 Yoseph A. Mekori1,2* and Alon Y. Hershko1,2
Yoseph A. Mekori1,2* and Alon Y. Hershko1,2
- 1 Laboratory of Allergy and Clinical Immunology, Department of Medicine, The Herbert Center of Mast Cell Disorders, Meir Medical Center, Kfar Saba, Israel
- 2 Sackler School of Medicine, Tel Aviv University, Tel Aviv, Israel
Close physical proximity between mast cells and T cells has been demonstrated in several T cell mediated inflammatory processes such as rheumatoid arthritis and sarcoidosis. However, the way by which mast cells are activated in these T cell-mediated immune responses has not been fully elucidated. We have identified and characterized a novel mast cell activation pathway initiated by physical contact with activated T cells, and showed that this pathway is associated with degranulation and cytokine release. The signaling events associated with this pathway of mast cell activation have also been elucidated confirming the activation of the Ras mitogen-activated protein kinase systems. More recently, we hypothesized and demonstrated that mast cells may also be activated by microparticles released from activated T cells that are considered as miniature version of a cell. By extension, microparticles might affect the activity of mast cells, which are usually not in direct contact with T cells at the inflammatory site. Recent works have also focused on the effects of regulatory T cells (Treg) on mast cells. These reports highlighted the importance of the cytokines IL-2 and IL-9, produced by mast cells and T cells, respectively, in obtaining optimal immune suppression. Finally, physical contact, associated by OX40–OX40L engagement has been found to underlie the down-regulatory effects exerted by Treg on mast cell function.
Introduction
Most commonly known for their role in the elicitation of IgE-mediated allergic inflammation, mast cells have been implicated in a range of other non-allergic inflammatory processes. Observations such as the close physical proximity between mast cells and T cells in inflamed tissues and the capability of the former to release a wide range of immunomodulatory mediators and to express surface molecules important in costimulation in both adaptive and innate immunity, have led investigators to propose a functional bi-directional relationship between these two cell populations (Mekori, 2004; Bachelet et al., 2006; Kalesnikoff and Galli, 2008). Indeed, morphologic studies have documented an increase in the local density of mast cells and their activation during T cell-mediated inflammatory processes, as observed in cutaneous delayed-type hypersensitivity, graft-versus-host reactions, sarcoidosis, Crohn’s disease, rheumatoid arthritis, and fibrosis (Mekori, 2004; Bachelet et al., 2006; Kalesnikoff and Galli, 2008; Dudeck et al., 2011). Both in vitro and in vivo studies have demonstrated that mast cells or their products are pivotal in mediating leukocyte recruitment into inflammatory sites, are capable of presenting antigens to T cells, interact directly with and affect the function of cells of the adaptive immune system, and mediate tissue remodeling (Mekori, 2004; Bachelet et al., 2006; Kalesnikoff and Galli, 2008; Dudeck et al., 2011). For instance, by using W/Wv mice reconstituted with mast cells obtained from TNF−/− mice, it could be shown in vivo that TNF-α and MIP-2 (the functional murine analog of human IL-8) were essential for appropriate neutrophil recruitment during T cell-induced cutaneous delayed hypersensitivity reactions. Both cytokines were dependent on the presence of mast cells (Biedermann et al., 2000). The combination of these two mediators is crucial for cell recruitment because TNF-α and MIP-2 provide two qualitatively different but synergistic signals. The induction of MIP-2 and TNF-α were strictly dependent on the presence of mast cells and local activation of memory T cells, indicating that the infiltrating T cells deliver signals that induced both TNF-α and MIP-2 production by mast cells (Biedermann et al., 2000). However, the way by which T cells activate mast cells in T cell-mediated immune responses have not yet been fully elucidated.
The understanding of T cell–mast cell interactions is further complicated by the fact that these two cell types have been shown to be both pro- and anti-inflammatory, depending on the immunological setting. In the case of T cells, it is well established that both effector and regulatory types exist (Shevach, 2006). Since the mid 1990s, a subset of CD4+CD25+ cells have been rigorously characterized as pivotal players in dampening immune responses (Shevach, 2004). This regulatory role is dependent on the expression of the transcription factor Foxp3. Likewise, there is strong evidence that mast cells, traditionally recognized as enhancers of inflammation, can also suppress certain disease models, thus suggesting the concept of “regulatory mast cells” (Frossi et al., 2010). However, contrary to regulatory T cells (Treg), the phenotypic features, and mode of action are considerably less understood in suppressor mast cells. Recent studies on T cell effects on mast cell function in the regulatory context are discussed in the present communication.
Mast Cell Activation by Heterotypic Adhesion to T Cells
We have previously reported on the effects of direct contact between mast cells and T lymphocytes on mast cell activation and mediator release. Both murine and human mast cells could be activated to both release granule-associated mediators, such as histamine and matrix metalloproteinase-9 (MMP-9), and to produce several cytokines (i.e., TNF-α, IL-4, IL-6, and IL-8) upon physical contact with activated, but not resting, T cells (Inamura et al., 1998; Baram et al., 2001; Salamon et al., 2005, 2008). Furthermore, the expression and release of these mediators, were also induced when mast cells were incubated with cell membranes isolated from activated, but not resting, T cells (Baram et al., 2001; Salamon et al., 2005, 2008). Gene expression profiling validated by qRT-PCR has demonstrated the expression and production of cytokines (oncostatin M) and enzymes (MMP-9) that were specifically induced by this novel here-to-fore unknown pathway of activation (Salamon et al., 2008). Studies with murine mast cells and myristate 13-acetate (PMA) – or anti-CD3-activated T cells attributed the T cell-induced mast cell activation to interactions of surface molecules, such as intercellular adhesion molecule 1 and lymphotoxin-β receptor, with their respective ligands (Baram et al., 2001; Stopfer et al., 2004). Thus, direct contact between surface molecules on mast cells and on activated T cells was found to provide the stimulatory signal in mast cells necessary for degranulation and cytokine release independent of T cell intracellular function, and in the absence of demonstrable soluble mediators. Indeed, separation of the two cell populations by a semipermeable porous membrane prevented this pathway of mast cell activation (Bhattacharyya et al., 1998; Baram et al., 2001).
The kinetics of the heterotypic adhesion-induced mast cell activation was found to be slow in nature. For example, MMP-9 release, found to be granule-associated, was first detected at 6 h and peaked at 22 h of incubation with activated T cell membranes, while TNF-α release peaked after only 6 h. Anti-TNF-α mAb inhibited the T cell membrane-induced MMP-9 expression and release, indicating a possible autocrine regulation of MMP release by mast cell TNF-α (Baram et al., 2001; Figure 1). When analyzing the signaling events that are associated with this novel pathway of mast cell activation we found that it is associated with phosphorylation of the mitogen-activated protein kinases (MAPKs) extracellular signal-regulated kinase (ERK) and p38 (Brill et al., 2004; Shefler et al., 2008). In this context, it has been found that cytokines such as IL-8 and oncostatin M are released from human mast cells after contact with activated T cells and that this process is subject to MAPK inhibition (Salamon et al., 2008). The best characterized upstream regulator of the MAPK system is the small guanosine triphosphate (GTP)-binding protein Ras. This protein has critical functions in many cell types, however, in mast cells its functions are not entirely understood. By investigating the spatiotemporal pattern of Ras activation in mast cells stimulated by activated T cell membranes, we could demonstrate that this stimulation resulted in N-Ras activation through the activation of the guanine exchange factor RasGRP-1 (Shefler et al., 2008).
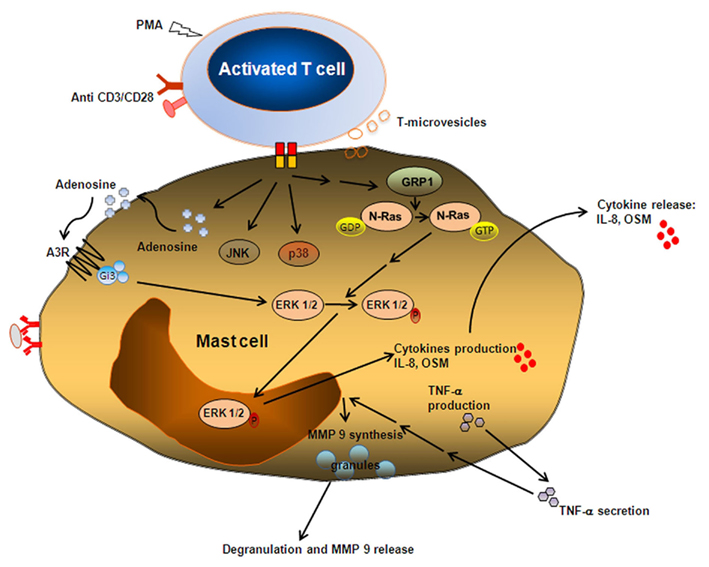
Figure 1. Heterotypic adhesion-induced mast cell activation. Mast cells can be activated by physical contact with activated, but not resting, T cells and/or by microvesicles released from the latter. This contact involves adhesion molecules such as integrins, lymphotoxin β receptor, and other yet unspecified surface molecules. Contact with membranes, derived from activated T cells, stimulate ERK1/2 activation, causing the expression of cytokines, chemokines, and growth factors. On contact with T cell membranes, adenosine is formed and released. By binding to its specific A3 receptors, adenosine initiates a complex signaling cross-talk, whereby the A3R, by coupling to the G-protein Gi3, eventually contributes to ERK1/2 signaling. N-Ras but not K Ras may be involved in this pathway or be directly activated by a yet undisclosed pathway and results in sustained ERK activation that is associated with increased dwell time at the nucleus and with cytokine release. Adhesion to activated T cells also results in an early (preformed) TNF-α release that affects in an autocrine manner the expression and production of the granule-associated enzyme MMP-9.
We further explored the downstream events associated with Ras activation. Thus, ERK activation in human mast cells activated by either contact with T cells or by FcεRI crosslinking was studied. Stimulation of human mast cells by contact with activated T cells resulted in sustained ERK activation. Furthermore, sustained ERK activation in these cells was associated with increased dwell time at the nucleus and with IL-8 release. Interestingly, when mast cells were stimulated by FcεRI crosslinking, ERK activation was transient. ERK activation was associated with a shorter dwell time at the nucleus and with TNF-α release (Mor et al., 2010). Thus, retaining ERK in the nucleus may be a mechanism utilized by human mast cells to generate different cytokines from a single signaling cascade. When analyzing upstream signaling events, it has been shown that contact with activated T cell membranes results in increased levels of endogenous adenosine that binds to and activates the A3R, culminating in Gi3-mediated signaling. Specifically, the majority of ERK1/2 signaling initiated by contact with activated T cell membranes was found to be mediated by Gi3 (Baram et al., 2010; Figure 1).
The biological relevance of this here-to-fore unrecognized pathway of mast cell activation can be envisaged from our findings with oncostatin M, a known fibrogenic cytokine, where we demonstrated that both oncostatin M mRNA and protein are induced in human mast cells specifically by means of heterotypic adhesion to activated T cells. Mast cell-derived oncostatin was found to induce the proliferation of lung fibroblasts and its presence was demonstrated in mast cells in the lungs of patients with sarcoidosis, a disease known to culminate in fibrosis (Salamon et al., 2008). These results suggest that human mast cells might contribute to T cell-mediated fibrotic inflammatory processes by means of local release of oncostatin M after direct activation by T cells. Contact-dependent bi-directional interactions have also been demonstrated between CD8+ T cells and mast cells, wherein mast cells supported T cell effector functions and survival, while T cells induced expression of co-stimulatory molecules and cytokine release by mast cells (Stelekati et al., 2009).
Mast Cell Activation by T Cell-Derived Microvesicles
Based on the above information, and due to the fact that mast cells usually do not reside in direct contact with T cells at the inflammatory sites, we further hypothesized that mast cells may be activated not only by heterotypic adhesion to T cells, but also by other components that may reside in the supernatants of activated T cells and are of high molecular weight that does not allow their migration through the porous membranes. Possible candidates are membrane vesicles that are secreted by T cells (Al-Nedawwi et al., 2009; Thery et al., 2009). Many reports have described how portions of cell membranes can be transferred between cells, either after direct cell–cell contact or through the secretion of membrane vesicles. The functional consequences of such membrane transfers include the induction, amplification, and/or modulation of immune responses, as well as the acquisition of new functional properties by recipient cells (Al-Nedawwi et al., 2009; Thery et al., 2009). Cells can generate membrane vesicles that are secreted into the extracellular space; such vesicles can form either at the plasma membrane or in the lumen of internal compartments. Irrespective of their origin, these vesicles contain cytosol and expose the extracellular side of the membrane they form from at their outer surface. Because their membrane orientation is the same as that of the donor cell, they can be considered to be miniature versions of a cell (Thery et al., 2009). Thus, membrane transfer is a mode of intercellular communication that may also involve T cell-induced mast cell activation within inflammatory sites in which both cell populations have been shown to be involved, such as rheumatoid arthritis, Crohn’s disease, and sarcoidosis (Shefler et al., 2011).
Consequently, we were able to show that high molecular weight fractions (MW > 100000 Da) of supernatants obtained from T cells, that were activated by either short incubation with PMA or with anti-CD3 anti-CD28 mAb-coated beads, contained microvesicles (100–800 nm in diameter) that expressed the adhesion molecule LFA-1/CD11a and bound Annexin V-FITC indicating that phosphatidylserine was exposed at their surface. Isolated microvesicles induced degranulation and cytokine (IL-8 and oncostatin M) release from human mast cells (Shefler et al., 2010). The results, including kinetics of the microvesicle-induced mediator release, were similar when mast cells were stimulated by activated fixed T cells or by whole membranes of the latter. This suggests that microvesicles carry similar mast cell-activating factors to cells from which they originate. Thus, by releasing microvesicles, T cells might convey surface molecules similar to those involved in the activation of mast cells by cellular contact. Consequently, through the generation of microvesicles, activated T cells may facilitate distant contact-mediated activation of mast cells which are not in direct contact with T cells at the inflammatory sites (Shefler et al., 2011). Blocking the release of microparticles and their interaction with mast cells, as well as downstream activation events, may serve as a therapeutic approach for T cell-mediated inflammatory processes in which mast cells are involved.
Regulatory T Cell Effect on Mast Cell Function
The activation of mast cells by T cells, as described herein, can be regarded as a mechanism underlying the propagation of T cell-mediated inflammatory processes. However, one should bear in mind that a significant proportion of T cells specialize in regulation and suppression of the inflammatory process. This sub-population, known as Treg, may appear in several forms, mostly characterized by the expression CD4, CD25, and the transcription factor Foxp3 (Kayshap et al., 2008). Naturally occurring Treg may account for as much as 10% of total CD4+ T cells in mice. They have been described as pivotal players in suppressing autoimmunity (Shevach, 2006), allergic responses, and inflammatory responses to tumors, in various systems. An intriguing question that has been investigated in recent years is whether these T cells can also interact with mast cells, in order to further support suppression (Hershko and Rivera, 2010).
In a mouse model of chronic allergic dermatitis, Treg were shown to require mast cell recruitment in order to obtain optimal suppression of inflammation (Hershko et al., 2011). In this model, mast cells migrated from the site of inflammation (skin) to the spleen, where they secrete IL-2, a critical cytokine for Treg proliferation and function (Figure 2). Following this process, disease was more effectively suppressed and the proportion of Treg in the skin increased. Mast cell accumulation in the spleen was shown to be IgE dependent. Furthermore, inhibition of Treg activity by anti-CD25 monoclonal antibodies enhanced IgE production thereby facilitating mast cell numbers in the spleen. Interestingly, mast cell accumulation was independent of the high-affinity IgE receptor (FcεRI). However, it was abrogated following blockade of the low affinity IgE receptor, CD23, which is not expressed on mast cells, thereby implying the involvement of another cell-type, as mediators of mast cell recruitment.
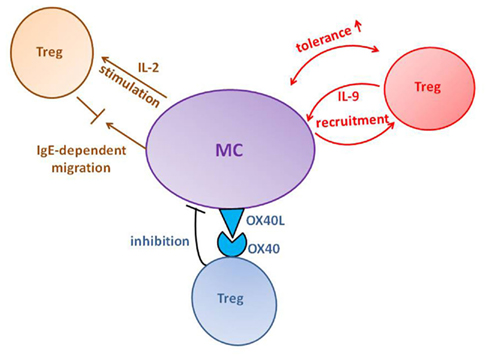
Figure 2. Proposed paradigms of Treg–mast cell (MC) interaction. Several modes of interaction have been described for Treg effects on MC activation and for the cross-talk between the two cells (from top left clockwise): Failure of Treg-mediated IgE suppression enhances MC migration and IL-2 production in the spleen which, in turn, boosts Treg activity; Treg–IL-9 production recruits MC to skin allografts where the two cells collaborate to create tolerance; engagement of OX40L and OX40 results in inhibition of MC degranulation and dampening of anaphylaxis.
The collaboration between Treg and mast cells in suppressing inflammatory responses has been suggested by other reports as well. The study of several disease models revealed that Treg can attract mast cells by producing considerable amounts of IL-9, a mast cell growth, and activation factor. This paradigm was first presented in a study of a skin allograft model, whose tolerance depended on Treg activity (Lu et al., 2006). However, Treg were not sufficient to prevent rejection, and required recruitment of mast cells to the graft. The functional link between the two cell types was shown to be Treg-derived IL-9. Neutralization of this cytokine by monoclonal antibodies abrogated mast cell recruitment and inhibited tolerance. The role of Treg-derived IL-9 in mast cell recruitment and disease suppression has been confirmed in subsequent reports. For example, in a murine model of nephrotoxic serum nephritis (NTS), Treg were shown to effectively inhibit disease development, and this protective effect was profoundly compromised in mast cell-deficient mice (Eller et al., 2011). Furthermore, both blocking of IL-9 by monoclonal antibodies and adoptive transfer of IL-9 deficient Treg resulted in failure of mast cell accumulation and impaired protection. The Treg, IL-9, and mast cell connection was similarly implicated in immune suppression in B-cell non-Hodgkin’s lymphoma in human patients (Feng et al., 2011) as well as in murine lymphoma. It is not entirely clear, however, from these reports what is the mechanism underlying the contribution of IL-9 mediated mast cell recruitment to immune suppression. This issue was more recently addressed in a study on a murine hepatocarcinoma model (Yang et al., 2010). In this work, mast cells were shown to support the mobilization and infiltration of myeloid-derived suppressor cells (MDSCs) to the tumor and to induce production of IL-17 by these cells. MDSC’s, in turn, attracted Treg and stimulated them to produce IL-9, which supported mast cell survival and pro-tumor effect. Therefore, similar to our work on the immunosuppressive effect of mast cells in chronic dermatitis, it has been suggested that mast cells play a role in promoting Treg recruitment in the hepatocellular model.
The effects that Treg exert on mast cells, however, appear to be more complex than collaboration in suppressing immune responses. In a model of intestinal polyposis mast cells play a critical role in disease progression. Adoptively transferred Treg suppressed focal mastocytosis in vivo, and thus inhibited disease (Gounaris et al., 2009). Nevertheless, endogenous Foxp3+ Treg populating the polyps could not counteract mastocytosis and polyposis. Endogenous Treg displayed defective IL-10 secretion as well as IL-17 production and were implicated in tumor growth through enhancement of a mast cell-mediated inflammation.
The paradigm of mast cells as targets of Treg-mediated suppression has been rigorously investigated in the setting of immediate hypersensitivity-allergic responses. The study of this aspect of Treg–mast cell interaction has yielded interesting mechanistical data. Mast cells were shown in vitro to have the capacity to recruit both regulatory and conventional T cells (Kayshap et al., 2008). This processes led to Treg-mediated inhibition of FcεRI expression on mast cells, requiring contact between the two cell types. However, the effects on mast cell-production of inflammatory mediators were somewhat more complex. Both regulatory and conventional T cell suppressed leukotriene C4 production and enhanced cytokine secretion, along with FcεRI-induced Stat5 phosphorylation. Along this line Treg were also found to directly inhibit the FcεRI-dependent degranulation of mast cells (Gri et al., 2008). This effect was achieved by Treg–mast cell contact leading to OX40–OX40L engagement, respectively (Figure 2). Suppression was associated with increased cAMP and decreased Ca2+ influx, unrelated to PLC-γ2 or intracellular Ca2+ stores. Furthermore, abrogation of cAMP increase essentially reversed the impact of Treg on mast cells. The physiological outcome of mast cell suppression was confirmed in vivo by the observation that interference with Treg function resulted in exacerbation of anaphylaxis. An elegant mechanistical insight was provided in a subsequent work, in which time-lapse video microscopy and transmission electron microscopy were used to record interactions on a single-cell level (Frossi et al., 2011). Co-cultures of mast cells with wild type, but not OX40-deficient, Treg resulted in morphological changes that indicated defective exocytosis, along with inhibition of degranulation and of Ca2+ mobilization. Thus, an “Immunological synapse” between the two cell types was convincingly visualized. Intriguingly, these observations have led to the finding that soluble OX40 is sufficient to mimic the inhibitory effects of Treg (Sibilano et al., 2011), providing a potentially novel therapeutic approach in allergic diseases.
In summary, we would like to suggest that mast cells can be utilized by T cells in order to enhance the physiological effects of the latter. Although T cells demonstrate considerable mast cell-independent activity, it seems that mast cells are required to obtain an optimal response. This conclusion is based on data provided by studies on both effector and Treg. Nevertheless, it should be noted, that the role of these cell types may be contradictory, thus leading to effects such as T (regulatory) cell counteracting mast cell function.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Alon Y. Hershko is the recipient of the Morasha Program fellowship of the Israel Science Foundation (grant No. 1084/10). Yoseph A. Mekori received a research grant form the Israel Science Foundation. Yoseph A. Mekori is the incumbent of the Frederick Reiss Chair in Dermatology at Tel Aviv University. The authors wish to thank Irit Shefler Ph.D., and Pazit Salamon MS for their technical assistance.
References
Bachelet, I., Levi-Schaffer, F., and Mekori, Y. A. (2006). Mast cells, not only in allergy. Immunol. Allergy Clin. North Am. 26, 407–426.
Baram, D., Dekel, O., Mekori, Y. A., and Sagi-Eisenberg, R. (2010). Activation of mast cells by trimeric G protein Gi3; coupling to the A3 adenosine receptor directly, and upon T cell contact. J. Immunol. 184, 3677–3688.
Baram, D., Vaday, G. G., Salamon, P., Drucker, I., Hershkovis, R., and Mekori, Y. A. (2001). Human mast cells release metalloproteinase-9 on contact with activated T cells: juxtacrine regulation by TNF-α. J. Immunol. 167, 4008–4016.
Bhattacharyya, S. P., Drucker, I., Reshef, T., Kirshenbaum, A. S., Metcalfe, D. D., and Mekori, Y. A. (1998). Activated lymphocytes induce degranulation and cytokine production by human mast cells following cell to cell contact. J. Leukoc. Biol. 63, 337–341.
Biedermann, T., Kneilling, M., Mailhammer, R., Maier, K., Sander, C. A., Kollias, G., Kunkel, S. L., Hultner, L., and Rocken, M. (2000). Mast cells controlneutrophil recruitment during T cell mediateddelayed-type hypersensitivity. J. Exp. Med. 192, 1441–1452.
Brill, A., Baram, D., Sela, U., Salamon, P., Mekori, Y. A., and Hershkovis, R. (2004). Induction of mast cell interactions with blood vessel wall components by direct contact with intact T cells or T cell membranes in vitro. Clin. Exp. Allergy 34, 1725–1731.
Dudeck, A., Dudeck, J., Scholten, J., Petzold, A., Surianarayanan, S., Kohler, A., Peshcke, K., Vohringer, D., Wascow, C., Krieg, T., Muller, W., Waismann, A., Hartmann, K., Gunzer, M., and Roers, A. (2011). Mast cells are key promoters of contact allergy that mediate the adjuvant effects of haptens. Immunity 34, 973–984.
Eller, K., Wolf, D., Huber, J. M., Metz, M., Mayer, G., McKenzie, A. N., Maurer, M., Rosenkranz, A. R., and Wolf, A. M. (2011). IL-9 production by regulatory T cells recruits mast cells that are essential for regulatory T cell-induced immune suppression. J. Immunol. 186, 83–91.
Feng, L. L., Gao, J. M., Li, P. P., and Wang, X. (2011). IL-9 contributes to immunosuppression mediated by regulatory T cells and mast cells in B-cell non-Hodgkin’s lymphoma. J. Clin. Immunol. 31, 1084–1094.
Frossi, B., D’Inca, F., Crivellato, E., Sibilano, R., Gri, G., Mongillo, M., Danelli, L., Maggi, L., and Pucillo, C. E. (2011). Single-cell dynamics of mast cell-CD4+CD25+ regulatory T cell interactions. Eur. J. Immunol. 41, 1872–1882.
Frossi, B., Gri, G., Tripodo, C., and Pucillo, C. (2010). Exploring a regulatory role for mast cells: “MCregs?” Trends. Immunol. 3, 97–102.
Gounaris, E., Blatner, N. R., Dennis, K., Magnusson, F., Gurish, M. F., Strom, T. B., Beckhove, P., Gounari, F., and Khazaie, K. (2009). T-regulatory cells shift from a protective anti-inflammatory to a cancer-promoting pro-inflammatory phenotype in polyposis. Cancer Res. 69, 5490–5497.
Gri, G., Piconese, S., Frossi, B., Manfroi, V., Merluzzi, S., Tripodo, C., Viola, A., Odom, S., Rivera, J., Colombo, M. P., and Pucillo, C. E. (2008). CD4+CD25+ regulatory T cells suppress mast cell degranulation and allergic responses through OX40-OX40L interaction. Immunity 29, 771–781.
Hershko, A. Y., and Rivera, J. (2010). Mast cell and T cell communication; amplification and control of adaptive immunity. Immunol. Lett. 128, 98–104.
Hershko, A. Y., Suzuki, R., Charles, N., Alvarez-Errico, D., Sargent, J., Laurence, A., and Rivera, J. (2011). Mast cell interleukin-2 production contributes to suppression of chronic allergic dermatitis. Immunity 35, 562–571.
Inamura, N., Mekori, Y. A., Bhattacaryya, S. P., Bianchine, P. J., and Metcalfe, D. D. (1998). Induction and enhancement of FCεRI-dependent mast cell degranulation following coculture with activated T cells: dependency on ICAM-1 and leukocyte function-associated antigen (LFA)-1-mediated heterotypic aggregation. J. Immunol. 160, 4026–4033.
Kalesnikoff, J., and Galli, S. J. (2008). New developments in mast cell biology. Nat. Immunol. 9, 1215–1223.
Kayshap, M., Thornton, A. M., Norton, S. K., Barnstein, B., Macey, M., Brenzovich, J., Shevach, E., Leonard, W. J., and Ryan, J. J. (2008). Cutting edge: CD4 T cell-mast cell interactions alter IgE receptor expression and signaling. J. Immunol. 180, 2039–2043.
Lu, L. F., Lind, E. F., Gondek, D. C., Bennett, K. A., Gleeson, M. W., Pino-Lagos, K., Scott, Z. A., Coyle, A. J., Reed, J. L., Van Snick, J., Strom, T. B., Zheng, X. X., and Noelle, R. J. (2006). Mast cells are essentials intermediaries in regulatory T-cell tolerance. Nature 442, 997–1002.
Mekori, Y. A. (2004). The mastocyte: the other inflammatory cell in immunopathogenesis. J. Allergy Clin. Immunol. 114, 52–57.
Mor, A., Shefler, I., Salamon, P., Kloog, Y., and Mekori, Y. A. (2010). Characterization of ERK activation by human mast cells stimulated by contact with activated T cells. Inflammation 33, 119–125.
Salamon, P., Shoham, N. G., Gavrieli, R., Wolach, B., and Mekori, Y. A. (2005). Human mast cells release interleukin-8 and induce neutrophil chemotaxis on contact with activated T cells. Allergy 60, 1316–1319.
Salamon, P., Shoham, N. G., Puxeddu, I., Paitan, Y., Levi-schaffer, F., and Mekori, Y. A. (2008). Human mast cells release oncostatin M on contact with activated T cells: possible biologic relevance. J. Allergy Clin. Immunol. 121, 448–455.
Shefler, I., Mekori, Y. A., and Mor, A. (2008). Stimulation of human mast cells by activated T cells leads to N-Ras activation through Ras guanine nucleotide releasing protein 1. J. Allergy Clin. Immunol. 122, 1222–1225.
Shefler, I., Salamon, P., Hershko, A. Y., and Mekori, Y. A. (2011). Mast cells as sources and targets of membrane microvesicles. Curr. Pharm. Des. 17, 3797–3804.
Shefler, I., Salamon, P., Mor, A., and Mekori, Y. A. (2010). T cell induced mast cell activation: a role for microparticles released from activated T cells. J. Immunol. 185, 4206–4212.
Shevach, E. M. (2004). Regulatory/suppressor T cells in health and disease. Arthritis Rheum. 50, 2721–2724.
Shevach, E. M. (2006). From vanilla to 28 flavors: multiple varieties of T regulatory cells. Immunity 25, 195–201.
Sibilano, R., Gri, G., Frossi, B., Tripodo, C., Suzuki, R., Rivera, J., Macdonald, A. S., and Pucillo, C. E. (2011). Technical advance: soluble OX40 molecule mimics regulatory T cell modulatory activity on FCeRI-dependent mast cell degranulation. J. Leukoc. Biol. 90, 831–838.
Stelekati, E., Bahri, R., DwOrlando, O., Orinska, Z., Mittrucker, H.-W., Langenhaun, R., Glatzel, M., Bollinger, A., Paus, R., and Bulfone-Paus, S. (2009). Mast cell-mediated antigen presentation regulates CD8+ T cell effector functions. Immunity 31, 665–676.
Stopfer, P., Mannel, D. N., and Hehlgans, T. (2004). Lymphotoxin-beta receptor activation by T cells induces cytokine release from mouse bone marrow-derived mast cells. J. Immunol. 172, 7459–7465
Thery, C., Ostrowski, M., and Segura, E. (2009). Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 9, 581–593.
Keywords: T cells, mast cells, microvesicles, heterotypic adhesion
Citation: Mekori YA and Hershko AY (2012) T cell-mediated modulation of mast cell function: heterotypic adhesion-induced stimulatory or inhibitory effects. Front. Immun. 3:6. doi: 10.3389/fimmu.2012.00006
Received: 25 October 2011;
Accepted: 09 January 2012;
Published online: 30 January 2012.
Edited by:
Toshiaki Kawakami, La Jolla Institute for Allergy and Immunology, USAReviewed by:
Ralf J. Ludwig, University of Lübeck, GermanyMasato Kubo, Tokyo University of Science, Japan
Copyright: © 2012 Mekori and Hershko. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Yoseph A. Mekori, Department of Medicine, Meir Medical Center, Kfar Saba 44281, Israel. e-mail:eW1la29yaUBjbGFsaXQub3JnLmls