 Julia Jellusova
Julia Jellusova Lars Nitschke*
Lars Nitschke*- Chair of Genetics, University of Erlangen, Erlangen, Germany
B cell antigen receptor (BCR) engagement can lead to many different physiologic outcomes. To achieve an appropriate response, the BCR signal is interpreted in the context of other stimuli and several additional receptors on the B cell surface participate in the modulation of the signal. Two members of the Siglec (sialic acid-binding immunoglobulin-like lectin) family, CD22 and Siglec-G have been shown to inhibit the BCR signal. Recent findings indicate that the ability of these two receptors to bind sialic acids might be important to induce tolerance to self-antigens. Sialylated glycans are usually absent on microbes but abundant in higher vertebrates and might therefore provide an important tolerogenic signal. Since the expression of the specific ligands for Siglec-G and CD22 is tightly regulated and since Siglecs are not only able to bind their ligands in trans but also on the same cell surface this might provide additional mechanisms to control the BCR signal. Although both Siglec-G and CD22 are expressed on B cells and are able to inhibit BCR mediated signaling, they also show unique biological functions. While CD22 is the dominant regulator of calcium signaling on conventional B2 cells and also seems to play a role on marginal zone B cells, Siglec-G exerts its function mainly on B1 cells and influences their lifespan and antibody production. Both Siglec-G and CD22 have also recently been linked to toll-like receptor signaling and may provide a link in the regulation of the adaptive and innate immune response of B cells.
Introduction to the Siglec Family
B cells are an important part of the immune system and through production of antibodies crucially contribute to the humoral immune response. Recognition of antigen by the B cell antigen receptor (BCR) is the main event in the B cell immune response and signaling through the BCR complex is tightly regulated by several different co-receptors. CD22 and Siglec-G are two members of the Siglec (sialic acid-binding immunoglobulin-like lectins) family and have been shown to negatively regulate B cell signaling. The review focuses on the role of CD22 and Siglec-G in B cells and discusses their possible biological functions.
Siglecs are a family of sialic acid-binding proteins expressed on cells of the immune system (Crocker et al., 2007) with the exception of myelin associated glycoprotein (MAG) which is expressed on myelinating glia cells, suggesting a role in the nervous system (Quarles, 2007). Also, few other human Siglecs have been detected on other tissues in addition to cells of the immune system. The expression of Siglec-6 has been observed on B cells and placental trophoblasts (Patel et al., 1999; Brinkman-Van der Linden et al., 2007) and Siglec-11 can be detected on macrophages and brain microglia (Angata et al., 2002). However, the majority of Siglecs seems to play a role mainly in the immune system in both human and mouse. Some Siglecs show a very restricted expression pattern (Table 1). CD22 and Sialoadhesin have been found only on B cells or macrophages (Crocker et al., 1994, 2007), respectively. Other Siglecs are expressed more broadly. For example, Siglec-9 can be found on B cells, neutrophils, monocytes, and a fraction of NK and T cells (Zhang et al., 2000). Murine Siglec-G is mainly expressed on B cells, however it might be also expressed on other cell types such as dendritic cells (Ding et al., 2007). The human ortholog Siglec-10 has been detected on B cells, NK cells, monocytes, dendritic cells, and eosinophils (Munday et al., 2001).
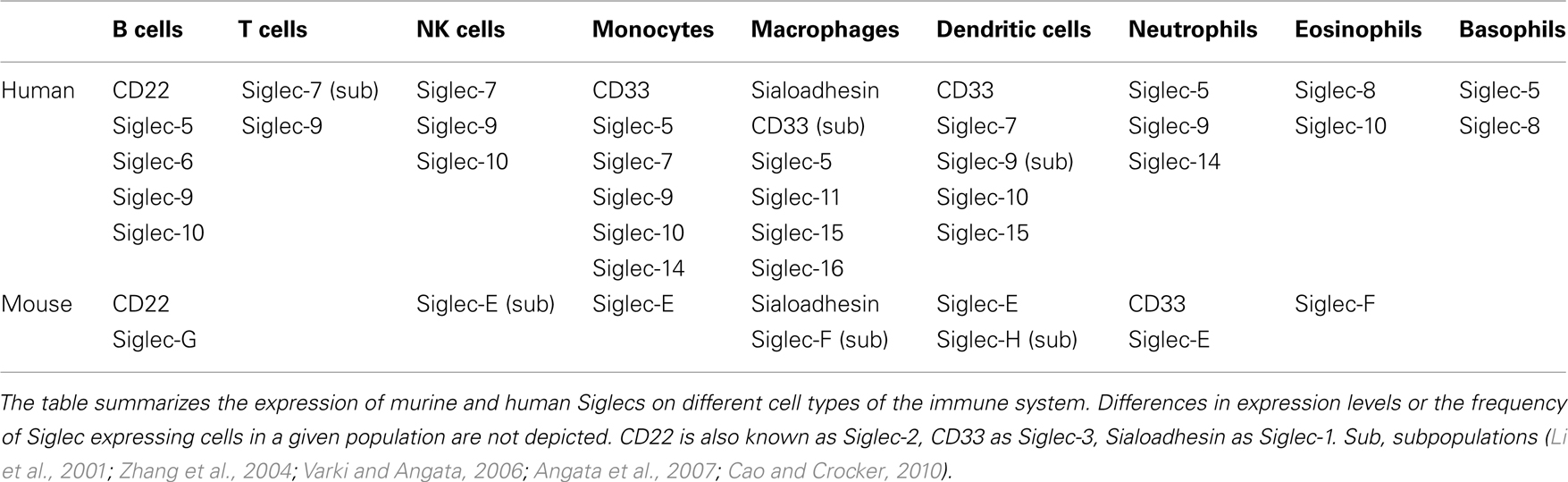
Table 1. Expression pattern of murine and human Siglecs on the cells of the immune system.
Members of the Siglec family are expressed in different vertebrates and are present in all investigated mammalian species (Angata, 2006). However, only four members of the Siglec family-Sialoadhesin (Siglec-1), CD22 (Siglec-2), MAG, and Siglec-15 are highly conserved throughout mammalian evolution. In contrast, the group of CD33 related Siglecs is rapidly evolving and often no clear orthologs can be assigned between different mammalian species (Angata et al., 2004). Humans express a larger set of CD33 related Siglecs (CD33/Siglec-3, Siglec-4, -5, -6, -7, -8, -9, -10, -11, -14, -16) than mice (CD33/Siglec-3, Siglec-E, -F, -G, -H) and the distribution on different cell types seems to be unique for each species as well. Interestingly Siglec-G is one of the few CD33 related Siglecs with a clear human ortholog – Siglec-10. In addition to sequence homology, both Siglec-G and Siglec-10 can be detected on B cells suggesting that they might also share some functional similarity (Munday et al., 2001).
All Siglecs share some structural features. They are transmembrane proteins with a variable number of immunoglobulin-like domains in their extracellular portions and a cytoplasmic part at the carboxy terminus. Located within the cytoplasmic tail are sites for possible tyrosine phosphorylation. These tyrosines are often part of the consensus sequence of immunoreceptor tyrosine based inhibitory motifs (ITIMs). After phosphorylation, ITIMs are able to recruit Src homology 2 (SH2) domain containing signaling molecules such as the tyrosine phosphatases SHP-1 or SHP-2. Interaction with SHP-1 could be demonstrated for CD22 (Doody et al., 1995), Siglec-7, Siglec-9 (Ikehara et al., 2004), and Siglec-10 (Whitney et al., 2001). ITIMs are sequence motifs present in many well-characterized inhibitory receptors such as CD72, PIR-B, or FcγRIIB (Nitschke et al., 1997) and therefore ITIM containing Siglecs are expected to play an inhibitory role in cell signaling. Indeed, several Siglecs have been reported to inhibit different signaling pathways. Siglec-7 and -9 can inhibit TCR mediated signaling (Ikehara et al., 2004), Siglec-G and CD22 are inhibitors of the BCR signal (Nitschke et al., 1997; Hoffmann et al., 2007), Siglec-E probably plays an inhibitory role in Toll-like receptor (TLR)-mediated IFNβ production (Boyd et al., 2009). However, some Siglecs lack ITIM motifs, associate with activating receptors and are therefore thought to be activatory transmembrane proteins (Crocker et al., 2007).
Unlike most other proteins from the immunoglobulin superfamily, Siglecs do not bind protein determinants but recognize exclusively sialylated carbohydrates. Sialylation is a common modification of glycoproteins in vertebrates and higher invertebrates, but relatively rare outside the Deuterostomia lineage (Angata and Varki, 2002). Sialic acids are a family of 9-carbon monosaccharides derived from neuraminic acid. These sugars are frequently found at the terminal position of oligosaccharide chains attached to numerous proteins creating a broad array of possible Siglec ligands. The substrate specificity of different Siglecs is very diverse and binding affinity can be influenced by the type of the sialic acid, the type of linkage to the underlying oligosaccharide chain and also the presence of other proximal sugars (Angata, 2006). Some Siglecs show strict ligand requirements and some recognize a broader range of sialic acids. Given the complex expression pattern of Siglecs on different cells of the immune system, the plasticity of possible interactions and the prominence of sialic acids on cell surfaces, it is not surprising that Siglecs are believed to participate in many different biological processes. Different members of the Siglec family have been suggested to play a role in cell–cell adhesion (Schadee-Eestermans et al., 2000), regulation of responses to tissue damage (Chen et al., 2009), controlling allergic reactions of eosinophils (Zhang et al., 2007), and maintenance of tolerance (Duong et al., 2010). Yet, because of the complexity of Siglec biology and due to recent discoveries of new Siglecs the exact biological function of many Siglecs remains incompletely understood.
Inhibition of BCR Signaling by Siglec-G and CD22
Siglec-G and CD22 are the only two Siglecs reported to be expressed on murine B cells so far. While CD22 appears to be expressed exclusively on B cells, Siglec-G is expressed predominantly on B cells, but is possibly also present on dendritic cells and on other cell types as well (Ding et al., 2007). Both Siglecs seem to be able to associate with the BCR (Peaker and Neuberger, 1993; Zhang and Varki, 2004 and our unpublished observation) and negatively regulate BCR mediated signaling. Siglec-G has been shown to be an important inhibitor on B1a cells, since Siglec-G-deficient B1a cells show increased calcium signaling after anti-IgM stimulation (Hoffmann et al., 2007). Siglec-G is also able to dampen the BCR signal when transfected into DT40 cells, a B cell line lacking endogenous Siglecs and widely used to study components of the BCR signaling cascade. However, conventional B2 cells show normal calcium signaling in Siglec-G-deficient mice, indicating a redundant function of Siglec-G in this B cell population (Hoffmann et al., 2007). The dominant inhibitor in conventional (B2) cells seems to be CD22 and CD22-deficient mice show increased calcium signaling in these cells. While the exact biochemical mechanism for the inhibition mediated by Siglec-G is mostly unknown, comprehensive studies have clarified signaling pathways downstream of CD22.
CD22 contains six tyrosines in its cytoplasmic tail, three of which are found within conventional ITIMs (Y783, Y843, Y863), one is part of an ITIM-like motif (Y817), and one is needed for the recruitment of Grb2 (Y828; Otipoby et al., 2001). CD22 is rapidly phosphorylated after BCR crosslinking and docking sites for different signaling molecules are created. The Src kinase Lyn plays a central role in this process (Smith et al., 1998). B cells from Lyn-deficient mice fail to induce CD22 phosphorylation after BCR stimulation and as a consequence SHP-1 recruitment to CD22 is abolished. SHP-1 is a phosphatase also involved in negative signaling from other receptors and believed to be the central signaling molecule mediating CD22 inhibition. CD22 is unable to inhibit BCR signaling in DT40 cells lacking SHP-1, but does not seem to require SHIP for its inhibitory function (Chen et al., 2004). The role of Grb2 binding to the tail of CD22 is not known. However, Grb2−/− mice show enhanced Ca2+ responses of their splenic B cells, similar to CD22−/− mice and decreased CD22 phosphorylation, suggesting a CD22-dependent mechanism (Ackermann et al., 2011; Jang et al., 2011).
The increase of free cytosolic calcium in B cells after BCR stimulation is the product of two processes. First, the BCR signaling cascade leads to the phosphorylation and activation of different signaling molecules such as Btk, BLNK/SLP65, and PLCγ2. PLCγ2 mediates the production of inositol-1,4,5-trisphosphate (IP3). This signaling molecule binds to IP3 receptor channels on the endoplasmic reticulum and initiates the release of calcium from intracellular calcium stores into the cytosol (Figure 1). The second mechanism contributing to the elevation of intracellular calcium levels is the regulation of calcium transport across the plasma membrane. Activation of calcium release activated calcium (CRAC) channels allows extracellular calcium to enter the cytoplasm. This process can be antagonized by calcium ATPase pumps (PMCA; Scharenberg et al., 2007). CD22 seems to affect both, the early events leading to the release of calcium and also the late events affecting the duration of the calcium signal. The phosphorylation of proteins involved in the calcium cascade such as CD19 (Fujimoto et al., 1999) and BLNK/SLP65 (Gerlach et al., 2003) is increased in B cells from CD22-deficient mice after anti-IgM stimulation. Furthermore, CD22 has been shown to interact with the calcium pump PMCA4 resulting in the activation of PMCA4 and augmented calcium efflux. This interaction is dependent on activation induced CD22 phosphorylation and recruitment of SHP-1 (Figure 1; Chen et al., 2004).
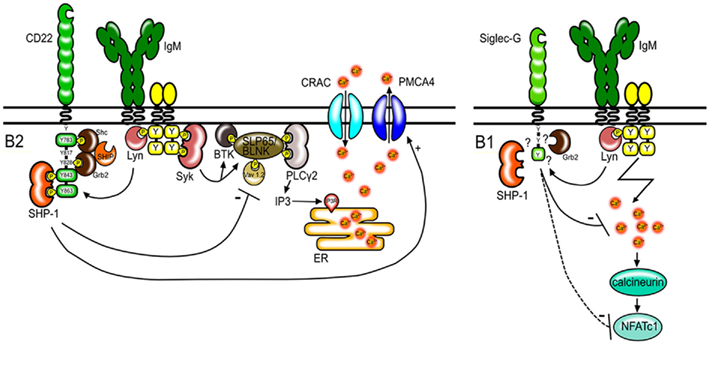
Figure 1. Regulation of BCR signaling by CD22 and Siglec-G. CD22 regulates BCR signaling in conventional B2 cells. After antigen stimulation, CD22 is tyrosine-phosphorylated by Lyn and docking sites for SH2 domain containing proteins are created. CD22 has six tyrosines in its intracellular tail, of which three form ITIM motifs (shown as green boxes). SHP-1 binds to two phosphorylated ITIMs. SHP-1 interferes with signaling pathways leading to the depletion of calcium stores from the endoplasmic reticulum (ER) and promotes calcium efflux through PMCA4. Shc and Grb2 bind to the indicated tyrosines and form a complex with SHIP. However the role of these molecules in CD22 transmitted inhibitory signals is not known. Siglec-G also has one ITIM motif (green box) and dampens the calcium signal on B1 cells thereby possibly influencing the activity and expression levels of the transcription factor NFATc1.
It has been convincingly shown that CD22 can inhibit BCR mediated signaling if the BCR is of the IgM isotype. During an immune response B cells are induced to switch to express IgG BCRs. Signals transmitted through the IgG BCR seem to differ from those mediated by IgM and the different signaling properties have been attributed to the differences in the cytoplasmic tail. BCRs with an IgG tail segment have been shown to induce stronger calcium signal than IgM BCRs (Horikawa et al., 2007). After initial reports that CD22 phosphorylation is impaired in IgG-expressing B cell lines (Wakabayashi et al., 2002), it has been assumed that CD22 is unable to regulate IgG transmitted signals. However, in vivo studies have challenged this model (Horikawa et al., 2007; Waisman et al., 2007). Knock-in mice expressing only membrane IgG1 show a strong calcium signal, yet the signal can be further increased if CD22 is absent. Furthermore, CD22 phosphorylation and association with SHP-1 after BCR stimulation are normal. This indicates that CD22 might also play a role in IgG mediated signaling (Waisman et al., 2007).
Although it has been well established that CD22 functions mainly as a negative regulator, further complexity arises by the ability of CD22 to associate with other signaling molecules otherwise involved in positive BCR signaling. Among others, PLCγ2, Syk, and PI3K have been reported to be recruited to CD22 (Law et al., 1996; Yohannan et al., 1999). However, the biological role of these interactions remains unclear.
Siglec-G is a recently discovered protein and therefore the exact biochemical function of this protein in B cells is not well defined. The phosphorylation of several signaling molecules involved in BCR mediated signaling such as PLCγ, Btk, and BLNK is normal in Siglec-G-deficient B cells (Hoffmann et al., 2007). However, Siglec-G-deficient B1a cells have been shown to contain increased levels of the transcription factor NFATc1 (Jellusova et al., 2010a). NFATc1 is known to be activated by the phosphatase calcineurin in a calcium dependent manner. The expression of the short isoform NFATc1/A can be promoted in an autoregulatory manner due to a NFAT binding site in the P1 promoter (Chuvpilo et al., 2002). Therefore, increased calcium signals in Siglec-G-deficient B1a cells could lead to increased expression of NFATc1 (Figure 1). Siglec-G has also been proposed to regulate activation of the NFκB pathway in B1 cells (Ding et al., 2007). However, samples used in the study of Ding et al. (2007) were prepared from total peritoneal washouts and likely contained other cells apart from B1 cells. Since the frequency of B1 cells in the lymphocyte population is significantly higher in the peritoneal cavity from Siglec-G-deficient mice than in wild type mice, the results from these experiments might be misleading. Enhanced NFκB activation in Siglec-G-deficient B1 cells could not be confirmed in our experiments with purified B1 cells (Jellusova et al., 2010a).
Siglecs and TLR Signaling
So far most studies have focused on the role of Siglec-G and CD22 in regulation of BCR-mediated signaling. However, recent evidence suggests that both Siglecs may play a role in TLR signaling as well. TLRs recognize specific bacterial or viral products such as dsRNA (TLR3), LPS (TLR4), dsDNA (TLR9), ssRNA (TLR7), or Flagellin (TLR5). TLR2 is able to bind a wide range of microbial structures including peptidoglycan and lipoteichoic acid. TLR1 and TLR6 can form heterodimers with TLR2 and discriminate between triacyl- and diacyl-lipopeptides (Akira and Takeda, 2004). TLRs are expressed by cells of the innate immune system such as dendritic cells and monocytes, but are also present on B cells. Siglec-G-deficient B cells show increased proliferation after stimulation with LPS (TLR4 ligand), R848 (TLR7 ligand), or CpG (TLR9 ligand; Jellusova et al., 2010b). Also CD22-deficient B cells exhibit hyperproliferation when stimulated with these ligands as well as after stimulation with polyI:C (TLR3 ligand; Jellusova et al., 2010b; Kawasaki et al., 2011). The expression levels of TLR3, TLR4, and TLR9 on CD22-deficient cells do not exceed the levels in wild type cells and the sequestration of CD22 by immobilized anti-CD22 antibodies results in enhanced proliferation after TLR stimulation, indicating that CD22 might affect downstream events in TLR signaling (Kawasaki et al., 2011; Figure 2). Ectopic expression of CD22 in a TLR4 reporter cell line, was found to dampen NFκB transcriptional activity (Kawasaki et al., 2011). How Siglec-G and CD22 interfere with TLR-transmitted signals is not known. However, TLR induced expression of SOCS1 and SOCS3 is reduced in CD22-deficient B cells (Kawasaki et al., 2011). SOCS1 and SOCS3 are known to play a negative role in cytokine and TLR-mediated signaling (Naka et al., 2005) and reduced expression of these molecules might result in a prolonged TLR signal.
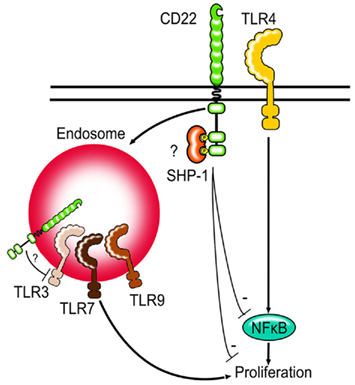
Figure 2. Siglecs and TLRs. Siglec-G and CD22-deficient B cells show increased proliferation after TLR stimulation. First evidence suggests that CD22 might be able to regulate NFκB activity after TLR stimulation. CD22 can be internalized by endocytosis and may be in direct contact to TLRs found in endosomes. Whether Siglec-G can limit TLR signaling via modulating NFκB activity in B cells is not known.
It is interesting to note, that unlike TLR4, TLR7, and TLR9 are not expressed on the cell surface, but are instead localized to the endosomes. CD22 is known to constantly recycle between the cell surface and the endosomes und can therefore possibly colocalize with the TLRs (Figure 2). It would be interesting to study whether the proximity to the TLRs is necessary for Siglec-G and CD22 to inhibit TLR-mediated proliferation or whether the inhibition is mediated in a more indirect manner. Other Siglecs have also been shown to inhibit TLR-mediated signaling (Blasius and Colonna, 2006; Boyd et al., 2009) suggesting that the ability to modulate TLR signals might be a function shared by the whole Siglec family. Siglec-E has been shown to recruit SHP-1 and SHP-2 after LPS stimulation and to inhibit TLR-mediated NFκB activation and cytokine production (Boyd et al., 2009).
The possible involvement of Siglec-G and CD22 in TLR signaling is interesting in several ways. Stimulation of B cells with TLR ligands can lead to proliferation, upregulation of the plasma cell marker Syndecan-1 and antibody secretion in vitro. Since TLRs are germline encoded and the presence of the corresponding ligand will activate all B cells irrespective of their BCR specificity, TLR signaling has been suggested to represent an innate-like immune response in B cells. This aspect of B cell biology has attracted less attention so far than BCR mediated signaling and how TLR signals are modulated on B cells is incompletely understood. Furthermore a combined BCR and TLR signal can lead to the activation of some auto-reactive B cells (Viglianti et al., 2003; Lau et al., 2005) and enhanced TLR signaling can lead to the development of autoimmunity (Deane et al., 2007). Therefore a better understanding of TLR signals on B cells could be relevant to better understand how tolerance can be broken. Siglec-G and CD22 could provide a link between the adaptive and innate immune responses of B cells and it would be interesting to study how BCR- and TLR-transmitted signals can influence each other.
B Cell Development and Differentiation in Siglec-G and CD22-Deficient Mice
Although both Siglec-G and CD22 are expressed from the pre B cell stage onward, neither seems to play a vital role in B cell development in the bone marrow since no defects have been observed in Siglec-G or CD22-deficient mice (Otipoby et al., 1996; O’Keefe et al., 1996; Sato et al., 1996; Nitschke et al., 1997; Hoffmann et al., 2007). Mature B cells are present in normal numbers in CD22-deficient mice but show an increased apoptosis rate after anti-IgM stimulation in vitro and a shortened lifespan in vivo. Decreased survival after anti-IgM stimulation, however does not reflect an altered immune response, since CD22-deficient mice show a normal production of antigen specific antibodies after immunization with thymus-dependent antigen (Otipoby et al., 1996; Sato et al., 1996; Nitschke et al., 1997). This is probably due to other signals that can rescue survival of activated CD22-deficient B cells.
Nevertheless, CD22 deficiency does affect the dynamic of a thymus-dependent response to some degree. A recent study indicates that the initial proliferative burst after a thymus-dependent immune response is stronger if CD22 is absent. Also the formation of germinal centers and development of plasma blasts seems to be more rapid in the initial phase of the immune response, however the number of antibody producing cells decreases at later time points (Onodera et al., 2008).
Although follicular B cells in lymph nodes and spleen are not affected in their cell numbers, mature recirculating B cells are lacking in the bone marrow of CD22-deficient mice (Otipoby et al., 1996; O’Keefe et al., 1996; Sato et al., 1996; Nitschke et al., 1997). Mice expressing CD22 with a mutated ligand binding domain display a similar phenotype (Poe et al., 2004). Notably, bone marrow sinusoidal epithelium has been shown to express CD22 ligands and injecting wild type mice with CD22–Fc to mask CD22 ligands leads to a reduction of mature B cell numbers in the bone marrow (Nitschke et al., 1999). This suggests that CD22 interactions with its ligands are essential for the maintenance of recirculating B cells in the bone marrow and that CD22 might possess specific functions as an adhesion molecule (Figure 4).
Another B cell population strongly affected by the loss of CD22 is the marginal zone B cells. Marginal zone B cells reside in close proximity to the marginal sinus in the spleen and are believed to play a crucial role in thymus-independent immune response (Martin and Kearney, 2002). This population of B cells is strongly reduced in CD22-deficient mice (Samardzic et al., 2002). In agreement with this finding CD22-deficient mice show a reduced immune response to thymus-independent antigen (Otipoby et al., 1996; Nitschke et al., 1997). It has been suggested that a strong BCR signal will favor the development of follicular B cells (and B1 cells) while a weak signal will bias the development toward the marginal zone B cell lineage (Pillai et al., 2004). Knock-in mice expressing CD22 with a mutated ligand binding domain show a similar reduction of marginal zone B cells but display weak calcium signaling after anti-IgM stimulation (Poe et al., 2004). This would indicate that the function of CD22 as an adhesive molecule is crucial for placing B cells in the marginal zone. However, knock-in mice expressing CD22 with mutated ITIM motifs show a similar MZ B cell defect (I. Obermeier, J., Müller, M., Wöhner, L., Nitschke, unpublished results). Thus it is unclear whether direct ligand binding or modulation of signaling by CD22 determines MZ B cell numbers. It is also possible that altered survival or proliferation of CD22-deficient B cells affects different B cell populations to a different degree and therefore mainly marginal zone B cells are affected.
Another B cell subset involved mainly in the immune response to thymus-independent antigens are the B1 cells. Unlike the marginal zone B cells, B1 cells seem to need a strong BCR signal for their generation and/or maintenance. Several gene targeted mouse lines with decreased BCR signaling show a diminished B1 cell population. On the other hand enhanced BCR mediated signaling due to the loss of signaling molecules known to dampen the BCR signal or over expression of positive regulators is often associated with increased numbers of B1a cells (Berland and Wortis, 2002). Surprisingly, the absence of CD22 does not seem to affect B1 cell numbers strongly. A slightly increased population of B1 cells has been observed in two out of four independent CD22 knockout lines generated (O’Keefe et al., 1996; Sato et al., 1998). CD22-deficient mice on a pure C57BL/6 background show normal B1 cell numbers (Nitschke et al., 1997). Although CD22 is present on B1 cells, these results suggest that CD22 is not the dominant inhibitor in BCR-transmitted signaling in B1 cells. In contrast, the loss of Siglec-G is not counterbalanced by other proteins in B1 cells from Siglec-G-deficient mice and thus Siglecg−/− mice show highly increased B1 cell numbers, as well as highly increased calcium mobilization in their B1 cells, when compared to wild type B1 cells (Hoffmann et al., 2007).
Siglec-G is a direct target of the transcription factor Pax5 (Schebesta et al., 2007) and expressed throughout the B cell lineage with the highest expression on B1 cells (Hoffmann et al., 2007). Most B cell populations do not seem to be significantly affected by the loss of Siglec-G since Siglec-G-deficient mice show normal development of follicular and marginal zone B cells and a largely normal immune response to thymus-dependent antigen. In contrast, the population of B1 cells is highly enlarged in Siglec-G-deficient mice. B1 cells differ from conventional B cells through their surface phenotype, localization, function, and ontogenesis. B1 cells are the main producers of natural IgM and in agreement with this, serum IgM levels are strongly increased in Siglec-G-deficient mice (Hoffmann et al., 2007). B1 cells can be found in a low percentage in the spleen, but are the dominant B cell population in the peritoneal cavity. Two distinct B1 subsets have been identified in the peritoneum – the B1a (CD5+, Mac1+, CD43+) and B1b (CD5−, Mac1+, CD43+) cells. All B1 cell subsets are found in increased numbers in Siglec-G-deficient mice with the largest expansion seen in the B1a cell population in the peritoneal cavity. This expansion is cell intrinsic as has been shown by adoptive transfer of mixed bone marrow cells (Hoffmann et al., 2007). The development of B1a cells in wild type mice is still a matter of discussion. Most B1a cells originate from the fetal liver and stable numbers are maintained by self-renewal. But the adult bone marrow has not lost the capability to generate B1a cells completely. Precursor cells have been identified that preferentially give raise to B1 cells and not to conventional B2 cells (Montecino-Rodriguez et al., 2006). These precursors are abundant in fetal liver and less frequent in adult bone marrow. However the mechanisms leading to the development of B1 cells are probably more plastic since there is evidence that the common lymphoid progenitor can also give rise to B1 cells (Esplin et al., 2009). As discussed, the strength of BCR signaling dramatically influences B1 cell numbers and an enhanced BCR signal leads to increased B1 cell numbers. This is consistent with the phenotype observed in Siglec-G-deficient mice. A recent study by Brenner et al. has provided the first evidence, that the BCR signal indeed might play a significant role in the observed B1a cell expansion. Siglec-G-deficient mice crossed to IgM hypomorphic mice show a normal B1a cell population. Due to the decreased levels of surface IgM the B1a population is nearly absent in IgM hypomorphic mice. When crossed to Siglec-G-deficient mice the effect of a low IgM expression is probably compensated for by the loss of negative regulation through Siglec-G and the resulting phenotype is intermediate between Siglec-G-deficient and IgM hypomorphic mice leading to approximately wildtype B1 numbers (Brenner et al., 2011).
Siglec-G-deficient B1a cells show prolonged survival in vitro and also in vivo when adoptively transferred into Rag1-deficient mice. This might lead to an accumulation of the cells and thereby explain the enlarged population observed in Siglec-G-deficient mice. The exact biochemical pathway leading to prolonged survival of Siglec-G-deficient mice is not known but might include the over expression of the transcription factor NFATc1. NFATc1 has been suggested to play a protective role against anti-IgM induced apoptosis in B cells. Furthermore, the loss of NFATc1 in gene targeted mice specifically suppresses the development of B1 cells but not B2 cells (Berland and Wortis, 2003; Bhattacharyya et al., 2011). Therefore it is tempting to speculate, that increased calcium signaling in Siglec-G-deficient mice results in higher levels of NFATc1 and subsequently in a prolonged lifespan. However, experiments probing the contribution of increased calcium signaling to the prolonged survival are needed to verify this hypothesis.
Another factor that may contribute to increase B1a cell numbers in Siglec-G-deficient mice is an altered selection of B cells into the pool of B1 cells. Fetal liver derived B1a cells show a distinct VDJ segment usage and the repertoire of produced antibodies is dominated by specificities for common bacterial structures (Baumgarth et al., 2005). Analysis of the VDJ recombination in Siglec-G-deficient B1a cells and studies of the secreted antibodies have shown that the BCR repertoire of Siglec-G-deficient B1a cells differs significantly from the repertoire of wild type B1a cells (Jellusova et al., 2010a). Similarities in the VDJ segment usage between Siglec-G-deficient B1a cells and the VDJ segment usage reported for B cells derived from the adult bone marrow led to the speculation that the fate decision of developing B cells is altered in Siglec-G-deficient mice and that more bone marrow-derived B cells are selected into the B1a cell pool as in wild type mice.
Mature follicular B cells show only a mild phenotype in either Siglec-G or CD22-deficient mice. Since both Siglecs are expressed on this population it is possible that Siglec-G and CD22 might have partly redundant functions. Indeed, Siglec-G × CD22 double-deficient mice display phenotypical features not observed in either Siglec-G or CD22 single-deficient mice. The B1 cell population in Siglec-G × CD22 double-deficient mice is enlarged, even to a higher extent than in Siglec-G single-deficient mice. Also, conventional B2 cells show a pre-activated phenotype and express higher levels of CD5, B7.2, and lower levels of CD23 (Jellusova et al., 2010b). Surprisingly in contrast to CD22 single-deficient mice, Siglec-G × CD22 double-deficient mice display an increased population of marginal zone B cells. Serum IgM levels in Siglec-G × CD22 double-deficient mice are highly elevated and exceed even those observed in Siglec-G single-deficient mice. While neither Siglec-G nor CD22 single-deficient mice show a defect in the immune response to thymus-dependent antigens, Siglec-G × CD22 double-deficient mice show a delayed and weaker production of antibodies after an immunization with NP-ovalbumin. This immunodeficient phenotype is surprising since all B cell populations (B1, follicular B, and MZ B cells) show enhanced calcium signaling after anti-IgM stimulation. A similar phenotype is observed in mice with a B cell specific SHP-1 deletion (Pao et al., 2007). These mice also show an enlarged population of B1 cells, increased BCR mediated calcium signaling in this population and a weak immune response to thymus-dependent antigen.
CD22 and Siglec-G Ligand Binding
While it is well established that a crucial function of CD22 and Siglec-G is to inhibit B cell signaling, the important question remains under which physiological situations the inhibitory signal is needed and how the inhibitory role of CD22 and Siglec-G is regulated. Recent studies suggest that ligand binding might play a crucial role. Both CD22 and Siglec-G contain an N-terminal V-set Ig domain, which mediates binding to α2–6 linked sialic acids (Powell et al., 1993; Engel et al., 1995; Varki and Angata, 2006). Siglec-G can additionally bind sialic acids with an α2–3 linkage (Duong et al., 2010,). Sialic acids with an α2–6 linkage are present on different plasma proteins such as haptoglobin or soluble IgM (Hanasaki et al., 1995; Adachi et al., 2012) and sialic acids are also abundantly expressed on the surface of many cells. Cells expressing sialic acid residues include T cells (Bi and Baum, 2009), cytokine activated endothelial cells (Hanasaki et al., 1994), monocytes (Stamatos et al., 2004), erythrocytes (Aminoff et al., 1977), and also B cells (Engel et al., 1993). Siglec-G and CD22 can therefore engage in many different interactions and bind ligands in trans on adjacent cell surfaces or in cis on the same cell surface. However, probably not all possible ligand interactions are relevant in physiological context.
The abundant expression of CD22 ligands on the cell surface of B cells appears to “mask” most of the CD22 molecules and limits its availability to trans ligands. Resting B cells only bind high affinity exogenous ligands in vitro and need to be treated with sialidase to bind ligands of lower affinity (Razi and Varki, 1998). Masking by endogenous ligands has been also shown for other Siglecs and therefore seems to be a general property of the Siglec family (Razi and Varki, 1999; Nakamura et al., 2002; Avril et al., 2006). Several proteins on B cells carry Sia residues and hence could be possible cis ligands. IgM, CD45, and CD22 itself are among the sialylated proteins. CD22 has been shown to bind IgM and CD45 and the proximity of CD22 to the B cell receptor seems to be crucial for its inhibitory function (Doody et al., 1995). However this interaction does not seem to depend on sialic acid-binding and is rather a product of protein protein interactions (Zhang and Varki, 2004). Instead CD22 itself seems to be the prominent cis ligand and CD22 homomultimers can be found on resting B cells (Figure 3; Han et al., 2005).
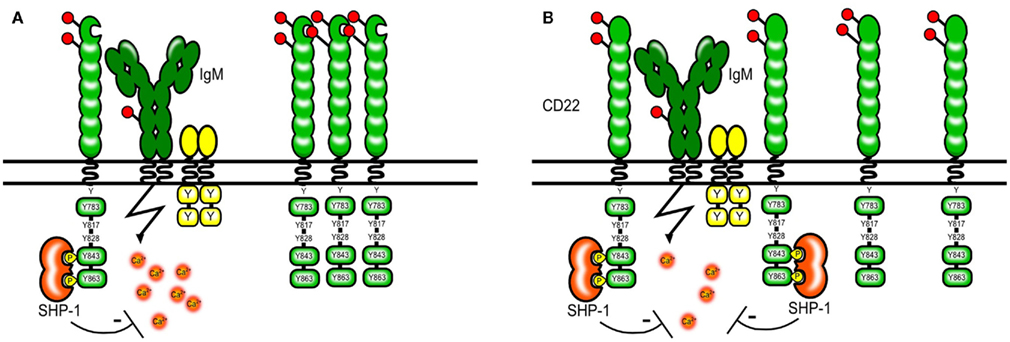
Figure 3. Model of CD22 cis interactions. Several proteins on the B cell surface are sialylated, however a large part of the CD22 molecules seems to interact via sialic acid-binding mainly with other CD22 molecules [shown in (A)]. The formation of homomultimers is disrupted in B cells from mice expressing CD22 that cannot bind sialic acids. This makes CD22 more available to interactions with the BCR and subsequently leads to a stronger inhibition of the BCR signal [shown in (B)]. A similar situation might arise in ST6Gal-deficient mice lacking CD22 ligands.
Even though the availability of CD22 for trans interactions on resting mature B cells seems to be very limited, there are some specific B cell populations with constantly unmasked CD22. A small part of freshly isolated splenic cells can be stained with a streptavidin-based sialylated artificial CD22 ligand without sialidase pretreatment. The frequency of B cells with unmasked CD22 is higher in marginal zone B cells, transitional B cells, and peritoneal B1a compared to mature splenic B cells (Danzer et al., 2003). B cells with unmasked CD22 are also enriched in the bone marrow (Floyd et al., 2000). It is tempting to speculate that unmasked CD22 indicates an activated state of these B cells, since unmasked murine splenic B cells have been shown to express higher levels of the activation markers CD69, MHCII, and B7.2 (Danzer et al., 2003) and in subpopulations of human B cells CD22 becomes unmasked after anti-IgM and CD40 stimulation (Razi and Varki, 1998).
To study whether ligand binding contributes to the inhibitory function of CD22 on BCR mediated signaling CD22 mutant mice were generated that either completely lack the ligand binding domain or harbor mutations in R130 and R137 which are critical amino acids for CD22–sialic acid interactions (Poe et al., 2004). Studies using these knock-in mice have shown that Sia binding modifies CD22 activity, however in a – at the first sight – unexpected way. It has been shown that simultaneous engagement of CD22 and the BCR with a sialylated antigen effectively inhibits the BCR signal (Courtney et al., 2009). This might happen in a natural environment when antigens binding to the BCR contain also sialylated structures and are therefore recognized by CD22. Therefore, the ability of CD22 to bind sialic acids appears to be crucial for its inhibitory function. This has been at first confirmed by studies using cell lines from Cd22−/− mice transfected with CD22 mutants. These cell lines with CD22 mutants lacking the sia binding domain or with R130A and R137E point mutations showed increased calcium signaling after anti-IgM stimulation (Jin et al., 2002). Also, treatment of B cell lines with high affinity artificial CD22 ligands led to increased BCR-induced Ca2+ (Kelm et al., 2002). Surprisingly, these findings could not be confirmed in gene targeted mice carrying similar mutations. Freshly isolated splenic B cells from CD22 mutant mice with disrupted ligand binding showed a normal or even a lower calcium signal after anti-IgM stimulation than wildtype mice (Poe et al., 2004; I. Obermeier, J., Müller, M., Wöhner, L., Nitschke, unpublished results). Several models explaining this finding have been proposed but none of them can unify the results obtained from cell lines and mouse primary B cells. The current model holds that in unstimulated B cells, CD22 forms homomultimers and is not associated with the BCR. Through photoaffinity crosslinking Han et al. (2005) have shown that even though multiple proteins on the B cell surface are sialylated and can be bound by CD22–Fc molecules in vitro, CD22 binds sialic acids only on other CD22 molecules in situ. Disrupting 2,6 Sia-binding and therefore the formation of multimers might free CD22 and change the distribution of the molecules so that more of them are now available for interactions with the BCR (Figure 3.) This would explain why the calcium signal is normal or decreased in B cells from CD22 knock-in mice with disrupted ligand binding. However, it is also possible that our understanding of the biological changes in these B cells is not complete since they show a pre-activated phenotype with lower levels of surface IgM and the responsiveness to anti-IgM might be affected also by other factors.
In addition to cis-binding, CD22 has also been suggested to engage in B cell–T cell or B cell–B cell interactions. B cell activation by antigen displayed on the surface of a target cell can be suppressed when the target cell co-expresses a 2,6 linked sialic acids. This interaction with target cells which co-express antigen and 2,6 Sia may induce co-ligation of CD22 and the BCR, leading to enhanced CD22-mediated inhibition (Lanoue et al., 2002; Figure 4.) In homotypic interactions, CD22 colocalizes with the BCR in clustered B cells at the site of contact and is believed to bind IgM on opposing B cells (Ramya et al., 2010). B cells deficient in ST6Gal, an enzyme creating CD22 ligands, show an equal CD22 distribution on the cell surface, however if in contact with wild type cells distribution to the site of contact is restored. In contrast, wild type B cells fail to do so when in contact with ST6Gal-deficient B cells showing that it is the ligand on the neighboring cell that recruits CD22 to the site of contact (Collins et al., 2004). This indicates that ligand binding by CD22 is a dynamic process and even though CD22 molecules appear to be masked on resting B cells they are not completely unavailable to trans interactions. Another CD22 trans ligand could be CD45 and CD22 has been reported to recognize CD45 on T cells (Sgroi et al., 1995). However the physiological relevance of CD22-mediated T cell–B cell and B cell–B cell interactions remains elusive. As already mentioned, the sinusoidal endothelium of the bone marrow also contains CD22 ligands and these ligands seem to be important for the retention of mature recirculating B cells (Nitschke et al., 1999). It is possible that the role of CD22 as an adhesion molecule is independent of its role as a signaling molecule in the BCR cascade and therefore studies addressing these functions in separate models are needed. The interaction of CD22 with cells might be even more complex and exceed α2,6 Sia mediated binding. One study reports cell contact dependent but sia independent inhibition of B cell proliferation by bone marrow dendritic cells (Santos et al., 2008).
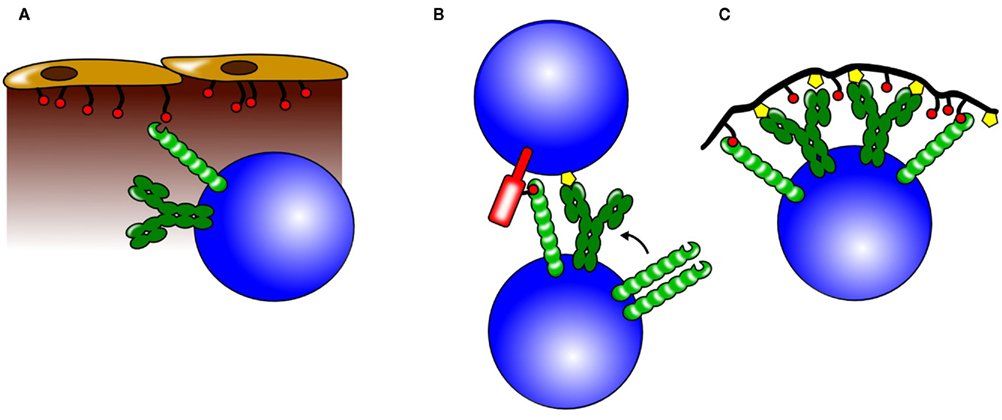
Figure 4. Possible roles for CD22 trans interactions. CD22 can bind ligands on neighboring cells and also on soluble proteins and thereby influence different biological processes. (A) Through binding of α2,6 sialic acids on sinusoidal endothelium, CD22 is believed to play a crucial role in the retention or homing of mature B cells to the bone marrow. (B) CD22 can participate in cell –cell interactions by binding to sialylated ligands on the opposing cell. Increased colocalization of CD22 and BCR molecules could lead to a stronger CD22-mediated inhibition. (C) Antigens engaging the BCR (yellow symbols), but also containing sialic acids (red circles) recruit CD22 to the BCR. This leads to a stronger inhibition of the BCR signal and might be one of the mechanisms how tolerance to self-antigens containing CD22 ligands is maintained.
Apart from interactions with proteins expressed on the cell surface, CD22 can also bind secreted proteins. Soluble immunoglobulins can contain sialylated epitopes and can be recognized by CD22 under certain conditions. Treatment of NP-specific B cells with NP ligated to IgM, simulating an antigen–antibody complex, resulted in higher calcium if sialylation of the complex was removed by neuraminidase treatment. Relative inhibition of the calcium signal depending on the sialylation status of the complex was not observed in CD22-deficient control B cells. Thus the sialylated residues linked to serum IgM might serve as a trans ligand for CD22 and trigger an inhibitory signal if the BCR is simultaneously activated (Adachi et al., 2012).
During an immune response B cells are induced to switch to produce antibodies of the IgG isotype. IgG antibodies can mediate pro-inflammatory but also anti-inflammatory functions resulting from differential sialylation of the Fc domain (Kaneko et al., 2006). An example for medical use of the anti-inflammatory properties of IgG is intravenous immunoglobulin (IVIG). IVIG consists mostly of human polyspecific IgG prepared from the plasma of several thousand donors. It is widely used to treat humans suffering from different autoimmune diseases including systemic lupus erythematosus (SLE). The exact mechanism how IVIG influences immune functions is still not clearly defined but may be a combination of several different processes. IVIG has been shown to reduce the viability of tonsilar B cells. Binding of IVIG was shown to modulate signaling after anti-IgM stimulation leading to cell cycle arrest and apoptosis of the cells. The effects of IVIG co-treatment included increased CD22 phosphorylation and decreased phosphorylation of CD19 and BLNK/SLP65 (Seite et al., 2010). These effects were only observed with the fraction enriched in sialylated immunoglobulins but not with the fraction free of sialic acid. Furthermore, IVIG was shown to colocalize with CD22 on the B cell surface and this colocalization was dependent on the presence of sialic acid. It is therefore possible that IVIG can modulate B cell functions by binding to CD22, which might lead to inhibition of cell cycle progression and result in apoptosis of the cells (Seite et al., 2010).
Sialyltransferases Involved in the Synthesis of CD22 Ligands
Since binding of ligands seems to be important for several CD22-mediated functions, further insight into the role of CD22 can be gained by studying mice deficient in specific enzymes creating these sugar structures. Sialic acids are attached to glycans via α2,3, α2,6, and α2,8 linkages and these glycan modifications are mediated by distinct enzymes. Four types of β-galactoside α2,3 sialyltransferases (ST3Gal) have been identified in mice and α2,3 linked sialic acids can be found on a great variety of cells (Kono et al., 1997). Alpha 2,6 linked sialic acids are less common and can be generated by several different enzymes (Takashima, 2008).
CD22 requires α2,6 linked sialic acid to galactose as a ligand and the ST6Gal I sialyltransferase is unique in its function of producing these ligands (Collins et al., 2002) and has been found to be expressed in hematopoietic cells and liver.
ST6GalI-deficient mice lack CD22 ligands and phenocopy CD22 knock-in mice with a mutated ligand binding domain in several ways. Isolated splenic B cells show lower calcium mobilization after IgM engagement than wildtype cells, the expression of CD22 and IgM on the cell surface is reduced and the B cells show suboptimal BCR-induced proliferation (Hennet et al., 1998; Poe et al., 2004). Since ST6GalI-deficient mice display a global change in sialylation and therefore other proteins might be affected in their function, one could argue that decreased calcium signaling could be a byproduct of these alterations and not dependent on CD22. However St6GalI × CD22 double-deficient mice show a calcium flux comparable with CD22-deficient mice. This indicates that the loss of CD22 function is dominant and affects the signal even in ST6GalI-deficient B cells (Collins et al., 2006b; Ghosh et al., 2006). It is tempting to speculate that decreased calcium signaling in St6GalI-deficient B cells as well as in CD22 knock-in cells is a product of an enhanced CD22 signal rather than a diminished. ST6GalI-deficient B cells show increased association of CD22 with the BCR (Collins et al., 2006b) and a stronger IgM induced CD22 phosphorylation.
B cell development in ST6GalI-deficient mice is normal, but the population of recirculating mature B cells in the bone marrow and the marginal zone B cells in the spleen are reduced (Collins et al., 2006b; Ghosh et al., 2006). This is in agreement with the phenotype observed in CD22 knock-in and CD22-deficient mice and supports the model, that CD22–Sia interactions are important for those two B cell subsets. Unlike CD22-deficient mice, ST6GalI-deficient mice show severely impaired immune responses to both thymus-independent and thymus-dependent antigens (Hennet et al., 1998). The exact mechanism leading to this phenotype is not clarified yet. It is possible that the stronger inhibition by CD22 renders the B cells irresponsive to activation. However, it is interesting that Siglec-G × CD22 double-deficient mice also respond only weakly to both thymus-dependent and thymus-independent immunization even though they show a very strong calcium signal after anti-IgM stimulation (Jellusova et al., 2010b). Siglec-G binds α2,6 Sia ligands, as well as a2,3 Sia ligands, and therefore a substantial part of its ligands might be missing in St6gal−/− mice.
Another enzyme associated with the generation of CD22 ligands is CMP-Neu5Ac hydroxylase (Cmah). In mice, Sia can be found in two main forms – Neu5Ac and Neu5Gc. Both forms differ only in one oxygen molecule added to C5 to form Neu5Gc. The enzyme Cmah is responsible for this modification and changes Neu5Ac to Neu5Gc. Humans lost the enzyme during evolution and therefore express exclusively Neu5Ac (Varki, 2009). While human CD22 can bind both forms of sialic acids, murine CD22 shows preferential binding to Neu5Gc. Splenic cells from Cmah-deficient mice show substantially weaker binding of a chimeric mCD22–Fc protein (a fusion protein consisting of the N-terminal domain of mouse CD22 and the Fc region of human IgG1) than wildtype cells, demonstrating strong reduction of CD22 ligands in these cells (Naito et al., 2007). Interestingly, the rat monoclonal antibody GL7 has been found to specifically bind the Neu5Ac sialic acid form linked to the sugar backbone in an α2,6 linkage. GL7 is widely used to identify germinal center B cells. Also LPS activated B cells and some B cells in the bone marrow can be stained with GL7 (Cervenak et al., 2001). This indicates that the expression of CD22 ligands changes with the activation status of B cells. Indeed the expression of Cmah is strongly reduced in germinal center B cells and mCD22–Fc binding is diminished (Naito et al., 2007). Most in vitro experiments performed to study the biochemical pathways influenced by CD22 have been conducted with naïve splenic B cells. It could be interesting to extend these studies to germinal center B cells, since the lack of CD22 ligands indicates that interaction between CD22 and other proteins might be different in this specific B cell population.
Cmah-deficient mice show a subtle phenotype reminiscent of CD22-deficient mice rather than St6Gal-deficient mice. The immune response to a thymus-independent antigen is increased, and normal to thymus-dependent antigen. Calcium signaling after anti-IgM stimulation is enhanced and the marginal zone and recirculating B cell populations are decreased (Naito et al., 2007).
Another common sialic acid modification is O-acetylation on the C9 position. In vitro studies have shown that CD22 binding to acetylated sialic acids on the C9 position is impaired (Sjoberg et al., 1994). The enzyme Sialate: O-acetyl esterase (Siae) is capable of removing acetyl groups from the 9-OH position of α2,6 sialic acids. This enzyme is upregulated in mature B cells, suggesting a role in B cell biology. The phenotype of Siae-deficient mice is again similar to the phenotype of CD22-deficient mice. Calcium signaling after BCR stimulation is elevated, the populations of marginal zone and recirculating B cells are decreased in their numbers (Cariappa et al., 2009).
Regulation of CD22 Surface Expression and Endocytosis
CD22 functionality can be regulated by cis-ligand binding of CD22 which affects association to the BCR, but also the expression levels of CD22 on the cell surface can be modulated. CD22 surface expression on conventional B2 cells is decreased after IgM ligation, but increased after LPS or anti-CD40 stimulation. The down regulation of surface CD22 after BCR crosslinking is probably a result of the decreased CD22 mRNA levels observed after 24 h of cell culture (Lajaunias et al., 2002). Furthermore, CD22 expression on the cell surface can be regulated by modulating CD22 internalization. CD22 can be recruited to clathrin-rich domains and internalized after stimulation with monoclonal antibodies or high affinity sialosides (Chan et al., 1998; Collins et al., 2006a). CD22 has been shown to interact with the adapter protein AP50, a subunit of the AP-2 complex. This complex is known to mediate recruitment of different receptors into clathrin coated pits. CD22 is believed to interact with AP50 via its tyrosine motifs and the removal of the cytoplasmic tail abolishes CD22 endocytosis. Also, while the treatment of B cells with monoclonal anti-CD22 antibodies promotes the internalization of CD22, BCR stimulation and subsequent CD22 phosphorylation transiently inhibit CD22 endocytosis (John et al., 2003). After internalization CD22 is able to release bound ligands and recycle back to the cell surface. In contrast, monoclonal antibodies bound to CD22 are believed to recycle together with CD22 to the cell surface (O’Reilly et al., 2011). This finding is important when constructs are designed to target CD22 in B cell lymphomas.
Role of CD22 in B Cell Proliferation and Apoptosis
B cell antigen receptor mediated signaling can lead to apoptosis or proliferation depending on the developmental status of B cells and other costimulatory or inhibitory signals. Equally, the impact of CD22 signaling on B cell proliferation and apoptosis varies depending on the context of other signals. CD22-deficient B cells show decreased proliferation to anti-IgM stimulation, but normal or increased proliferation to TLR or CD40 stimulation (Nitschke et al., 1997). The observed differences could be a consequence of a missing CD22 signal and might show a direct role of CD22 signaling in these signaling pathways. However, it is also possible that altered proliferative responses are an indirect product of CD22 deficiency, since CD22-deficient B cells show a pre-activated phenotype. Nevertheless, CD22 crosslinking with monoclonal antibodies can change the proliferative and survival properties of B cells depending on additional signals provided, the type of B cells and even the type of the chosen antibody. Antibodies blocking the ligand binding domain of human CD22 such as the monoclonal antibody HB22.7 have been shown to induce apoptosis in B cell lymphoma cell lines (Tuscano et al., 1999). This effect is not seen with antibodies targeting other parts of the CD22 molecule. However, how this stimulation relates to physiological functions of CD22 is unclear. Interestingly, mutated CD22 lacking a large part of the cytoplasmic tail was found in infants diagnosed with B-precursor leukemia (Uckun et al., 2010). Leukemic cells expressing CD22 in this truncated form were resistant to apoptosis induction by the HB22.7 monoclonal antibody and caused leukemia when injected into SCID mice. Transgenic mice expressing human CD22 with deleted exon 12 developed B cell hyperplasia. This suggests that dysregulated CD22 signaling can have proliferation-promoting effects on B cells (Uckun et al., 2010).
The Role of CD22 and Siglec-G in Tolerance and Autoimmunity
B cell activation needs to be tightly regulated to avoid autoimmunity while maintaining an adequate immune response to pathogens. Hyperresponsiveness of B cells due to defective inhibition of the BCR signal often results in the development of autoimmunity.
Mice lacking the inhibitory receptor FcγRIIb on a C57BL/6 background develop anti-nuclear autoantibodies leading to glomerulonephritis and increased mortality (Bolland and Ravetch, 2000). Also mice deficient in cytosolic signaling molecules involved in negative signaling pathways such as SHP-1 and Lyn (Chan et al., 1997) show signs of autoimmunity as well. Both CD22 and Siglec-G are negative regulators of the BCR signal, therefore it was expected that the loss of either of these proteins would predispose to autoimmunity. Several studies have linked CD22 deficiency to the development of autoimmunity. In one out of four separately generated Cd22−/− lines autoantibodies could be detected in aging mice (O’Keefe et al., 1999). When CD22-deficient mice were crossed to Y linked autoimmune accelerator (YAA) mice even heterozygosity would lead to the production of IgG autoantibodies (Mary et al., 2000). However, caution is needed when interpreting these results. These studies have been performed using a CD22 line generated with the aid of 129 embryonic stem cells and subsequently crossed to the C57BL/6 background. It has recently been shown that several 129-derived loci contribute to the development of autoimmunity on a C57/BL6 background (Bygrave et al., 2004; Heidari et al., 2006). CD22 is known to be in close proximity to one of those loci on the murine chromosome 7. Since this locus is linked to the CD22 locus they might be inherited together in CD22-deficient mice. In contrast, this locus might be lost in the process of backcrossing in wild type controls since the gene for wild type CD22 will be derived from the C57BL/6 background. Therefore mice generated with this breeding strategy might show a higher production of autoantibodies in CD22-deficient animals compared to the control animals irrespective of the CD22 deficiency. Indeed, mice generated with C57BL/6 embryonic stem cells do not show any signs of autoimmunity. Therefore the loss of CD22 alone does not seem to be enough for spontaneous development of autoantibodies. However, CD22 deficiency might increase the susceptibility to autoimmune disorders on a background where other factors predispose to autoimmunity. Notable, several mouse strains prone to develop autoimmunity such as NZW, MRL, and BXSB carry different CD22 alleles compared to the mouse strains C57BL/6 and BALB/c (Lajaunias et al., 1999; Mary et al., 2000). The Cd22a allele and the related Cd22c allele associated with lupus prone genetic backgrounds lead to the expression of aberrant CD22 proteins. The truncated CD22 protein in Cd22a strains is constitutively unmasked and is possibly impaired in its ability to bind natural ligands. Alternative splicing of the Cd22a mRNA can lead to deletions in the ligand binding domain (Mary et al., 2000). To assess the functional effect of the aberrant Cd22a allele on a normal genetic background B6.CD22 congenic mice have been generated. B cells from these mice show a similar phenotype to CD22 knock-in mice with a mutated ligand binding domain. They show decreased surface IgM expression and increased expression of MHCII. Calcium signaling after anti-IgM stimulation seems to be normal (Nitschke et al., 2006).
Siglec-G-deficient mice show an increased population of B1a cells and elevated serum IgM levels. Multiple autoimmune-prone mouse strains (e.g., NZB/W, NZB) have enlarged populations of B1 cells. Also in several gene targeted mice autoimmunity correlates with increased B1 cell numbers. Although controversially discussed, B1 cells might play a direct role in the development of autoimmunity. The BCR repertoire of B1 cells is skewed toward polyreactive, weakly auto-reactive antibodies. These antibodies are usually of the IgM isotype and not pathogenic under normal conditions. However dysregulated B cell signaling could possibly lead to isotype switching and to the development of high affinity IgG autoantibodies. Also, B1 cells might contribute to the development of autoimmune pathologies through antigen presentation or secretion of cytokines (Duan and Morel, 2006). However Siglec-G-deficient mice do not develop high affinity IgG autoantibodies and also no signs of renal dysfunction are observed. The only potentially pathogenic autoantibodies found in aged Siglec-G-deficient mice are Rhesus factor IgM and anti-erythrocyte IgM (Hoffmann et al., 2007). However no differences in survival or general well-being have been observed between wild type and Siglec-G-deficient mice. Both types of autoantibodies found are of the IgM isotype and probably reflect the overall increased levels of serum IgM. This demonstrates that an enlarged B1a cell population and hyperactive B1 cells are not sufficient to spontaneously develop autoimmunity.
Neither Siglec-G deficiency nor CD22 ablation alone seem to result in spontaneous production of autoantibodies. However, as already mentioned Siglec-G and CD22 seem to have partly redundant functions. It is therefore possible that the expression of either one of those receptors is sufficient to maintain tolerance and only the ablation of both molecules would have a more profound effect. Indeed, Siglec-G × CD22 double-deficient mice develop significant levels of IgG autoantibodies with specificities including ssDNA, dsDNA, and RNA (Jellusova et al., 2010b). In some of these mice deposition of antibodies in the kidneys could be detected, accompanied by mild glomerular nephritis. Furthermore, Siglecg−/− Cd22+/− and Siglecg+/− Cd22−/− mice also developed autoantibodies although at later time points than Siglec-G × CD22 double-deficient mice (Jellusova et al., 2010b). This suggests that Siglec-G and CD22 are essential for the maintenance of tolerance and also that a possible gene dosage effect could take place since mice with three mutated alleles show development of autoantibodies unlike Siglec-G or CD22 single-deficient mice. An interesting model explaining how Siglec-G and CD22 contribute to the maintenance of tolerance postulates that antigen containing sialylated epitopes in trans will bring Siglec-G and CD22 in close proximity to the BCR and therefore inhibit the immune response. Since sialic acids are abundant in vertebrates, but less frequent in bacteria, sialylated structures might therefore provide a suitable signal to distinguish between self-antigen and foreign structures (Figure 4; Duong et al., 2010).
There is no convincing link between CD22 polymorphisms in humans and autoimmune diseases. Genome-wide association studies have not detected human CD22 or SIGLEC10 as susceptibility loci, e.g., in SLE (Criswell, 2008). However, functionally defective rare variants in the human SIAE gene have been detected as being associated with several human autoimmune diseases (Surolia et al., 2010). A similar phenotype was observed in SIAE-deficient mice. This finding is relevant to Siglec biology, as the enzyme SIAE modifies CD22- and Siglec-G-ligands on B cells, thus affects the same pathway.
Conclusion
Siglec-G and CD22 are two members of the Siglec family expressed on B cells. They share the ability to bind sialic acids and inhibit BCR mediated signaling. They might possess some redundant function as shown with the aid of Siglec-G × CD22 double-deficient mice. The loss of both proteins results in an activated B cell phenotype, increased calcium signaling after anti-IgM stimulation, a weaker immune response and spontaneous production of autoantibodies. Interestingly, Siglec-G and CD22 also play unique roles on specific B cell populations and the loss of either receptor is not functionally compensated through the other in these cells. CD22 seems to play a crucial role in marginal zone B cells in the spleen and mature recirculating B cells in the bone marrow since both B cell populations are diminished in CD22-deficient mice. CD22 seems to be also the dominant inhibitor of calcium signaling in conventional B2 cells. Siglec-G on the other hand is crucial for the inhibition of BCR mediated signaling in B1 cells and also regulates their survival and antibody repertoire. An interesting aspect of the Siglec function is their ability to bind sialic acids and to couple the signal to their regulatory role of the BCR-transmitted signal. Some valuable insight has been already gained in the biological function of ligand binding through CD22, however certain issues still remain unresolved. The role of Siglec-G ligand binding is not characterized yet and would be interesting to study in future.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the DFG for funding this research through SFB643 and NI549/6-2.
References
Ackermann, J. A., Radtke, D., Maurberger, A., Winkler, T. H., and Nitschke, L. (2011). Grb2 regulates B-cell maturation, B-cell memory responses and inhibits B-cell Ca2+ signalling. EMBO J. 30, 1621–1633.
Adachi, T., Harumiya, S., Takematsu, H., Kozutsumi, Y., Wabl, M., Fujimoto, M., and Tedder, T. F. (2012). CD22 serves as a receptor for soluble IgM. Eur. J. Immunol. 42, 241–247.
Aminoff, D., Bruegge, W. F., Bell, W. C., Sarpolis, K., and Williams, R. (1977). Role of sialic acid in survival of erythrocytes in the circulation: interaction of neuraminidase-treated and untreated erythrocytes with spleen and liver at the cellular level. Proc. Natl. Acad. Sci. U.S.A. 74, 1521–1524.
Angata, T. (2006). Molecular diversity and evolution of the Siglec family of cell-surface lectins. Mol. Divers. 10, 555–566.
Angata, T., Kerr, S. C., Greaves, D. R., Varki, N. M., Crocker, P. R., and Varki, A. (2002). Cloning and characterization of human Siglec-11. A recently evolved signaling molecule that can interact with SHP-1 and SHP-2 and is expressed by tissue macrophages, including brain microglia. J. Biol. Chem. 277, 24466–24474.
Angata, T., Margulies, E. H., Green, E. D., and Varki, A. (2004). Large-scale sequencing of the CD33-related Siglec gene cluster in five mammalian species reveals rapid evolution by multiple mechanisms. Proc. Natl. Acad. Sci. U.S.A. 101, 13251–13256.
Angata, T., Tabuchi, Y., Nakamura, K., and Nakamura, M. (2007). Siglec-15: an immune system Siglec conserved throughout vertebrate evolution. Glycobiology 17, 838–846.
Angata, T., and Varki, A. (2002). Chemical diversity in the sialic acids and related alpha-keto acids: an evolutionary perspective. Chem. Rev. 102, 439–469.
Avril, T., North, S. J., Haslam, S. M., Willison, H. J., and Crocker, P. R. (2006). Probing the cis interactions of the inhibitory receptor Siglec-7 with alpha2,8-disialylated ligands on natural killer cells and other leukocytes using glycan-specific antibodies and by analysis of alpha2,8-sialyltransferase gene expression. J. Leukoc. Biol. 80, 787–796.
Baumgarth, N., Tung, J. W., and Herzenberg, L. A. (2005). Inherent specificities in natural antibodies: a key to immune defense against pathogen invasion. Springer Semin. Immunopathol. 26, 347–362.
Berland, R., and Wortis, H. H. (2002). Origins and functions of B-1 cells with notes on the role of CD5. Annu. Rev. Immunol. 20, 253–300.
Berland, R., and Wortis, H. H. (2003). Normal B-1a cell development requires B cell-intrinsic NFATc1 activity. Proc. Natl. Acad. Sci. U.S.A. 100, 13459–13464.
Bhattacharyya, S., Deb, J., Patra, A. K., Thuy Pham, D. A., Chen, W., Vaeth, M., Berberich-Siebelt, F., Klein-Hessling, S., Lamperti, E. D., Reifenberg, K., Jellusova, J., Schweizer, A., Nitschke, L., Leich, E., Rosenwald, A., Brunner, C., Engelmann, S., Bommhardt, U., Avots, A., Muller, M. R., Kondo, E., and Serfling, E. (2011). NFATc1 affects mouse splenic B cell function by controlling the calcineurin – NFAT signaling network. J. Exp. Med. 208, 823–839.
Bi, S., and Baum, L. G. (2009). Sialic acids in T cell development and function. Biochim. Biophys. Acta 1790, 1599–1610.
Blasius, A. L., and Colonna, M. (2006). Sampling and signaling in plasmacytoid dendritic cells: the potential roles of Siglec-H. Trends Immunol. 27, 255–260.
Bolland, S., and Ravetch, J. V. (2000). Spontaneous autoimmune disease in Fc(gamma)RIIB-deficient mice results from strain-specific epistasis. Immunity 13, 277–285.
Boyd, C. R., Orr, S. J., Spence, S., Burrows, J. F., Elliott, J., Carroll, H. P., Brennan, K., Ni Gabhann, J., Coulter, W. A., Jones, C., Crocker, P. R., Johnston, J. A., and Jefferies, C. A. (2009). Siglec-E is up-regulated and phosphorylated following lipopolysaccharide stimulation in order to limit TLR-driven cytokine production. J. Immunol. 183, 7703–7709.
Brenner, S., Drewel, D., Steinbart, T., Weisel, F., Hartel, E., Potzsch, S., Welzel, H., Brandl, A., Yu, P., Mudde, G. C., Schweizer, A., Nitschke, L., and Winkler, T. H. (2011). A hypomorphic IgH-chain allele affects development of B-cell subsets and favours receptor editing. EMBO J. 30, 2705–2718.
Brinkman-Van der Linden, E. C., Hurtado-Ziola, N., Hayakawa, T., Wiggleton, L., Benirschke, K., Varki, A., and Varki, N. (2007). Human-specific expression of Siglec-6 in the placenta. Glycobiology 17, 922–931.
Bygrave, A. E., Rose, K. L., Cortes-Hernandez, J., Warren, J., Rigby, R. J., Cook, H. T., Walport, M. J., Vyse, T. J., and Botto, M. (2004). Spontaneous autoimmunity in 129 and C57BL/6 mice-implications for autoimmunity described in gene-targeted mice. PLoS Biol. 2, e243. doi:10.1371/journal.pbio.0020243
Cao, H., and Crocker, P. R. (2010). Evolution of CD33-related siglecs: regulating host immune functions and escaping pathogen exploitation? Immunology 132, 18–26.
Cariappa, A., Takematsu, H., Liu, H., Diaz, S., Haider, K., Boboila, C., Kalloo, G., Connole, M., Shi, H. N., Varki, N., Varki, A., and Pillai, S. (2009). B cell antigen receptor signal strength and peripheral B cell development are regulated by a 9-O-acetyl sialic acid esterase. J. Exp. Med. 206, 125–138.
Cervenak, L., Magyar, A., Boja, R., and Laszlo, G. (2001). Differential expression of GL7 activation antigen on bone marrow B cell subpopulations and peripheral B cells. Immunol. Lett. 78, 89–96.
Chan, C. H., Wang, J., French, R. R., and Glennie, M. J. (1998). Internalization of the lymphocytic surface protein CD22 is controlled by a novel membrane proximal cytoplasmic motif. J. Biol. Chem. 273, 27809–27815.
Chan, V. W., Meng, F., Soriano, P., Defranco, A. L., and Lowell, C. A. (1997). Characterization of the B lymphocyte populations in Lyn-deficient mice and the role of Lyn in signal initiation and down-regulation. Immunity 7, 69–81.
Chen, G. Y., Tang, J., Zheng, P., and Liu, Y. (2009). CD24 and Siglec-10 selectively repress tissue damage-induced immune responses. Science 323, 1722–1725.
Chen, J., Mclean, P. A., Neel, B. G., Okunade, G., Shull, G. E., and Wortis, H. H. (2004). CD22 attenuates calcium signaling by potentiating plasma membrane calcium-ATPase activity. Nat. Immunol. 5, 651–657.
Chuvpilo, S., Jankevics, E., Tyrsin, D., Akimzhanov, A., Moroz, D., Jha, M. K., Schulze-Luehrmann, J., Santner-Nanan, B., Feoktistova, E., Konig, T., Avots, A., Schmitt, E., Berberich-Siebelt, F., Schimpl, A., and Serfling, E. (2002). Autoregulation of NFATc1/A expression facilitates effector T cells to escape from rapid apoptosis. Immunity 16, 881–895.
Collins, B. E., Blixt, O., Bovin, N. V., Danzer, C. P., Chui, D., Marth, J. D., Nitschke, L., and Paulson, J. C. (2002). Constitutively unmasked CD22 on B cells of ST6Gal I knockout mice: novel sialoside probe for murine CD22. Glycobiology 12, 563–571.
Collins, B. E., Blixt, O., Desieno, A. R., Bovin, N., Marth, J. D., and Paulson, J. C. (2004). Masking of CD22 by cis ligands does not prevent redistribution of CD22 to sites of cell contact. Proc. Natl. Acad. Sci. U.S.A. 101, 6104–6109.
Collins, B. E., Blixt, O., Han, S., Duong, B., Li, H., Nathan, J. K., Bovin, N., and Paulson, J. C. (2006a). High-affinity ligand probes of CD22 overcome the threshold set by cis ligands to allow for binding, endocytosis, and killing of B cells. J. Immunol. 177, 2994–3003.
Collins, B. E., Smith, B. A., Bengtson, P., and Paulson, J. C. (2006b). Ablation of CD22 in ligand-deficient mice restores B cell receptor signaling. Nat. Immunol. 7, 199–206.
Courtney, A. H., Puffer, E. B., Pontrello, J. K., Yang, Z. Q., and Kiessling, L. L. (2009). Sialylated multivalent antigens engage CD22 in trans and inhibit B cell activation. Proc. Natl. Acad. Sci. U.S.A. 106, 2500–2505.
Criswell, L. A. (2008). The genetic contribution to systemic lupus erythematosus. Bull. NYU. Hosp. Jt. Dis. 66, 176–183.
Crocker, P. R., Mucklow, S., Bouckson, V., Mcwilliam, A., Willis, A. C., Gordon, S., Milon, G., Kelm, S., and Bradfield, P. (1994). Sialoadhesin, a macrophage sialic acid binding receptor for haemopoietic cells with 17 immunoglobulin-like domains. EMBO J. 13, 4490–4503.
Crocker, P. R., Paulson, J. C., and Varki, A. (2007). Siglecs and their roles in the immune system. Nat. Rev. Immunol. 7, 255–266.
Danzer, C. P., Collins, B. E., Blixt, O., Paulson, J. C., and Nitschke, L. (2003). Transitional and marginal zone B cells have a high proportion of unmasked CD22: implications for BCR signaling. Int. Immunol. 15, 1137–1147.
Deane, J. A., Pisitkun, P., Barrett, R. S., Feigenbaum, L., Town, T., Ward, J. M., Flavell, R. A., and Bolland, S. (2007). Control of toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity 27, 801–810.
Ding, C., Liu, Y., Wang, Y., Park, B. K., Wang, C. Y., and Zheng, P. (2007). Siglecg limits the size of B1a B cell lineage by down-regulating NFkappaB activation. PLoS ONE 2, e997. doi:10.1371/journal.pone.0000997
Doody, G. M., Justement, L. B., Delibrias, C. C., Matthews, R. J., Lin, J., Thomas, M. L., and Fearon, D. T. (1995). A role in B cell activation for CD22 and the protein tyrosine phosphatase SHP. Science 269, 242–244.
Duong, B. H., Tian, H., Ota, T., Completo, G., Han, S., Vela, J. L., Ota, M., Kubitz, M., Bovin, N., Paulson, J. C., and Nemazee, D. (2010). Decoration of T-independent antigen with ligands for CD22 and Siglec-G can suppress immunity and induce B cell tolerance in vivo. J. Exp. Med. 207, 173–187.
Engel, P., Nojima, Y., Rothstein, D., Zhou, L. J., Wilson, G. L., Kehrl, J. H., and Tedder, T. F. (1993). The same epitope on CD22 of B lymphocytes mediates the adhesion of erythrocytes, T and B lymphocytes, neutrophils, and monocytes. J. Immunol. 150, 4719–4732.
Engel, P., Wagner, N., Miller, A. S., and Tedder, T. F. (1995). Identification of the ligand-binding domains of CD22, a member of the immunoglobulin superfamily that uniquely binds a sialic acid-dependent ligand. J. Exp. Med. 181, 1581–1586.
Esplin, B. L., Welner, R. S., Zhang, Q., Borghesi, L. A., and Kincade, P. W. (2009). A differentiation pathway for B1 cells in adult bone marrow. Proc. Natl. Acad. Sci. U.S.A. 106, 5773–5778.
Floyd, H., Nitschke, L., and Crocker, P. R. (2000). A novel subset of murine B cells that expresses unmasked forms of CD22 is enriched in the bone marrow: implications for B-cell homing to the bone marrow. Immunology 101, 342–347.
Fujimoto, M., Bradney, A. P., Poe, J. C., Steeber, D. A., and Tedder, T. F. (1999). Modulation of B lymphocyte antigen receptor signal transduction by a CD19/CD22 regulatory loop. Immunity 11, 191–200.
Gerlach, J., Ghosh, S., Jumaa, H., Reth, M., Wienands, J., Chan, A. C., and Nitschke, L. (2003). B cell defects in SLP65/BLNK-deficient mice can be partially corrected by the absence of CD22, an inhibitory coreceptor for BCR signaling. Eur. J. Immunol. 33, 3418–3426.
Ghosh, S., Bandulet, C., and Nitschke, L. (2006). Regulation of B cell development and B cell signalling by CD22 and its ligands alpha2,6-linked sialic acids. Int. Immunol. 18, 603–611.
Han, S., Collins, B. E., Bengtson, P., and Paulson, J. C. (2005). Homomultimeric complexes of CD22 in B cells revealed by protein-glycan cross-linking. Nat. Chem. Biol. 1, 93–97.
Hanasaki, K., Powell, L. D., and Varki, A. (1995). Binding of human plasma sialoglycoproteins by the B cell-specific lectin CD22. Selective recognition of immunoglobulin M and haptoglobin. J. Biol. Chem. 270, 7543–7550.
Hanasaki, K., Varki, A., Stamenkovic, I., and Bevilacqua, M. P. (1994). Cytokine-induced beta-galactoside alpha-2,6-sialyltransferase in human endothelial cells mediates alpha 2,6-sialylation of adhesion molecules and CD22 ligands. J. Biol. Chem. 269, 10637–10643.
Heidari, Y., Bygrave, A. E., Rigby, R. J., Rose, K. L., Walport, M. J., Cook, H. T., Vyse, T. J., and Botto, M. (2006). Identification of chromosome intervals from 129 and C57BL/6 mouse strains linked to the development of systemic lupus erythematosus. Genes Immun. 7, 592–599.
Hennet, T., Chui, D., Paulson, J. C., and Marth, J. D. (1998). Immune regulation by the ST6Gal sialyltransferase. Proc. Natl. Acad. Sci. U.S.A. 95, 4504–4509.
Hoffmann, A., Kerr, S., Jellusova, J., Zhang, J., Weisel, F., Wellmann, U., Winkler, T. H., Kneitz, B., Crocker, P. R., and Nitschke, L. (2007). Siglec-G is a B1 cell-inhibitory receptor that controls expansion and calcium signaling of the B1 cell population. Nat. Immunol. 8, 695–704.
Horikawa, K., Martin, S. W., Pogue, S. L., Silver, K., Peng, K., Takatsu, K., and Goodnow, C. C. (2007). Enhancement and suppression of signaling by the conserved tail of IgG memory-type B cell antigen receptors. J. Exp. Med. 204, 759–769.
Ikehara, Y., Ikehara, S. K., and Paulson, J. C. (2004). Negative regulation of T cell receptor signaling by Siglec-7 (p70/AIRM) and Siglec-9. J. Biol. Chem. 279, 43117–43125.
Jang, I. K., Cronshaw, D. G., Xie, L. K., Fang, G., Zhang, J., Oh, H., Fu, Y. X., Gu, H., and Zou, Y. (2011). Growth-factor receptor-bound protein-2 (Grb2) signaling in B cells controls lymphoid follicle organization and germinal center reaction. Proc. Natl. Acad. Sci. U.S.A. 108, 7926–7931.
Jellusova, J., Duber, S., Guckel, E., Binder, C. J., Weiss, S., Voll, R., and Nitschke, L. (2010a). Siglec-G regulates B1 cell survival and selection. J. Immunol. 185, 3277–3284.
Jellusova, J., Wellmann, U., Amann, K., Winkler, T. H., and Nitschke, L. (2010b). CD22 x Siglec-G double-deficient mice have massively increased B1 cell numbers and develop systemic autoimmunity. J. Immunol. 184, 3618–3627.
Jin, L., Mclean, P. A., Neel, B. G., and Wortis, H. H. (2002). Sialic acid binding domains of CD22 are required for negative regulation of B cell receptor signaling. J. Exp. Med. 195, 1199–1205.
John, B., Herrin, B. R., Raman, C., Wang, Y. N., Bobbitt, K. R., Brody, B. A., and Justement, L. B. (2003). The B cell coreceptor CD22 associates with AP50, a clathrin-coated pit adapter protein, via tyrosine-dependent interaction. J. Immunol. 170, 3534–3543.
Kaneko, Y., Nimmerjahn, F., and Ravetch, J. V. (2006). Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science 313, 670–673.
Kawasaki, N., Rademacher, C., and Paulson, J. C. (2011). CD22 regulates adaptive and innate immune responses of B cells. J. Innate Immun. 3, 411–419.
Kelm, S., Gerlach, J., Brossmer, R., Danzer, C. P., and Nitschke, L. (2002). The ligand-binding domain of CD22 is needed for inhibition of the B cell receptor signal, as demonstrated by a novel human CD22-specific inhibitor compound. J. Exp. Med. 195, 1207–1213.
Kono, M., Ohyama, Y., Lee, Y. C., Hamamoto, T., Kojima, N., and Tsuji, S. (1997). Mouse beta-galactoside alpha 2,3-sialyltransferases: comparison of in vitro substrate specificities and tissue specific expression. Glycobiology 7, 469–479.
Lajaunias, F., Ibnou-Zekri, N., Fossati Jimack, L., Chicheportiche, Y., Parkhouse, R. M., Mary, C., Reininger, L., Brighouse, G., and Izui, S. (1999). Polymorphisms in the Cd22 gene of inbred mouse strains. Immunogenetics 49, 991–995.
Lajaunias, F., Nitschke, L., Moll, T., Martinez-Soria, E., Semac, I., Chicheportiche, Y., Parkhouse, R. M., and Izui, S. (2002). Differentially regulated expression and function of CD22 in activated B-1 and B-2 lymphocytes. J. Immunol. 168, 6078–6083.
Lanoue, A., Batista, F. D., Stewart, M., and Neuberger, M. S. (2002). Interaction of CD22 with alpha2,6-linked sialoglycoconjugates: innate recognition of self to dampen B cell autoreactivity? Eur. J. Immunol. 32, 348–355.
Lau, C. M., Broughton, C., Tabor, A. S., Akira, S., Flavell, R. A., Mamula, M. J., Christensen, S. R., Shlomchik, M. J., Viglianti, G. A., Rifkin, I. R., and Marshak-Rothstein, A. (2005). RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J. Exp. Med. 202, 1171–1177.
Law, C. L., Sidorenko, S. P., Chandran, K. A., Zhao, Z., Shen, S. H., Fischer, E. H., and Clark, E. A. (1996). CD22 associates with protein tyrosine phosphatase 1C, Syk, and phospholipase C-gamma(1) upon B cell activation. J. Exp. Med. 183, 547–560.
Li, N., Zhang, W., Wan, T., Zhang, J., Chen, T., Yu, Y., Wang, J., and Cao, X. (2001). Cloning and characterization of Siglec-10, a novel sialic acid binding member of the Ig superfamily, from human dendritic cells. J. Biol. Chem. 276, 28106–28112.
Mary, C., Laporte, C., Parzy, D., Santiago, M. L., Stefani, F., Lajaunias, F., Parkhouse, R. M., O’Keefe, T. L., Neuberger, M. S., Izui, S., and Reininger, L. (2000). Dysregulated expression of the Cd22 gene as a result of a short interspersed nucleotide element insertion in Cd22a lupus-prone mice. J. Immunol. 165, 2987–2996.
Montecino-Rodriguez, E., Leathers, H., and Dorshkind, K. (2006). Identification of a B-1 B cell-specified progenitor. Nat. Immunol. 7, 293–301.
Munday, J., Kerr, S., Ni, J., Cornish, A. L., Zhang, J. Q., Nicoll, G., Floyd, H., Mattei, M. G., Moore, P., Liu, D., and Crocker, P. R. (2001). Identification, characterization and leucocyte expression of Siglec-10, a novel human sialic acid-binding receptor. Biochem. J. 355, 489–497.
Naito, Y., Takematsu, H., Koyama, S., Miyake, S., Yamamoto, H., Fujinawa, R., Sugai, M., Okuno, Y., Tsujimoto, G., Yamaji, T., Hashimoto, Y., Itohara, S., Kawasaki, T., Suzuki, A., and Kozutsumi, Y. (2007). Germinal center marker GL7 probes activation-dependent repression of N-glycolylneuraminic acid, a sialic acid species involved in the negative modulation of B-cell activation. Mol. Cell. Biol. 27, 3008–3022.
Naka, T., Fujimoto, M., Tsutsui, H., and Yoshimura, A. (2005). Negative regulation of cytokine and TLR signalings by SOCS and others. Adv. Immunol. 87, 61–122.
Nakamura, K., Yamaji, T., Crocker, P. R., Suzuki, A., and Hashimoto, Y. (2002). Lymph node macrophages, but not spleen macrophages, express high levels of unmasked sialoadhesin: implication for the adhesive properties of macrophages in vivo. Glycobiology 12, 209–216.
Nitschke, L., Carsetti, R., Ocker, B., Kohler, G., and Lamers, M. C. (1997). CD22 is a negative regulator of B-cell receptor signalling. Curr. Biol. 7, 133–143.
Nitschke, L., Floyd, H., Ferguson, D. J., and Crocker, P. R. (1999). Identification of CD22 ligands on bone marrow sinusoidal endothelium implicated in CD22-dependent homing of recirculating B cells. J. Exp. Med. 189, 1513–1518.
Nitschke, L., Lajaunias, F., Moll, T., Ho, L., Martinez-Soria, E., Kikuchi, S., Santiago-Raber, M. L., Dix, C., Parkhouse, R. M., and Izui, S. (2006). Expression of aberrant forms of CD22 on B lymphocytes in Cd22a lupus-prone mice affects ligand binding. Int. Immunol. 18, 59–68.
O’Keefe, T. L., Williams, G. T., Batista, F. D., and Neuberger, M. S. (1999). Deficiency in CD22, a B cell-specific inhibitory receptor, is sufficient to predispose to development of high affinity autoantibodies. J. Exp. Med. 189, 1307–1313.
O’Keefe, T. L., Williams, G. T., Davies, S. L., and Neuberger, M. S. (1996). Hyperresponsive B cells in CD22-deficient mice. Science 274, 798–801.
Onodera, T., Poe, J. C., Tedder, T. F., and Tsubata, T. (2008). CD22 regulates time course of both B cell division and antibody response. J. Immunol. 180, 907–913.
O’Reilly, M. K., Tian, H., and Paulson, J. C. CD22 is a recycling receptor that can shuttle cargo between the cell surface and endosomal compartments of B cells. (2011). J. Immunol. 186, 1554–1563.
Otipoby, K. L., Andersson, K. B., Draves, K. E., Klaus, S. J., Farr, A. G., Kerner, J. D., Perlmutter, R. M., Law, C. L., and Clark, E. A. (1996). CD22 regulates thymus-independent responses and the lifespan of B cells. Nature 384, 634–637.
Otipoby, K. L., Draves, K. E., and Clark, E. A. (2001). CD22 regulates B cell receptor-mediated signals via two domains that independently recruit Grb2 and SHP-1. J. Biol. Chem. 276, 44315–44322.
Pao, L. I., Lam, K. P., Henderson, J. M., Kutok, J. L., Alimzhanov, M., Nitschke, L., Thomas, M. L., Neel, B. G., and Rajewsky, K. (2007). B cell-specific deletion of protein-tyrosine phosphatase Shp1 promotes B-1a cell development and causes systemic autoimmunity. Immunity 27, 35–48.
Patel, N., Brinkman-Van Der Linden, E. C., Altmann, S. W., Gish, K., Balasubramanian, S., Timans, J. C., Peterson, D., Bell, M. P., Bazan, J. F., Varki, A., and Kastelein, R. A. (1999). OB-BP1/Siglec-6. A leptin- and sialic acid-binding protein of the immunoglobulin superfamily. J. Biol. Chem. 274, 22729–22738.
Peaker, C. J., and Neuberger, M. S. (1993). Association of CD22 with the B cell antigen receptor. Eur. J. Immunol. 23, 1358–1363.
Pillai, S., Cariappa, A., and Moran, S. T. (2004). Positive selection and lineage commitment during peripheral B-lymphocyte development. Immunol. Rev. 197, 206–218.
Poe, J. C., Fujimoto, Y., Hasegawa, M., Haas, K. M., Miller, A. S., Sanford, I. G., Bock, C. B., Fujimoto, M., and Tedder, T. F. (2004). CD22 regulates B lymphocyte function in vivo through both ligand-dependent and ligand-independent mechanisms. Nat. Immunol. 5, 1078–1087.
Powell, L. D., Sgroi, D., Sjoberg, E. R., Stamenkovic, I., and Varki, A. (1993). Natural ligands of the B cell adhesion molecule CD22 beta carry N-linked oligosaccharides with alpha-2,6-linked sialic acids that are required for recognition. J. Biol. Chem. 268, 7019–7027.
Quarles, R. H. (2007). Myelin-associated glycoprotein (MAG): past, present and beyond. J. Neurochem. 100, 1431–1448.
Ramya, T. N., Weerapana, E., Liao, L., Zeng, Y., Tateno, H., Yates, J. R. III, Cravatt, B. F., and Paulson, J. C. (2010). In situ trans ligands of CD22 identified by glycan-protein photocross-linking-enabled proteomics. Mol. Cell. Proteomics 9, 1339–1351.
Razi, N., and Varki, A. (1998). Masking and unmasking of the sialic acid-binding lectin activity of CD22 (Siglec-2) on B lymphocytes. Proc. Natl. Acad. Sci. U.S.A. 95, 7469–7474.
Razi, N., and Varki, A. (1999). Cryptic sialic acid binding lectins on human blood leukocytes can be unmasked by sialidase treatment or cellular activation. Glycobiology 9, 1225–1234.
Samardzic, T., Marinkovic, D., Danzer, C. P., Gerlach, J., Nitschke, L., and Wirth, T. (2002). Reduction of marginal zone B cells in CD22-deficient mice. Eur. J. Immunol. 32, 561–567.
Santos, L., Draves, K. E., Boton, M., Grewal, P. K., Marth, J. D., and Clark, E. A. (2008). Dendritic cell-dependent inhibition of B cell proliferation requires CD22. J. Immunol. 180, 4561–4569.
Sato, S., Miller, A. S., Inaoki, M., Bock, C. B., Jansen, P. J., Tang, M. L., and Tedder, T. F. (1996). CD22 is both a positive and negative regulator of B lymphocyte antigen receptor signal transduction: altered signaling in CD22-deficient mice. Immunity 5, 551–562.
Sato, S., Tuscano, J. M., Inaoki, M., and Tedder, T. F. (1998). CD22 negatively and positively regulates signal transduction through the B lymphocyte antigen receptor. Semin. Immunol. 10, 287–297.
Schadee-Eestermans, I. L., Hoefsmit, E. C., Van De Ende, M., Crocker, P. R., Van Den Berg, T. K., and Dijkstra, C. D. (2000). Ultrastructural localisation of sialoadhesin (siglec-1) on macrophages in rodent lymphoid tissues. Immunobiology 202, 309–325.
Scharenberg, A. M., Humphries, L. A., and Rawlings, D. J. (2007). Calcium signalling and cell-fate choice in B cells. Nat. Rev. Immunol. 7, 778–789.
Schebesta, A., Mcmanus, S., Salvagiotto, G., Delogu, A., Busslinger, G. A., and Busslinger, M. (2007). Transcription factor Pax5 activates the chromatin of key genes involved in B cell signaling, adhesion, migration, and immune function. Immunity 27, 49–63.
Seite, J. F., Cornec, D., Renaudineau, Y., Youinou, P., Mageed, R. A., and Hillion, S. (2010). IVIg modulates BCR signaling through CD22 and promotes apoptosis in mature human B lymphocytes. Blood 116, 1698–1704.
Sgroi, D., Koretzky, G. A., and Stamenkovic, I. (1995). Regulation of CD45 engagement by the B-cell receptor CD22. Proc. Natl. Acad. Sci. U.S.A. 92, 4026–4030.
Sjoberg, E. R., Powell, L. D., Klein, A., and Varki, A. (1994). Natural ligands of the B cell adhesion molecule CD22 beta can be masked by 9-O-acetylation of sialic acids. J. Cell Biol. 126, 549–562.
Smith, K. G., Tarlinton, D. M., Doody, G. M., Hibbs, M. L., and Fearon, D. T. (1998). Inhibition of the B cell by CD22: a requirement for Lyn. J. Exp. Med. 187, 807–811.
Stamatos, N. M., Curreli, S., Zella, D., and Cross, A. S. (2004). Desialylation of glycoconjugates on the surface of monocytes activates the extracellular signal-related kinases ERK 1/2 and results in enhanced production of specific cytokines. J. Leukoc. Biol. 75, 307–313.
Surolia, I., Pirnie, S. P., Chellappa, V., Taylor, K. N., Cariappa, A., Moya, J., Liu, H., Bell, D. W., Driscoll, D. R., Diederichs, S., Haider, K., Netravali, I., Le, S., Elia, R., Dow, E., Lee, A., Freudenberg, J., De Jager, P. L., Chretien, Y., Varki, A., Macdonald, M. E., Gillis, T., Behrens, T. W., Bloch, D., Collier, D., Korzenik, J., Podolsky, D. K., Hafler, D., Murali, M., Sands, B., Stone, J. H., Gregersen, P. K., and Pillai, S. (2010). Functionally defective germline variants of sialic acid acetylesterase in autoimmunity. Nature 466, 243–247.
Takashima, S. (2008). Characterization of mouse sialyltransferase genes: their evolution and diversity. Biosci. Biotechnol. Biochem. 72, 1155–1167.
Tuscano, J. M., Riva, A., Toscano, S. N., Tedder, T. F., and Kehrl, J. H. (1999). CD22 cross-linking generates B-cell antigen receptor-independent signals that activate the JNK/SAPK signaling cascade. Blood 94, 1382–1392.
Uckun, F. M., Goodman, P., Ma, H., Dibirdik, I., and Qazi, S. (2010). CD22 EXON 12 deletion as a pathogenic mechanism of human B-precursor leukemia. Proc. Natl. Acad. Sci. U.S.A. 107, 16852–16857.
Varki, A. (2009). Multiple changes in sialic acid biology during human evolution. Glycoconj. J. 26, 231–245.
Varki, A., and Angata, T. (2006). Siglecs – the major subfamily of I-type lectins. Glycobiology 16, 1R–27R.
Viglianti, G. A., Lau, C. M., Hanley, T. M., Miko, B. A., Shlomchik, M. J., and Marshak-Rothstein, A. (2003). Activation of autoreactive B cells by CpG dsDNA. Immunity 19, 837–847.
Waisman, A., Kraus, M., Seagal, J., Ghosh, S., Melamed, D., Song, J., Sasaki, Y., Classen, S., Lutz, C., Brombacher, F., Nitschke, L., and Rajewsky, K. (2007). IgG1 B cell receptor signaling is inhibited by CD22 and promotes the development of B cells whose survival is less dependent on Ig alpha/beta. J. Exp. Med. 204, 747–758.
Wakabayashi, C., Adachi, T., Wienands, J., and Tsubata, T. (2002). A distinct signaling pathway used by the IgG-containing B cell antigen receptor. Science 298, 2392–2395.
Whitney, G., Wang, S., Chang, H., Cheng, K. Y., Lu, P., Zhou, X. D., Yang, W. P., Mckinnon, M., and Longphre, M. (2001). A new siglec family member, siglec-10, is expressed in cells of the immune system and has signaling properties similar to CD33. Eur. J. Biochem. 268, 6083–6096.
Yohannan, J., Wienands, J., Coggeshall, K. M., and Justement, L. B. (1999). Analysis of tyrosine phosphorylation-dependent interactions between stimulatory effector proteins and the B cell co-receptor CD22. J. Biol. Chem. 274, 18769–18776.
Zhang, J. Q., Biedermann, B., Nitschke, L., and Crocker, P. R. (2004). The murine inhibitory receptor mSiglec-E is expressed broadly on cells of the innate immune system whereas mSiglec-F is restricted to eosinophils. Eur. J. Immunol. 34, 1175–1184.
Zhang, J. Q., Nicoll, G., Jones, C., and Crocker, P. R. (2000). Siglec-9, a novel sialic acid binding member of the immunoglobulin superfamily expressed broadly on human blood leukocytes. J. Biol. Chem. 275, 22121–22126.
Zhang, M., Angata, T., Cho, J. Y., Miller, M., Broide, D. H., and Varki, A. (2007). Defining the in vivo function of Siglec-F, a CD33-related Siglec expressed on mouse eosinophils. Blood 109, 4280–4287.
Keywords: Siglecs, B lymphocytes, BCR signaling, inhibitory signaling, TLR, autoimmunity, B1 cells
Citation: Jellusova J and Nitschke L (2012) Regulation of B cell functions by the sialic acid-binding receptors Siglec-G and CD22. Front. Immun. 2:96. doi: 10.3389/fimmu.2011.00096
Received: 17 November 2011; Paper pending published: 12 December 2011;
Accepted: 28 December 2011; Published online: 11 January 2012.
Edited by:
Anthony L. DeFranco, University of California San Francisco, USAReviewed by:
Pablo Engel, University of Barcelona, SpainI-Hsin Su, Nanyang Technological University, Singapore
Copyright: © 2012 Jellusova and Nitschke. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Lars Nitschke, Chair of Genetics, University of Erlangen, Staudtstr. 5, 91058 Erlangen, Germany. e-mail:bml0c2Noa2VAYmlvbG9naWUudW5pLWVybGFuZ2VuLmRl