D. M., and Gong, J. Q. (2010b). mRNA expression of chemokine receptors on peripheral blood mononuclear cells and correlation with clinical features in systemic lupus erythematosus patients. Chin. Med. Sci. J. 25, 162–168.
Lin, J., Ziring, D., Desai, S., Kim, S., Wong, M., Korin, Y., Braun, J., Reed, E., Gjertson, D., and Singh, R. R. (2008). TNFalpha blockade in human diseases: an overview of efficacy and safety. Clin. Immunol. 126, 13–30.
Liuzzo, G., Vallejo, A. N., Kopecky, S. L., Frye, R. L., Holmes, D. R., Goronzy, J. J., and Weyand, C. M. (2001). Molecular fingerprint of interferon-gamma signaling in unstable angina. Circulation 103, 1509–1514.
Marasini, B., Cossutta, R., Selmi, C., Pozzi, M. R., Gardinali, M., Massarotti, M., Erario, M., Battaglioli, L., and Biondi, M. L. (2005). Polymorphism of the fractalkine receptor CX3CR1 and systemic sclerosis-associated pulmonary arterial hypertension. Clin. Dev. Immunol. 12, 275–279.
Marchesi, F., Locatelli, M., Solinas, G., Erreni, M., Allavena, P., and Mantovani, A. (2010). Role of CX3CR1/CX3CL1 axis in primary and secondary involvement of the nervous system by cancer. J. Neuroimmunol. 224, 39–44.
Matsumiya, T., Ota, K., Imaizumi, T., Yoshida, H., Kimura, H., and Satoh, K. (2010). Characterization of synergistic induction of CX3CL1/fractalkine by TNF-alpha and IFN-gamma in vascular endothelial cells: an essential role for TNF-alpha in post-transcriptional regulation of CX3CL1. J. Immunol. 184, 4205–4214.
Matsunawa, M., Isozaki, T., Odai, T., Yajima, N., Takeuchi, H. T., Negishi, M., Ide, H., Adachi, M., and Kasama, T. (2006). Increased serum levels of soluble fractalkine (CX3CL1) correlate with disease activity in rheumatoid vasculitis. Arthritis Rheum. 54, 3408–3416.
Moon, S. O., Kim, W., Sung, M. J., Lee, S., Kang, K. P., Kim, D. H., Lee, S. Y., So, J. N., and Park, S. K. (2006). Resveratrol suppresses tumor necrosis factor-alpha-induced fractalkine expression in endothelial cells. Mol. Pharmacol. 70, 112–119.
Nakatani, K., Yoshimoto, S., Iwano, M., Asai, O., Samejima, K., Sakan, H., Terada, M., Hasegawa, H., Nose, M., and Saito, Y. (2010). Fractalkine expression and CD16+ monocyte accumulation in glomerular lesions: association with their severity and diversity in lupus models. Am. J. Physiol. Renal Physiol. 299, F207–F216.
Nakayama, T., Watanabe, Y., Oiso, N., Higuchi, T., Shigeta, A., Mizuguchi, N., Katou, F., Hashimoto, K., Kawada, A., and Yoshie, O. (2010). Eotaxin-3/CC chemokine ligand 26 is a functional ligand for CX3CR1. J. Immunol. 185, 6472–6479.
Nanki, T., Imai, T., Nagasaka, K., Urasaki, Y., Nonomura, Y., Taniguchi, K., Hayashida, K., Hasegawa, J., Yoshie, O., and Miyasaka, N. (2002). Migration of CX3CR1-positive T cells producing type 1 cytokines and cytotoxic molecules into the synovium of patients with rheumatoid arthritis. Arthritis Rheum. 46, 2878–2883.
Nanki, T., Urasaki, Y., Imai, T., Nishimura, M., Muramoto, K., Kubota, T., and Miyasaka, N. (2004). Inhibition of fractalkine ameliorates murine collagen-induced arthritis. J. Immunol. 173, 7010–7016.
Nishimura, M., Umehara, H., Nakayama, T., Yoneda, O., Hieshima, K., Kakizaki, M., Dohmae, N., Yoshie, O., and Imai, T. (2002). Dual functions of fractalkine/CX3C ligand 1 in trafficking of perforin+/granzyme B+ cytotoxic effector lymphocytes that are defined by CX3CR1 expression. J. Immunol. 168, 6173–6180.
Odai, T., Matsunawa, M., Takahashi, R., Wakabayashi, K., Isozaki, T., Yajima, N., Miwa, Y., and Kasama, T. (2009). Correlation of CX3CL1 and CX3CR1 levels with response to infliximab therapy in patients with rheumatoid arthritis. J. Rheumatol. 36, 1158–1165.
Okamoto, H., Yamamura, M., Morita, Y., Harada, S., Makino, H., and Ota, Z. (1997). The synovial expression and serum levels of interleukin-6, interleukin-11, leukemia inhibitory factor, and oncostatin M in rheumatoid arthritis. Arthritis Rheum. 40, 1096–1105.
Paludan, S. R. (2000). Synergistic action of pro-inflammatory agents: cellular and molecular aspects. J. Leukoc. Biol. 67, 18–25.
Pirvulescu, M. M., Gan, A. M., Stan, D., Simion, V., Calin, M., Butoi, E., Tirgoviste, C. I., and Manduteanu, I. (2011). Curcumin and a morus alba extract reduce pro-inflammatory effects of resistin in human endothelial cells. Phytother. Res. 25, 1737–1742.
Pope, R. M. (2002). Apoptosis as a therapeutic tool in rheumatoid arthritis. Nat. Rev. Immunol. 2, 527–535.
Ramos-Casals, M., Tzioufas, A. G., Stone, J. H., Siso, A., and Bosch, X. (2010). Treatment of primary Sjogren syndrome: a systematic review. JAMA 304, 452–460.
Ruth, J. H., Volin, M. V., Haines, G. K. III, Woodruff, D. C., Katschke, K. J. Jr., Woods, J. M., Park, C. C., Morel, J. C., and Koch, A. E. (2001). Fractalkine, a novel chemokine in rheumatoid arthritis and in rat adjuvant-induced arthritis. Arthritis Rheum. 44, 1568–1581.
Sato, E., Iikuni, N., Yoshio, T., Minota, S., Kamatani, N., and Okamoto, H. (2006). Soluble fractalkine in the cerebrospinal fluid of patients with neuropsychiatric lupus. Ann. Rheum. Dis. 65, 1257–1259.
Sawai, H., Park, Y. W., He, X., Goronzy, J. J., and Weyand, C. M. (2007). Fractalkine mediates T cell-dependent proliferation of synovial fibroblasts in rheumatoid arthritis. Arthritis Rheum. 56, 3215–3225.
Schroder, K., Hertzog, P. J., Ravasi, T., and Hume, D. A. (2004). Interferon-gamma: an overview of signals, mechanisms and functions. J. Leukoc. Biol. 75, 163–189.
Sciume, G., Soriani, A., Piccoli, M., Frati, L., Santoni, A., and Bernardini, G. (2010). CX3CR1/CX3CL1 axis negatively controls glioma cell invasion and is modulated by transforming growth factor-beta1. Neuro-oncology 12, 701–710.
Scott, D. L., and Kingsley, G. H. (2006). Tumor necrosis factor inhibitors for rheumatoid arthritis. N. Engl. J. Med. 355, 704–712.
Sicinska, J., Gorska, E., Cicha, M., Kuklo-Kowalska, A., Hamze, V., Stepien, K., Wasik, M., and Rudnicka, L. (2008). Increased serum fractalkine in systemic sclerosis. Down-regulation by prostaglandin E1. Clin. Exp. Rheumatol. 26, 527–533.
Tanaka, Y., Adams, D. H., Hubscher, S., Hirano, H., Siebenlist, U., and Shaw, S. (1993). T-cell adhesion induced by proteoglycan-immobilized cytokine MIP-1 beta. Nature 361, 79–82.
Tsubota, K., Nishiyama, T., Mishima, K., Inoue, H., Doi, T., Hattori, Y., Kodama, T., Higuchi, A., Hayashi, Y., and Saito, I. (2009). The role of fractalkine as an accelerating factor on the autoimmune exocrinopathy in mice. Invest. Ophthalmol. Vis. Sci. 50, 4753–4760.
Turner, S. L., Mangnall, D., Bird, N. C., Blair-Zajdel, M. E., and Bunning, R. A. (2010). Effects of pro-inflammatory cytokines on the production of soluble fractalkine and ADAM17 by HepG2 cells. J. Gastrointestin. Liver Dis. 19, 265–271.
Umehara, H., Bloom, E., Okazaki, T., Domae, N., and Imai, T. (2001). Fractalkine and vascular injury. Trends Immunol. 22, 602–607.
Umehara, H., Bloom, E. T., Okazaki, T., Nagano, Y., Yoshie, O., and Imai, T. (2004). Fractalkine in vascular biology: from basic research to clinical disease. Arterioscler. Thromb. Vasc. Biol. 24, 34–40.
Umehara, H., Tanaka, M., Sawaki, T., Jin, Z. X., Huang, C. R., Dong, L., Kawanami, T., Karasawa, H., Masaki, Y., Fukushima, T., Hirose, Y., and Okazaki, T. (2006). Fractalkine in rheumatoid arthritis and allied conditions. Mod. Rheumatol. 16, 124–130.
Volin, M. V., Huynh, N., Klosowska, K., Chong, K. K., and Woods, J. M. (2007). Fractalkine is a novel chemoattractant for rheumatoid arthritis fibroblast-like synoviocyte signaling through MAP kinases and Akt. Arthritis Rheum. 56, 2512–2522.
Volin, M. V., Woods, J. M., Amin, M. A., Connors, M. A., Harlow, L. A., and Koch, A. E. (2001). Fractalkine: a novel angiogenic chemokine in rheumatoid arthritis. Am. J. Pathol. 159, 1521–1530.
Wan, Y., and Evans, R. M. (2010). Rosiglitazone activation of PPARgamma suppresses fractalkine signaling. J. Mol. Endocrinol. 44, 135–142.
Wang, W., Wang, Y. L., Chen, X. Y., Li, Y. T., Hao, W., Jin, Y. P., and Han, B. (2011). Dexamethasone attenuates development of monocrotaline-induced pulmonary arterial hypertension. Mol. Biol. Rep. 38, 3277–3284.
Wiener, J. J., Sun, S., and Thurmond, R. L. (2010). Recent advances in the design of cathepsin S inhibitors. Curr. Top. Med. Chem. 10, 717–732.
Wildenberg, M. E., Van Helden-Meeuwsen, C. G., Drexhage, H. A., and Versnel, M. A. (2008). Altered fractalkine cleavage potentially promotes local inflammation in NOD salivary gland. Arthritis Res. Ther. 10, R69.
Yajima, N., Kasama, T., Isozaki, T., Odai, T., Matsunawa, M., Negishi, M., Ide, H., Kameoka, Y., Hirohata, S., and Adachi, M. (2005). Elevated levels of soluble fractalkine in active systemic lupus erythematosus: potential involvement in neuropsychiatric manifestations. Arthritis Rheum. 52, 1670–1675.
Yoneda, O., Imai, T., Goda, S., Inoue, H., Yamauchi, A., Okazaki, T., Imai, H., Yoshie, O., Bloom, E. T., Domae, N., and Umehara, H. (2000). Fractalkine-mediated endothelial cell injury by NK cells. J. Immunol. 164, 4055–4062.
Yoshida, H., Imaizumi, T., Fujimoto, K., Matsuo, N., Kimura, K., Cui, X., Matsumiya, T., Tanji, K., Shibata, T., Tamo, W., Kumagai, M., and Satoh, K. (2001). Synergistic stimulation, by tumor necrosis factor-alpha and interferon-gamma, of fractalkine expression in human astrocytes. Neurosci. Lett. 303, 132–136.
Keywords: fractalkine/CX3CL1, rheumatic disorders, proteolytic shedding, ADAM17/TACE, ADAM10
Citation: Jones B, Koch AE and Ahmed S (2012) Pathological role of fractalkine/CX3CL1 in rheumatic diseases: a unique chemokine with multiple functions. Front. Immun. 2:82. doi: 10.3389/fimmu.2011.00082
Received: 18 October 2011; Accepted: 06 December 2011;
Published online: 02 January 2012.
Edited by:
Mauro Martins Teixeira, Universidade Federal de Minas Gerais, BrazilReviewed by:
José C. Alves-Filho, University of São Paulo, BrazilGustavo B. Menezes, Universidade Federal de Minas Gerais, Brazil
Hideki Nakano, National Institute of Environmental Health Science, USA
Copyright: © 2012 Jones, Koch and Ahmed. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Salahuddin Ahmed, Department of Pharmacology, College of Pharmacy and Pharmaceutical Sciences, The University of Toledo, Health Education Building, Room 282F, Mail Stop 1015, 3000 Arlington Avenue, Toledo, OH 43614, USA. e-mail:c2FsYWguYWhtZWRAdXRvbGVkby5lZHU=