 Irena Slavuljica
Irena Slavuljica
- Department of Histology and Embryology, Faculty of Medicine, University of Rijeka, Rijeka, Croatia
NKG2D is a potent activating receptor expressed on NK cells, NKT cells, γδ T cells, and CD8 T cells. NKG2D recognizes cell surface molecules structurally related to MHC class I proteins induced by infection or other type of cellular stress. The engagement of NKG2D leads to NK cell cytotoxicity and cytokine secretion or to a co-stimulation of CD8 T cells. Both human and mouse cytomegalovirus (CMV) have evolved numerous mechanisms to evade NKG2D-mediated immune response. This review describes the mechanisms used by CMV to inhibit NKG2D ligand expression and the recent advances in exploiting the NKG2D recognition pathway for mounting efficient and long-lasting immune response.
NK cells are lymphocytes of innate immunity which target virally infected and tumor cells. Their activity is determined by a delicate balance of signals from a sophisticated repertoire of inhibitory and activating receptors expressed on their cell surface (reviewed in Diefenbach and Raulet, 2001; Lanier, 2005). Inhibitory receptors gauge the expression of MHC class I molecules, which are constitutively expressed by healthy cells but may be downregulated under conditions of cellular stress. Therefore, inhibitory receptors provide a mean for NK cells to exhibit cytotoxicity toward infected or transformed cells via the “missing-self” axis. In contrast, “induced-self” recognition involves the engagement of various activating NK cell receptors by proteins expressed under cellular stress, thus allowing the immune response to proceed. One of the most potent activating receptors which mediate the “induced-self” axis is NKG2D, a receptor shared by both the cells of innate and adaptive immunity.
NKG2D Receptor
NKG2D is a type-2 transmembrane glycoprotein expressed as a disulfide linked homodimer on the cell surface (Diefenbach et al., 2002; Jamieson et al., 2002). It is encoded by KLRK1 (killer cell lectin-like receptor subfamily K, member 1), a single gene with little polymorphism located on the mouse chromosome 6 and in the syntenic position on human chromosome 12 (Houchins et al., 1991). In mice, alternative RNA splicing results in two NKG2D isoforms – long (NKG2D-L) and short (NKG2D-S) – that differ in 13 amino acids (Diefenbach et al., 2002; Gilfillan et al., 2002). Resting mouse NK cells express very little NKG2D-S, but its expression is induced after NK cell activation. Neither isoform can be detected in resting mouse CD8 T cells but after T cell receptor (TCR) stimulation the expression of both isoforms is upregulated. Within its intracellular domain NKG2D has no signaling motif but it associates with signal-transducing proteins through charged residues in the transmembrane region. The NKG2D-L isoform pairs with the DAP10 signaling molecule, while NKG2D-S associates with either DAP10 or DAP12. However, because CD8 T cells do not express DAP12, the two NKG2D isoforms that are expressed by activated T cells can interact with DAP10 only, whereas activated NK cells can transmit signals through both DAP10 and DAP12 (Figure 1). The only isoform expressed in humans corresponds to the long form in mouse and it interacts with DAP10 (Bauer et al., 1999; Wu et al., 1999; Rosen et al., 2004).
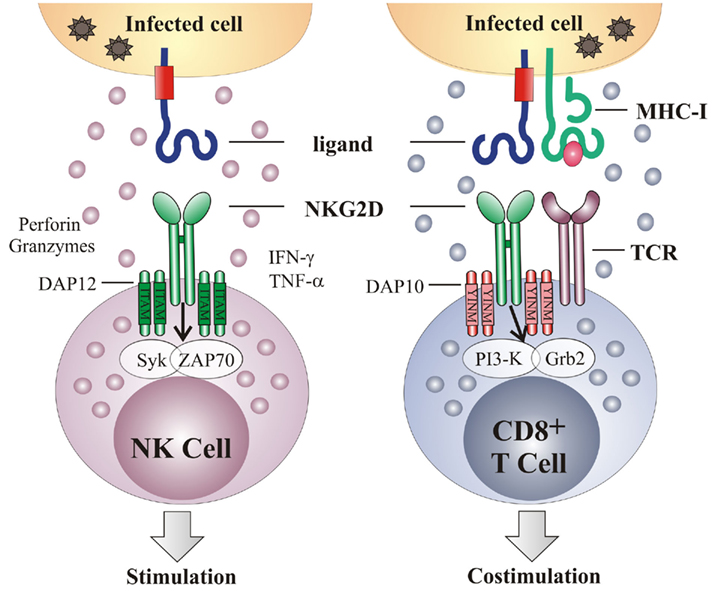
Figure 1. NKG2D acts as an activating and a co-stimulatory receptor. NKG2D is a type-2 transmembrane homodimer that signals via association with adapter molecules DAP10 or DAP12. Association with DAP12 leads to phosphorylation of the ITAM and triggering of the Syk and/or Zap70 signaling cascade. Association with DAP10 results in tyrosine phosphorylation on the YINM motif and recruitment of the PI3K and Grb2 cascade. On NK cells, NKG2D serves as an activation receptor, such that NKG2D engagement is sufficient to trigger NK cell-mediated cytotoxicity and cytokine production. In contrast, NKG2D acts as a co-stimulatory receptor on CD8 T cells, requiring TCR-mediated signaling for their full activation. Grb2, growth factor receptor-bound protein 2; IFN-γ, interferon γ; ITAM, immunoreceptor tyrosine-based activating motif; PI3K, phosphoinositide 3-kinase; Syk, spleen tyrosine kinase; TCR, T cell receptor; TNF-α, tumor necrosis factor α; Zap70, zeta-chain associated protein kinase 70.
NKG2D is expressed by all NK cells, most NKT cells, a subset of γδ T cells, all human CD8 T cells, activated mouse CD8 T cells and a subset of CD4 T cells (Bauer et al., 1999; Diefenbach et al., 2000, 2002; Girardi et al., 2001; Jamieson et al., 2002; Ehrlich et al., 2005). On NK cells NKG2D serves as a primary activating receptor, meaning that the engagement of NKG2D can override inhibitory signals in the absence of the “missing-self” recognition (Cerwenka et al., 2001; Figure 1). In addition to cytotoxicity, activation of NK cells via NKG2D triggers the production of different cytokines, including IFN-γ, TNF-α, and MIP-1β. Via this mechanism NK cells are involved in the regulation of adaptive immunity. The engagement of the NKG2D receptor on CD8 T cells is insufficient to generate a T cell response (Bauer et al., 1999; Ehrlich et al., 2005). Instead, NKG2D acts as a co-stimulatory receptor which augments TCR-induced responses (Groh et al., 2001; Maasho et al., 2005; Markiewicz et al., 2005). The ability of NKG2D to co-stimulate T cells may be determined by additional factors, such as the activation state of the T cells or the cellular environment (Roberts et al., 2001; Meresse et al., 2004; Verneris et al., 2004).
NKG2D Ligands
NKG2D ligands are distantly related homologs of the MHC-I proteins and are characterized by a striking structural diversity, different expression patterns, and regulation mechanisms. Human NKG2D ligands are MHC class-I-related protein A (MICA), MICB, and UL16-binding proteins (ULBP1-6). MICA and MICB, encoded by the genes within human MHC (Groh et al., 1996; Bauer et al., 1999), are the only NKG2D ligands containing three immunoglobulin-like domains (α1, α2, and α3), but unlike MHC molecules, they do not associate with β2 microglobulin nor bind antigenic peptides. All other NKG2D ligands are related to MHC-I molecules but contain only α1 and α2 domains. Although named by their ability to bind human CMV (HCMV) protein UL16, only the first two identified ULBP proteins ULBP1, ULBP2, and the subsequently described ULBP6 bind this viral protein (Cosman et al., 2001; Radosavljevic et al., 2002; Bacon et al., 2004; Eagle et al., 2009). Like MIC proteins, ULBP5 and ULBP6 are transmembrane proteins, while proteins ULBP1-3 are anchored to the membrane via glycosylphosphatidylinositol (GPI; Eleme et al., 2004). ULBP family is also known as the retinoic acid early transcript 1 (RAET-1) family, since they show sequence homology to the mouse retinoic acid early 1 (RAE-1) proteins (Nomura et al., 1994; Cerwenka et al., 2000; Diefenbach et al., 2001). Five members of the RAE-1 family (RAE-1α–ε) are highly related to each other (>85% amino acid homology) and differentially expressed in various mouse strains. A second family of mouse NKG2D ligands is the H60 family that comprises three members H60 (a, b, and c) of which H60a is a minor histocompatibility antigen (Malarkannan et al., 1998; Takada et al., 2008; Whang et al., 2009). Finally, murine UL16 protein-like transcript 1 (MULT-1) is the sole member of the third family of mouse NKG2D ligands (Carayannopoulos et al., 2002; Diefenbach et al., 2003).
NKG2D ligands are highly polymorphic, in particular MICA and MICB for which 70 and 31 alleles with different affinities for NKG2D have been identified, respectively (Steinle et al., 2001). There is also some degree of polymorphism in RAET-1 genes and mouse H60 and RAE-1 genes (Eagle et al., 2006; Romphruk et al., 2009). The reason for such a high degree of polymorphism is unknown at the present but it may have evolved to counter the selective pressure formed by pathogens and their evasion strategies, in order to maintain a proper recognition of infected cells by NK cells.
Molecular Mechanisms in Control of NKG2D Ligand Expression
NKG2D ligand expression is induced by various stress conditions, including cellular stress, malignant transformation or infection. The exact mechanism by which this occurs is, however, mostly unknown. NKG2D ligands are constitutively expressed on many tumor cell lines and actively growing epithelial and hematological tumors (reviewed in Nausch and Cerwenka, 2008). The expression of NKG2D ligands makes tumors susceptible to killing by NK cells in vitro and is associated with a decreased tumor progression and a decreased capacity to form metastases in vivo (Bauer et al., 1999; Cerwenka et al., 2000, 2001; Diefenbach et al., 2000, 2001). NKG2D ligands are also induced on cells infected with bacteria and viruses, including cells infected with cytomegalovirus (CMV; Tieng et al., 2002; Siren et al., 2004; Vankayalapati et al., 2005; Draghi et al., 2007; Ward et al., 2007, 2009; Fang et al., 2008; Walsh et al., 2008). The control of NKG2D ligands expression includes both transcriptional and posttranscriptional mechanisms. The promoter regions of MIC genes contain heat shock elements similar to those found in the HSP70 gene, which bind heat shock factor-1 and induce MIC transcription in stressed cells (Jinushi et al., 2003; Venkataraman et al., 2007). Similarly, the retinoic acid-inducible element is mapped in the RAE-1γ promoter thus suggesting that gene expression is directly regulated by the retinoic acid (Nomura et al., 1994, 1996). Both human and mouse cells upregulate NKG2D ligands following treatment with DNA-damaging agents (Gasser et al., 2005) that activate a major DNA damage pathway initiated by ATM (Ataxia telangiectasia, mutated) or ATR (ATM- and Rad3-related) protein kinases (Bartkova et al., 2005; Gorgoulis et al., 2005). Oncogenes, such as the adenovirus E1A, induce transcription NKG2D ligands (Routes et al., 2005). These findings are the proof that there is a link between the constitutive activity of the DNA damage pathway and oncogenes in tumors, and a frequent upregulation of NKG2D ligands by transformed cells. Recent findings suggest that activation of the phosphatidylinositol-3-kinase pathway induced by murine CMV (MCMV) infection or tumor transformation is required for the induction of the RAE-1 family of mouse NKG2D ligands (Tokuyama et al., 2011). Finally, the expression of HCMV immediate early gene (IE1 and IE2) displaces histone deacetylases and induces transcription of MIC proteins in infected cells (Venkataraman et al., 2007).
Cytokines and toll-like receptor (TLR) signaling differentially influence NKG2D ligand expression in different cell types and environments. TGF-β downmodulates the expression of RAE-1 proteins, which might be one of the immune suppressive mechanisms mediated by this cytokine (Routes et al., 2005; Eisele et al., 2006). IFN-γ decreases the expression of MICA and ULBP2 on melanoma and glioma cells, while IFN-γ and IFN-α selectively downregulate H60a on sarcoma cell lines (Jinushi et al., 2003; Schwinn et al., 2009). The downregulation of NKG2D ligands by the proinflammatory cytokine IFN-γ may be unexpected, but, on the other hand, it may represent a mechanism to prevent sustained triggering of the NKG2D receptor or, in the context of tumor cells, a strategy to escape from immune surveillance. Furthermore, IFN-γ-inducible small non-coding microRNAs (miRNAs) mediate downregulation of MICA (Yadav et al., 2009). On the other hand, the treatment of peritoneal macrophages or dendritic cells (DC) with TLR agonists upregulates the transcription of ULBP genes that may participate in the activation of NK and T cells (Hamerman et al., 2004; Draghi et al., 2007; Ebihara et al., 2007; Nedvetzki et al., 2007; Kloss et al., 2008).
In addition, various posttranscriptional mechanisms prevent the expression of NKG2D ligands on healthy cells. Although MICA and MICB genes are continuously transcribed, proteins are not expressed on the cell surface because their translation is held in check by cellular miRNAs. Cellular stress, such as a heat shock or viral infection, induces MICA and MICB transcription to levels that exceed the repressive ability of cellular miRNAs and allow MIC proteins to be quickly expressed on the cell surface (Stern-Ginossar et al., 2008b; Yadav et al., 2009). In healthy cells, MULT-1 protein undergoes ubiquitination and subsequent lysosomal degradation preventing its surface expression. In response to cellular stress, such as ultraviolet irradiation or heat shock, ubiquitin-dependent degradation of MULT-1 is reduced, leading to expression of MULT-1 protein at the cell surface (Nice et al., 2009). Among other NKG2D ligands, MICA, MICB, and ULBP5 are transmembrane proteins containing lysine residues within their cytoplasmic tails and therefore might be regulated by ubiquitination as well. Collectively, multiple checkpoints operating at different levels of NKG2D ligand synthesis facilitate the expression of NKG2D ligands to a broad range of cellular emergencies.
Manipulation of NKG2D Ligand Expression by Cytomegalovirus
Infection with both HCMV and MCMV upregulates the transcription of NKG2D ligands, which can result in NKG2D-mediated lysis of infected cells by NK cells. Viruses have developed different evasive mechanisms to prevent the expression of NKG2D ligands on the cell surface (Wilkinson et al., 2008; Figure 2). The HCMV protein UL16 binds MICB, ULBP1, ULBP2, and ULBP6 in the endoplasmatic reticulum (ER) and retains these ligands inside the cell (Cosman et al., 2001; Dunn et al., 2003; Rolle et al., 2003; Vales-Gomez et al., 2003; Welte et al., 2003; Wu et al., 2003). The structure of UL16 revealed that this viral protein mimics a central binding motif of otherwise structurally unrelated NKG2D, thus enabling the virus to evade several diverse NKG2D ligands (Muller et al., 2010). Another HCMV immunoevasin, UL142, retains newly synthesized full-length MICA in the cis-Golgi compartment (Chalupny et al., 2006; Ashiru et al., 2009). Interestingly, the MICA*008 allele, which lacks the cytoplasmic domain, is resistant to the action of UL142. Since MICA*008 is frequently found in humans, this suggests that during co-evolution HCMV exerts selective pressure to the host and thus drives diversity and polymorphism of NKG2D ligands. In addition to targeting already synthesized proteins, HCMV employs miRNAs to prevent the translation of ligands for NKG2D. HCMV encodes miRNA–UL112 that competes with endogenous miRNA for binding to the 3′-UTR (untranslated region) of the MICA transcript, thus repressing the translation of this NKG2D ligand (Stern-Ginossar et al., 2007, 2008a).
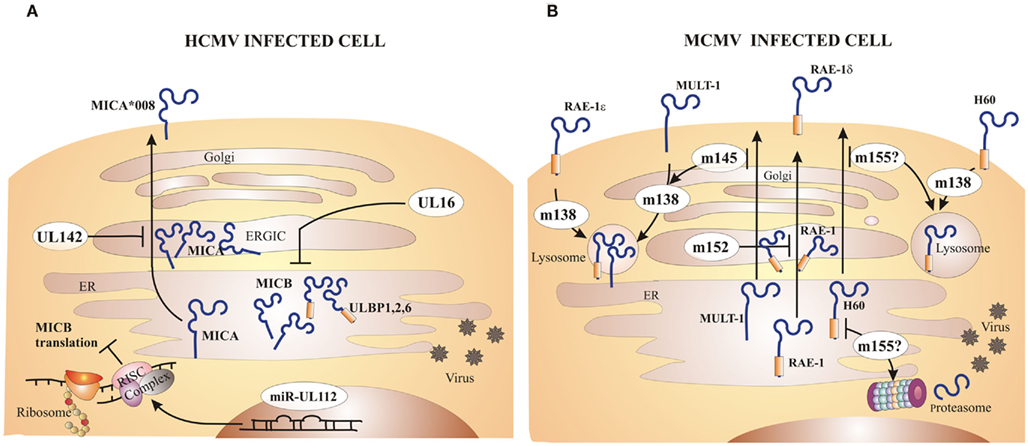
Figure 2. Major CMV inhibitors of NKG2D ligands. Both HCMV and MCMV encode immunoevasins that inhibit expression of NKG2D ligands on the surface of infected cells. (A) The HCMV protein UL16 inhibits MICB, ULBP1, ULBP2, and ULBP6 expression by retaining them inside the cell. UL142 prevents MICA expression via retention in the cis-Golgi compartment. Certain alleles of MICA, such as MICA*008, are resistant to inhibition by HCMV due to truncation of the cytoplasmic domain. HCMV microRNA (miR)–UL112 forms RISC complex that binds to the MICB transcript and represses its translation. (B) MCMV encodes four immunoevasins that redundantly downmodulate all mouse NKG2D ligands. The m152 product retains RAE-1 family of proteins in ER/cis-Golgi compartment. Not all RAE-1 proteins are equally susceptible to inhibition by MCMV, namely RAE-1δ is more resistant than other RAE-1 proteins. The m145 product interferes with MULT-1 expression by making it more susceptible to the inhibition by the other immunoevasin, m138. The m155 product redirects H60 for lysosomal and/or proteasomal degradation. Surface RAE-1ε, MULT-1, and H60 are additionally downmodulated by m138 which interferes with their recycling.
Most wild-derived and laboratory mouse strains fail to control MCMV during the first days post infection in spite of intact NK cells expressing the NKG2D receptor. The explanation for this phenomenon came with the discovery of MCMV inhibitors that prevent the expression of NKG2D ligands on the surface of infected cells (Figure 2). The deletion of one or more of these viral immunoevasins results in an attenuated MCMV replication in vivo during early days post infection (Lenac et al., 2008; Figure 3). However, if mice are depleted of NK cells or an NKG2D blocking antibody is administered prior to the viral challenge, the replication of MCMV can be restored. gp40, encoded by m152 gene, was the first MCMV protein found to inhibit NKG2D. This finding was unexpected, because m152 was known to be involved in the maturation of MHC-I molecules and thus expected to enhance, rather than inhibit, the NK cell response (Ziegler et al., 1997, 2000; Krmpotic et al., 1999). The m152 protein targets RAE-1 ligands for downregulation, thus promoting viral escape from NK cell control (Lodoen et al., 2003; Arapovic et al., 2009). In this case, a single viral gene product acts in evasion of both NK cell and T cell antiviral immune responses. The m152 retains RAE-1 proteins in the ERGIC/cis-Golgi compartment (Krmpotic et al., 2002; Lodoen et al., 2003; Arapovic et al., 2009). However, not all RAE-1 isoforms are equally susceptible to MCMV regulation. RAE-1δ is more resistant to MCMV than other RAE-1 isoforms which is, at least in part, due to the absence of the PLWY motif in the RAE-1δ intracellular domain (Arapovic et al., 2009). Subsequent studies have identified three more MCMV proteins involved in the regulation of NKG2D ligands. The product of m155 gene downmodulates the expression of H60 (Lodoen et al., 2004; Hasan et al., 2005), although the underlying molecular mechanism is not entirely understood. Lodoen et al. (2004) reported that H60 is degraded in MCMV-infected cells in a proteasome-dependent pathway. Our results, however, show that the acquisition of endo-b-N-acetylglucosaminodase H resistance and the half-life of the protein are not affected by the virus, indicating that m155 affects H60 after it leaves the ER (Hasan et al., 2005). The product of the m145 gene binds MULT-1 after it leaves ER (Krmpotic et al., 2005) and makes it more susceptible to another viral inhibitor. Namely, MCMV proteins involved in downregulation of NKG2D ligands are not restricted to m145 family; m138 gene product also downmodulates MULT-1, as well as H60 (Lenac et al., 2006). The m138 was originally described as a viral protein that binds the Fc portion of immunoglobulin G and was designated as fcr-1 (Thale et al., 1994). Since the deletion of this gene results in viral attenuation in vivo, it was assumed that the attenuation is caused by enhanced susceptibility of the virus to antibody dependant cellular cytotoxicity. However, m138 deletion mutant virus was also attenuated in B cell-deficient mice, suggesting additional function for this protein (Crnkovic-Mertens et al., 1998). A subsequent study demonstrated that m138/fcr-1 targets MULT-1 and H60 for internalization and thus contributes to viral resistance to NK cell-mediated attack in vivo (Lenac et al., 2006). Likely, additional virus-encoded proteins or miRNAs that inhibit expression of human and mouse NKG2D ligands will be discovered in the future.
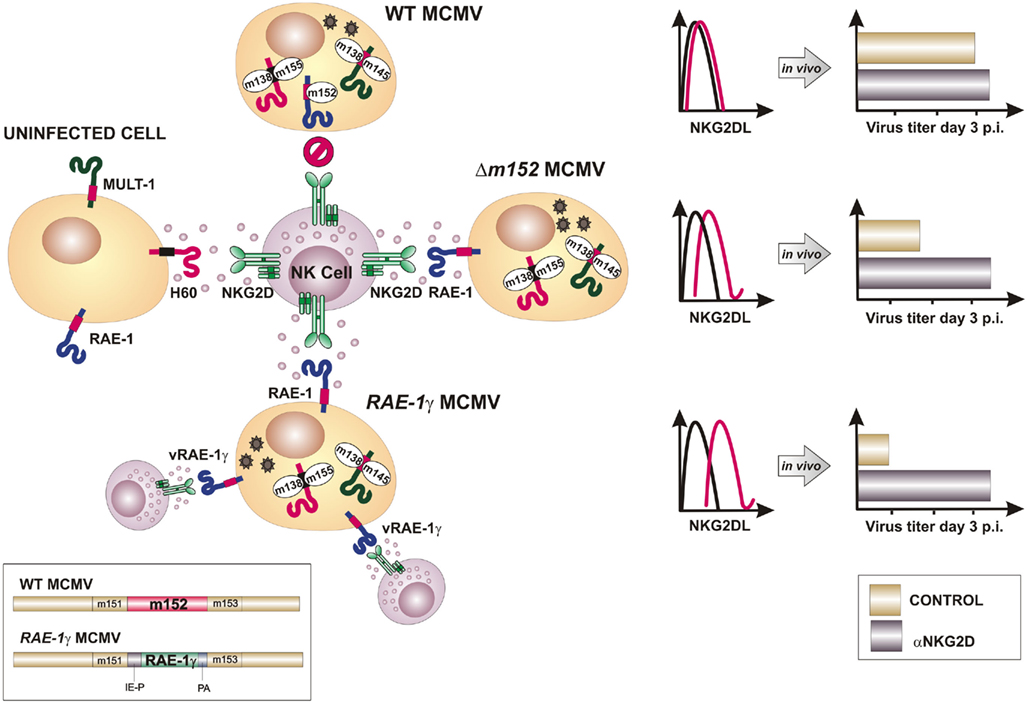
Figure 3. Insertion of NKG2D ligand RAE-1γ into the MCMV genome overcomes viral immunoevasins of NK cells. To avoid lysis of infected cell by the NK cells, MCMV encodes four immunoevasins (m138, m145, m152, and m155) that downmodulate cell surface expression of mouse NKG2D ligands. Consequently, blocking of NKG2D has little or no effect on virus growth in vivo (upper part). Deletion of either of NKG2D immunoevasins from the MCMV genome (e.g., m152) results in the reconstitution of the surface expression of its respective target and attenuated phenotype in vivo that can be restored by NKG2D blocking (in the middle). Recombinant virus RAE-1γMCMV was engineered by replacing m152 gene, whose product inhibits RAE-1 expression, with a cassette expressing RAE-1γ under HCMV immediate early 1 promoter (IE-P). Infection with RAE-1γMCMV results in high surface expression of cellular and virally encoded RAE-1γ (vRAE-1γ) and profoundly attenuated phenotype in vivo (bottom part). PA, polyadenylation sequence.
Despite so many immunoevasion strategies targeting NKG2D, immunocompetent hosts succeed in the containment of CMV infection. This raises the question of why would the virus evolve such a robust immunosubversive potential if the host is able to cope with primary infection. One of the possible explanations is that NKG2D inhibitors, by facilitating virus escape from immune surveillance, may ensure virus dissemination to various organs, including the salivary glands, and aid in the horizontal virus spread. Furthermore, by increasing the titer of the replicating virus, the load of the latent viral genome could be increased, thus representing an advantage for the virus to reactivate from latency and spread.
The Role of NKG2D in Shaping Virus Specific CD8 T Cell Response
In striking contrast to the well-defined role of NK cells in control of early MCMV infection, very little is known regarding the contribution of these innate effector cells in the regulation of a specific immune response and long-term immunosurveillance of viral infection. We previously investigated the contribution of specific lymphocyte subsets in the control of MCMV latency (Polic et al., 1998). Recurrent infection was measured upon ablation of CD4+ T cells, CD8+ T cells, and NK cells individually and in combination. These studies revealed that control of CMV latency and prevention of recurrent infection is organized in a redundant and hierarchical fashion. Importantly, recurrence did not occur, or occurred only rarely upon depletion of any individual subset. However, depletion of NK cells led to a low (0–25%) or high frequency (80–87%) of recurrence when performed in conjunction with depletion of CD4+ or CD8+ T cells, respectively. This study provides strong evidence demonstrating that innate immune responses mediated by NK cells also operate in control of latent MCMV infection, but the ultimate mechanism(s) involved remained unsettled. Related to this, the role of viral evasion of NK cells with regard to their role in control of chronic/latent infection remains largely undefined. In addition, since some NK cell receptors are also expressed on cells of specific immune responses, the outcome of viral regulation of these receptors may also have a direct role in shaping the quality of virus specific immune response. For instance, NKG2D acts as a co-stimulatory receptor on CD8 T cells (Groh et al., 2001; Maasho et al., 2005; Markiewicz et al., 2005), which are primary effector cells in the resolution of a productive MCMV infection and establishment of latency (Krmpotic et al., 2003). It is therefore feasible that viral inhibitors of NKG2D ligands may have a direct impact on antiviral T cell priming and effector functions.
To modify MCMV in a way that it overrides inhibitory signals delivered to NK cells by self MHC-I molecules and potentiated by viral immunoevasins, we have introduced an NKG2D ligand to the MCMV genome (Slavuljica et al., 2010). The ligand employed was a member of the RAE-1 family, RAE-1γ, which was inserted in place of its viral inhibitor m152 (Lodoen et al., 2003; Krmpotic et al., 2005; Figure 3). The insertion of RAE-1γ into the MCMV genome resulted in a recombinant virus with profoundly attenuated phenotype in vivo, even in immunologically immature newborn mice and transgenic mice lacking type I interferon receptor, as well as in mice immunologically suppressed by total body γ-irradiation. Despite an attenuated phenotype, the MCMV expressing the NKG2D ligand efficiently primed and maintained an adaptive immune response, including the CD8 T cell response that was comparable to the one induced by the wild-type MCMV (Figure 4). In addition, the adoptive transfer of CD8+ T cells from mice vaccinated with MCMV expressing RAE-1γ to immunodepleted and infected host mice resulted in the protection against CMV disease. Intriguingly, in a challenge infection, mice immunized with MCMV expressing RAE-1γ resisted challenge with virulent salivary gland-derived MCMV, with improved survival compared with mice initially infected with wild-type MCMV, suggesting that expression of RAE-1γ in the context of the MCMV infection provides an immune stimulus that results in enhanced effectiveness of the adaptive immune response.
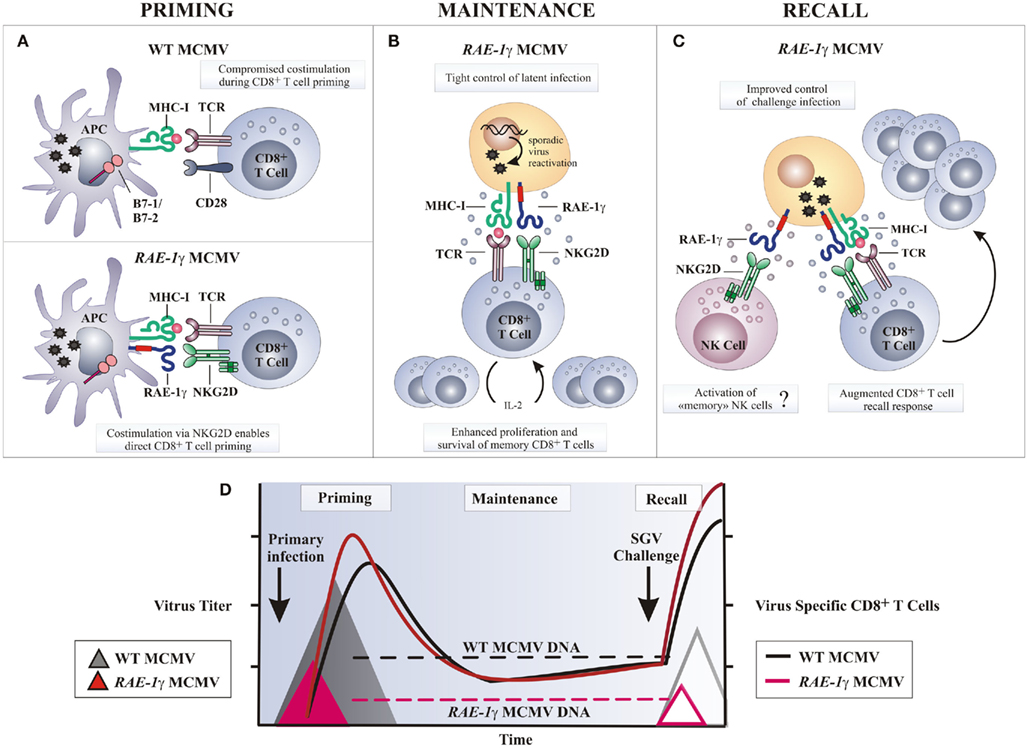
Figure 4. Potential mechanisms of amplified CD8 T cell response following RAE-1γMCMV infection. (A) Naive CD8 T cells require engagement of the TCR by MHC-I molecules presenting antigen along with co-stimulatory signals, such as CD28 ligation by molecules of B7 family, to be fully activated. Wild-type MCMV (WT MCMV) dowmodulates B7.1 and B7.2 from the surface of infected APCs resulting in compromised T cell priming. Following RAE-1γMCMV infection co-stimulation via RAE-1γ–NKG2D interaction may replace B7-CD28 interaction and allow optimal CD8 T cell priming. (B) Stochastic MCMV reactivation during latency enables memory CD8 T cells co-stimulation via NKG2D–RAE-1γ interaction. Consequently, memory CD8 T cells are maintained or even inflated over time despite significantly reduced viral DNA load. (C) Challenge infection or any other serious stress leads to reactivation of latent RAE-1γMCMV and enhanced virus specific T cell response as a consequence of NKG2D ligand expression on cells undergoing virus reactivation. (D) Despite profoundly attenuated replication in vivo and reduced latent viral DNA load as compared to the WT MCMV, RAE-1γMCMV efficiently primes and maintains virus specific CD8 T cell response. In a challenge infection, mice immunized with the RAE-1γMCMV (red line triangle) resist salivary gland virus (SGV) challenge better as compared to mice initially infected with the WT MCMV (gray line triangle). APC, antigen presenting cell.
What could be the mechanism behind a more robust CD8 T cell response after the infection with a recombinant MCMV expressing the NKG2D ligand RAE-1γ instead of its viral inhibitor m152? DCs can prime the CD8 T cell response in two ways; by direct presentation of endogenous peptides, or alternatively, exogenously acquired antigens may be presented, this function known as cross-presentation (Bevan, 2006). Although MCMV can infect DCs (Andrews et al., 2001), thus providing viral peptides for direct presentation, the majority of MCMV-specific CD8 T cells are not directly primed (Snyder et al., 2010). It is a current belief that viral immune evasion genes, which inhibit antigen presentation and modulate the maturation and function of infected DCs, profoundly impair direct priming of CD8 T cells (Andrews et al., 2001; Basta et al., 2002; Loewendorf et al., 2004; Munks et al., 2007; Benedict et al., 2008). MCMV encodes three immunoevasion proteins m04, m06, and m152 that interfere with MHC-I expression and severely impair CD8 T cells recognition of infected cells (Pinto and Hill, 2005; Lemmermann et al., 2011). Since m152 gene is absent from the genome of MCMV expressing RAE-1γ, the density of antigen-MHC-I complexes on the surface of infected DCs may be higher and therefore strengthen antigenic signals delivered to T cells. In T cells, co-stimulation is largely provided by the interaction of CD28 and CD40L on the CD8 T cells with B7 molecules and CD40 on the DCs, respectively. MCMV targets the co-stimulation signals by altering the ratio of positive and negative co-signaling molecules on the DC surface. The MCMV encoded m138, m147.5, and m155 downmodulate expression of B7.1 (Loewendorf et al., 2004), B7.2 (Mintern et al., 2006), and CD40 (Loewendorf et al., 2011), respectively, and therefore compromise T cell priming (Figure 4). Moreover, MCMV selectively upregulates and retains inhibitory ligand PD-L1 on the surface of infected DCs to ensure the induction of T cell anergy, tolerance, or deletion (Benedict et al., 2008). Since co-stimulation of CD8 T cells by the NKG2D is in many ways similar to co-stimulation by the CD28 molecule and leads to increased proliferation, cytotoxicity, and cytokine response (Markiewicz et al., 2005), MCMV expressing RAE-1γ may tip the signaling balance of infected DCs toward positive ones and thus promote T cell priming. It might be possible that the engagement of NKG2D on CD8 T cells by RAE-1γ on infected DCs, and perhaps on other antigen presenting cells, provides a potent co-stimulatory signal during T cell priming able to override viral immunoevasins and allow direct CD8 T cell priming to take place (Figure 4).
Cytokines, such as IL-2, IL-12, and IFN-α/β, stimulate CD8 T expansion and differentiation and are required for optimal T cell responses. Early in the course of an MCMV infection, IFN-α/β is mainly produced by the activated plasmacytoid DCs (pDCs; Dalod et al., 2002, 2003) and is critical for host survival (Presti et al., 1998; Strobl et al., 2005). It mediates a number of immunoregulatory functions that vary depending upon the amount produced, with high IFN-α/β levels promoting immune suppression (Biron, 2001; Theofilopoulos et al., 2005). MCMV infection of BALB/c mice induces a very high systemic production of IFN-α/β resulting in the ablation of CD8α DCs and delayed CD8 T cell responses (Andrews et al., 2003; Robbins et al., 2007). In contrast, an efficient virus control by NK cells after infection with MCMV expressing RAE-1γ decreases the intensity and the duration of pDC activation and limits the production of IFN-α/β (Slavuljica et al., 2010). In this manner NK cells prevent cytokine-mediated loss of splenic CD8α DCs and potentially promote T cell priming.
RAE-1γ–NKG2D co-signals may also positively regulate the CD8 T cell response at later times after priming. The expansion of the antiviral CD8 T cells in a latently infected host, also known as “memory inflation,” is a feature of the immune response to MCMV (Holtappels et al., 2002; Karrer et al., 2003). Based on the detection of viral transcripts in tissue during latency, it has been proposed that it is driven by the stochastic episodes of reactivation of a limited number of viral genes (Reddehase et al., 2008). In support of this, inflationary CD8 T cells display an effector memory phenotype suggestive of repeated antigen stimulation (Sierro et al., 2005; Munks et al., 2006). The accelerated clearance of productive infection with MCMV expressing RAE-1γ leading to a reduced latent viral genome load may also reduce latency-associated viral transcription and compromise “memory inflation” (Reddehase et al., 1994). However, the number of antiviral CD8 T cells increases, rather than fades over time, suggesting that the NKG2D co-stimulation may augment TCR signal during latency and substitute for a reduced latent viral genome load (Figure 4). Recently, it has been recognized that NK cells can be long-lived and mount an antigen-specific recall response (O’Leary et al., 2006; Cooper et al., 2009; Sun et al., 2009; Paust et al., 2010). Whether or not infection with MCMV expressing RAE-1γ induces memory NK cells to mount responses during a subsequent encounter with MCMV remains merely a speculation.
There is an urgent need to develop new therapeutic and preventive antiviral regimens, because at present no effective vaccine for CMV is available. In this regard, the mouse model of infection is currently the best system to experimentally define critical parameters affecting recurrent herpes virus infections. Both HCMV and MCMV employed numerous strategies to downregulate the expression of NKG2D ligands, pointing to the importance of NKG2D signaling in virus surveillance. The MCMV engineered to express an NKG2D ligand shows an outstanding safety profile, due to improved innate immune clearance following inoculation, but also because of being equally or even more immunogenic than the wild-type MCMV, leading to a more efficient adaptive immune response. Therefore, it is possible that an approach like the one described here might be used to generate an attenuated vaccine or a CMV-based vaccine vector.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Drs. Antonella Viola, Tamara Jonjic, and Luka Traven for critical reading of the manuscript. Stipan Jonjic is supported by NIH grant 1R01AI083201-01 and The Croatian Science Foundation grant 04/16.
References
Andrews, D. M., Andoniou, C. E., Granucci, F., Ricciardi-Castagnoli, P., and Degli-Esposti, M. A. (2001). Infection of dendritic cells by murine cytomegalovirus induces functional paralysis. Nat. Immunol. 2, 1077–1084.
Andrews, D. M., Scalzo, A. A., Yokoyama, W. M., Smyth, M. J., and Degli-Esposti, M. A. (2003). Functional interactions between dendritic cells and NK cells during viral infection. Nat. Immunol. 4, 175–181.
Arapovic, J., Lenac, T., Antulov, R., Polic, B., Ruzsics, Z., Carayannopoulos, L. N., Koszinowski, U. H., Krmpotic, A., and Jonjic, S. (2009). Differential susceptibility of RAE-1 isoforms to mouse cytomegalovirus. J. Virol. 83, 8198–8207.
Ashiru, O., Bennett, N. J., Boyle, L. H., Thomas, M., Trowsdale, J., and Wills, M. R. (2009). NKG2D ligand MICA is retained in the cis-Golgi apparatus by human cytomegalovirus protein UL142. J. Virol. 83, 12345–12354.
Bacon, L., Eagle, R. A., Meyer, M., Easom, N., Young, N. T., and Trowsdale, J. (2004). Two human ULBP/RAET1 molecules with transmembrane regions are ligands for NKG2D. J. Immunol. 173, 1078–1084.
Bartkova, J., Horejsi, Z., Koed, K., Kramer, A., Tort, F., Zieger, K., Guldberg, P., Sehested, M., Nesland, J. M., Lukas, C., Orntoft, T., Lukas, J., and Bartek, J. (2005). DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 434, 864–870.
Basta, S., Chen, W., Bennink, J. R., and Yewdell, J. W. (2002). Inhibitory effects of cytomegalovirus proteins US2 and US11 point to contributions from direct priming and cross-priming in induction of vaccinia virus-specific CD8(+) T cells. J. Immunol. 168, 5403–5408.
Bauer, S., Groh, V., Wu, J., Steinle, A., Phillips, J. H., Lanier, L. L., and Spies, T. (1999). Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 285, 727–729.
Benedict, C. A., Loewendorf, A., Garcia, Z., Blazar, B. R., and Janssen, E. M. (2008). Dendritic cell programming by cytomegalovirus stunts naive T cell responses via the PD-L1/PD-1 pathway. J. Immunol. 180, 4836–4847.
Biron, C. A. (2001). Interferons alpha and beta as immune regulators – a new look. Immunity 14, 661–664.
Carayannopoulos, L. N., Naidenko, O. V., Fremont, D. H., and Yokoyama, W. M. (2002). Cutting edge: murine UL16-binding protein-like transcript 1: a newly described transcript encoding a high-affinity ligand for murine NKG2D. J. Immunol. 169, 4079–4083.
Cerwenka, A., Bakker, A. B., Mcclanahan, T., Wagner, J., Wu, J., Phillips, J. H., and Lanier, L. L. (2000). Retinoic acid early inducible genes define a ligand family for the activating NKG2D receptor in mice. Immunity 12, 721–727.
Cerwenka, A., Baron, J. L., and Lanier, L. L. (2001). Ectopic expression of retinoic acid early inducible-1 gene (RAE-1) permits natural killer cell-mediated rejection of a MHC class I-bearing tumor in vivo. Proc. Natl. Acad. Sci. U.S.A. 98, 11521–11526.
Chalupny, N. J., Rein-Weston, A., Dosch, S., and Cosman, D. (2006). Down-regulation of the NKG2D ligand MICA by the human cytomegalovirus glycoprotein UL142. Biochem. Biophys. Res. Commun. 346, 175–181.
Cooper, M. A., Elliott, J. M., Keyel, P. A., Yang, L., Carrero, J. A., and Yokoyama, W. M. (2009). Cytokine-induced memory-like natural killer cells. Proc. Natl. Acad. Sci. U.S.A. 106, 1915–1919.
Cosman, D., Mullberg, J., Sutherland, C. L., Chin, W., Armitage, R., Fanslow, W., Kubin, M., and Chalupny, N. J. (2001). ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity 14, 123–133.
Crnkovic-Mertens, I., Messerle, M., Milotic, I., Szepan, U., Kucic, N., Krmpotic, A., Jonjic, S., and Koszinowski, U. H. (1998). Virus attenuation after deletion of the cytomegalovirus Fc receptor gene is not due to antibody control. J. Virol. 72, 1377–1382.
Dalod, M., Hamilton, T., Salomon, R., Salazar-Mather, T. P., Henry, S. C., Hamilton, J. D., and Biron, C. A. (2003). Dendritic cell responses to early murine cytomegalovirus infection: subset functional specialization and differential regulation by interferon alpha/beta. J. Exp. Med. 197, 885–898.
Dalod, M., Salazar-Mather, T. P., Malmgaard, L., Lewis, C., Asselin-Paturel, C., Briere, F., Trinchieri, G., and Biron, C. A. (2002). Interferon alpha/beta and interleukin 12 responses to viral infections: pathways regulating dendritic cell cytokine expression in vivo. J. Exp. Med. 195, 517–528.
Diefenbach, A., Hsia, J. K., Hsiung, M. Y., and Raulet, D. H. (2003). A novel ligand for the NKG2D receptor activates NK cells and macrophages and induces tumor immunity. Eur. J. Immunol. 33, 381–391.
Diefenbach, A., Jamieson, A. M., Liu, S. D., Shastri, N., and Raulet, D. H. (2000). Ligands for the murine NKG2D receptor: expression by tumor cells and activation of NK cells and macrophages. Nat. Immunol. 1, 119–126.
Diefenbach, A., Jensen, E. R., Jamieson, A. M., and Raulet, D. H. (2001). Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature 413, 165–171.
Diefenbach, A., and Raulet, D. H. (2001). Strategies for target cell recognition by natural killer cells. Immunol. Rev. 181, 170–184.
Diefenbach, A., Tomasello, E., Lucas, M., Jamieson, A. M., Hsia, J. K., Vivier, E., and Raulet, D. H. (2002). Selective associations with signaling proteins determine stimulatory versus costimulatory activity of NKG2D. Nat. Immunol. 3, 1142–1149.
Draghi, M., Pashine, A., Sanjanwala, B., Gendzekhadze, K., Cantoni, C., Cosman, D., Moretta, A., Valiante, N. M., and Parham, P. (2007). NKp46 and NKG2D recognition of infected dendritic cells is necessary for NK cell activation in the human response to influenza infection. J. Immunol. 178, 2688–2698.
Dunn, C., Chalupny, N. J., Sutherland, C. L., Dosch, S., Sivakumar, P. V., Johnson, D. C., and Cosman, D. (2003). Human cytomegalovirus glycoprotein UL16 causes intracellular sequestration of NKG2D ligands, protecting against natural killer cell cytotoxicity. J. Exp. Med. 197, 1427–1439.
Eagle, R. A., Traherne, J. A., Ashiru, O., Wills, M. R., and Trowsdale, J. (2006). Regulation of NKG2D ligand gene expression. Hum. Immunol. 67, 159–169.
Eagle, R. A., Traherne, J. A., Hair, J. R., Jafferji, I., and Trowsdale, J. (2009). ULBP6/RAET1L is an additional human NKG2D ligand. Eur. J. Immunol. 39, 3207–3216.
Ebihara, T., Masuda, H., Akazawa, T., Shingai, M., Kikuta, H., Ariga, T., Matsumoto, M., and Seya, T. (2007). Induction of NKG2D ligands on human dendritic cells by TLR ligand stimulation and RNA virus infection. Int. Immunol. 19, 1145–1155.
Ehrlich, L. I., Ogasawara, K., Hamerman, J. A., Takaki, R., Zingoni, A., Allison, J. P., and Lanier, L. L. (2005). Engagement of NKG2D by cognate ligand or antibody alone is insufficient to mediate costimulation of human and mouse CD8+ Tcells. J. Immunol. 174, 1922–1931.
Eisele, G., Wischhusen, J., Mittelbronn, M., Meyermann, R., Waldhauer, I., Steinle, A., Weller, M., and Friese, M. A. (2006). TGF-beta and metalloproteinases differentially suppress NKG2D ligand surface expression on malignant glioma cells. Brain 129, 2416–2425.
Eleme, K., Taner, S. B., Onfelt, B., Collinson, L. M., Mccann, F. E., Chalupny, N. J., Cosman, D., Hopkins, C., Magee, A. I., and Davis, D. M. (2004). Cell surface organization of stress-inducible proteins ULBP and MICA that stimulate human NK cells and T cells via NKG2D. J. Exp. Med. 199, 1005–1010.
Fang, M., Lanier, L. L., and Sigal, L. J. (2008). A role for NKG2D in NK cell-mediated resistance to poxvirus disease. PLoS Pathog. 4, e30. doi:10.1371/journal.ppat.0040030
Gasser, S., Orsulic, S., Brown, E. J., and Raulet, D. H. (2005). The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 436, 1186–1190.
Gilfillan, S., Ho, E. L., Cella, M., Yokoyama, W. M., and Colonna, M. (2002). NKG2D recruits two distinct adapters to trigger NK cell activation and costimulation. Nat. Immunol. 3, 1150–1155.
Girardi, M., Oppenheim, D. E., Steele, C. R., Lewis, J. M., Glusac, E., Filler, R., Hobby, P., Sutton, B., Tigelaar, R. E., and Hayday, A. C. (2001). Regulation of cutaneous malignancy by gammadelta T cells. Science 294, 605–609.
Gorgoulis, V. G., Vassiliou, L. V., Karakaidos, P., Zacharatos, P., Kotsinas, A., Liloglou, T., Venere, M., Ditullio, R. A. Jr., Kastrinakis, N. G., Levy, B., Kletsas, D., Yoneta, A., Herlyn, M., Kittas, C., and Halazonetis, T. D. (2005). Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 434, 907–913.
Groh, V., Bahram, S., Bauer, S., Herman, A., Beauchamp, M., and Spies, T. (1996). Cell stress-regulated human major histocompatibility complex class I gene expressed in gastrointestinal epithelium. Proc. Natl. Acad. Sci. U.S.A. 93, 12445–12450.
Groh, V., Rhinehart, R., Randolph-Habecker, J., Topp, M. S., Riddell, S. R., and Spies, T. (2001). Costimulation of CD8alphabeta T cells by NKG2D via engagement by MIC induced on virus-infected cells. Nat. Immunol. 2, 255–260.
Hamerman, J. A., Ogasawara, K., and Lanier, L. L. (2004). Cutting edge: toll-like receptor signaling in macrophages induces ligands for the NKG2D receptor. J. Immunol. 172, 2001–2005.
Hasan, M., Krmpotic, A., Ruzsics, Z., Bubic, I., Lenac, T., Halenius, A., Loewendorf, A., Messerle, M., Hengel, H., Jonjic, S., and Koszinowski, U. H. (2005). Selective down-regulation of the NKG2D ligand H60 by mouse cytomegalovirus m155 glycoprotein. J. Virol. 79, 2920–2930.
Holtappels, R., Thomas, D., Podlech, J., and Reddehase, M. J. (2002). Two antigenic peptides from genes m123 and m164 of murine cytomegalovirus quantitatively dominate CD8 T-cell memory in the H-2d haplotype. J. Virol. 76, 151–164.
Houchins, J. P., Yabe, T., Mcsherry, C., and Bach, F. H. (1991). DNA sequence analysis of NKG2, a family of related cDNA clones encoding type II integral membrane proteins on human natural killer cells. J. Exp. Med. 173, 1017–1020.
Jamieson, A. M., Diefenbach, A., Mcmahon, C. W., Xiong, N., Carlyle, J. R., and Raulet, D. H. (2002). The role of the NKG2D immunoreceptor in immune cell activation and natural killing. Immunity 17, 19–29.
Jinushi, M., Takehara, T., Kanto, T., Tatsumi, T., Groh, V., Spies, T., Miyagi, T., Suzuki, T., Sasaki, Y., and Hayashi, N. (2003). Critical role of MHC class I-related chain A and B expression on IFN-alpha-stimulated dendritic cells in NK cell activation: impairment in chronic hepatitis C virus infection. J. Immunol. 170, 1249–1256.
Karrer, U., Sierro, S., Wagner, M., Oxenius, A., Hengel, H., Koszinowski, U. H., Phillips, R. E., and Klenerman, P. (2003). Memory inflation: continuous accumulation of antiviral CD8+ Tcells over time. J. Immunol. 170, 2022–2029.
Kloss, M., Decker, P., Baltz, K. M., Baessler, T., Jung, G., Rammensee, H. G., Steinle, A., Krusch, M., and Salih, H. R. (2008). Interaction of monocytes with NK cells upon Toll-like receptor-induced expression of the NKG2D ligand MICA. J. Immunol. 181, 6711–6719.
Krmpotic, A., Bubic, I., Polic, B., Lucin, P., and Jonjic, S. (2003). Pathogenesis of murine cytomegalovirus infection. Microbes Infect. 5, 1263–1277.
Krmpotic, A., Busch, D. H., Bubic, I., Gebhardt, F., Hengel, H., Hasan, M., Scalzo, A. A., Koszinowski, U. H., and Jonjic, S. (2002). MCMV glycoprotein gp40 confers virus resistance to CD8+ Tcells and NK cells in vivo. Nat. Immunol. 3, 529–535.
Krmpotic, A., Hasan, M., Loewendorf, A., Saulig, T., Halenius, A., Lenac, T., Polic, B., Bubic, I., Kriegeskorte, A., Pernjak-Pugel, E., Messerle, M., Hengel, H., Busch, D. H., Koszinowski, U. H., and Jonjic, S. (2005). NK cell activation through the NKG2D ligand MULT-1 is selectively prevented by the glycoprotein encoded by mouse cytomegalovirus gene m145. J. Exp. Med. 201, 211–220.
Krmpotic, A., Messerle, M., Crnkovic-Mertens, I., Polic, B., Jonjic, S., and Koszinowski, U. H. (1999). The immunoevasive function encoded by the mouse cytomegalovirus gene m152 protects the virus against T cell control in vivo. J. Exp. Med. 190, 1285–1296.
Lemmermann, N. A., Bohm, V., Holtappels, R., and Reddehase, M. J. (2011). In vivo impact of cytomegalovirus evasion of CD8 T-cell immunity: facts and thoughts based on murine models. Virus Res. 157, 161–174.
Lenac, T., Arapovic, J., Traven, L., Krmpotic, A., and Jonjic, S. (2008). Murine cytomegalovirus regulation of NKG2D ligands. Med. Microbiol. Immunol. 197, 159–166.
Lenac, T., Budt, M., Arapovic, J., Hasan, M., Zimmermann, A., Simic, H., Krmpotic, A., Messerle, M., Ruzsics, Z., Koszinowski, U. H., Hengel, H., and Jonjic, S. (2006). The herpesviral Fc receptor fcr-1 down-regulates the NKG2D ligands MULT-1 and H60. J. Exp. Med. 203, 1843–1850.
Lodoen, M., Ogasawara, K., Hamerman, J. A., Arase, H., Houchins, J. P., Mocarski, E. S., and Lanier, L. L. (2003). NKG2D-mediated natural killer cell protection against cytomegalovirus is impaired by viral gp40 modulation of retinoic acid early inducible 1 gene molecules. J. Exp. Med. 197, 1245–1253.
Lodoen, M. B., Abenes, G., Umamoto, S., Houchins, J. P., Liu, F., and Lanier, L. L. (2004). The cytomegalovirus m155 gene product subverts natural killer cell antiviral protection by disruption of H60-NKG2D interactions. J. Exp. Med. 200, 1075–1081.
Loewendorf, A., Kruger, C., Borst, E. M., Wagner, M., Just, U., and Messerle, M. (2004). Identification of a mouse cytomegalovirus gene selectively targeting CD86 expression on antigen-presenting cells. J. Virol. 78, 13062–13071.
Loewendorf, A. I., Steinbrueck, L., Peter, C., Busche, A., Benedict, C. A., and Kay-Jackson, P. C. (2011). The mouse cytomegalovirus glycoprotein m155 inhibits CD40 expression and restricts CD4 T cell responses. J. Virol. 85, 5208–5212.
Maasho, K., Opoku-Anane, J., Marusina, A. I., Coligan, J. E., and Borrego, F. (2005). NKG2D is a costimulatory receptor for human naive CD8+ Tcells. J. Immunol. 174, 4480–4484.
Malarkannan, S., Shih, P. P., Eden, P. A., Horng, T., Zuberi, A. R., Christianson, G., Roopenian, D., and Shastri, N. (1998). The molecular and functional characterization of a dominant minor H antigen, H60. J. Immunol. 161, 3501–3509.
Markiewicz, M. A., Carayannopoulos, L. N., Naidenko, O. V., Matsui, K., Burack, W. R., Wise, E. L., Fremont, D. H., Allen, P. M., Yokoyama, W. M., Colonna, M., and Shaw, A. S. (2005). Costimulation through NKG2D enhances murine CD8+ CTL function: similarities and differences between NKG2D and CD28 costimulation. J. Immunol. 175, 2825–2833.
Meresse, B., Chen, Z., Ciszewski, C., Tretiakova, M., Bhagat, G., Krausz, T. N., Raulet, D. H., Lanier, L. L., Groh, V., Spies, T., Ebert, E. C., Green, P. H., and Jabri, B. (2004). Coordinated induction by IL15 of a TCR-independent NKG2D signaling pathway converts CTL into lymphokine-activated killer cells in celiac disease. Immunity 21, 357–366.
Mintern, J. D., Klemm, E. J., Wagner, M., Paquet, M. E., Napier, M. D., Kim, Y. M., Koszinowski, U. H., and Ploegh, H. L. (2006). Viral interference with B7-1 costimulation: a new role for murine cytomegalovirus fc receptor-1. J. Immunol. 177, 8422–8431.
Muller, S., Zocher, G., Steinle, A., and Stehle, T. (2010). Structure of the HCMV UL16-MICB complex elucidates select binding of a viral immunoevasin to diverse NKG2D ligands. PLoS Pathog. 6, e1000723. doi:10.1371/journal.ppat.1000723
Munks, M. W., Cho, K. S., Pinto, A. K., Sierro, S., Klenerman, P., and Hill, A. B. (2006). Four distinct patterns of memory CD8 T cell responses to chronic murine cytomegalovirus infection. J. Immunol. 177, 450–458.
Munks, M. W., Pinto, A. K., Doom, C. M., and Hill, A. B. (2007). Viral interference with antigen presentation does not alter acute or chronic CD8 T cell immunodominance in murine cytomegalovirus infection. J. Immunol. 178, 7235–7241.
Nedvetzki, S., Sowinski, S., Eagle, R. A., Harris, J., Vely, F., Pende, D., Trowsdale, J., Vivier, E., Gordon, S., and Davis, D. M. (2007). Reciprocal regulation of human natural killer cells and macrophages associated with distinct immune synapses. Blood 109, 3776–3785.
Nice, T. J., Coscoy, L., and Raulet, D. H. (2009). Posttranslational regulation of the NKG2D ligand Mult1 in response to cell stress. J. Exp. Med. 206, 287–298.
Nomura, M., Takihara, Y., and Shimada, K. (1994). Isolation and characterization of retinoic acid-inducible cDNA clones in F9 cells: one of the early inducible clones encodes a novel protein sharing several highly homologous regions with a Drosophila polyhomeotic protein. Differentiation 57, 39–50.
Nomura, M., Zou, Z., Joh, T., Takihara, Y., Matsuda, Y., and Shimada, K. (1996). Genomic structures and characterization of Rae1 family members encoding GPI-anchored cell surface proteins and expressed predominantly in embryonic mouse brain. J. Biochem. 120, 987–995.
O’Leary, J. G., Goodarzi, M., Drayton, D. L., and Von Andrian, U. H. (2006). T cell- and B cell-independent adaptive immunity mediated by natural killer cells. Nat. Immunol. 7, 507–516.
Paust, S., Gill, H. S., Wang, B. Z., Flynn, M. P., Moseman, E. A., Senman, B., Szczepanik, M., Telenti, A., Askenase, P. W., Compans, R. W., and Von Andrian, U. H. (2010). Critical role for the chemokine receptor CXCR6 in NK cell-mediated antigen-specific memory of haptens and viruses. Nat. Immunol. 11, 1127–1135.
Pinto, A. K., and Hill, A. B. (2005). Viral interference with antigen presentation to CD8+ Tcells: lessons from cytomegalovirus. Viral Immunol. 18, 434–444.
Polic, B., Hengel, H., Krmpotic, A., Trgovcich, J., Pavic, I., Luccaronin, P., Jonjic, S., and Koszinowski, U. H. (1998). Hierarchical and redundant lymphocyte subset control precludes cytomegalovirus replication during latent infection. J. Exp. Med. 188, 1047–1054.
Presti, R. M., Pollock, J. L., Dal Canto, A. J., O’Guin, A. K., and Virgin, H. W. T. (1998). Interferon gamma regulates acute and latent murine cytomegalovirus infection and chronic disease of the great vessels. J. Exp. Med. 188, 577–588.
Radosavljevic, M., Cuillerier, B., Wilson, M. J., Clement, O., Wicker, S., Gilfillan, S., Beck, S., Trowsdale, J., and Bahram, S. (2002). A cluster of ten novel MHC class I related genes on human chromosome 6q24.2-q25.3. Genomics 79, 114–123.
Reddehase, M. J., Balthesen, M., Rapp, M., Jonjic, S., Pavic, I., and Koszinowski, U. H. (1994). The conditions of primary infection define the load of latent viral genome in organs and the risk of recurrent cytomegalovirus disease. J. Exp. Med. 179, 185–193.
Reddehase, M. J., Simon, C. O., Seckert, C. K., Lemmermann, N., and Grzimek, N. K. (2008). Murine model of cytomegalovirus latency and reactivation. Curr. Top. Microbiol. Immunol. 325, 315–331.
Robbins, S. H., Bessou, G., Cornillon, A., Zucchini, N., Rupp, B., Ruzsics, Z., Sacher, T., Tomasello, E., Vivier, E., Koszinowski, U. H., and Dalod, M. (2007). Natural killer cells promote early CD8 T cell responses against cytomegalovirus. PLoS Pathog. 3, e123. doi:10.1371/journal.ppat.0030123
Roberts, A. I., Lee, L., Schwarz, E., Groh, V., Spies, T., Ebert, E. C., and Jabri, B. (2001). NKG2D receptors induced by IL-15 costimulate CD28-negative effector CTL in the tissue microenvironment. J. Immunol. 167, 5527–5530.
Rolle, A., Mousavi-Jazi, M., Eriksson, M., Odeberg, J., Soderberg-Naucler, C., Cosman, D., Karre, K., and Cerboni, C. (2003). Effects of human cytomegalovirus infection on ligands for the activating NKG2D receptor of NK cells: up-regulation of UL16-binding protein (ULBP)1 and ULBP2 is counteracted by the viral UL16 protein. J. Immunol. 171, 902–908.
Romphruk, A. V., Romphruk, A., Naruse, T. K., Raroengjai, S., Puapairoj, C., Inoko, H., and Leelayuwat, C. (2009). Polymorphisms of NKG2D ligands: diverse RAET1/ULBP genes in northeastern Thais. Immunogenetics 61, 611–617.
Rosen, D. B., Araki, M., Hamerman, J. A., Chen, T., Yamamura, T., and Lanier, L. L. (2004). A structural basis for the association of DAP12 with mouse, but not human, NKG2D. J. Immunol. 173, 2470–2478.
Routes, J. M., Ryan, S., Morris, K., Takaki, R., Cerwenka, A., and Lanier, L. L. (2005). Adenovirus serotype 5 E1A sensitizes tumor cells to NKG2D-dependent NK cell lysis and tumor rejection. J. Exp. Med. 202, 1477–1482.
Schwinn, N., Vokhminova, D., Sucker, A., Textor, S., Striegel, S., Moll, I., Nausch, N., Tuettenberg, J., Steinle, A., Cerwenka, A., Schadendorf, D., and Paschen, A. (2009). Interferon-gamma down-regulates NKG2D ligand expression and impairs the NKG2D-mediated cytolysis of MHC class I-deficient melanoma by natural killer cells. Int. J. Cancer 124, 1594–1604.
Sierro, S., Rothkopf, R., and Klenerman, P. (2005). Evolution of diverse antiviral CD8+ Tcell populations after murine cytomegalovirus infection. Eur. J. Immunol. 35, 1113–1123.
Siren, J., Sareneva, T., Pirhonen, J., Strengell, M., Veckman, V., Julkunen, I., and Matikainen, S. (2004). Cytokine and contact-dependent activation of natural killer cells by influenza A or sendai virus-infected macrophages. J. Gen. Virol. 85, 2357–2364.
Slavuljica, I., Busche, A., Babic, M., Mitrovic, M., Gasparovic, I., Cekinovic, D., Markova Car, E., Pernjak Pugel, E., Cikovic, A., Lisnic, V. J., Britt, W. J., Koszinowski, U., Messerle, M., Krmpotic, A., and Jonjic, S. (2010). Recombinant mouse cytomegalovirus expressing a ligand for the NKG2D receptor is attenuated and has improved vaccine properties. J. Clin. Invest. 120, 4532–4545.
Snyder, C. M., Allan, J. E., Bonnett, E. L., Doom, C. M., and Hill, A. B. (2010). Cross-presentation of a spread-defective MCMV is sufficient to prime the majority of virus-specific CD8+ Tcells. PLoS ONE 5, e9681. doi:10.1371/journal.pone.0009681
Steinle, A., Li, P., Morris, D. L., Groh, V., Lanier, L. L., Strong, R. K., and Spies, T. (2001). Interactions of human NKG2D with its ligands MICA, MICB, and homologs of the mouse RAE-1 protein family. Immunogenetics 53, 279–287.
Stern-Ginossar, N., Elefant, N., Zimmermann, A., Wolf, D. G., Saleh, N., Biton, M., Horwitz, E., Prokocimer, Z., Prichard, M., Hahn, G., Goldman-Wohl, D., Greenfield, C., Yagel, S., Hengel, H., Altuvia, Y., Margalit, H., and Mandelboim, O. (2007). Host immune system gene targeting by a viral miRNA. Science 317, 376–381.
Stern-Ginossar, N., Gur, C., Biton, M., Horwitz, E., Elboim, M., Stanietsky, N., Mandelboim, M., and Mandelboim, O. (2008a). Human microRNAs regulate stress-induced immune responses mediated by the receptor NKG2D. Nat. Immunol. 9, 1065–1073.
Stern-Ginossar, N., Gur, C., Biton, M., Horwitz, E., Elboim, M., Stanietsky, N., Mandelboim, M., and Mandelboim, O. (2008b). Human microRNAs regulate stress-induced immune responses mediated by the receptor NKG2D. Nat. Immunol. 9, 1065–1073.
Strobl, B., Bubic, I., Bruns, U., Steinborn, R., Lajko, R., Kolbe, T., Karaghiosoff, M., Kalinke, U., Jonjic, S., and Muller, M. (2005). Novel functions of tyrosine kinase 2 in the antiviral defense against murine cytomegalovirus. J. Immunol. 175, 4000–4008.
Sun, J. C., Beilke, J. N., and Lanier, L. L. (2009). Adaptive immune features of natural killer cells. Nature 457, 557–561.
Takada, A., Yoshida, S., Kajikawa, M., Miyatake, Y., Tomaru, U., Sakai, M., Chiba, H., Maenaka, K., Kohda, D., Fugo, K., and Kasahara, M. (2008). Two novel NKG2D ligands of the mouse H60 family with differential expression patterns and binding affinities to NKG2D. J. Immunol. 180, 1678–1685.
Thale, R., Lucin, P., Schneider, K., Eggers, M., and Koszinowski, U. H. (1994). Identification and expression of a murine cytomegalovirus early gene coding for an Fc receptor. J. Virol. 68, 7757–7765.
Theofilopoulos, A. N., Baccala, R., Beutler, B., and Kono, D. H. (2005). Type I interferons (alpha/beta) in immunity and autoimmunity. Annu. Rev. Immunol. 23, 307–336.
Tieng, V., Le Bouguenec, C., Du Merle, L., Bertheau, P., Desreumaux, P., Janin, A., Charron, D., and Toubert, A. (2002). Binding of Escherichia coli adhesin AfaE to CD55 triggers cell-surface expression of the MHC class I-related molecule MICA. Proc. Natl. Acad. Sci. U.S.A. 99, 2977–2982.
Tokuyama, M., Lorin, C., Delebecque, F., Jung, H., Raulet, D. H., and Coscoy, L. (2011). Expression of the RAE-1 family of stimulatory NK-cell ligands requires activation of the PI3K pathway during viral infection and transformation. PLoS Pathog. 7, e1002265. doi:10.1371/journal.ppat.1002265
Vales-Gomez, M., Browne, H., and Reyburn, H. T. (2003). Expression of the UL16 glycoprotein of human cytomegalovirus protects the virus-infected cell from attack by natural killer cells. BMC Immunol. 4, 4. doi:10.1186/1471-2172-4-4
Vankayalapati, R., Garg, A., Porgador, A., Griffith, D. E., Klucar, P., Safi, H., Girard, W. M., Cosman, D., Spies, T., and Barnes, P. F. (2005). Role of NK cell-activating receptors and their ligands in the lysis of mononuclear phagocytes infected with an intracellular bacterium. J. Immunol. 175, 4611–4617.
Venkataraman, G. M., Suciu, D., Groh, V., Boss, J. M., and Spies, T. (2007). Promoter region architecture and transcriptional regulation of the genes for the MHC class I-related chain A and B ligands of NKG2D. J. Immunol. 178, 961–969.
Verneris, M. R., Karami, M., Baker, J., Jayaswal, A., and Negrin, R. S. (2004). Role of NKG2D signaling in the cytotoxicity of activated and expanded CD8+ Tcells. Blood 103, 3065–3072.
Walsh, K. B., Lanier, L. L., and Lane, T. E. (2008). NKG2D receptor signaling enhances cytolytic activity by virus-specific CD8+ Tcells: evidence for a protective role in virus-induced encephalitis. J. Virol. 82, 3031–3044.
Ward, J., Bonaparte, M., Sacks, J., Guterman, J., Fogli, M., Mavilio, D., and Barker, E. (2007). HIV modulates the expression of ligands important in triggering natural killer cell cytotoxic responses on infected primary T-cell blasts. Blood 110, 1207–1214.
Ward, J., Davis, Z., Dehart, J., Zimmerman, E., Bosque, A., Brunetta, E., Mavilio, D., Planelles, V., and Barker, E. (2009). HIV-1 Vpr triggers natural killer cell-mediated lysis of infected cells through activation of the ATR-mediated DNA damage response. PLoS Pathog. 5, e1000613. doi:10.1371/journal.ppat.1000613
Welte, S. A., Sinzger, C., Lutz, S. Z., Singh-Jasuja, H., Sampaio, K. L., Eknigk, U., Rammensee, H. G., and Steinle, A. (2003). Selective intracellular retention of virally induced NKG2D ligands by the human cytomegalovirus UL16 glycoprotein. Eur. J. Immunol. 33, 194–203.
Whang, M. I., Guerra, N., and Raulet, D. H. (2009). Costimulation of dendritic epidermal gammadelta T cells by a new NKG2D ligand expressed specifically in the skin. J. Immunol. 182, 4557–4564.
Wilkinson, G. W., Tomasec, P., Stanton, R. J., Armstrong, M., Prod’homme, V., Aicheler, R., Mcsharry, B. P., Rickards, C. R., Cochrane, D., Llewellyn-Lacey, S., Wang, E. C., Griffin, C. A., and Davison, A. J. (2008). Modulation of natural killer cells by human cytomegalovirus. J. Clin. Virol. 41, 206–212.
Wu, J., Chalupny, N. J., Manley, T. J., Riddell, S. R., Cosman, D., and Spies, T. (2003). Intracellular retention of the MHC class I-related chain B ligand of NKG2D by the human cytomegalovirus UL16 glycoprotein. J. Immunol. 170, 4196–4200.
Wu, J., Song, Y., Bakker, A. B., Bauer, S., Spies, T., Lanier, L. L., and Phillips, J. H. (1999). An activating immunoreceptor complex formed by NKG2D and DAP10. Science 285, 730–732.
Yadav, D., Ngolab, J., Lim, R. S., Krishnamurthy, S., and Bui, J. D. (2009). Cutting edge: down-regulation of MHC class I-related chain A on tumor cells by IFN-gamma-induced microRNA. J. Immunol. 182, 39–43.
Ziegler, H., Muranyi, W., Burgert, H. G., Kremmer, E., and Koszinowski, U. H. (2000). The luminal part of the murine cytomegalovirus glycoprotein gp40 catalyzes the retention of MHC class I molecules. EMBO J. 19, 870–881.
Keywords: NKG2D, cytomegalovirus, immune evasion
Citation: Slavuljica I, Krmpotić A and Jonjić S (2011) Manipulation of NKG2D ligands by cytomegaloviruses: impact on innate and adaptive immune response. Front. Immun. 2:85. doi: 10.3389/fimmu.2011.00085
Received: 28 October 2011; Paper pending published: 08 November 2011;
Accepted: 12 December 2011; Published online: 28 December 2011.
Edited by:
Linda F. Van Dyk, University of Colorado Denver School of Medicine, USAReviewed by:
Ofer Mandelboim, The Hebrew University of Jerusalem, IsraelKlaus Früh, Oregon Health and Science University, USA
Copyright: © 2011 Slavuljica, Krmpotić and Jonjić. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Stipan Jonjić, Department of Histology and Embryology, Faculty of Medicine, University of Rijeka, B. Branchetta 20, 51 000 Rijeka, Croatia. e-mail:anN0aXBhbkBtZWRyaS5ocg==