 Alfonso Colombatti1,2,3*
Alfonso Colombatti1,2,3* Paola Spessotto1 Roberto Doliana1 Maurizio Mongiat1 Giorgio Maria Bressan4
Paola Spessotto1 Roberto Doliana1 Maurizio Mongiat1 Giorgio Maria Bressan4 Gennaro Esposito2,3
Gennaro Esposito2,3
- 1 Experimental Oncology 2, Centro di Riferimento Oncologico, Istituto di Ricerca e Cura a Carattere Scientifico, Aviano, Italy
- 2 Department of Biomedical Science and Technology, University of Udine, Udine, Italy
- 3 Microgravity, Ageing, Training, Immobility Excellence Center, University of Udine, Udine, Italy
- 4 Department of Histology Microbiology and Medical Biotechnologies, University of Padova, Padova, Italy
Elastin microfibrillar interface proteins (EMILINs) and Multimerins (EMILIN1, EMILIN2, Multimerin1, and Multimerin2) constitute a four member family that in addition to the shared C-terminus gC1q domain typical of the gC1q/TNF superfamily members contain a N-terminus unique cysteine-rich EMI domain. These glycoproteins are homotrimeric and assemble into high molecular weight multimers. They are predominantly expressed in the extracellular matrix and contribute to several cellular functions in part associated with the gC1q domain and in part not yet assigned nor linked to other specific regions of the sequence. Among the latter is the control of arterial blood pressure, the inhibition of Bacillus anthracis cell cytotoxicity, the promotion of cell death, the proangiogenic function, and a role in platelet hemostasis. The focus of this review is to highlight the multiplicity of functions and domains of the EMILIN/Multimerin family with a particular emphasis on the regulatory role played by the ligand–receptor interactions of the gC1q domain. EMILIN1 is the most extensively studied member both from the structural and functional point of view. The structure of the gC1q of EMILIN1 solved by NMR highlights unique characteristics compared to other gC1q domains: it shows a marked decrease of the contact surface of the trimeric assembly and while conserving the jelly-roll topology with two β-sheets of antiparallel strands it presents a nine-stranded β-sandwich fold instead of the usual 10-stranded fold. This is likely due to the insertion of nine residues that disrupt the ordered strand organization and forma a highly dynamic protruding loop. In this loop the residue E933 is the site of interaction between gC1q and the α4β1 and α9β1 integrins, and contrary to integrin occupancy that usually upregulates cell growth, when gC1q is ligated by the integrin the cells reduce their proliferative activity.
Introduction
The elastin microfibrillar interface protein (EMILIN) and Multimerin family of glycoproteins is part of the large superfamily of collagenous and non-collagenous proteins containing the gC1q signature (Shapiro and Scherer, 1998; Doliana et al., 1999; Kishore et al., 2004). A phylogenetic analysis of gC1q sequences of the 32 proteins present in the human genome carrying this domain (Tom Tang et al., 2005; Ghai et al., 2007), show that EMILINs/Multimerins form a small family well distinct from the rest of the superfamily. Comparison of the coding sequences of EMILIN/Multimerin family components against Drosophila melanogaster and other invertebrate genome database gives no matches, indicating that this gene family emerged during vertebrate evolution, first appearing in lower cordate, where orthologs of EMILIN1, EMILIN2, and MMRN2 have been identified (Mei and Gui, 2008; Milanetto et al., 2008). MMRN1 is present starting from birds, indicating that this member of the family originated much later during vertebrate evolution. The members of the EMILIN/Multimerin family exert a diverse range of functions directly linked to their gC1q domains although several important functions depend on interactions of soluble proteins and/or cellular receptors with other domains. Structure, function, and physiological and pathological consequences of the activity and/or lack of activity of family members will be summarized.
Fundamental Notions about ECM and Integrin Receptors
The cells of the microenvironment are embedded in a supporting network of extracellular matrix (ECM) constituents that include collagens, elastin, proteoglycans, and glycoproteins (Li et al., 2007; Zhang and Huang, 2011). The ECM does not constitute a mere structural scaffold but it elicits profound influences on cell behavior and affects cell growth, differentiation, motility, and viability (Bissell et al., 2005; Marastoni et al., 2008; Hynes, 2009; Cukierman and Bassi, 2010). The complexity of the microenvironment is further magnified by the property of ECM molecules to function as reservoirs of growth factors, cytokines, matrix metalloproteinases (MMPs), and processing enzymes (Sternlicht and Werb, 2001). The relative availability of these elements may be affected when the ECM rearranges during wound healing or tumor progression.
The cells sense their microenvironment through interactions with specific receptors, the integrins. Integrins, a large family of αβ heterodimeric cell membrane receptors, are key sensory molecules that translate chemical and physical cues from the dynamic constituents of ECM into biochemical signals that regulate many interrelated cellular processes (Legate et al., 2009). Integrins function as mechanotransducers transforming mechanical forces created by the ECM or the cytoskeleton into cell movement (Chen et al., 2004; Hynes, 2009; Schwartz, 2010). Eighteen α and eight β subunits have been so far discovered in mammals and at least 24 different combinations of α/β subunits have been characterized. Integrins display overlapping ligand specificity and many cells express multiple integrin receptors for the same ligand. Ligation of different integrin receptors by a certain ligand may induce different cellular effects in response to ECM ligation. Following integrin ligation a variety of downstream signaling events ensue: for instance, integrins activate survival pathways via the PI3K and MAPK pathways and act as essential cofactors for growth factors stimulation. Elevated integrin expression in the absence of appropriate ligands, or in the presence of natural or synthetic antagonists, promotes apoptosis under otherwise permissive growth conditions. Moreover, integrins coordinate survival or death responses or cell behavior as a function not only of ECM composition but also of its relative stiffness (Levental et al., 2009; Provenzano and Keely, 2011). This dual function provides an elegant mechanism through which tissue-remodeling events may regulate cell death or survival in a temporal, ECM-governed manner (Dupont et al., 2011). Thus, growth factor shortage and deficiency of pro-survival stimuli or absence of integrin activation leads to intracellular signals, mainly Akt down-regulation, which in turn activate a peculiar type of apoptosis in substrate-adherent cells, anoikis (Gilmore, 2005). Similar balances are at work if one considers the other cellular processes regulated by the many integrin–ECM relationships.
The EMILIN/Multimerin Family of Proteins
Proteins containing the EMI domain, a cysteine-rich sequence of approximately 80 amino acids, define the EMI domain endowed (EDEN) superfamily in mammals (Braghetta et al., 2004). This superfamily comprises seven genes, which can be subdivided into three families on the basis of the major protein domains. The first family is formed by Emu1 and Emu2 genes (Leimeister et al., 2002) that except for the presence of an EMI domain do not share structural similarities with the other EDEN members. The second family comprises only one gene EMILIN-truncated (EMILIN-T) with a structure similar to the third larger family, but lacking the C-terminal globular domain of C1q (gC1q). The third is the EMILIN/Multimerin family. It is characterized by the N-terminal EMI domain, a central part of the molecule formed by a long (approximately 700 amino acids) region with high probability for coiled-coil structures, and a region homologous to the gC1q domain (Figure 1). The family includes EMILIN1 (Colombatti et al., 1985; Bressan et al., 1993; Doliana et al., 1999), EMILIN2 (Doliana et al., 2001), Multimerin1 (MMRN1; Hayward et al., 1991, 1995), and Multimerin2 (MMRN2; Sanz-Moncasi et al., 1994; Christian et al., 2001). Apart from MMRN1 that is deposited in the ECM but for the most part is sequestered in megakaryocytes, endothelial cells (ECs), and platelets granules (Adam et al., 2005), the members of the EMILIN family are constituents of the ECM Under normal conditions the expression of each individual gene overlaps with those of some other members of the family; the only exception is apparently the central nervous system, where only EMILIN2 is detected outside blood vessels, at least at the mRNA level (Braghetta et al., 2004). Moreover, EMILIN2 is specifically and abundantly expressed in mouse cochlear basilar membrane (Amma et al., 2003).
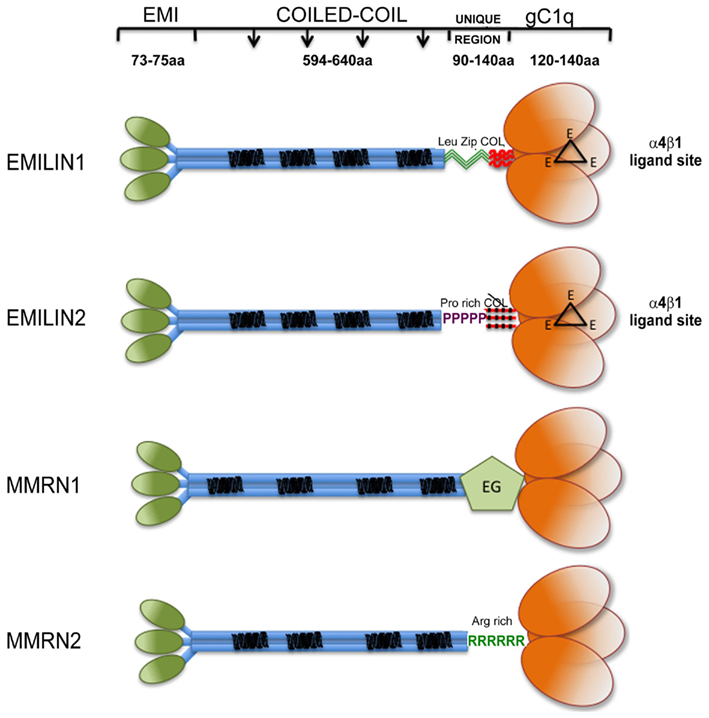
Figure 1. Graphical view of EMILIN/Multimerin family members. Note that: (i) EMI domain: there is some experimental evidence suggesting that EMI domains might self-interact; however, since it is not clear if they form trimers they are depicted separated; (ii) Coiled-coil regions: the similarity between the central regions of the family members is only structural, the heptad repeats are placed in different positions along the primary sequence of the different members of the family. (iii) the so called “UNIQUE REGION” is represented in EMILIN1 by two leucine zippers followed by a functional 17 triplets long collagenic sequence; in EMILIN2 by a proline rich sequence followed by a non-functional 17 triplets long collagenic sequence; in MMRN1 by an arginine rich sequence; in MMRN2 by an EGF-like domain; (iv); the gC1q of EMILIN1 and EMILIN2 possess three unstructured loops (black triangles) each bearing a glutamic acid (E) able to interact with the α4β1 integrin.
Domain Organization
The EMI domain (Doliana et al., 2000a) is located at the N-terminus in all the proteins in which it is expressed and displays a high sequence homology ranging from 60 to 70% between the family members. Cysteine-rich repeat modules are present in various ECM proteins, including several constituents or proteins associated with the elastic fiber (Pereira et al., 1993; Sinha et al., 1998; Doliana et al., 2000a). The EMI domain is rather unique: in fact, while most of the cysteine-rich domains described to date contain either six or eight cysteine residues, it has seven cysteine residues located at regular positions, with the exception of MMRN1 in which the second cysteine residue is missing. The specific spacing of cysteines and a number of strongly conserved aromatic and hydrophobic residues represent a diagnostic feature of this sequence. The consensus WRCCPG(Y/F)xGxxC toward the C-terminus of the domain is highly preserved and apparently unique of the EMI domain. Together with the invariable N-terminal position in all proteins discovered to date it clearly distinguishes the EMI domain from other cysteine-rich domain as, for example, the EGF-like that are often embedded within the protein sequence. In the last decade on the basis of reiterated sequence homology searches other proteins including Drosophila CED-1, fasciclin, and periostin have been proposed to harbor an EMI domain (Callebaut et al., 2003). However, after careful comparison refined by eye, it appears that these cysteine-rich domains very scarcely conform to the EMI domain consensus motif and should be classified as novel entities.
A few ECM proteins present long domains in which the 3–4–3–4 spacing of hydrophobic residues (heptad repeats) predicts that they form α-helical coiled-coils which result in a rigid rod (Berger et al., 1995; Wolf et al., 1997). The large central regions of all the EMILIN/Multimerin family members shows a very low or undetectable homology at the primary sequence level; however, when analyzed by specific algorithms it reveals a common structural homology as all the members contain several regions in which the probability for coiled-coil formation is very high.
Between the long central coiled-coil part of the molecules and the gC1q C-terminal domain each component of the family presents different sequences that could confer specific structural and/or functional properties. EMILIN1 presents a 90 residues long sequence, including two sequences corresponding to structures referred to as “leucine zipper.” Although its precise function has not been determined yet it might contribute to the coiled-coil association in the C- to N-terminal direction for the assembly of a proper EMILIN1 trimer (see below). Right downstream the leucine zipper there is a short collagenic region formed by 17 GXY triplets organized in a trypsin-resistant triple helix (Mongiat et al., 2000). In place of the leucine zipper motif EMILIN2 harbors a unique proline-rich motif of 53 residues that is highly hydrophilic and exposed to the solvent and thus available for potential interactions with other ligands or provide some flexibility that is not present in EMILIN1. Just upstream of the gC1q domain EMILIN2 also presents a collagenous sequence consisting of 17 triplets like EMILIN1, but with four interruptions, which makes it very unlikely that this sequence could form a stable triple helix. MMRN1 and MMRN2 do not harbor any collagenous sequence. On the contrary, in the corresponding position MMRN1 presents an EGF-like domain and MMRN2 an arginine rich sequence, whose functions are still undisclosed.
The gC1q domain lengths vary between 131 (C1q-C chain of C1q) and 151 (EMILIN1) residues with a high level of conservation of several hydrophobic and uncharged residues. Long before the first crystal structure was resolved Fourier-transform infrared spectroscopy and structure prediction of 15 gC1q sequences suggested a β-sheet secondary structure arrangement for this domain (Smith et al., 1994). The structure was then formally demonstrated by the analysis of the ACRP30/AdipoQ C1q crystal (Shapiro and Scherer, 1998) followed by the solution of the crystal structures of the gC1q of human collagen X (Bogin et al., 2002), mouse collagen VIII (Kvansakul et al., 2003), and human complement gC1q (Gaboriaud et al., 2003). All these crystals show similar quaternary structures obtained by the non-covalent association of three polypeptides whose tertiary structure adopts invariability a beta sandwich topology formed by 10 beta strands organized in two beta sheets. We were then the first to determine the three dimensional NMR solution structure of the human EMILIN1 gC1q homotrimer (Verdone et al., 2004, 2008, 2009). This domain exhibits a striking homology to the gC1q domains of several other members of the C1q/TNF superfamily but, at the same time, displays very peculiar characteristics (Figure 2). The quaternary structure of the complex is formed by three mostly identical subunits, apart from very minor differences inherently associated with the molecular dynamic simulations (Verdone et al., 2008, 2009). Each protomer conserves the typical jelly-roll topology of two β-sheets of antiparallel strands. However, in contrast to all the gC1q structures solved to date, it presents a nine-stranded β-sandwich fold instead of the 10-stranded fold. By sequence homology alignment of C1q family members, the lack of the F beta strand in EMILIN1 gC1q may be due to the insertion of nine residues in the middle of strand F that disrupt its secondary structure. As an important consequence, the highly ordered and rigid strand is substituted by a 19-residues long segment spanning from Y927 to G945. This sequence is unfolded, highly dynamic and highly accessible to solvent, with 10–11 residues protruding from the main globular structure making this region a good candidate for hosting an interaction site. Indeed, by site-directed mutagenesis experiments focused on this segment, we mapped the acidic residue E933 as the site of interaction between gC1q and the α4β1 integrin. This loop is not present in any of the already determined gC1q structures, and seems the only region of gC1q where single mutation or even deletions do not alter the overall domain structure. Interestingly, the quaternary structure of EMILIN1 gC1q assembly locates the unstructured loop at the apex of the homotrimer (Figure 2D). Another important information derived from the NMR solution structure of the EMILIN1 gC1q is a marked decrease of the monomer contact surface in the trimeric assembly (3,000 Å2), compared to ACRP (5,324 Å2), complement (5,490 Å2), Type VIII (6,150 Å2), and type X collagen (7,360 Å2) gC1q domains. Since the conserved residues are nearly identical in the different gC1q domains, it seems reasonable that the lower thermal stability detected in some of the domains, namely ACRP30 and EMILIN1 depends on the partly different neighboring residues among the various domains. Moreover, the decrease of the buried surface may in part be due to the missing contribution of unstructured element, or even to a contact disturbing activity, as suggested by the decrease of trimer thermal stability observed in some mutants (Doliana et al., unpublished data). Despite various attempts, we were unable to express a soluble form of EMILIN2 gC1q domain and hence to determine its structure, but, based on the sequence homology and functional data, it is conceivable that also the gC1q of EMILIN2 presents the same unstructured loop able to interact with the integrin receptor (see below). None of the other gC1q of the superfamily possess the described unstructured insertion and there are no data on a possible interaction with the α4β1 integrin. It is of note that the E993 residue is the only site of interaction with an integrin localized to date in a gC1q domain.
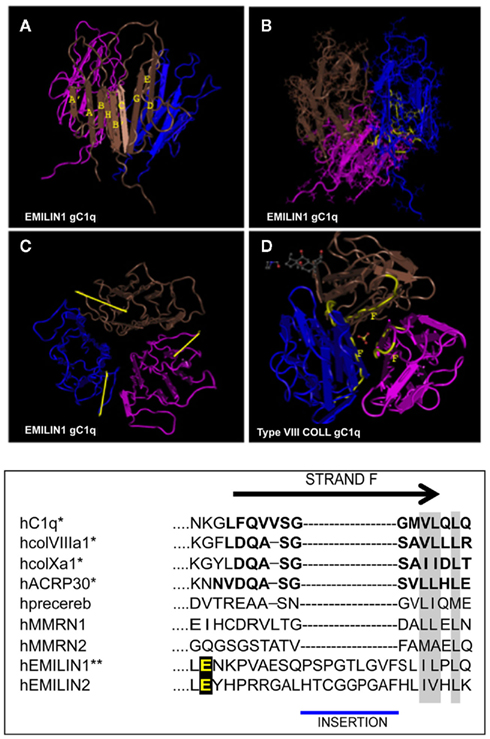
Figure 2. Top: NMR solution structure of the homotrimeric EMILIN1 gC1q domain. The structure was downloaded from database of protein structures maintained at NCBI site (http://www.ncbi.nlm.nih.gov/Structure/mmdb/mmdbsrv.cgi?uid = 68072). Ribbon representation of the assembly, as side view, is presented in (A) the three protomers in the trimer are shown in different colors (pink, blue, and brown). Each monomer has a nine-stranded folding topology, with strands labeled according to the gC1q/tumor necrosis factor superfamily nomenclature. (B) The residues highly conserved in all the members of the gC1q superfamily and essential for a correct domain folding are shown in yellow only in one monomer for clarity. (C) Top view of EMILIN1 gC1q domain. The yellow bar highlights the solvent exposed position of the unstructured segment Tyr927–Gly945 (D) the X-ray structure of type VIII collagen gC1q, http://www.ncbi.nlm.nih.gov/Structure/mmdb/mmdbsrv.cgi?uid = 25284, showing the buried position of strand F. The same position of strand F is present in the X-ray solution structures of ACRP, type X collagen and Complement gC1q domains. Bottom: Sequence alignment between strand F (residues in bold) of representative members of the C1q superfamily and EMILINs gC1q. Asterisks indicate proteins for which the structure has been solved. Similar residues that are conserved in all proteins are shaded in gray, and the glutamic acid residue interacting whit α4β1 is in yellow.
Genomic Organization
The EMI domain of EMILIN1 is contained in exons 2 and 3 (Doliana et al., 2000b) and the organization of this domain is remarkably similar to that of EMILIN2 (Doliana et al., 2001). Exon 4 (almost 2000 bp in length) encodes for four coiled-coil structures and two leucine zippers which may represent a sort of functional unit. The short collagenous region of EMILIN1 is encoded by two very short exons (5, 6) and part of exon 7. Finally, the gC1q domain is encoded by part of exon 7 and exon 8. Similarly to EMILIN1 also the EMILIN2 gene consists of eight exons and seven introns. However, whereas the exon structure is remarkably similar between the two genes, several introns of EMILIN2 are much larger than those of EMILIN1 resulting in an overall gene size of about 40 kb as opposed to the remarkably compact EMILIN1 gene (8 kb). Nevertheless, the exon size pattern and location of introns in the coding sequence are very well conserved between the two genes likely indicating that they evolved from a common ancestor. Whereas most of gC1q domains are encoded within one exon, with the notable exception of cerebellin family where the coding sequence is splitted in three exons, those of EMILIN1 and EMILIN2 are encoded by two exons. MMRN1 and MMRN2 genes have a similar organization with EMILINs genes only limited to the EMI domain and the large exon coding for the coiled-coil structures. At variance, MMRNs present two additional short exons interposed between the EMI domain coding exons and the coiled-coil coding exon. In both genes the gC1q domain is encoded within a single exon.
Before describing in more detail the functions of EMILIN1/Multimerin family formally associated with the gC1q domain, a short summary of the various functions of EMILIN/Multimerin proteins not directly dependent gC1q will follow (Figure 3).
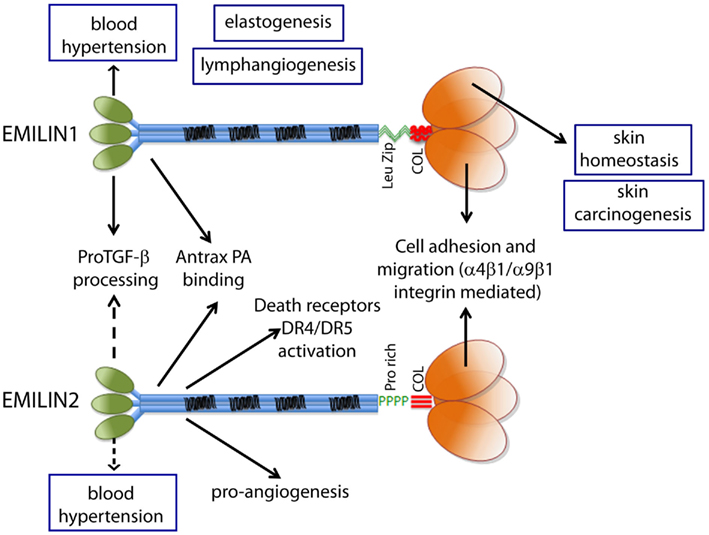
Figure 3. The functions ascribed to the gC1q domain and to the other domains of EMILIN1 and EMILIN2 are summarized to show the versatility of these glycoproteins. The regulation of elastogenesis and of lymphangiogenesis have not yet been pinpointed to a specific domain although it is tempting to assume that gC1q plays an important role.
gC1q-Independent Functions
EMILIN1 and Blood Pressure Control
Arterial blood pressure is a function of the vasculature resistance and of the cardiac output, i.e., the amount of blood pumped out by the heart. The integrity and elasticity of the vessels and the modulation of blood pressure are determined by smooth muscle cells and ECs lining the vascular walls and by their relationship with ECM. EMILIN1 is intimately associated with elastic fibers and microfibrils in blood vessels (Colombatti et al., 1985, 2000). EMILIN1 is implicated in elastogenesis and in maintenance of blood vascular cell morphology (Zanetti et al., 2004). Emilin1−/− mice display elevated systemic blood pressure independent of cardiac output without defects in vascular contractility or mechanical properties. They have significantly narrower arteries causing the hypertensive phenotype. EMILIN1 binds to proTGF-β1 prior to the cleavage of LAP and upstream of the furin convertases and prevents its processing (Zacchigna et al., 2006). The increased TGF-β in the absence of EMILIN1 as it occurs in Emilin1−/− mice leads to reduced vascular cell proliferation and results in narrower blood vessels and increased peripheral resistance, thereby causing hypertension. Also Emilin2−/− and MMRN2−/− mice mice display hypertensive phenotypes but the underlying mechanisms are different and do not affect proTGF-β1 processing (G. M. Bressan et al., unpublished data). While the pathogenetic mechanism of blood hypertension is largely explained by the interaction between the EMI domain and proTGF-β, EMILIN1 may also be involved in the hypertensive phenotype with other domains; for instance, in anchoring smooth muscle cells to elastic fibers or in the process of vessel assembly. In fact, in the media of arteries there are specific sites of interaction between smooth muscle cells and elastic fibers (Daga-Gordini et al., 1987). At these sites the cell basement membranes are interrupted and the cell surface is in direct contact with the periphery of the amorphous elastin core, a region enriched in fibrillin (Sakai et al., 1986) and EMILIN1 (Bressan et al., 1993). The finding that EMILIN1 gC1q is an adhesive substrate (see below) and provides anchorage and/or homeostatic control of cell proliferation (see below) suggests that it might also contribute to affect the cell number and the size of smooth muscle cells in arterial walls.
Defense Function of EMILIN/Multimerin Family Members
The informational spectrum method (ISM), a virtual spectroscopy approach, is a fast and simple structure analysis of proteins and their functionally important domains (Veljkovic and Metlas, 1988; Shepherd et al., 2003; Wen et al., 2005). By applying the ISM to the human sequence database all EMILIN/Multimerin family members were predicted to interact with anthrax protective antigen (PA). The interaction with PA was then formally proven for EMILIN1 and EMILIN2 by solid phase assays using recombinant proteins (Doliana et al., 2008). The interacting part of EMILIN2 resides in the first N-terminal 108 residues. Residue D425 of PA is important in mediating Bacillus anthracis cell toxicity (Ahuia et al., 2003) and its deletion results in a PA variant which still assembles but forms non-functional complexes leading to a complete inhibition of the channel forming ability of PA (Sellman et al., 2001). The deletion mutant (del425) of PA binds to EMILIN1 with very low efficiency suggesting that EMILIN1 interacts with this residue to prevent cell toxicity. The interaction is independent of the presence of divalent cations and does not involve PA aspartic residue at 683, a critical residue in cell surface receptor binding. The functional consequences of the specific interaction of PA in vitro and the inhibition of cell cytotoxicity by the LF–PA complex in the presence of EMILIN1 suggests that EMILINs/Multimerins, along with the LDL receptor-related protein LRP6 (Mayer et al., 2001), represent additional targets and/or binding proteins potentially useful for countermeasures against B. anthracis toxin lethality.
EMILIN2 and Cell Death Promotion
Among the few ECM molecules that impair cell viability (Shichiri and Hirata, 2001; Todorovicç et al., 2005; Seidler et al., 2006; Ramakrishnan et al., 2007; Tai and Tang, 2008; Juric et al., 2009) EMILIN2 adopts an entirely different mechanism, i.e., the activation of the extrinsic apoptotic pathway through a direct binding to death receptor 4 (DR4) and 5 (DR5; Pan et al., 1997; LeBlanc and Ashkenazi, 2003). This binding, in analogy with the downstream events that occur after ligation of the death ligand TRAIL to DR4 or DR5, is followed by receptor clustering, co-localization with lipid rafts, DISC assembly, and caspase activation (Mongiat et al., 2007). Thus, EMILIN2 mimics the activity of the known death receptor ligands (Walczak et al., 1997; Ashkenazi, 2008; Johnstone et al., 2008). This pro-apoptotic function resides in a homotrimeric 90 residues long region toward the N-terminus of the molecule, positioned right after the EMI domain in the coiled-coil domain (Mongiat et al., 2010). Since normal cells do not express DR4 or DR5 it can be speculated that EMILIN2 or specific peptides encompassing the pro-apoptotic region might be used as anti-tumor agents in combination with anti-angiogenic drugs such as bevacizumab (Mongiat et al., 2010) and in cases where TRAIL resistance is involved (Hengartner, 2000; Fulda, 2009).
MMRN2, EMILIN2, and Angiogenesis
MMRN2 is found along the blood vessels in close contact with ECs in normal and tumor vasculature (Sanz-Moncasi et al., 1994; Christian et al., 2001; Koperek et al., 2007) and contributes to the maintenance of blood vessels’ homeostasis by affecting the VEGF-A/VEGFR2 axis (Lorenzon et al., 2011). Neither proliferation nor apoptotic rate are affected. By preventing the interaction of VEGF-A with VEGFR1 and VEGFR2, MMRN2 reduces their activation and impaires the non-integrin dependent motility of ECs leading to a decreased intra-tumoral angiogenesis and consequent impaired tumor growth. It is conceivable that MMRN2 attains a high EC pericellular concentration and thus competes for the binding of VEGF-A to its receptors. MMRN2 may thus represent a novel key homeostatic molecule indispensable for the maintenance of blood vessel integrity.
Despite the strong reduction of tumor growth induced by the pro-apoptotic function of EMILIN2 (Mongiat et al., 2007, 2010), EMILIN2 also increases the overall intratumor blood vessel density. As a consequence, added bevacizumab is able to further inhibit tumor growth (Mongiat et al., 2010).
MMRN1 and Platelet Function
MMRN1 is a soluble S–S linked homopolymer stored in platelets, megakaryocytes, and ECs and deposited in ECM (Hayward, 1997; Adam et al., 2005). It supports the adhesion of platelets, neutrophils, and ECs via integrin αvβ3 and αIIbβ3 (Adam et al., 2005). It binds to collagen and it is able to enhance von Willebrand factor-dependent platelet adhesion to collagen thus supporting thrombus formation. MMRN1 has a high affinity for factor V (Jeimi et al., 2008) and this facilitates the co-storage in platelet α-granules. When MMRN1 is released from platelets during platelet activation it regulates thrombin formation limiting thrombus formation. This protein is playing an important homeostatic control in platelets aggregation and its consequences. Unfortunately, all these multiple functions have not been assigned to any of the domains of MMRN1 apart from cell adhesion that depends on an RGD peptide at the N-terminus (Adam et al., 2005).
gC1q-Dependent Functions
Homotrimer Association
Several members of the C1q/TNF superfamily associate into trimers. The chain composition of these different superfamily members varies from heterotrimers formed of three distinct chains as in C1q, or of homotrimers as in collagen types VIII (Rosenblum, 1996; Illidge et al., 1998; Greenhill et al., 2000), and X (Thomas et al., 1991; Reichenberger et al., 1992; Chan et al., 1996; Frischholtz et al., 1998), ACRP30 (Hu et al., 1996; Shapiro and Scherer, 1998), MMRN1 (Hayward et al., 1995). The potential self-interaction of the EMILIN1 gC1q domain was formally demonstrated with the use of two-hybrid system approaches, yeast mating and cotransformation that both confirm the self-association of the single protomers (Mongiat et al., 2000).
Supramolecular Assembly
The smallest EMILIN1 protomer under reducing conditions is a trimer held together by strong non-covalent bonds. Likely the formation of the gC1q homotrimer acts as a nucleation in a C- to N-terminal direction, facilitating the formation of the triple helix. Several of the gC1q domain-containing molecules assemble to high order S–S bonded structures composed of 9–12 polypeptides multimers, as in ACRP (Scherer et al., 1995), 18 polypeptides as in C1q (Reid, 1983; Kishore and Reid, 2000), collagens type X (Kwan et al., 1991), and probably also type VIII (Sawada et al., 1990). The sizes of MMRN1 multimers are well above 24 polypeptides (Hayward et al., 1991). EMILIN1 multimers can reach sizes of several million daltons, corresponding to S–S bonded complexes composed of several dozens of polypeptides (Mongiat et al., 2000). We have preliminary evidence that secreted recombinant EMILIN2 is assembled in smaller multimers compared to EMILIN1.
At present, the mechanistic basis for the formation of EMILINs supramolecular aggregates is unknown. While covalent S–S bonds are involved in the association it is possible that also non-covalent interactions participate to allow supramolecular assembly. In addition, it is not clear whether each EMILIN exclusively forms homotypic macro-aggregates consisting of only one member or also heterotypic macro-aggregates consisting of more members. This latter possibility is supported, at least for EMILIN1 and EMILIN2, by the partial overlapping gene expression pattern in vivo in many tissues (Braghetta et al., 2002), and by the codistribution of the secreted deposited proteins in in vitro cell cultures (Doliana et al., 2001). In situations where both EMILINs are expressed simultaneously, it is theoretically possible that each EMILIN forms either separate homotypic or that they may form heterotypic assemblies. The isolation of EMILIN2 from expression libraries using the gC1q domain of EMILIN1 as a bite proved that gC1q domains from distinct but similar molecules could interact at least in the two-hybrid system (Doliana et al., 2001). Then, also homo- and hetero-interactions between gC1q and EMI domains were determined by qualitative and quantitative two-hybrid assays and by immunochemical evidence of co-expression in yeast human cell extracts (Bot et al., in preparation). It is possible, at least in vitro, that gC1q and EMI domains from the same or different EMILINs interact in a head-to-tail fashion. The question remains whether there is the need for adapter molecules that favor this interaction or whether this interaction is direct and EMILIN1 and EMILIN2 can co-polymerize head-to-tail to form heterotypic non-covalent multimers in vivo.
Interaction of gC1q with α4β1 Integrin
α4β1 integrin binds to the cell surface vascular cell adhesion molecule-1 (VCAM-1) on activated endothelium (Hamann et al., 1994) and to the ECM molecule fibronectin (FN; Humphries et al., 1986; Komoriya et al., 1991). The key VCAM-1 binding site is the tripeptide motif sequence IDS. This sequence is homologous to the LDV active motif on the CS-1 peptide of FN, suggesting that α4β1 may interact with FN and VCAM-1 through a similar mechanism. EMILIN1 is an adhesive ligand for α4β1 (Spessotto et al., 2003, 2006; Verdone et al., 2008; Danussi et al., 2011) and this function is fully accounted for by the gC1q domain (Spessotto et al., 2003). All polypeptides with integrin binding capability display exposed aspartic or glutamic acid residues located in mobile loops protruding from the main core of the ligand (Leahy et al., 1996; Casasnovas et al., 1998). These residues are critical for integrin recognition. The three dimensional conformation of the active site on ligand–receptor binding is extensively demonstrated (in particular for RGD containing ligands; Humphries, 1990; Leahy et al., 1996; Arnaout et al., 2007; Barczyk et al., 2010). The aspartic-acid-based sequences (e.g., RGD, LDV, KGD, RTD, and KQAGD) bind to the majority of integrins (Yamada, 1991) and mutations around the aspartic acid residue, i.e., G in RGD or L in LDV, greatly affect integrin recognition (Pierschbacher and Ruoslahti, 1984; Komoriya et al., 1991; Cherny et al., 1993). Other integrins interact with ligands that contain glutamic-acid-based sequences (Michishita et al., 1993). However, it is likely that the scenario in the EMILIN1–α4β1 interaction is more complex than in conventional α4β1 binding to short linear peptide consensus sequences, representing the common feature of ECM integrin ligands (Humphries, 1990; Humphries et al., 2006). A gC1q domain in which the glutamic acid at position 933 (E933) was substituted with an alanine residue is no longer functional in cell adhesion assays and it is not recognized by α4β1 integrin suggesting that this residue plays a major role in this interaction. We exclude that the loss of activity of the E933A mutant or of deletion mutants encompassing this residue is due to a global folding change of the protein (Verdone et al., 2008). Thus, the gC1q–α4β1 interaction provides a further example of the functional relevance of disordered regions in proteins (Radivojac et al., 2007).
How would an homotrimeric molecule such as gC1q mechanistically interact with the α4β1 integrin? The explanation that a linear tripeptide from one single monomer as in CS-1 peptide of FN could represent the α4β1 ligand binding site on gC1q is apparently excluded by the finding that the L932A mutant does not loose its cell adhesion binding property. Several mutant residues around E933 are still functional and this indicates that the α4β1 integrin binding site involves only E933. Thus, this residue plays an absolutely central role in α4 integrin mediated interaction, whereas several residues close to E933 do not seem to participate in this interaction (Verdone et al., 2008). The NMR solved structure locates residue E933 in the flexible loop at the apex of the EMILIN1 gC1q domain assembled into a symmetric trimer. The stoichiometry of three integrin molecules each recognizing one E933 residue on the unstructured loops of the gC1q trimer is ruled out for steric reasons. Thus, the interaction pattern with α4β1 might well entail the binding of the three E933 carboxylate groups to a single integrin molecule. An example of an integrin–ligand interaction that requires two structural elements residing on different polypeptide chains has been already reported: to be recognized by αvβ3 integrin the LG1–3 globular domain of laminin α chain requires the intact heterotrimeric C-terminal portion of the coiled-coil domain, including the most distant C-terminal region of the β and γ chains of laminin (Künneken et al., 2004). In that case it has been hypothesized that the C-terminus of the γ chain is assembled together with a specific region of LG1–3, leading to the formation of an interchain functional ligand tripeptide. The gC1q–α4 interaction could represent the first example of an integrin binding site located on a homotrimeric assembly that employs a different mode of integrin engagement and prospects a new model for the interaction geometry recognition between an integrin binding site and a functional ligand located on a homotrimeric assembly. In this scenario we hypothesize the involvement of two/three E933 residues for a single integrin molecule engagement for a proper interaction able to fit the α4 pocket in the βMIDAS region. One possibility to tackle this question is the use of bicistronic vectors coding for wild type and non-functional mutant E933A gC1q in order to obtain trimers with a single copy or two copies of the wild type sequence on average in the homotrimer. In this systems the mutant and wild type monomers are randomly assembled into trimers to form a heterogeneous population and can be isolated by affinity chromatography. Preliminary data suggests that cell adhesion to heterogeneous gC1q trimers composed by both wild type and E933A mutants, is negatively affected implying the need of all three wild type monomers to achieve a proper integrin ligation with the homotrimer (Capuano et al., in preparation).
Functional Consequences of the gC1q/Integrin Ligation
Cell Adhesion
It is known that the adhesion of α5β1 or αvβ3 integrins to FN and vitronectin (VN) promotes a profound cytoskeletal reorganization (Ruoslahti and Pierschbacher, 1987; Defilippi et al., 1999; Ruoslahti, 1999; Zamir and Geiger, 2001; Campbell, 2008). On the contrary, the α4β1-dependent interactions extensively studied in hematopoietic cells (Morimoto et al., 1998; Steeber et al., 2005; Rose et al., 2007) as well as in transfected adherent cells (Pinco et al., 2002) prompt only the initial and intermediate stages of cell adhesion, i.e., attachment and spreading, whereas focal adhesion and stress fiber formation, characterizing strong cell adhesion, are rarely if ever observed (Iida et al., 1995).
Contrary to common beliefs that consider α4β1 an exclusive leukocyte integrin, there is a wealth of data available that demonstrates that this integrin is widely expressed, although at lower levels, in normal tissues including brain, heart, kidney, lung, muscle, liver prostate, skin, and ovary as well as their tumoral counterparts; α4β1 is also expressed in colon, bladder, breast, cervix, and melanoma tumor cells (Holzmann et al., 1998). In addition, its expression is upregulated by inflammatory cytokines (Weber et al., 1996; Milner and Campbell, 2003). This data makes α4β1 an integrin which is potentially involved in multiple and extensive interactions under several biological conditions including tumor spread.
Cells attached to EMILIN1 display a pattern of actin and paxillin distribution with accumulation of ruffles-inducing signals and lack of polarization and of stress fiber formation (Spessotto et al., 2003). Recognition of the gC1q domain does not require exogenous activation of α4β1 nor the addition of Mn2+ nor anti-β1-activating mAbs; instead the presence of Ca2+ or Mg2+ is necessary since EDTA treatment fully abolishes cell adhesion (Spessotto et al., 2003). Similar results are observed upon binding to EMILIN2 (Spessotto et al., unpublished observations). On the contrary, MMRN1 that possesses a RGD site binds to αvβ3 and αIIbβ3 integrins on platelets (Adam et al., 2005).
Cell Migration
Among the numerous cell types that migrate on EMILIN1 trophoblasts are particularly interesting. The finding that trophoblast cells could attach to and very efficiently migrate and haptoractically move toward EMILIN1 using the α4β1 integrin without any prior artificial cellular activation was rather unexpected (Spessotto et al., 2006) because directional haptotaxis was much more efficient toward EMILIN1 compared with FN. In addition, under several conditions the cooperation of MMPs with integrins plays a contributing role in order to better direct cellular migration (Deryugina et al., 2001). Accordingly, MT1-MMP is strongly upregulated when trophoblasts cells are co-cultured with EMILIN1-producing decidual stromal cells. The finding that the general MMP inhibitor GM6001 is more effective and affects cell migration toward EMILIN1 at 1-h migration, whereas at 4 h the inhibitory effect is stronger when cells move in an α5β1 integrin dependent mode toward FN (Spessotto et al., 2006), suggests that both ligands might be involved in the uterine wall invasion process but with a sequential role. Deposited EMILIN1 in the uterine tissue could thus favor the initial α4β1 dependent invasion by trophoblast cells to be assisted at later times by other ECM constituents such as FN and different integrins.
Cell Proliferation
It is generally known that integrin engagement positively regulates cell growth (Clark and Brugge, 1995; Walker and Assoian, 2005; Gilcrease, 2007; Streuli, 2009). The finding that EMILIN1 by the direct engagement of the gC1q domain modulates skin cell proliferation points out a novel function of α4β1 as well as the structurally and functionally similar α9β1 integrin (Danussi et al., 2011). The lack of integrin occupancy by EMILIN1, as it occurs in Emilin1−/− mice, results in increased Ki67 positive cells in epidermis and dermis. This is accompanied by reduction of PTEN phosphatase and strong upregulation of pErk1/2. Accordingly, when HT1080 and CaCo-2 cells expressing α4β1 or α9β1 integrins, respectively, adhere to gC1q PTEN levels increase. Even the addition of soluble gC1q to cells attached to plastic upregulates PTEN and the addition of TGF-β further increases PTEN levels. This is accompanied by decreased pErk1/2 levels and suggests a link between PTEN and α4/α9 integrin engagement: when α4/α9 integrins are not ligated by gC1q PTEN is down-regulated determining the activation of proliferative pathways such as pAkt and pErk1/2. Mechanistically, the increased pErk1/2 reduces TGF-β signaling by the phosphorylation of Smad2 at inhibitory Ser245/250/255 residues. It seems likely that these downstream changes concur in vivo to the decreased proliferation of basal keratinocytes and dermal fibroblasts (Figure 4).
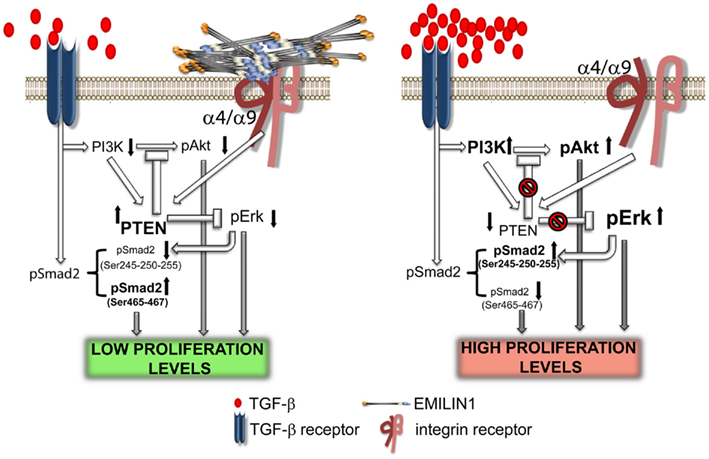
Figure 4. Elastin microfibrillar interface protein 1 in skin homeostasis. This illustration summarizes the proposed molecular mechanism underlying the regulatory role of EMILIN1 in skin. When EMILIN1 is expressed (left) TGF-β triggers cytostatic signals via pSmad2 (Ser465/467), down-modulates PI3K/Akt and hence PTEN expression is increased also because EMILIN1 binds to α4/α9β1 integrins. PTEN activation then inhibits the pro-proliferative activity of pErk1/2. When EMILIN1 is not present (right) the increased levels of mature TGF-β and the lack of α4/α9β1 integrin specific engagement downregulates PTEN determining the activation of the pro-proliferative pAkt and pErk1/2 pathways. In addition, TGF-β signaling is reduced since Erk1/2-phosphorylates the inhibitory Ser245/250/255 residues of Smad2.
Role of EMILIN1 gC1q in Physiological and Pathological Conditions
Targeted inactivation of the Emilin1 gene in the mouse induces multiple phenotypes characterized by: decreased diameter of arterial vessels and systemic hypertension (Zacchigna et al., 2006); a lymphatic phenotype with a significant reduction of anchoring filaments (AFs) and enlargement of lymphatic vessels leading to a mild lymphatic dysfunction (Danussi et al., 2008); dermal and epidermal hyperproliferation (Danussi et al., 2011). Only the last phenotype is strictly dependent on the lack of gC1q.
EMILIN1 in Lymphatic Vessel Function
The connections between ECM and lymphatic endothelial cells (LECs) that critically affect lymphatic vasculature function are mediated by AFs (Swartz and Skobe, 2001). These fibrillin rich structures, which are part of a hierarchical fibrillar elastic apparatus (Gerli et al., 2000), are highly sensitive to interstitial stresses (Swartz and Skobe, 2001) and exert a significant tension on LECs: their main function is to widen the capillary lumen and open the overlapping cell junctions facilitating lymph formation (Cueni and Detmar, 2006). Abnormalities of AFs may reduce adsorption from interstitium and propulsion of lymph and cells and promote pathological conditions such as lymphedema or diseases related to impaired immune responses (De Cock et al., 2006). Dysfunctions may also be induced by extensive and chronic degradation of the ECM that renders lymphatic vessels non-responsive to the changes in the interstitium (Pelosi et al., 2007).
Consistent with the absence of a basement membrane in vivo (Pepper and Skobe, 2003), LECs secrete very little ECM (Farnsworth et al., 2006). EMILIN1 is produced by LECs in vitro and is present at the abluminal side of lymphatic vessels. It forms fibers directed from LECs to the surrounding ECM. Therefore, the detection of abundant EMILIN1 suggested the involvement of this protein in the structure and function of lymphatic vessels (Danussi et al., 2008). In fact, a comparative study between wild type and Emilin1−/− mice highlighted that Emilin1−/− mice display hyperplasia and enlargement of lymphatic vessels compared to wt mice. Morphological alterations of the lymphatic vessels of Emilin1−/− mice also are evident at the ultrastructural level. Structural abnormalities such as the reduction in the number of AFs and the formation of extensive multiple overlapping junctions of LECs are among the most evident alterations. LECs in Emilin1−/− mice frequently appear detached from the underlying ECM and from each other and result in the formation of irregular and multiple intraluminal flaps suggesting that EMILIN1 is involved in the process of lymphatic remodeling observed in these mice (Danussi et al., 2008). These alterations are associated with inefficient lymph drainage, enhanced lymph leakage, and lymphedema. However, in contrast to other recently described lymphatic-lineage gene targeting in mouse models, that in most cases result embryonic or perinatal lethal (Tammela et al., 2005), homozygous disruption of Emilin1 gene induces only a mild phenotype. Notably, this is the first abnormal lymphatic phenotype associated with the deficiency of an ECM protein. Finally, the valve defects observed in collecting vessels are grossly similar to those of α9 integrin null mice (Bazigou et al., 2009), suggesting that the interaction of EMILIN1 and α9 integrin demonstrated with basal skin keratinocytes (Danussi et al., 2011) may also be important in valve morphogenesis in LECs.
EMILIN1 in Skin Homeostasis
The phenotype of the Emilin1−/− mouse skin revealed increased thickness of epidermis and dermis.
Elastin microfibrillar interface protein 1 interacts with α4β1 (expressed on fibroblasts) and the closely related α9β1 (expressed on keratinocytes) to provide an important external cue for the maintenance of a correct homeostasis between proliferation and differentiation (Danussi et al., 2011). In this context, signals emanating from EMILIN1-ligated α4/α9 integrins are anti-proliferative (Danussi et al., 2008, 2011). But how does EMILIN1 brings about the control of cell proliferation? EMILIN1 is produced only by skin fibroblasts and it is deposited in the dermis where it is abundantly expressed. However, EMILIN1-positive fibrils depart from near the BM and reach the basal keratinocyte layer, thus connecting the epidermis to the underlying dermal layer. In vitro co-cultures of EMILIN1-producing fibroblasts and keratinocytes provided the formal demonstration that the contact between deposited EMILIN1 and α4/α9 integrins directly regulates cell proliferation: only “contact” and not “transwell” (so that the two cell types are physically separated) co-cultures of keratinocytes with wild type EMILIN1-producing fibroblasts inhibits proliferation. Consequently, silencing of EMILIN1 stimulates the proliferation of co-cultured keratinocytes. Proliferative and anti-proliferative signals occur simultaneously and the latter, EMILIN1/gC1q ligation in this case, can override the pro-proliferative signals only when they reach a certain threshold (Müller et al., 2008). It is likely that under certain circumstances the pericellular concentration of EMILIN1 in basal keratinocytes (and fibroblasts) exceeds this threshold and stops cell proliferation. Our findings open new perspectives in the molecular mechanisms of quiescence versus proliferation in basal keratinocytes. In addition, given the high expression of α9β1 integrin on these cells, the demonstration that EMILIN1 is a novel ligand for this integrin (Danussi et al., 2011) supports a scenario in which one of the functional consequences is the integration of EMILIN1 into the complex connections of basal keratinocyte turn over and the cross talks between basal keratinocytes, underlying ECM and stromal cells.
EMILIN1 in Cancer Development
In Emilin1−/− mice subjected to a skin carcinogenesis protocol tumor appearance was significantly accelerated and the number and size of tumors compared to wild type mice increased (Danussi et al., submitted). It seems likely that the aberrant skin homeostasis generated by EMILIN1 deficiency in these mice (Danussi et al., 2011) induced a pro-tumorigenic environment. Functional studies support the hypothesis that PTEN is a critical tumor suppressor for skin cancer in humans and in mice (Di et al., 1998; Segrelles et al., 2002; Suzuki et al., 2003; Komazawa et al., 2004) by negatively regulating signal pathways involved in cell proliferation (Schindler et al., 2009). In Emilin1−/− mice the lack of EMILIN1 ligation of α4β1/α9β1 integrins reduces the expression of PTEN also in the skin tumor environment. One attractive hypothesis assumes that the mechanisms controlling the homeostasis of cell proliferation and hence the enhanced tumor development when EMILIN1 expression is genetically or functionally knocked down depends on the unique mode of gC1q–α4β1/α9β1 integrin interaction.
We are confident that the elucidation of the fine molecular interactions between α4β1/α9β1 integrins and the E933 residue(s) on the unstructured loops of the gC1q trimer and the downstream signaling events will help clarify the numerous gC1q-dependent cellular responses.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The work was supported by grants from AIRC (Associazione Italiana per la Ricerca sul Cancro; IG 10119), ACC2 (Alleanza Concro il Cancro) WP5/5, PRIN (Progetti di Ricerca di Interesse Nazionale; 20074S758W_002), and FIRB (Fondo per gli Investimenti della Ricerca di Base; RBRN07BMCT).
References
Adam, F., Zheng, S., Joshi, N., Kelton, D. S., Sandhu, A., Suehiro, Y., Jeimy, S. B., Santos, A. V., Massé, J. M., Kelton, J. G., Cramer, E. M., and Hayward, C. P. (2005). Analyses of cellular multimerin 1 receptors: in vitro evidence of binding mediated by alphaIIbbeta3 and alphavbeta3. Thromb. Haemost. 94, 1004–1011.
Ahuia, N., Kumar, P., Alam, S., Gupta, M., and Bhatnagar, R. (2003). Deletion mutants of protective antigen that inhibit anthrax toxin both in vitro and in vivo. Biochem. Biophys. Res. Commun. 37, 446–450.
Amma, L. L., Goodyear, R., Faris, J. S., Jones, I., Ng, L., Richardson, G., and Forrest, D. (2003). An emilin family extracellular matrix protein identified in the cochlear basilar membrane. Mol. Cell. Neurosci. 23, 460–472.
Arnaout, M. A., Goodman, S. L., and Xiong, J. P. (2007). Structure and mechanics of integrin-based cell adhesion. Curr. Opin. Cell Biol. 19, 495–507.
Ashkenazi, A. (2008). Targeting the extrinsic apoptosis pathway in cancer. Cytokine Growth Factor Rev. 19, 325–331.
Bazigou, E., Xie, S., Chen, C., Weston, A., Miura, N., Sorokin, L., Adams, R., Muro, A. F., Sheppard, D., and Makinen, T. (2009). Integrin-alpha9 is required for fibronectin matrix assembly during lymphatic valve morphogenesis. Dev. Cell 17, 175–186.
Berger, B., Wilson, D. B., Wolf, E., Tonchev, T., Milla, M., and Kim, P. S. (1995). Predicting coiled coils by use of pairwise residue correlations. Proc. Natl. Acad. Sci. U.S.A. 92, 8259–8263.
Bissell, M. J., Kenny, P. A., and Radisky, D. C. (2005). Microenvironmental regulators of tissue structure and function also regulate tumor induction and progression: the role of extracellular matrix and its degrading enzymes. Cold Spring Harb. Symp. Quant. Biol. 70, 343–356.
Bogin, O., Kvansakul, M., Rom, E., Singer, J., Yayon, A., and Hohenester, E. (2002). Insight into Schmid metaphyseal chondrodysplasia from the crystal structure of the collagen X NC1 domain trimer. Structure 10, 165–173.
Braghetta, P., Ferrari, A., de Gemmis, P., Zanetti, M., Volpin, D., Bonaldo, P., and Bressan, G. M. (2002). Expression of the EMILIN-1 gene during mouse development. Matrix Biol. 21, 603–609.
Braghetta, P., Ferrari, A., de Gemmis, P., Zanetti, M., Volpin, D., Bonaldo, P., and Bressan, G. M. (2004). Overlapping, complementary and site-specific expression pattern of genes of the EMILIN/multimerin family. Matrix Biol. 22, 549–556.
Bressan, G. M., Daga-Gordini, D., Colombatti, A., Castellani, I., Marigo, V., and Volpin, D. (1993). Emilin, a component of elastic fibers preferentially located at the elastin-microfibrils interface. J. Cell Biol. 121, 201–212.
Callebaut, I., Mignotte, V., Souchet, M., and Mornon, J. P. (2003). EMI domains are widespread and reveal the probable orthologs of the Caenorhabditis elegans CED-1 protein. Biochem. Biophys. Res. Commun. 300, 619–623.
Casasnovas, J. M., Stehle, T., Liu, J. H., Wang, J. H., and Springer, T. A. (1998). A dimeric crystal structure for the N-terminal two domains of intercellular adhesion molecule-1. Proc. Natl. Acad. Sci. U.S.A. 95, 4134–4139.
Chan, D., Weng, Y. M., Hocking, A. M., Golub, S., McQuillan, D. J., and Bateman, J. F. (1996). Site-directed mutagenesis of human type X collagen. Expression of alpha1(X) NC1, NC2, and helical mutations in vitro and in transfected cells. J. Biol. Chem. 271, 13566–13572.
Chen, C. S., Tan, J., and Tien, J. (2004). Mechanotransduction at cell-matrix and cell-cell contacts. Annu. Rev. Biomed. Eng. 6, 275–302.
Cherny, R. C., Honan, M. A., and Thiagarajan, P. (1993). Site-directed mutagenesis of the arginine-glycine-aspartic acid in vitronectin abolishes cell adhesion. J. Biol. Chem. 268, 9725–9729.
Christian, S., Ahorn, H., Novatchkova, M., Garin-Chesa, P., Park, J. E., and Weber, G. (2001). Molecular cloning and characterization of EndoGlyx-1, an EMILIN-like multisubunit glycoprotein of vascular endothelium. J. Biol. Chem. 276, 48588–48595.
Clark, E. A., and Brugge, J. S. (1995). Integrins and signal transduction pathways: the road taken. Science 268, 233–239.
Colombatti, A., Bressan, G. M., Castellani, I., and Volpin, D. (1985). Glycoprotein 115, a glycoprotein isolated from chick blood vessels, is widely distributed in connective tissue. J. Cell Biol. 100, 18–26.
Colombatti, A., Doliana, R., Bot, S., Canton, A., Mongiat, M., Mungiguerra, G., Paron-Cilli, S., and Spessotto, P. (2000). The EMILIN protein family. Matrix Biol. 19, 289–301.
Cueni, L. N., and Detmar, M. (2006). New insights into the molecular control of the lymphatic vascular system and its role in disease. J. Invest. Dermatol. 126, 2167–2177.
Cukierman, E., and Bassi, D. E. (2010). Physico-mechanical aspects of extracellular matrix influences on tumorigenic behaviors. Semin. Cancer Biol. 20, 139–145.
Daga-Gordini, D., Bressan, G. M., Castellani, I., and Volpin, D. (1987). Fine mapping of tropoelastin-derived components in the aorta of developing chick embryo. Histochem. J. 19, 623–632.
Danussi, C., Petrucco, A., Wassermann, B., Pivetta, E., Modica, T. M., Del Bel Belluz, L., Colombatti, A., and Spessotto, P. (2011). EMILIN1-α4/α9 integrin interaction inhibits dermal fibroblast and keratinocyte proliferation. J. Cell Biol. 195, 131–145.
Danussi, C., Spessotto, P., Petrucco, A., Wassermann, B., Sabatelli, P., Montesi, M., Doliana, R., Bressan, G. M., and Colombatti, A. (2008). Emilin1 deficiency causes structural and functional defects of lymphatic vasculature. Mol. Cell. Biol. 28, 4026–4039.
De Cock, H. E., Affolter, V. K., Farver, T. B., Van Brantegem, L., Scheuch, B., and Ferraro, G. L. (2006). Measurement of skin desmosine as an indicator of altered cutaneous elastin in draft horses with chronic progressive lymphedema. Lymphat. Res. Biol. 4, 67–72.
Defilippi, P., Olivo, C., Venturino, M., Dolce, L., Silengo, L., and Tarone, G. (1999). Actin cytoskeleton organization in response to integrin-mediated adhesion. Microsc. Res. Tech. 47, 67–78.
Deryugina, E. I., Ratnikov, B., Monosov, E., Postnova, T. I., DiScipio, R., Smith, J. W., and Strongin, A. Y. (2001). MT1-MMP initiates activation of pro-MMP-2 and integrin alphavbeta3 promotes maturation of MMP-2 in breast carcinoma cells. Exp. Cell Res. 263, 209–223.
Di, C. A., Pesce, B., Cordon-Cardo, C., and Pandolfi, P. P. (1998). Pten is essential for embryonic development and tumour suppression. Nat. Genet. 19, 348–355.
Doliana, R., Bot, S., Bonaldo, P., and Colombatti, A. (2000a). EMI, a novel cysteine-rich domain of EMILINs and other extracellular proteins, interacts with the gC1q domains and participates in multimerization. FEBS Lett. 484, 164–168.
Doliana, R., Canton, A., Bucciotti, F., Mongiat, M., Bonaldo, P., and Colombatti, A. (2000b). Structure, chromosomal localization, and promoter analysis of the human elastin microfibril interfase located protein (EMILIN) gene. J. Biol. Chem. 275, 785–792.
Doliana, R., Bot, S., Mungiguerra, G., Canton, A., Cilli, S. P., and Colombatti, A. (2001). Isolation and characterization of EMILIN-2, a new component of the growing EMILINs family and a member of the EMI domain-containing superfamily. J. Biol. Chem. 276, 12003–12011.
Doliana, R., Mongiat, M., Bucciotti, F., Giacomello, E., Deutzmann, R., Volpin, D., Bressan, G. M., and Colombatti, A. (1999). EMILIN, a component of the elastic fiber and a new member of the C1q/tumor necrosis factor superfamily of proteins. J. Biol. Chem. 274, 16773–16781.
Doliana, R., Veljkovic, V., Prljic, J., Veljkovic, N., De Lorenzo, E., Mongiat, M., Ligresti, G., Marastoni, S., and Colombatti, A. (2008). EMILINs interact with anthrax protective antigen and inhibit toxin action in vitro. Matrix Biol. 27, 96–106.
Dupont, S., Morsut, L., Aragona, M., Enzo, E., Giulitti, S., Cordenonsi, M., Zanconato, F., Le Digabel, J., Forcato, M., Bicciato, S., Elvassore, N., and Piccolo, S. (2011). Role of YAP/TAZ in mechanotransduction. Nature 474, 179–183.
Farnsworth, R. H., Achen, M. G., and Stacker, S. A. (2006). Lymphatic endothelium: an important interactive surface for malignant cells. Pulm. Pharmacol. Ther. 19, 51–60.
Frischholtz, S., Beier, F., Girkontaite, I., Wagner, K., Poschl, E., Turnay, J., Mayer, U., and von der Mark, K. (1998). Characterization of human type X procollagen and its NC-1 domain expressed as recombinant proteins in HEK293 cells. J. Biol. Chem. 273, 4547–4555.
Gaboriaud, C., Juanhuix, J., Gruez, A., Lacroix, M., Darnault, C., Pignol, D., Verger, D., Fontecilla-Camps, J. C., and Arlaud, G. J. (2003). The crystal structure of the globular head of complement protein C1q provides a basis for its versatile recognition properties. J. Biol. Chem. 278, 46974–46982.
Gerli, R., Solito, R., Weber, E., and Aglianó, M. (2000). Specific adhesion molecules bind anchoring filaments and endothelial cells in human skin initial lymphatics. Lymphology 33, 148–157.
Ghai, R., Waters, P., Roumenina, L. T., Gadjeva, M., Kojouharova, M. S., Reid, K. B., Sim, R. B., and Kishore, U. (2007). C1q and its growing family. Immunobiology 212, 253–266.
Greenhill, N. S., Ruger, B. M., Hasan, Q., and Davis, P. F. (2000). The alpha1 (VIII) and alpha2 (VIII) collagen chains form two distinct homotrimeric proteins in vivo. Matrix Biol. 19, 9–28.
Hamann, A., Andrei, D. P., Jablonski-Westrich, D., Holzmann, B., and Butcher, E. C. (1994). Role of alpha 4-integrins in lymphocyte homing to mucosal tissues in vivo. J. Immunol. 152, 3282–3293.
Hayward, C. P. (1997). Multimerin: a bench-to-bedside chronology of a unique platelet and endothelial cell protein-from discovery to function to abnormalities in disease. Clin. Invest. Med. 20, 176–187.
Hayward, C. P., Hassell, J. A., Denomme, G. A., Rachubinski, R. A., Brown, C., and Kelton, J. G. (1995). The cDNA sequence of human endothelial cell multimerin. A unique protein with RGDS, coiled-coil, and epidermal growth factor-like domains and a carboxyl terminus similar to the globular domain of complement C1q and collagens type VIII and X. J. Biol. Chem. 270, 18246–18251.
Hayward, C. P., Smith, J. W., Horsewood, P., Warkentin, T. E., and Kelton, J. G. (1991). p-155, a multimeric platelet protein that is expressed on activated platelets. J. Biol. Chem. 266, 7114–7120.
Holzmann, B., Gosslar, U., and Bitter, M. (1998). Alpha 4 integrins and tumor metastasis. Curr. Top. Microbiol. Immunol. 231, 125–141.
Hu, E., Liang, P., and Spiegelman, B. M. (1996). AdipoQ is a novel adipose-specific gene dysregulated in obesity. J. Biol. Chem. 271, 10697–10703.
Humphries, J. D., Byron, A., and Humphries, M. J. (2006). Integrin ligands at a glance. J. Cell Sci. 119, 3901–3903.
Humphries, M. J. (1990). The molecular basis and specificity of integrin-ligand interactions. J. Cell Sci. 97, 585–592.
Humphries, M. J., Akiyama, S. K., Komoriya, A., Olden, K., and Yamada, K. M. (1986). Identification of an alternatively spliced site in human plasma fibronectin that mediates cell type-specific adhesion. J. Cell Biol. 103, 2637–2647.
Iida, J., Meijne, A. M., Spiro, R. C., Roos, E., Furcht, L. T., and McCarthy, J. B. (1995). Spreading and focal contact formation of human melanoma cells in response to the stimulation of both melanoma-associated proteoglycan (NG2) and alpha 4 beta 1 integrin. Cancer Res. 55, 2177–2185.
Illidge, C., Kielty, C., and Shuttleworth, A. (1998). The alpha1 (VIII) and alpha2 (VIII) chains of type VIII collagen can form stable homotrimeric molecules. J. Biol. Chem. 273, 22091–22095.
Jeimi, S. B., Fuller, N., Tasneem, S., Segers, S., Stafford, A. R., Weitz, J. I., Camire, R. M., Nicolaes, G. A., and Hayward, C. P. (2008). Multimerin 1 binds factor V and activated factor V with high affinity and inhibits thrombin generation. Thromb. Haemost. 100, 1058–1067.
Johnstone, R. W., Frew, A. J., and Smyth, M. J. (2008). The TRAIL apoptotic pathway in cancer onset, progression and therapy. Nat. Rev. Cancer 8, 782–798.
Juric, V., Chen, C. C., and Lau, L. F. (2009). Fas-mediated apoptosis is regulated by the extracellular matrix protein CCN1 (CYR61) in vitro and in vivo. Mol. Cell. Biol. 29, 3266–3279.
Kishore, U., Gaboriaud, C., Waters, P., Shrive, A. K., Greenhough, T. J., Reid, K. B. M., Sim, R. B., and Arlaud, G. J. (2004). C1q and tumor necrosis factor superfamily: modularity and versatility. Trends Immunol. 25, 551–561.
Kishore, U., and Reid, K. B. M. (2000). C1q: structure, function, and receptors. Immunopharmacology 49, 159–170.
Komazawa, N., Suzuki, A., Sano, S., Horie, K., Matsuura, N., Mak, T. W., Nakano, T., Takeda, J., and Kondoh, G. (2004). Tumorigenesis facilitated by Pten deficiency in the skin: evidence of p53-Pten complex formation on the initiation phase. Cancer Sci. 95, 639–643.
Komoriya, A., Green, L. J., Mervic, M., Yamada, S. S., Yamada, K. M., and Humphries, M. J. (1991). The minimal essential sequence for a major cell type-specific adhesion site (CS1) within the alternatively spliced type III connecting segment domain of fibronectin is leucine-aspartic acid-valine. J. Biol. Chem. 266, 15075–15079.
Koperek, O., Scheuba, C., Puri, C., Birner, P., Haslinger, C., Rettig, W., Niederle, B., Kaserer, K., and Garin Chesa, P. (2007). Molecular characterization of the desmoplastic tumor stroma in medullary thyroid carcinoma. Int. J. Oncol. 31, 59–67.
Künneken, K., Pohlentz, G., Schmidt-Hederich, A., Odenthal, U., Smyth, N., Peter-Katalinic, J., Bruckner, P., and Eble, J. A. (2004). Recombinant human laminin-5 domains. Effects of heterotrimerization, proteolytic processing, and N-glycosylation on alpha3beta1 integrin binding. J. Biol. Chem. 279, 5184–5193.
Kvansakul, M., Bogin, O., Hohenester, E., and Yayon, A. (2003). Crystal structure of the collagen alpha1 (VIII) NC1 trimer. Matrix Biol. 22, 145–152.
Kwan, A. P., Cummings, C. E., Chapman, J. A., and Grant, M. E. (1991). Macromolecular organization of chicken type X collagen in vitro. J. Cell Biol. 114, 597–604.
Leahy, D. J., Aukhil, I., and Erickson, H. P. (1996). 2.0 A crystal structure of a four-domain segment of human fibronectin encompassing the RGD loop and synergy region. Cell 84, 155–164.
LeBlanc, H. N., and Ashkenazi, A. (2003). Apo2L/TRAIL and its death and decoy receptors. Cell Death Differ. 10, 66–75.
Legate, K. R., Wickstrom, S. A., and Fassler, R. (2009). Genetic and cell biological analysis of integrin outside-in signaling. Genes Dev. 23, 397–418.
Leimeister, C., Steidl, C., Schumacher, N., Erhard, S., and Gessler, M. (2002). Developmental expression and biochemical characterization of Emu family members. Dev. Biol. 249, 204–218.
Levental, R., Yu, H., Kass, L., Lakins, J. N., Egeblad, M., Erler, J. T., Fong, S. F., Csiszar, K., Giaccia, A., Weninger, W., Yamauchi, M., Gasser, D. L., and Weaver, V. M. (2009). Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 139, 891–906.
Li, H., Fan, X., and Houghton, J. M. (2007). Tumor microenvironment: the role of the tumor stroma in cancer. J. Cell. Biochem. 101, 805–815.
Lorenzon, E., Colladel, R., Andreuzzi, E., Marastoni, S., Todaro, F., Schiappacassi, M., Ligresti, G., Colombatti, A., and Mongiat, M. (2011). MULTIMERIN2 impairs tumor angiogenesis and growth by interfering with VEGF-A/VEGFR2 pathway. Oncogene. doi: 10.1038/onc.2011.487
Marastoni, S., Ligresti, G., Lorenzon, E., Colombatti, A., and Mongiat, M. (2008). Extracellular matrix: a matter of life and death. Connect. Tissue Res. 49, 203–206.
Mayer, T. A., Bersoff-Matcha, S., Murphy, C., Earls, J., Harper, S., Pauze, D., Nguyen, M., Rosenthal, J., Cerva, D. Jr., Druckenbrod, G., Hanfling, D., Fatteh, N., Napoli, A., Nayyar, A., and Berman, E. L. (2001). Clinical presentation of inhalational anthrax following bioterrorism exposure: report of 2 surviving patients. JAMA 286, 2549–2553.
Mei, J., and Gui, J. (2008). Bioinformatic identification of genes encoding C1q-domain-containing proteins in zebrafish. J. Genet. Genomics 35, 17–24.
Michishita, M., Videm, V., and Arnaout, A. M. (1993). A novel divalent cation-binding site in the a domain of the β2 integrin CR3 (CD11b/CD18) is essential for ligand binding. Cell 72, 857–867.
Milanetto, M., Tiso, N., Braghetta, P., Volpin, D., Argenton, F., and Bonaldo, P. (2008). EMILIN genes are duplicated and dynamically expressed during zebrafish embryonic development. Dev. Dyn. 237, 222–232.
Milner, R., and Campbell, I. L. (2003). The extracellular matrix and cytokines regulate microglial integrin expression and activation. J. Immunol. 170, 3850–3858.
Mongiat, M., Ligresti, G., Marastoni, S., Lorenzon, E., Doliana, R., and Colombatti, A. (2007). Regulation of the extrinsic apoptotic pathway by the extracellular matrix glycoprotein EMILIN2. Mol. Cell. Biol. 27, 7176–7187.
Mongiat, M., Marastoni, S., Ligresti, G., Lorenzon, E., Schiappacassi, M., Perris, R., Frustaci, S., and Colombatti, A. (2010). The extracellular matrix glycoprotein elastin microfibril interface located protein 2: a dual role in the tumor microenvironment. Neoplasia 12, 294–304.
Mongiat, M., Mungiguerra, G., Bot, S., Mucignat, M. T., Giacomello, E., Doliana, R., and Colombatti, A. (2000). Self-assembly and supramolecular organization of EMILIN. J. Biol. Chem. 275, 25471–25480.
Morimoto, C., Iwata, S., and Tachibana, K. (1998). VLA-4-mediated signaling. Curr. Top. Microbiol. Immunol. 231, 1–22.
Müller, E. J., Williamson, L., Kolly, C., and Suter, M. M. (2008). Outside-in signaling through integrins and cadherins: a central mechanism to control epidermal growth and differentiation? J. Invest. Dermatol. 128, 501–516.
Pan, G., O’Rourke, K., Chinnaiyan, A. M., Gentz, R., Ebner, R., Ni, J., and Dixit, V. M. (1997). The receptor for the cytotoxic ligand TRAIL. Science 276, 111–113.
Pelosi, P., Rocco, P. R., Negrini, D., and Passi, A. (2007). The extracellular matrix of the lung and its role in edema formation. An. Acad. Bras. Cienc. 79, 285–297.
Pepper, M. S., and Skobe, M. (2003). Lymphatic endothelium: morphological, molecular and functional properties. J. Cell Biol. 163, 209–213.
Pereira, L., D’Alessio, M., Ramirez, F., Lynch, J. R., Sykes, B., Pangilinan, T., and Bonadio, J. (1993). Genomic organization of the sequence coding for fibrillin, the defective gene product in Marfan syndrome. Hum. Mol. Genet. 2, 961–968.
Pierschbacher, M. D., and Ruoslahti, E. (1984). Variants of the cell recognition site of fibronectin that retain attachment-promoting activity. Proc. Natl. Acad. Sci. U.S.A. 81, 5985–5988.
Pinco, K. A., He, W., and Yang, J. T. (2002). Alpha4beta1 integrin regulates lamellipodia protrusion via a focal complex/focal adhesion-independent mechanism. Mol. Biol. Cell 13, 3203–3217.
Provenzano, P. P., and Keely, P. J. (2011). Mechanical signaling through the cytoskeleton regulates cell proliferation by coordinated focal adhesion and Rho GTPase signaling. J. Cell Sci. 124, 1195–1205.
Radivojac, P., Iakoucheva, L. M., Oldfield, C. J., Obradovic, Z., Uversky, V. N., and Dunker, A. K. (2007). Intrinsic disorder and functional proteomics. Biophys. J. 92, 1439–1456.
Ramakrishnan, S., Nguyen, T. M., Subramanian, I. V., and Kelekar, A. (2007). Autophagy and angiogenesis inhibition. Autophagy 3, 512–515.
Reichenberger, E., Beier, F., LuValle, P., Olsen, B. R., von der Mark, K., and Bertling, W. M. (1992). Genomic organization and full-length cDNA sequence of human collagen X. FEBS Lett. 311, 305–310.
Reid, K. B. M. (1983). Proteins involved in the activation and control of the two pathways of human complement. Biochem. Soc. Trans. 11, 1–12.
Rose, D. M., Alon, R., and Ginsberg, M. H. (2007). Integrin modulation and signaling in leukocyte adhesion and migration. Immunol. Rev. 218, 126–134.
Rosenblum, N. D. (1996). Recombinant alpha 1(VIII) collagen chains form homotrimers in vitro. Biochem. Biophys. Res. Commun. 227, 205–210.
Ruoslahti, E., and Pierschbacher, M. D. (1987). New perspectives in cell adhesion: RGD and integrins. Science 238, 491–497.
Sakai, L. Y., Keene, D. R., and Engvall, E. (1986). Fibrillin, a new 350-kD glycoprotein, is a component of extracellular microfibrils. J. Cell Biol. 103, 2499–2509.
Sanz-Moncasi, M. P., Garin-Chesa, P., Stockert, E., Jaffe, E. A., Old, L. J., and Rettig, W. J. (1994). Identification of a high molecular weight endothelial cell surface glycoprotein, endoGlyx-1, in normal and tumor blood vessels. Lab. Invest. 71, 366–373.
Sawada, H., Konomi, H., and Hirosawa, K. (1990). Characterization of the collagen in the hexagonal lattice of Descemet’s membrane: its relation to type VIII collagen. J. Cell Biol. 110, 219–227.
Scherer, P. E., Williams, S., Fogliano, M., Baldini, G., and Lodish, H. F. (1995). A novel serum protein similar to C1q, produced exclusively in adipocytes. J. Biol. Chem. 270, 26746–26749.
Schindler, E. M., Hindes, A., Gribben, E. L., Burns, C. J., Yin, Y., Lin, M. H., Owen, R. J., Longmore, G. D., Kissling, G. E., Arthur, J. S., and Efimova, T. (2009). p38delta mitogen-activated protein kinase is essential for skin tumor development in mice. Cancer Res. 69, 4648–4655.
Schwartz, M. A. (2010). Integrins and extracellular matrix in mechanotransduction. Cold Spring Harb. Perspect. Biol. 2, a005066.
Segrelles, C., Ruiz, S., Perez, P., Murga, C., Santos, M., Budunova, I. V., Martínez, J., Larcher, F., Slaga, T. J., Gutkind, J. S., Jorcano, J. L., and Paramio, J. M. (2002). Functional roles of Akt signaling in mouse skin tumorigenesis. Oncogene 21, 53–64.
Seidler, D. G., Goldoni, S., Agnew, C., Cardi, C., Thakur, M. L., Owens, R. T., McQuillan, D. J., and Iozzo, R. V. (2006). Decorin protein core inhibits in vivo cancer growth and metabolism by hindering epidermal growth factor receptor function and triggering apoptosis via caspase-3 activation. J. Biol. Chem. 281, 26408–26418.
Sellman, B. R., Mourez, M., and Collier, R. J. (2001). Dominant-negative mutants of a toxin subunit: an approach to therapy of anthrax. Science 292, 695–697.
Shapiro, L., and Scherer, P. E. (1998). The crystal structure of a complement-1q family protein suggests an evolutionary link to tumor necrosis factor. Curr. Biol. 8, 335–338.
Shepherd, A. J., Gorse, D., and Thornton, J. M. (2003). A novel approach to the recognition of protein architecture from sequence using Fourier analysis and neural networks. Proteins 50, 290–302.
Sinha, S., Nevett, C., Shuttleworth, C. A., and Kielty, C. M. (1998). Cellular and extracellular biology of the latent transforming growth factor-beta binding proteins. Matrix Biol. 17, 529–545.
Smith, K. F., Haris, P. I., Chapman, D., Reid, K. B. M., and Perkins, S. J. (1994). Beta-sheet secondary structure of the trimeric globular domain of C1q of complement and collagen types VIII and X by Fourier-transform infrared spectroscopy and averaged structure predictions. Biochem. J. 301, 249–256.
Spessotto, P., Bulla, R., Danussi, C., Radillo, O., Cervi, M., Monami, G., Bossi, F., Tedesco, F., Doliana, R., and Colombatti, A. (2006). EMILIN1 represents a major stromal element determining human trophoblast invasion of the uterine wall. J. Cell Sci. 119, 4574–4584.
Spessotto, P., Cervi, M., Mucignat, M. T., Mungiguerra, G., Sartoretto, I., Doliana, R., and Colombatti, A. (2003). Beta 1 integrin-dependent cell adhesion to EMILIN-1 is mediated by the gC1q domain. J. Biol. Chem. 278, 6170–6177.
Steeber, D. A., Venturi, G. M., and Tedder, T. F. (2005). A new twist to the leukocyte adhesion cascade: intimate cooperation is key. Trends Immunol. 26, 9–12.
Sternlicht, M. D., and Werb, Z. (2001). How matrix metalloproteinases regulate cell behavior. Annu. Rev. Cell Dev. Biol. 17, 463–516.
Suzuki, A., Itami, S., Ohishi, M., Hamada, K., Inoue, T., Komazawa, N., Senoo, H., Sasaki, T., Takeda, J., Manabe, M., Mak, T. W., and Nakano, T. (2003). Keratinocyte-specific Pten deficiency results in epidermal hyperplasia, accelerated hair follicle morphogenesis and tumor formation. Cancer Res. 63, 674–681.
Swartz, M. A., and Skobe, M. (2001). Lymphatic function, lymphangiogenesis, and cancer metastasis. Microsc. Res. Tech. 55, 92–99.
Tai, I. T., and Tang, M. J. (2008). SPARC in cancer biology: its role in cancer progression and potential for therapy. Drug Resist. Updat. 11, 231–246.
Tammela, T., Petrova, T. V., and Alitalo, K. (2005). Molecular lymphangiogenesis: new players. Trends Cell Biol. 15, 434–441.
Thomas, J. T., Cresswell, C. J., Rash, B., Nicolai, H., Jones, T., Solomon, E., Grant, M. E., and Boot-Handford, R. P. (1991). The human collagen X gene. Complete primary translated sequence and chromosomal localization. Biochem. J. 280, 617–623.
Todorovicç, V., Chen, C. C., Hay, N., and Lau, L. F. (2005). The matrix protein CCN1 (CYR61) induces apoptosis in fibroblasts. J. Cell Biol. 171, 559–568.
Tom Tang, Y., Hu, T., Arterburn, M., Boyle, B., Bright, J. M., Palencia, S., Emtage, P. C., and Funk, W. D. (2005). The complete complement of C1q-domain-containing proteins in Homo sapiens. Genomics 86, 100–111.
Veljkovic, V., and Metlas, R. (1988). Identification of nanopeptide from HTLV-III, ARV-2 and LAV envelope gp120 determining binding to T4 cell surface protein. Cancer Biochem. Biophys. 10, 191–206.
Verdone, G., Colebrooke, S. A., Boyd, J., Viglino, P., Corazza, A., Doliana, R., Mungiguerra, G., Colombatti, A., Esposito, G., and Campbell, I. D. (2004). Sequence-specific backbone NMR assignments for the C-terminal globular domain of EMILIN-1. J. Biomol. NMR 29, 91–92.
Verdone, G., Corazza, A., Colebrooke, S. A., Cicero, D., Eliseo, T., Boyd, J., Doliana, R., Fogolari, F., Viglino, P., Colombatti, A., Campbell, I. D., and Esposito, G. (2009). NMR-based homology model for the solution structure of the C-terminal globular domain of EMILIN1. J. Biomol. NMR 43, 79–96.
Verdone, G., Doliana, R., Corazza, A., Colebrooke, S. A., Spessotto, P., Bot, S., Bucciotti, F., Capuano, A., Silvestri, A., Viglino, P., Campbell, I. D., Colombatti, A., and Esposito, G. (2008). The solution structure of EMILIN1 globular C1q domain reveals a disordered insertion necessary for interaction with the alpha4beta1 integrin. J. Biol. Chem. 283, 18947–18956.
Walczak, H., degli-Esposti, M. A., Johnson, R. S., Smolak, P. J., Waugh, J. Y., Boiani, N., Timour, M. S., Gerhart, M. J., Schooley, K. A., Smith, C. A., Goodwin, R. G., and Rauch, C. T. (1997). TRAIL-R2: a novel apoptosis-mediating receptor for TRAIL. EMBO J. 16, 5386–5397.
Walker, J. L., and Assoian, R. K. (2005). Integrin-dependent signal transduction regulating cyclin D1 expression and G1 phase cell cycle progression. Cancer Metastasis Rev. 24, 383–393.
Weber, C., Alon, R., Moser, B., and Springer, T. A. (1996). Sequential regulation of alpha 4 beta 1 and alpha 5 beta 1 integrin avidity by CC chemokines in monocytes: implications for transendothelial chemotaxis. J. Cell Biol. 134, 1063–1073.
Wen, Z., Wang, K., Li, F., Nie, F., and Yang, Y. (2005). Analyzing functional similarity of protein sequences with discrete wavelet transform Comput. Biol. Chem. 29, 220–228.
Wolf, E., Kim, P. S., and Berger, B. (1997). MultiCoil: a program for predicting two- and three-stranded coiled coils. Protein Sci. 6, 1179–1189.
Zacchigna, L., Vecchione, C., Notte, A., Cordenonsi, M., Dupont, S., Maretto, S., Cifelli, G., Ferrari, A., Maffei, A., Fabbro, C., Braghetta, P., Marino, G., Selvetella, G., Aretini, A., Colonnese, C., Bettarini, U., Russo, G., Soligo, S., Adorno, M., Bonaldo, P., Volpin, D., Piccolo, S., Lembo, G., and Bressan, G. M. (2006). Emilin1 links TGF-beta maturation to blood pressure homeostasis. Cell 124, 929–942.
Zamir, E., and Geiger, B. (2001). Molecular complexity and dynamics of cell-matrix adhesions. J. Cell Sci. 114, 3583–3590.
Zanetti, M., Braghetta, P., Sabatelli, P., Mura, I., Doliana, R., Colombatti, A., Volpin, D., Bonaldo, P., and Bressan, G. M. (2004). EMILIN-1 deficiency induces elastogenesis and vascular cell defects. Mol. Cell. Biol. 24, 638–650.
Keywords: α4β1 integrin, gC1q-dependent cell adhesion, cell migration, gC1q NMR solution structure, EMI domain, extracellular matrix, skin homeostasis
Citation: Colombatti A, Spessotto P, Doliana R, Mongiat M, Bressan GM and Esposito G (2012) The EMILIN/multimerin family. Front. Immun. 2:93. doi: 10.3389/fimmu.2011.00093
Received: 30 November 2011; Accepted: 21 December 2011;
Published online: 06 January 2012.
Edited by:
Uday Kishore, Brunel University, UKReviewed by:
Uday Kishore, Brunel University, UKKenneth Reid, Green Templeton College University of Oxford, UK
Copyright: © 2012 Colombatti, Spessotto, Doliana, Mongiat, Bressan and Esposito. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Alfonso Colombatti, Division of Experimental Oncology 2, Centro di Riferimento Oncologico, Istituto di Ricerca e Cura a Carattere Scientifico, 33081 Aviano, Italy. e-mail:YWNvbG9tYmF0dGlAY3JvLml0