 Omer Dushek1,2*
Omer Dushek1,2*- 1 Sir William Dunn School of Pathology, University of Oxford, Oxford, UK
- 2 Centre for Mathematical Biology, University of Oxford, Oxford, UK
The mechanism by which antigen binding to the T cell antigen receptor (TCR) generates intracellular signaling, a process termed TCR triggering, is incompletely understood. A large body of experimental evidence has implicated multiple biophysical/biochemical effects and multiple molecules in the process of TCR triggering, which likely reflect the uniquely demanding role of the TCR in recognizing diverse antigenic ligands. In this perspective, I propose that breaking down the process of TCR triggering into tractable elementary steps may be a useful approach in building mechanistic TCR triggering models. Once these elementary steps are understood, they can be recombined to build a unified model of TCR triggering.
Cell surface receptors initiate and regulate intracellular signaling cascades that lead to a variety of functional cellular outcomes. Many receptor families have been extensively studied in the hope that a refined knowledge of how ligand binding initiates intracellular signaling, a process termed receptor triggering, can be exploited for the design of therapeutics that modulate the activity of specific receptors. The triggering of the T cell antigen receptor (TCR) on the surface of T cells by peptide bound major histocompatibility complexes (pMHC) on the surface of antigen presenting cells (APC) has been extensively studied but remains controversial van der Merwe and Dushek (2011). The triggering of the TCR is thought to be tightly regulated because TCR-induced signaling may lead to T cell activation and the initiation of an adaptive immune response. In this perspective, I propose that a combination of experiments and mathematical modeling that aim to breakdown the process of TCR triggering into elementary steps may provide a mechanistic understanding of TCR triggering.
The TCR is a multi-subunit receptor that contains a ligand binding and signal transducing subunits. The signal transducing subunits contain a total of 20 tyrosine residues, which are distributed on 10 immunoreceptor tyrosine-based activation motifs (ITAMs) Love and Hayes (2010). The TCR does not contain intrinsic catalytic domains and is instead regulated by a membrane-anchored kinase (Lck) and phosphatase (CD45). It is expected that the relative concentration of active Lck and CD45 (i.e., [Lck]/[CD45]) will determine the concentration of phosphorylated TCR (Figure 1A, solid black line). The exact relationship between the kinase-phosphatase ratio and the phosphorylation state of the substrate has been extensively studied in generic mathematical models Salazar and Höfer (2007), Thomson and Gunawardena (2009), Salazar and Höfer (2009), Wang et al. (2010) and it has recently been predicted that membrane-anchored substrates with multiple phosphorylation sites, as is the case for the TCR, may exhibit switch-like or ultrasensitive responses (Figure 1A, dashed gray line; Dushek et al., 2011). In this way, TCR triggering can be viewed as a reversible enzymatic module, whereby ITAMs serve as the substrate for Lck and CD45. In the absence of pMHC, the TCR ITAMs are not phosphorylated (or the total phosphorylation is kept low) and upon pMHC binding, by a process that is presently unclear, the ITAMs become phosphorylated. Note that by defining TCR triggering in this way, discussion of downstream signaling and feedback may be omitted but it is important to note that triggering a single TCR does not necessarily lead to T cell activation.
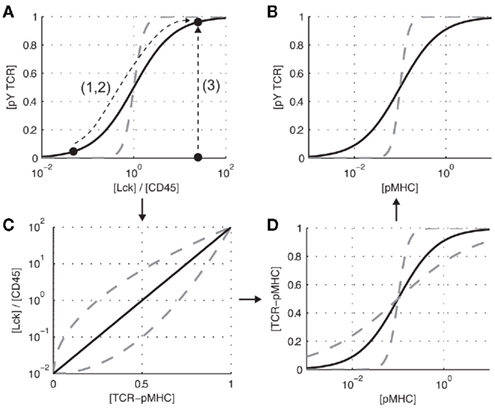
Figure 1. Elementary steps in TCR triggering. (A) The relative concentrations of active kinase to phosphatase ([Lck]/[CD45]) is expected to modulate the concentration of phosphorylated TCR ([pY TCR]). (C) The kinase-phosphatase ratio itself is expected to be related to the concentration of TCR-pMHC complexes ([TCR-pMHC]). (D) The concentration of pMHC ([pMHC]) determines the concentration of TCR-pMHC complexes. (B) These elementary relationship are proposed to underlie TCR triggering (the increase in TCR phosphorylation in response to pMHC binding). Dashed gray lines indicate alternate relationships between the variables. Note that all panels depict schematics and are not the result of experiments or mathematical modeling.
In the language of Figure 1A, TCR triggering is the process by which pMHC binding perturbs this enzymatic module so that the TCR becomes phosphorylated. We can recast the major models of TCR triggering by how they perturb the module:
1. Phosphatase exclusion. The kinetic-segregation model of TCR triggering Davis and van der Merwe (2006) postulates that pMHC binding to the TCR will spatially segregate CD45 because it has a long ectodomain compared to the intermembrane length of the TCR/pMHC complex. The aggregation of TCR as a result of pMHC binding (e.g., as a result of a conformational change in the TCR-CD3 complex Reich et al., 1997; Alam et al., 1999; Gil et al., 2002) is also a proposed model of TCR triggering van der Merwe and Dushek (2011), and such aggregation may sterically exclude CD45. In these models, pMHC binding leads to a decrease in [CD45], and therefore an increase in [Lck]/[CD45], promoting TCR phosphorylation.
2. Kinase enrichment. The lipid raft model of TCR triggering proposes that pMHC binding to the TCR leads to the inclusion of the TCR-pMHC complex into lipid rafts, which are enriched in Lck (Zech et al., 2009; Harder and Sangani, 2009). A subset of aggregation models of TCR triggering predict an enrichment of Lck because Lck is associated with coreceptors which directly interact with pMHC van der Merwe and Dushek (2011). In these models, pMHC binding leads to a local increase in [Lck], and therefore an increase in [Lck]/[CD45], promoting TCR phosphorylation. Nika et al. (2010) have recently demonstrated that the total concentration of active Lck does not change following TCR triggering, suggesting that models based on a change in Lck activity following pMHC binding may be excluded.
3. Substrate availability. A subset of conformational change models of TCR triggering (e.g., Sun et al., 2001; Gil et al., 2002; Kuhns and Davis, 2008; Li et al., 2010; Ma and Finkel, 2010; and further reviewed in van der Merwe and Dushek, 2011) propose that the signaling chains of the TCR are not accessible to Lck or CD45 in the absence of pMHC but that pMHC binding induces a conformational change (e.g., by mechanical force Ma et al., 2008; Ma and Finkel, 2010) that makes the ITAMs accessible to Lck and CD45. Since this model does not rely on perturbations to the [Lck]-[CD45] ratio, it is expected that this ratio is constitutively large so that an increase in ITAM availability leads to their phosphorylation.
These three perturbations leading to TCR triggering are shown in Figure 1A. In reality, it is experimentally challenging to measure the concentration of active Lck and CD45 as a function of time in the vicinity of newly formed TCR-pMHC complexes at the T cell-APC interface. A more tractable experiment is to measure TCR phosphorylation as a function of pMHC concentration (Figure 1B, solid black line). The challenge then becomes relating the x-axis of Figures 1A,B (i.e., relating [pMHC] to [Lck]/[CD45]). It is expected that [Lck]/[CD45] will be related to the concentration of TCR-pMHC complexes (Figure 1C, solid black line), which itself is related to the concentration of pMHC (Figure 1D, solid black line). Thus, [Lck]/[CD45] is related to [pMHC] through at least one intermediate variable, namely [TCR-pMHC].
Experimentally determining the relationships in Figure 1 will be useful in constraining models of TCR triggering. The relationship depicted in Figure 1B can readily be determined by a dose-response assay. Similarly, determining the concentration of TCR-pMHC complexes as a function of the pMHC concentration is experimentally tractable and mathematical models of different binding stoichiometries (e.g., monovalent, bivalent, higher order valencies) will predict different relationships between [pMHC] and [TCR-pMHC] (Figure 1D, gray dashed lines). Perturbing the kinase/phosphatase ratio, using chemical inhibitors to Lck (e.g., PP2) and phosphatase inhibitors (e.g., vanadate), can be used to determine the relationship shown in Figure 1A. We have recently formulated a mathematical model of a kinase and phosphatase regulating multiple phosphorylation sites on membrane-anchored receptors Dushek et al. (2011). We found that multiple sites on the TCR signaling chains can generate switch-like responses making the phosphorylation state of the TCR highly sensitive to the relative concentration of Lck and CD45. Future experiments utilizing ITAM mutants and chemical inhibitors (e.g., PP2 to inhibit Lck) can be used to map out the relationship between ITAM phosphorylation and the relative concentration of active Lck and CD45.
The most difficult relationship to determine experimentally is between [TCR-pMHC] and [Lck]/[CD45] (Figure 1C). However, this relationship can be inferred by knowing the relationships in Figures 1A,D. To formulate mathematical models of the process, refined mechanistic hypotheses that are rooted in detailed biochemistry are needed. For lipid raft models, it would be useful to know the fraction of the cell membrane covered by rafts, the Lck concentration inside and outside rafts, and the effective affinity of the TCR to enter rafts when bound by pMHC. In the case of the kinetic-segregation model, a mathematical model that couples the elasticity of the membrane to the compressional stiffness of the CD45 ectodomain can be used to predict the redistribution of CD45 around newly formed TCR-pMHC complexes. We have recently formulated such a mathematical model (Allard et al., submitted), which is able to predict [CD45] following pMHC binding to the TCR. The model predicts that CD45 segregation is robust to parameter variation and occurs on a supra-diffusive timescale because CD45 is mechanically pushed away from TCR-pMHC complexes.
In formulating the elementary steps of TCR triggering I have focused on essential features for the purpose of clarity but it is important to note that many features not discussed can impact the schematics shown in Figure 1. For example, I have focused on the canonical enzymes regulating TCR phosphorylation, namely Lck and CD45, but additional molecules have been implicated in TCR proximal signaling Acuto et al. (2008) and others yet to be described may also be involved. A triggering mechanism based on a conformational change in TCR-CD3ε that recruits Nck has been proposed Gil et al. (2002). Once the mechanism relating Nck recruitment and ITAM phosphorylation is determined, this triggering mechanism can also be incorporated into the presented model. It will also be important to resolve individual phosphorylation sites on the TCR signaling chains Love and Hayes (2010) instead of simply focusing on total phosphorylation. It is also worth mentioning that insights into TCR triggering, and to T cell activation as a whole, have often come from experiments with soluble pMHC dimers and tetramers. Use of this powerful tool to investigate the elementary steps of TCR triggering may be possible but careful characterization of the TCR-pMHC binding stoichiometry is needed. Collectively, mathematical models can be formulated that incorporate these and other features of TCR proximal signaling.
In summary, I propose that TCR triggering can be viewed as a classic enzymatic module, whereby the kinase Lck and the phosphatase CD45 act on their substrates, the ITAM-bearing TCR signaling chains. Given the complexity of this enzymatic module, it is useful to break down the process of TCR triggering into elementary steps (Figure 1). For each elementary step, detailed hypotheses, that have well-posed biochemical underpinnings, can be mathematically formulated and directly compared to experimental data. In this way, it may be possible to obtain mechanistic models of each elementary step which can then be reintegrated into a unified model of TCR triggering. With detailed biochemical interactions, stoichiometry, and estimated parameters, mathematical models can be used to formulate these hypotheses and systematically reject mechanisms that are inconsistent with experimental data. This approach has been valuable in understanding triggering of heterotrimeric guanine nucleotide-binding protein (G protein)-coupled receptors (GPCRs; Colquhoun, 1998; Kenakin, 2004) and receptor tyrosine kinases (RTKs; Wofsy et al., 1992; Park et al., 2003; Macdonald and Pike, 2008). The TCR belongs to a larger family of cell surface receptors, which have been termed non-catalytic tyrosine-phosphorylated receptors (NTRs; Dushek et al., 2011) and other NTRs (e.g., Dectin-1 Goodridge et al., 2011; NKG2D; and KIR2DL1 Brzostek et al., 2010; Köhler et al., 2010) may share similar triggering mechanisms.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I wish to thank P. Anton van der Merwe, Oreste Acuto, Neil Barclay, Marion Brown, Shaun-Paul Cordoba, and present and former members of the van der Merwe laboratory for helpful discussions.
Abbreviations
APC, antigen presenting cell, ITAM, immunoreceptor tyrosine-based activation motif; TCR, T cell receptor.
References
Acuto, O., Bartolo, V. D., and Michel, F. (2008). Tailoring T-cell receptor signals by proximal negative feedback mechanisms. Nat. Rev. Immunol. 8, 699–712.
Alam, S. M., Davies, G. M., Lin, C. M., Zal, T., Nasholds, W., Jameson, S. C., Hogquist, K. A., Gascoigne, N. R., and Travers, P. J. (1999). Qualitative and quantitative differences in T cell receptor binding of agonist and antagonist ligands. Immunity 10, 227–237.
Brzostek, J., Chai, J.-G., Gebhardt, F., Busch, D. H., Zhao, R., van der Merwe, P. A., and Gould, K. G. (2010). Ligand dimensions are important in controlling NK-cell responses. Eur. J. Immunol. 40, 2050–2059.
Colquhoun, D. (1998). Binding, gating, affinity and efficacy: the interpretation of structure-activity relationships for agonists and of the effects of mutating receptors. Br. J. Pharmacol. 125, 924–947.
Davis, S. J., and van der Merwe, P. A. (2006). The kinetic-segregation model: TCR triggering and beyond. Nat. Immunol. 7, 803–809.
Dushek, O., van der Merwe, P. A., and Shahrezaei, V. (2011). Ultrasensitivity in multisite phosphorylation of membrane-anchored proteins. Biophys. J. 100, 1189–1197.
Gil, D., Schamel, W. W. A., Montoya, M., Sanchez-Madrid, F., and Alarcon, B. (2002). Recruitment of Nck by CD3 epsilon reveals a ligand-induced conformational change essential for T cell receptor signaling and synapse formation. Cell 109, 901–912.
Goodridge, H. S., Reyes, C. N., Becker, C. A., Katsumoto, T. R., Ma, J., Wolf, A. J., Bose, N., Chan, A. S. H., Magee, A. S., Danielson, M. E., Weiss, A., Vasilakos, J. P., and Underhill, D. M. (2011). Activation of the innate immune receptor dectin-1 upon formation of a ‘phagocytic synapse.’ Nature 472, 471–475.
Harder, T., and Sangani, D. (2009). Plasma membrane rafts engaged in T cell signalling: new developments in an old concept. Cell Commun. Signal 7, 21.
Köhler, K., Xiong, S., Brzostek, J., Mehrabi, M., Eissmann, P., Harrison, A., Cordoba, S.-P., Oddos, S., Miloserdov, V., Gould, K., Burroughs, N. J., van der Merwe, P. A., and Davis, D. M. (2010). Matched sizes of activating and inhibitory receptor/ligand pairs are required for optimal signal integration by human natural killer cells. PLoS ONE 5, e15374. doi:10.1371/journal.pone.0015374
Li, Y.-C., Chen, B.-M., Wu, P.-C., Cheng, T.-L., Kao, L.-S., Tao, M.-H., Lieber, A., and Roffler, S. R. (2010). Cutting edge: mechanical forces acting on T cells immobilized via the TCR complex can trigger TCR signaling. J. Immunol. 184, 5959–5963.
Love, P. E., and Hayes, S. M. (2010). ITAM-mediated signaling by the T-cell antigen receptor. Cold Spring Harb. Perspect. Biol. 2, a002485.
Ma, Z., Sharp, K. A., Janmey, P. A., and Finkel, T. H. (2008). Surface-anchored monomeric agonist pMHCs alone trigger TCR with high sensitivity. PLoS Biol. 6, e43. doi:10.1371/journal.pbio.0060043
Macdonald, J. L., and Pike, L. J. (2008). Heterogeneity in EGF-binding affinities arises from negative cooperativity in an aggregating system. Proc. Natl. Acad. Sci. U.S.A. 105, 112–117.
Nika, K., Soldani, C., Salek, M., Paster, W., Gray, A., Etzensperger, R., Fugger, L., Polzella, P., Cerundolo, V., Dushek, O., Höfer, T., Viola, A., and Acuto, O. (2010). Constitutively active Lck kinase in T cells drives antigen receptor signal transduction. Immunity 32, 766–777.
Park, C. S., Schneider, I. C., and Haugh, J. M. (2003). Kinetic analysis of platelet-derived growth factor receptor/phosphoinositide 3-kinase/Akt signaling in fibroblasts. J. Biol. Chem. 278, 37064–37072.
Reich, Z., Boniface, J. J., Lyons, D. S., Borochov, N., Wachtel, E. J., and Davis, M. M. (1997). Ligand-specific oligomerization of T-cell receptor molecules. Nature 387, 617–620.
Salazar, C., and Höfer, T. (2007). Versatile regulation of multisite protein phosphorylation by the order of phosphate processing and protein-protein interactions. FEBS J. 274, 1046–1061.
Salazar, C., and Höfer, T. (2009). Multisite protein phosphorylation–from molecular mechanisms to kinetic models. FEBS J. 276, 3177–3198.
Sun, Z. J., Kim, K. S., Wagner, G., and Reinherz, E. L. (2001). Mechanisms contributing to T cell receptor signaling and assembly revealed by the solution structure of an ectodomain fragment of the CD3 epsilon gamma heterodimer. Cell 105, 913–923.
Thomson, M., and Gunawardena, J. (2009). Unlimited multistability in multisite phosphorylation systems. Nature 460, 274–277.
van der Merwe, P. A., and Dushek, O. (2011). Mechanisms for T cell receptor triggering. Nat. Rev. Immunol. 11, 47–55.
Wang, L., Nie, Q., and Enciso, G. (2010). Nonessential sites improve phosphorylation switch. Biophys. J. 99, L41–L43.
Wofsy, C., Goldstein, B., Lund, K., and Wiley, H. S. (1992). Implications of epidermal growth factor (EGF) induced egf receptor aggregation. Biophys. J. 63, 98–110.
Keywords: T cell receptor, receptor triggering, kinase-phosphatase cycles, mathematical model, T cell activation
Citation: Dushek O (2012) Elementary steps in T cell receptor triggering. Front. Immun. 2:91. doi: 10.3389/fimmu.2011.00091
Received: 18 September 2011;
Accepted: 21 December 2011;
Published online: 10 January 2012.
Edited by:
Oreste Acuto, University of Oxford, UKReviewed by:
Balbino Alarcon, Consejo Superior de Investigaciones Científicas, SpainThomas Hofer, German Cancer Research Center (DKFZ), Germany
Copyright: © 2012 Dushek. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Omer Dushek, Sir William Dunn School of Pathology, University of Oxford, South Parks Road, Oxford OX1 3RE, UK. e-mail:b21lci5kdXNoZWtAcGF0aC5veC5hYy51aw==