 Jacob S. Lee
Jacob S. Lee Marina Cella and Marco Colonna*
Marina Cella and Marco Colonna*- Department of Pathology and Immunology, Washington University School of Medicine, St. Louis, MO, USA
Mucosal innate lymphoid cells (ILCs) are an emerging population of diverse and heterogeneous immune cells, all with the unique ability to mount a rapid response against invading pathogens. They are further divided into subsets based on their differing cell surface markers as well as in their functional specialization. In this review, we summarize recent reports describing the importance of the transcription factor aryl hydrocarbon receptor (AHR) in regulating the development of one of these subsets, the Type-17/22 ILCs, as well as in the organization of postnatal lymphoid structures. We discuss the mechanisms behind the AHR dependence for development in Type-17/22 ILCs as well as reviewing the proposed physiological ligands that are mediating this effect.
Introduction
Innate Lymphoid Cells: Heterogeneity and Functional Specialization
Mucosal innate lymphoid cells (ILCs) are a population of immune cells that has broad roles in host immunity as well as maintaining homeostasis between the host and its environment. As their name suggests, ILCs are a family of lymphoid cells from the innate immune system, and develop from hematopoietic precursors (Spits and Di Santo, 2011). Murine ILCs are heterogeneous and they include two major subsets that we will define here as Type-17/22 ILCs and Type-2 ILCs. These major subsets can be further divided to include subsets that differ in the expression of cell surface markers as well as in their functional specialization. Type-17/22 ILCs comprise NKp46+ ILCs (also known as NK-22 in humans; Satoh-Takayama et al., 2008; Cella et al., 2009; Luci et al., 2009; Sanos et al., 2009), LTi-like ILCs (CD4+, CD4− LTi-like cells; Takatori et al., 2009) and Thy1high Sca1+ ILCs (Buonocore et al., 2010). These populations collectively known as Type-17/22 ILCs, specialize in the production of IL-22 and/or IL-17. Type-2 ILCs include “natural helper cells” (NH; Moro et al., 2010), “nuocytes” (Neill et al., 2010), innate helper type-2 (Ih2) cells (Price et al., 2010), and multipotent progenitor type-2 cells (MPPtype2; Saenz et al., 2010) which specialize in the production of IL-5 and IL-13. Type-17/22 ILCs (Cella et al., 2009; Cupedo et al., 2009; Crellin et al., 2010) and Type-2 ILCs (Moro et al., 2010; Mjösberg et al., 2011; Monticelli et al., 2011) have also been described in humans. Most ILCs are ablated in Il2rc−/− mice (Satoh-Takayama et al., 2008; Koyasu and Moro, 2011), suggesting the requirement of a γc-associated cytokine receptor for their development and/or expansion. Interestingly, an IL-22 producing innate splenic population independent of Rag2 and the γc but resembling CD4− LTi-like ILCs have been described (Dumoutier et al., 2011). Whether these cells represent a precursor of intestinal IL-22 producing ILCs or its central counterpart has yet to be determined. Type-17/22 ILC subsets are developmentally related in their requirement for Id2 and RORγt expression, as Id2 (Yokota et al., 1999; Satoh-Takayama et al., 2010) and RORγt (Satoh-Takayama et al., 2008; Luci et al., 2009; Sanos et al., 2009) deficient animals have greatly reduced numbers of these ILCs. The key transcription factors that drive development of Type-2 ILCs are not well defined. However, they seem to develop independently of RORγt (Moro et al., 2010). In addition, while Type-17/22 ILCs are largely confined to gut and lung mucosa and found in low numbers in secondary lymphoid organs, Type-2 ILCs have some distinct tissue distribution. Similar to Type-17/22 ILCs, nuocytes, MPPtype2, and Ih2 were found in spleen, lymph nodes, small intestine, gut-associated lymphoid tissue, and liver; however and in contrast to Type-17/22 ILCs, NH ILCs are uniquely localized in “fat associated lymphoid clusters” (Moro et al., 2010). Although the exact relationship among the Type-2 ILC subsets is currently not as clear as the relationship among the Type-17/22 ILC subsets, it is likely that Type-2 ILC subsets are strictly related and may represent the same cell type with different tissue localization although it cannot be excluded that they derive from a common lymphoid precursor and represent different cell lineages (Yang et al., 2011).
One of the main features that characterize Type-17/22 ILCs is their ability to promptly respond to the inflammatory cytokine IL-23 by secreting IL-22 and/or IL-17 (Sonnenberg et al., 2011). IL-1β alongside IL-23 has been shown promote IL-17 and IL-22 production in murine invariant Natural Killer T cells (Doisne et al., 2011) and γδ T cells (Sutton et al., 2009), NKp46+ ILCs (Reynders et al., 2011), as well as sustaining IL-22 production in human ILCs cultured in vitro (Cella et al., 2010; Hughes et al., 2010). Interestingly, the lymphotoxin pathway has also been demonstrated to play an important role in controlling IL-22 production by Type-17/22 ILCs (Wang et al., 2010; Tumanov et al., 2011). Conversely, one of the primary characteristics of Type-2 ILCs is their capacity to quickly respond to the inflammatory cytokines IL-33 and/or IL-25 by producing IL-5 and IL-13. The unique ability to respond within hours to changing environmental cues has demonstrated increasing evidence that ILCs play a critical role in dictating appropriate tissue responses to incoming pathogens. ILCs are crucial for limiting tissue damage at mucosal sites and they provide the initial innate layer of protection while appropriate adaptive responses are developing. The intrinsic ability of ILC to quickly respond to cytokines closely resembles the long-ago recognized capacity of conventional NK cells to respond to IL-12 and IL-18 by rapidly producing IFN-γ (Takeda et al., 1998). In light of this unique attribute, IL-12 and IL-18 responding cells, including conventional NK cells, should be viewed as Type-1 ILCs. The characteristics of the ILC subsets are summarized in (Table 1). Considering the relevance of ILCs in disease and homeostasis, understanding the developmental pathways of ILCs is critical in their manipulation toward therapeutic exploitation. Therefore, increasing effort is ongoing into understanding how ILCs develop, expand, differentiate, and respond to a rapidly changing environment. One of the main focuses of recent research has been the attempt to elucidate transcriptional factors that are crucial for ILC differentiation. Here in this review, we will discuss recent findings on the role of the transcription factors aryl hydrocarbon receptor (AHR) and Notch in the development and function of Type-17/22 ILCs.
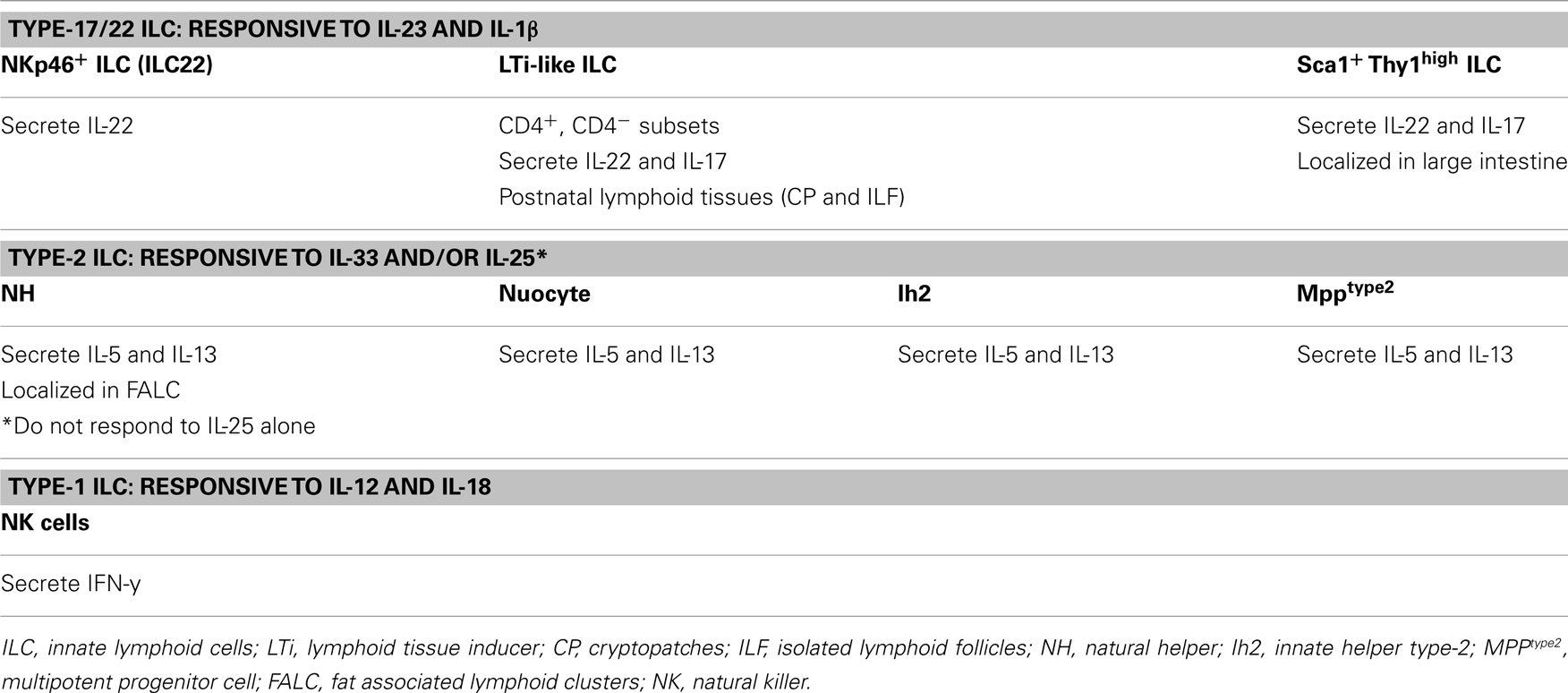
Table 1. Characteristics of ILC subsets.
The Aryl Hydrocarbon Receptor Pathway
Numerous studies have shown that AHR is a master regulator of drug metabolism and that it mediates the biochemical and toxic effects of dioxins, polyaromatic hydrocarbons, and related compounds and has been reviewed (Nebert et al., 1993; Wilson and Safe, 1998; Mimura and Fujii-Kuriyama, 2003; Nguyen and Bradfield, 2008). While AHR was initially identified as a key regulator of xenobiotic metabolism, it has since been shown to modulate the expression of certain genes and the activity of other transcription factors to influence other cellular functions such as apoptosis, proliferation, cell growth, and differentiation. AHR contains a basic helix–loop–helix (bHLH) domain similar to that present in many DNA-binding proteins, and is a member of the Per–Arnt–Sim (PAS) superfamily of proteins which contain proteins involved in detection of intracellular or environmental changes, sensing light, oxygen, and circadian rhythm (Gu et al., 2000). In the absence of agonists, AHR resides in the cytosol as an inactive complex including the chaperones heat shock protein (Hsp90; Perdew, 1988), aryl hydrocarbon receptor interacting protein (AIP; Carver and Bradfield, 1997), and p23 (Kazlauskas et al., 1999). The PAS domain on AHR is an important site for agonist binding, which leads to a conformational change in AHR. This change results in a modified interaction with its chaperones and exposes a nuclear localization signal on AHR. After migrating into the nucleus, AHR heterodimerizes with another bHLH–PAS protein known as the aryl hydrocarbon receptor nuclear translocator (ARNT; Reyes et al., 1992) and this complex binds specific enhancer sequences adjacent to target promoters called dioxin responsive elements (DRE), inducing transcription of multiple target genes. These genes include xenobiotic metabolizing monooxygenases such as the members of the Cytochrome P450 family Cyp1a1, Cyp1a2, and Cyp1b1, but also encompass a large number of other genes involved in important developmental and physiological processes (Nguyen and Bradfield, 2008).
Developmental Defects in AHR Deficient Mice
The hypothesis that AHR has a role in development independent of binding xenobiotics was greatly augmented through the generation of three independent AHR deficient animals (Fernandez-Salguero et al., 1995; Schmidt et al., 1996; Shimizu et al., 2000). Examination of AHR null mice revealed common developmental anomalies (Lahvis et al., 2000) including increased neonatal lethality, portal vascular hypertrophy, and patent ductus venosus (DV; Esser, 2009). Lymphocytes from the lymph nodes and spleen were greatly reduced in the mouse generated by Fernandez-Salguero et al. but a similar phenotype was not observed in the other two knockout mice. While the explanation of these contrasting phenotypes is still unclear, the common developmental defects in AHR deficient mice provides evidence supporting the hypothesis that AHR plays an important role in normal development and that AHR is regulating normal vascular or hematopoietic development. This important impact on development would suggest that AHR is more than simply a xenobiotic sensor, and thus it is of importance to identify and characterize the vast diversity of AHR ligands as well as elucidating the mechanism behind AHR regulated development.
AHR in the Immune System
While AHR biology has mainly been focused on environmental toxicology studies and more recently into its importance during normal physiology and development, AHR has found its way into mainstream immunology research in the past few years. Several studies have identified the importance of AHR in both adaptive and innate immune systems and have recently been reviewed (Stevens et al., 2009; Stockinger et al., 2011). The focus of this review will be centered on the emerging evidence connecting AHR, its ligands, and intestinal immune function.
AHR and Type-17/22 ILC
While the importance of AHR expression on the differentiation and function of Th17, Treg, and DC subsets has been described (Stockinger et al., 2011), recent reports have also demonstrated AHR expression in human and murine mucosal RORγt-dependent Type-17/22 ILCs (Kiss et al., 2011; Lee et al., 2011; Qiu et al., 2011; Figure 1). Driven by the hypothesis that similar to TH17 cells, AHR was necessary for the production of IL-22 in Type-17/22 ILCs, it was of great interest to test the impact of AHR deficiency on the function and/or development of these ILCs. Similar to the phenotype seen in AHR deficient TH17 cells, Peyer’s patches, and lamina propria cells isolated from the mucosal tissues of AHR deficient animals stimulated with IL-23 failed to produce IL-22. Further investigation demonstrated that surprisingly, AHR signaling in vivo was critical for the presence of Type-17/22 ILCs cells and not simply for their ability to secrete IL-22, as these cells were absent in Ahr−/− mice. AHR deficiency affected NKp46+ ILCs as well as the CD4+ and CD4− LTi-like ILCs. Histological examination of gut-associated secondary lymphoid structures found an interesting and unique phenotype in AHR deficient animals. While embryonically imprinted lymphoid structures, including the mesenteric lymph nodes, Peyer’s patches, and cecum-associated patches, remained largely intact in AHR deficient animals, the postnatally imprinted lymphoid structures were severely diminished. These included small intestine lamina propria cryptopatches (CPs) and isolated lymphoid follicles (ILFs), as well as colon associated lymphoid patches.
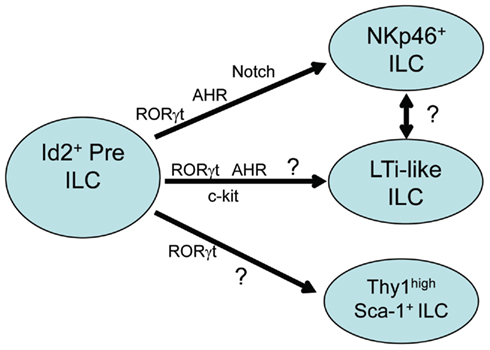
Figure 1. Transcription factors in type-17/22 ILC development. Type-17/22 ILCs which include NKp46+ ILC, LTi-like ILC, and Thy1 high Sca1+ ILC, are derived from hematopoietic precursors which require the expression of Id2. All subsets depend on the expression of RORγt for their development and recent reports have also shown the importance of AHR expression on the development of at least the NKp46+ and LTi-like ILC subsets. AHR expression on Thy1 high Sca1+ ILCs have as of yet not been described. Induction of genes downstream of AHR such as Notch and c-kit are also important in the development of NKp46+ and LTi-like ILC subsets respectively. Whether LTi-like ILCs and NKp46+ ILCs correspond to sequential developmental stages of the same cell type is unclear.
CPs and ILFs
Cryptopatches and ILFs are well-organized structures along the gastrointestinal tract that are thought to be functionally similar to Peyer’s patches in serving as important sites for the induction of protective mucosal immune responses (Lorenz and Newberry, 2004; Shikina et al., 2004). CPs are located in the lamina propria, and are composed of clusters of RORγt+ LTi-like ILCs scattered within the gastrointestinal tract. These structures are not present at birth, but start to develop about 2 weeks after birth. Their formation depends on factors necessary for the development and proper function of Type-17/22 ILCs such as IL-7 signaling (Adachi, 1998; Yoshida, 1999) and the expression of RORγt (Eberl et al., 2004) but seem to be independent of B and T cells (Hamada et al., 2002). Although original reports suggested that Type-17/22 ILCs required intestinal microbiota to develop (Satoh-Takayama et al., 2008; Sanos et al., 2009), subsequent investigation failed to confirm the need for microbiota (Lee et al., 2011; Reynders et al., 2011; Sawa et al., 2011). It is thought that inflammatory signals acting through Nod1 (Bouskra et al., 2008) as well as other TLRs during microbial colonization of the gut promote the transition of CPs into ILFs. Accordingly, germ-free animals have normal numbers of CPs (Kanamori et al., 1996) and medium-size ILFs (Hamada et al., 2002) but reduced number of large-size ILFs (Lorenz and Newberry, 2004). ILFs are thought to develop from CPs as CCR6+CXCR5+ B cells are recruited through the chemokines CCL20 and CXCL13 produced by epithelial cells and dendritic cells respectively (McDonald et al., 2010; van de Pavert and Mebius, 2010). These structures are quite dynamic and plastic in nature, representing a spectrum of varying maturity states, possibly influenced by the constant interaction between the host and its luminal contents.
As mentioned earlier, the development of CPs and ILFs requires factors involved in the proper functioning of Type-17/22 ILCs, with AHR now added to the growing list. It is important to clarify that AHR has broad expression patterns within different cell types including immune cells. Therefore it was important to determine whether AHR signaling was critical in Type-17/22 ILCs or was required in other subsets that orchestrate Type-17/22 ILC recruitment to CPs and ILFs or to gut lamina propria. Using various conditional deletion mutants for AHR, it was found that the requirement for AHR in the development of Type-17/22 ILCs (Kiss et al., 2011; Lee et al., 2011) as well as the organization of CPs and ILFs was indeed intrinsic to RORγt expressing cells. CPs and ILFs from AHR conditional deletion mutants driven by RORγt-Cre recombinase had the same phenotype as AHR null animals while these structures from AHR conditional deletion mutants driven by CD11c or Villin – Cre were unaffected (Kiss et al., 2011). These studies seem to indicate that while AHR is expressed in intestinal epithelial cells or other mucosal-associated immune populations, AHR signaling within RORγt expressing ILCs is critical for their development as well as the formation of CPs and ILFs.
Proposed Mechanisms for AHR Requirement in Type-17/22 ILC Development
C-kit
What is the mechanism behind the AHR-mediated development of Type-17/22 ILCs? Interestingly using similar approaches, two different mechanisms have been proposed. AHR has been shown to induce the transcription of many target genes, and it was important to determine which of these target genes were important for Type-17/22 ILC development. One study focused on the receptor tyrosine kinase c-kit as a downstream target of AHR signaling (Kiss et al., 2011). Type-17/22 ILCs are identified by their expression of c-kit, and c-kit was shown to regulate intestinal ILC numbers and lymph node organogenesis (Chappaz et al., 2010). Closer examination of the c-kit promoter uncovered two canonical xenobiotic responsive (XRE) elements and ChIP analysis revealed the specific interaction of AHR with these XRE elements within the c-kit promoter. It is noteworthy to point out that AHR-driven upregulation of c-kit has also been reported to be critical for the development of epidermal resident γδ T cells (Li et al., 2011). Activation of AHR with agonists in vitro demonstrated the induction of c-kit transcription and this AHR-mediated transcription of c-kit was abrogated when the XRE elements within the c-kit promoter were mutated. It was also found that Kitwv/wv mice, which express a receptor with impaired kinase activity thus severely reducing c-kit signaling, also had decreased numbers of Type-17/22 ILCs and severely diminished numbers of CPs and ILFs. Therefore the conclusion from this study was that AHR-mediated transcription stabilizes c-kit expression which is necessary for the stem cell factor-dependent maintenance of the Type-17/22 ILC pool numbers and subsequent formation of postnatal lymphoid follicles. Accordingly, this study showed that in AHR deficient mice Type-17/22 ILCs are found in normal numbers at early time points after birth, but because of the lack of AHR signaling and c-kit expression they do not survive in the AHR-devoid environment.
Along the same lines, a second study has also demonstrated normal frequencies of intestinal Type-17/22 ILCs in WT or AHR deficient mice at embryonic day 17.5 (Qiu et al., 2011). This study suggests that AHR is important for the survival and maintenance of Type-17/22 ILCs. Intestinal Type-17/22 ILCs had higher Annexin V staining as well as decreased expression of the antiapoptotic genes Bcl2 and Bcl2l1, perhaps through the reduction of IL-7 in the large intestines of AHR deficient mice. However the exact mechanisms behind the regulation of AHR, IL-7, and antiapoptotic genes need to be addressed further.
Notch
A third study has proposed the induction of Notch as the mechanism behind AHR-driven development of at least small intestine lamina propria NKp46+ ILCs (Lee et al., 2011). Notch signaling affects tissue renewal, maintenance, and cell differentiation decisions across a broad range of cell types and at different steps during cell lineage progression. Notch proteins are cell surface receptors, four of which have been described in mammals: Notch1–Notch4 (Kopan and Ilagan, 2009). The requirement for Notch signaling during T cell differentiation as well as activation is well characterized and has been reviewed (Osborne and Minter, 2007). Notch has also been implicated in the development of adult but not fetal RORγt+ ILCs in vitro (Possot et al., 2011) as well as in promoting the differentiation of TH17 cells (Mukherjee et al., 2009; Keerthivasan et al., 2011). Another interesting report suggested a potential Notch–AHR axis for IL-22 production through the Notch induced generation of as of yet unknown endogenous AHR ligands in CD4+ T cells (Alam et al., 2010). However a connection between AHR signaling and the upregulation of Notch have not been clearly demonstrated. AHR agonists administered in vivo resulted in the rapid induction of Notch genes in both the liver and in the lamina propria cells isolated from the small intestine. NK cell lines transfected with a constitutively active form of AHR also upregulated Notch2, demonstrating that AHR signaling in part, regulates the expression of Notch. Animals lacking RBP-J in hematopoietic cells partially resembled the phenotype of AHR deficient animals. RBP-J is the binding partner of all Notch proteins and necessary for Notch signaling (Tanigaki and Honjo, 2010). These animals showed a severe reduction of NKp46+ ILCs in the small intestine lamina propria. However, in contrast to AHR deficient animals, the CD4+ LTi-like ILCs were to some extent preserved. Subsequent histological examination of diffuse postnatal lymphoid follicles in the RBP-J deficient animals revealed intact CPs and ILFs, confirming the presence of functional LTi-like ILCs. It is possible that additional signals downstream of AHR partially compensate for the lack of Notch signaling in LTi-like ILCs. However, this dichotomy in the requirement for AHR by all Type-17/22 ILCs but the specific requirement of Notch signaling by NKp46+ ILCs, open new and interesting questions.
Fate mapping experiments of Type-17/22 ILCs have shown that the frequencies of RORγt+NKp46+ ILCs increase with time after birth, reaching a plateau 4–5 weeks after birth (Sawa et al., 2010). However, RORγt+CD4+ LTi-like ILCs have an opposite behavior, as their frequencies steadily decrease over time after birth, reaching a lower set point 2–3 weeks after birth. This evidence raises the possibility that indeed NKp46+ ILCs only generate from adult, most likely bone marrow derived, precursor cells through a pathway that is both AHR and Notch-dependent. Although some of these cells may localize into CPs and ILFs, they are mostly dispersed throughout the lamina propria and are irrelevant for CP and ILF formation. On the contrary, CD4+ LTi-like ILCs detectable during the first 2 weeks of age may still be of fetal origin, possibly from the fetal spleen or fetal liver hematopoietic stem cells (Mebius et al., 1997; Kim et al., 2008). These cells are AHR dependent but, as suggested by in vitro experiments, Notch-independent (Possot et al., 2011) for their development, and they may represent the critical clue in the generation of CP and ILF structures. Interestingly, during our bone marrow chimera experiments (Lee et al., 2011) when either Ahr−/− bone marrow or WT bone marrow were transferred into WT recipients, we noticed that some of the CD4+ LTi-like ILCs cells were still of host origin 11 weeks post-bone marrow transfer. This finding suggests that some of these cells are radioresistant. Therefore, it is possible that some of the CD4+ LTi-like ILCs found in the adult mouse derive from a putative fetal precursor that was seeded early on after birth and slowly proliferate in locus, while other CD4+ LTi-like ILCs derive from an adult precursor that depend on Notch signaling. This hypothesis would explain the partial dependence of CD4+ LTi-like ILCs on Notch signaling. Further investigation and generation of new tools that allow inducible ablation of Type-17/22 ILCs at different time points before and after birth will be required to determine whether this is indeed the case. Another interesting observation is that AHR seems to selectively impact the cell types responsible for the organization of CPs and ILFs which develop postnatally, but not Peyer’s patches and other peripheral lymph nodes which are imprinted prenatally. This finding not only suggests a temporal or environmental separation of these processes, but perhaps heterogeneity in the cell types responsible for their organogenesis. Even though the numbers and frequencies of both CD4+ and CD4− LTi-like ILCs are impacted by AHR deficiency, AHR seems to selectively effect the postnatal expansion of CD4− LTi-like ILCs (Kiss et al., 2011). However it is unclear from this study whether the observed expansion also included NKp46+ ILCs which would agree with previously published studies (Sawa et al., 2010). Whether RBP-J expression and Notch signaling also has selective effects on CD4− LTi-like ILCs has yet to be determined.
Exogenous and Endogenous Sources of AHR Ligands; Which Matters the Most?
What are the AHR ligands responsible for development and/or maintenance of Type-17/22 ILCs? Currently, known environmental ligands of AHR include halogenated aromatic hydrocarbons, polychlorinated biphenyls (PCBs), and polycyclic aromatic hydrocarbons (PAHs; Nguyen and Bradfield, 2008). Endogenous AHR ligands isolated from mammalian tissues have also been identified (Song et al., 2002), and are currently an intense area of research. Arachidonic acid metabolites, heme metabolites, and tryptophan metabolites (Veldhoen et al., 2009), have been identified as having AHR agonist activity and are well reviewed (Denison and Nagy, 2003; Nguyen and Bradfield, 2008). Natural dietary compounds are also an abundant source of AHR agonists. Indole-3-carbinol (I3C) is a phytochemical found in cruciferous plants including broccoli and Brussels sprouts. The acidic environment of the stomach produces I3C acid-condensation products that have potent AHR agonist activity (Bjeldanes et al., 1991). One such I3C derivative is indolo[3,2-b]carbazole (ICZ) which binds to AHR with high affinity (Chen et al., 1995). Natural flavonoids ubiquitously found in fruits and vegetables were also found to activate AHR, including chrysin, galangin, baicalein, genistein, daidzein, and apigenin (Amakura et al., 2003; Zhang et al., 2003).
Exogenous Dietary Sources of AHR Ligands
Two independent studies have focused on natural plant derived dietary compounds found in the conventional rodent diet as a source of AHR agonists (Kiss et al., 2011; Li et al., 2011). It was demonstrated that replacing the conventional plant derived diet with a synthetic, highly refined form of rodent diet seemingly devoid of plant products reduced the numbers of AHR dependent cells in the gut including Type-17/22 ILCs (Kiss et al., 2011) as well as intraepithelial (IEL) γδ T cells (Li et al., 2011). The addition of I3C into this synthetic diet restored normal numbers of these cell types, thus suggesting the importance of vegetable derived dietary components rich in AHR ligands especially early in life for the maintenance of immune populations within the gut mucosa. These studies demonstrated a mechanistic link between dietary compounds and the modulation of immune populations and their functions. However, our preliminary data on diet supplemented with AHR ligands are at odd with these results. It is possible that in our settings the existence of alternative and unexpected sources of AHR ligands (such as bedding or water), and slight variations in the formula of the synthetic diets, as well as variations between different suppliers might explain these discrepancies. Although recent publications have demonstrated Type-17/22 ILCs develop independently of the microbiota, it cannot be ruled out that the differences observed with purified synthetic diets may also be a result of variations between the microbiota in the mice housed among the various animal facilities and their contribution to AHR ligands. However, an alternative interpretation may be that, while exogenous dietary derived AHR ligands may be playing a role in certain circumstances, endogenous agonists are sufficient in a steady state condition to induce development or to maintain Type-17/22 ILCs.
Natural Endogenous Sources of AHR Ligands
While the physiologically relevant endogenous agonist has yet to be concretely established, some attractive candidates have been identified. Kynurenine, a tryptophan metabolite generated from the indoleamine-2,3 dioxygenase (IDO) pathway, has recently been demonstrated to induce Cyp1a1 in bone marrow derived dendritic cells (Mezrich et al., 2010; Nguyen et al., 2010), as well as in CD103+ mucosal dendritic cells (Matteoli et al., 2010). IDO expression as well as its induction has been shown to be dependent upon AHR signaling (Mezrich et al., 2010; Nguyen et al., 2010), and IDO enzymatic dependent and independent mechanisms of immunomodulation have been described (Pallotta et al., 2011). Many studies have suggested that AHR expression in DCs is critical to expand Treg through an IDO-dependent production of the tryptophan catabolite kynurenine, that acts as an endogenous AHR ligand (Veldhoen et al., 2009; Mezrich et al., 2010; Nguyen et al., 2010; Wu et al., 2011). This mechanism of Treg induction is most likely highly active in tumors, since it has been recently shown that kynurenine is abundantly expressed in many tumors (Opitz et al., 2011). In tumors, however, kynurenine is not produced by the tryptophan metabolizing enzymes IDO1 and/or IDO2, as in dendritic cells, but is uniquely generated by the enzyme tryptophan-2,3 dioxygenase (TDO; Opitz et al., 2011). Given the abundance of studies demonstrating the important role of tryptophan metabolism in modulating immune responses, further investigation into the AHR/kynurenine pathway in Type-17/22 ILCs is quite promising. While synthetic highly purified diets low in AHR agonists may reduce the environmental contribution of AHR ligands, it may be more difficult to distinguish among other factors such as nutrients, preservatives, as well as housing and bedding variances. New ways to partially block formation or deplete endogenous AHR ligands may be a promising avenue to dissect the contribution of natural endogenous versus exogenous compounds that drive the differentiation of Type-17/22 ILCs.
Conclusion
A recent string of publications connecting the classical dioxin sensor AHR with the development and maintenance of mucosal immune cell populations demonstrate an unexplored frontier in mucosal immunology. While teasing out the source of these exogenous and/or endogenous ligands is not clearly understood at this moment, the hypothesis that dietary derived compounds are intricately involved in the development of mucosal immune structures as well as the maintenance of immune cells in the gutis quite intriguing. Further studies will need to carefully characterize the exact mechanism by which AHR signaling seems to be exerting its effects, as well as identifying the natural ligands responsible; either from endogenous compounds or from exogenous dietary sources.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adachi, S. (1998). Essential role of IL-7 receptor alpha in the formation of Peyer’s patch anlage. Int. Immunol. 10, 1–6.
Alam, M. S., Maekawa, Y., Kitamura, A., Tanigaki, K., Yoshimoto, T., Kishihara, K., and Yasutomo, K. (2010). Notch signaling drives IL-22 secretion in CD4+ T cells by stimulating the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. U.S.A. 107, 5943–5948.
Amakura, Y., Tsutsumi, T., Nakamura, M., Kitagawa, H., Fujino, J., Sasaki, K., Toyoda, M., Yoshida, T., and Maitani, T. (2003). Activation of the aryl hydrocarbon receptor by some vegetable constituents determined using in vitro reporter gene assay. Biol. Pharm. Bull. 26, 532–539.
Bjeldanes, L. F., Kim, J. Y., Grose, K. R., Bartholomew, J. C., and Bradfield, C. A. (1991). Aromatic hydrocarbon responsiveness-receptor agonists generated from indole-3-carbinol in vitro and in vivo: comparisons with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Proc. Natl. Acad. Sci. U.S.A. 88, 9543–9547.
Bouskra, D., Brézillon, C., Bérard, M., Werts, C., Varona, R., Boneca, I. G., and Eberl, G. (2008). Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature 456, 507–510.
Buonocore, S., Ahern, P. P., Uhlig, H. H., Ivanov, I. I., Littman, D. R., Maloy, K. J., and Powrie, F. (2010). Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature 464, 1371–1375.
Carver, L. A., and Bradfield, C. A. (1997). Ligand-dependent interaction of the aryl hydrocarbon receptor with a novel immunophilin homolog in vivo. J. Biol. Chem. 272, 11452–11456.
Cella, M., Fuchs, A., Vermi, W., Facchetti, F., Otero, K., Lennerz, J. K. M., Doherty, J. M., Mills, J. C., and Colonna, M. (2009). A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature 457, 722–725.
Cella, M., Otero, K., and Colonna, M. (2010). Expansion of human NK-22 cells with IL-7, IL-2, and IL-1beta reveals intrinsic functional plasticity. Proc. Natl. Acad. Sci. U.S.A. 107, 10961–10966.
Chappaz, S., Gärtner, C., Rodewald, H.-R., and Finke, D. (2010). Kit ligand and Il7 differentially regulate Peyer’s patch and lymph node development. J. Immunol. 185, 3514–3519.
Chen, Y. H., Riby, J., Srivastava, P., Bartholomew, J., Denison, M., and Bjeldanes, L. (1995). Regulation of CYP1A1 by indolo[3,2-b]carbazole in murine hepatoma cells. J. Biol. Chem. 270, 22548–22555.
Crellin, N. K., Trifari, S., Kaplan, C. D., Cupedo, T., and Spits, H. (2010). Human NKp44+IL-22+ cells and LTi-like cells constitute a stable RORC+ lineage distinct from conventional natural killer cells. J. Exp. Med. 207, 281–290.
Cupedo, T., Crellin, N. K., Papazian, N., Rombouts, E. J., Weijer, K., Grogan, J. L., Fibbe, W. E., Cornelissen, J. J., and Spits, H. (2009). Human fetal lymphoid tissue-inducer cells are interleukin 17-producing precursors to RORC+ CD127+ natural killer-like cells. Nat. Immunol. 10, 66–74.
Denison, M. S., and Nagy, S. R. (2003). Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu. Rev. Pharmacol. Toxicol. 43, 309–334.
Doisne, J.-M., Soulard, V., Bécourt, C., Amniai, L., Henrot, P., Havenar-Daughton, C., Blanchet, C., Zitvogel, L., Ryffel, B., Cavaillon, J.-M., Marie, J. C., Couillin, I., and Benlagha, K. (2011). Cutting edge: crucial role of IL-1 and IL-23 in the innate IL-17 response of peripheral lymph node NK1.1-invariant NKT cells to bacteria. J. Immunol. 186, 662–666.
Dumoutier, L., de Heusch, M., Orabona, C., Satoh-Takayama, N., Eberl, G., Sirard, J.-C., Di Santo, J. P., and Renauld, J.-C. (2011). IL-22 is produced by γC-independent CD25+CCR6+ innate murine spleen cells upon inflammatory stimuli and contributes to LPS-induced lethality. Eur. J. Immunol. 41, 1075–1085.
Eberl, G., Marmon, S., Sunshine, M.-J., Rennert, P. D., Choi, Y., and Littman, D. R. (2004). An essential function for the nuclear receptor RORgamma(t) in the generation of fetal lymphoid tissue inducer cells. Nat. Immunol. 5, 64–73.
Esser, C. (2009). The immune phenotype of AhR null mouse mutants: not a simple mirror of xenobiotic receptor over-activation. Biochem. Pharmacol. 77, 597–607.
Fernandez-Salguero, P., Pineau, T., Hilbert, D. M., McPhail, T., Lee, S. S., Kimura, S., Nebert, D. W., Rudikoff, S., Ward, J. M., and Gonzalez, F. J. (1995). Immune system impairment and hepatic fibrosis in mice lacking the dioxin-binding Ah receptor. Science 268, 722–726.
Gu, Y. Z., Hogenesch, J. B., and Bradfield, C. A. (2000). The PAS superfamily: sensors of environmental and developmental signals. Annu. Rev. Pharmacol. Toxicol. 40, 519–561.
Hamada, H., Hiroi, T., Nishiyama, Y., Takahashi, H., Masunaga, Y., Hachimura, S., Kaminogawa, S., Takahashi-Iwanaga, H., Iwanaga, T., Kiyono, H., Yamamoto, H., and Ishikawa, H. (2002). Identification of multiple isolated lymphoid follicles on the antimesenteric wall of the mouse small intestine. J. Immunol. 168, 57–64.
Hughes, T., Becknell, B., Freud, A. G., McClory, S., Briercheck, E., Yu, J., Mao, C., Giovenzana, C., Nuovo, G., Wei, L., Zhang, X., Gavrilin, M. A., Wewers, M. D., and Caligiuri, M. A. (2010). Interleukin-1beta selectively expands and sustains interleukin-22+ immature human natural killer cells in secondary lymphoid tissue. Immunity 32, 803–814.
Kanamori, Y., Ishimaru, K., Nanno, M., Maki, K., Ikuta, K., Nariuchi, H., and Ishikawa, H. (1996). Identification of novel lymphoid tissues in murine intestinal mucosa where clusters of c-kit+ IL-7R+ Thy1+ lympho-hemopoietic progenitors develop. J. Exp. Med. 184, 1449–1459.
Kazlauskas, A., Poellinger, L., and Pongratz, I. (1999). Evidence that the co-chaperone p23 regulates ligand responsiveness of the dioxin (Aryl hydrocarbon) receptor. J. Biol. Chem. 274, 13519–13524.
Keerthivasan, S., Suleiman, R., Lawlor, R., Roderick, J., Bates, T., Minter, L., Anguita, J., Juncadella, I., Nickoloff, B. J., Le Poole, I. C., Miele, L., and Osborne, B. A. (2011). Notch signaling regulates mouse and human Th17 differentiation. J. Immunol. 187, 692–701.
Kim, M.-Y., Rossi, S., Withers, D., McConnell, F., Toellner, K.-M., Gaspal, F., Jenkinson, E., Anderson, G., and Lane, P. J. L. (2008). Heterogeneity of lymphoid tissue inducer cell populations present in embryonic and adult mouse lymphoid tissues. Immunology 124, 166–174.
Kiss, E. A., Vonarbourg, C., Kopfmann, S., Hobeika, E., Finke, D., Esser, C., and Diefenbach, A. (2011). Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science 334, 1561–1565.
Kopan, R., and Ilagan, M. X. G. (2009). The canonical Notch signaling pathway: unfolding the activation mechanism. Cell 137, 216–233.
Koyasu, S., and Moro, K. (2011). Innate Th2-type immune responses and the natural helper cell, a newly identified lymphocyte population. Curr. Opin. Allergy Clin. Immunol. 11, 109–114.
Lahvis, G. P., Lindell, S. L., Thomas, R. S., McCuskey, R. S., Murphy, C., Glover, E., Bentz, M., Southard, J., and Bradfield, C. A. (2000). Portosystemic shunting and persistent fetal vascular structures in aryl hydrocarbon receptor-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 97, 10442–10447.
Lee, J. S., Cella, M., McDonald, K. G., Garlanda, C., Kennedy, G. D., Nukaya, M., Mantovani, A., Kopan, R., Bradfield, C. A., Newberry, R. D., and Colonna, M. (2011). AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat. Immunol. 13, 144–151.
Li, Y., Innocentin, S., Withers, D. R., Roberts, N. A., Gallagher, A. R., Grigorieva, E. F., Wilhelm, C., and Veldhoen, M. (2011). Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell 147, 629–640.
Lorenz, R. G., and Newberry, R. D. (2004). Isolated lymphoid follicles can function as sites for induction of mucosal immune responses. Ann. N. Y. Acad. Sci. 1029, 44–57.
Luci, C., Reynders, A., Ivanov, I. I., Cognet, C., Chiche, L., Chasson, L., Hardwigsen, J., Anguiano, E., Banchereau, J., Chaussabel, D., Dalod, M., Littman, D. R., Vivier, E., and Tomasello, E. (2009). Influence of the transcription factor RORgammat on the development of NKp46+ cell populations in gut and skin. Nat. Immunol. 10, 75–82.
Matteoli, G., Mazzini, E., Iliev, I. D., Mileti, E., Fallarino, F., Puccetti, P., Chieppa, M., and Rescigno, M. (2010). Gut CD103+ dendritic cells express indoleamine 2,3-dioxygenase which influences T regulatory/T effector cell balance and oral tolerance induction. Gut 59, 595–604.
McDonald, K. G., McDonough, J. S., Dieckgraefe, B. K., and Newberry, R. D. (2010). Dendritic cells produce CXCL13 and participate in the development of murine small intestine lymphoid tissues. Am. J. Pathol. 176, 2367–2377.
Mebius, R. E., Rennert, P., and Weissman, I. L. (1997). Developing lymph nodes collect CD4+CD3-LTbeta+ cells that can differentiate to APC, NK cells, and follicular cells but not T or B cells. Immunity 7, 493–504.
Mezrich, J. D., Fechner, J. H., Zhang, X., Johnson, B. P., Burlingham, W. J., and Bradfield, C. A. (2010). An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 185, 3190–3198.
Mimura, J., and Fujii-Kuriyama, Y. (2003). Functional role of AhR in the expression of toxic effects by TCDD. Biochim. Biophys. Acta 1619, 263–268.
Mjösberg, J. M., Trifari, S., Crellin, N. K., Peters, C. P., van Drunen, C. M., Piet, B., Fokkens, W. J., Cupedo, T., and Spits, H. (2011). Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat. Immunol. 12, 1055–1062.
Monticelli, L. A., Sonnenberg, G. F., Abt, M. C., Alenghat, T., Ziegler, C. G. K., Doering, T. A., Angelosanto, J. M., Laidlaw, B. J., Yang, C. Y., Sathaliyawala, T., Kubota, M., Turner, D., Diamond, J. M., Goldrath, A. W., Farber, D. L., Collman, R. G., Wherry, E. J., and Artis, D. (2011). Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat. Immunol. 12, 1045–1054.
Moro, K., Yamada, T., Tanabe, M., Takeuchi, T., Ikawa, T., Kawamoto, H., Furusawa, J.-I., Ohtani, M., Fujii, H., and Koyasu, S. (2010). Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature 463, 540–544.
Mukherjee, S., Schaller, M. A., Neupane, R., Kunkel, S. L., and Lukacs, N. W. (2009). Regulation of T cell activation by Notch ligand, DLL4, promotes IL-17 production and Rorc activation. J. Immunol. 182, 7381–7388.
Nebert, D. W., Puga, A., and Vasiliou, V. (1993). Role of the Ah receptor and the dioxin-inducible [Ah] gene battery in toxicity, cancer, and signal transductiona. Ann. N. Y. Acad. Sci. 685, 624–640.
Neill, D. R., Wong, S. H., Bellosi, A., Flynn, R. J., Daly, M., Langford, T. K. A., Bucks, C., Kane, C. M., Fallon, P. G., Pannell, R., Jolin, H. E., and McKenzie, A. N. (2010). Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 464, 1367–1370.
Nguyen, L. P., and Bradfield, C. A. (2008). The search for endogenous activators of the aryl hydrocarbon receptor. Chem. Res. Toxicol. 21, 102–116.
Nguyen, N. T., Kimura, A., Nakahama, T., Chinen, I., Masuda, K., Nohara, K., Fujii-Kuriyama, Y., and Kishimoto, T. (2010). Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc. Natl. Acad. Sci. U.S.A. 107, 19961–19966.
Opitz, C. A., Litzenburger, U. M., Sahm, F., Ott, M., Tritschler, I., Trump, S., Schumacher, T., Jestaedt, L., Schrenk, D., Weller, M., Jugold, M., Guillemin, G. J., Miller, C. L., Lutz, C., Radlwimmer, B., Lehmann, I., von Deimling, A., Wick, W., and Platten, M. (2011). An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 478, 197–203.
Osborne, B. A., and Minter, L. M. (2007). Notch signalling during peripheral T-cell activation and differentiation. Nat. Rev. Immunol. 7, 64–75.
Pallotta, M. T., Orabona, C., Volpi, C., Vacca, C., Belladonna, M. L., Bianchi, R., Servillo, G., Brunacci, C., Calvitti, M., Bicciato, S., Mazza, E. M., Boon, L., Grassi, F., Fioretti, M. C., Fallarino, F., Puccetti, P., and Grohmann, U. (2011). Indoleamine 2,3-dioxygenase is a signaling protein in long-term tolerance by dendritic cells. Nat. Immunol. 12, 870–878.
Perdew, G. H. (1988). Association of the Ah receptor with the 90-kDa heat shock protein. J. Biol. Chem. 263, 13802–13805.
Possot, C., Schmutz, S., Chea, S., Boucontet, L., Louise, A., Cumano, A., and Golub, R. (2011). Notch signaling is necessary for adult, but not fetal, development of RORγt(+) innate lymphoid cells. Nat. Immunol. 12, 949–958.
Price, A. E., Liang, H.-E., Sullivan, B. M., Reinhardt, R. L., Eisley, C. J., Erle, D. J., and Locksley, R. M. (2010). Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc. Natl. Acad. Sci. U.S.A. 107, 11489–11494.
Qiu, J., Heller, J. J., Guo, X., Chen, Z. M., Fish, K., Fu, Y.-X., and Zhou, L. (2011). The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity 36, 92–104.
Reyes, H., Reisz-Porszasz, S., and Hankinson, O. (1992). Identification of the Ah receptor nuclear translocator protein (Arnt) as a component of the DNA binding form of the Ah receptor. Science 256, 1193–1195.
Reynders, A., Yessaad, N., Vu Manh, T.-P., Dalod, M., Fenis, A., Aubry, C., Nikitas, G., Escalière, B., Renauld, J. C., Dussurget, O., Cossart, P., Lecuit, M., Vivier, E., and Tomasello, E. (2011). Identity, regulation and in vivo function of gut NKp46+RORγt+ and NKp46+RORγt- lymphoid cells. EMBO J. 30, 2934–2947.
Saenz, S. A., Siracusa, M. C., Perrigoue, J. G., Spencer, S. P., Urban, J. F., Tocker, J. E., Budelsky, A. L., Kleinschek, M. A., Kastelein, R. A., Kambayashi, T., Bhandoola, A., and Artis, D. (2010). IL25 elicits a multipotent progenitor cell population that promotes T(H)2 cytokine responses. Nature 464, 1362–1366.
Sanos, S. L., Bui, V. L., Mortha, A., Oberle, K., Heners, C., Johner, C., and Diefenbach, A. (2009). RORgammat and commensal microflora are required for the differentiation of mucosal interleukin 22-producing NKp46+ cells. Nat. Immunol. 10, 83–91.
Satoh-Takayama, N., Lesjean-Pottier, S., Vieira, P., Sawa, S., Eberl, G., Vosshenrich, C. A. J., and Di Santo, J. P. (2010). IL-7 and IL-15 independently program the differentiation of intestinal CD3-NKp46+ cell subsets from Id2-dependent precursors. J. Exp. Med. 207, 273–280.
Satoh-Takayama, N., Vosshenrich, C. A. J., Lesjean-Pottier, S., Sawa, S., Lochner, M., Rattis, F., Mention, J.-J., Thiam, K., Cerf-Bensussan, N., Mandelboim, O., Eberl, G., and Di Santo, J. P. (2008). Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity 29, 958–970.
Sawa, S., Cherrier, M., Lochner, M., Satoh-Takayama, N., Fehling, H. J., Langa, F., Di Santo, J. P., and Eberl, G. (2010). Lineage relationship analysis of RORgammat+ innate lymphoid cells. Science 330, 665–669.
Sawa, S., Lochner, M., Satoh-Takayama, N., Dulauroy, S., Bérard, M., Kleinschek, M., Cua, D., Di Santo, J. P., and Eberl, G. (2011). RORγt+ innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nat. Immunol. 12, 320–326.
Schmidt, J. V., Su, G. H., Reddy, J. K., Simon, M. C., and Bradfield, C. A. (1996). Characterization of a murine Ahr null allele: involvement of the Ah receptor in hepatic growth and development. Proc. Natl. Acad. Sci. U.S.A. 93, 6731–6736.
Shikina, T., Hiroi, T., Iwatani, K., Jang, M. H., Fukuyama, S., Tamura, M., Kubo, T., Ishikawa, H., and Kiyono, H. (2004). IgA class switch occurs in the organized nasopharynx- and gut-associated lymphoid tissue, but not in the diffuse lamina propria of airways and gut. J. Immunol. 172, 6259–6264.
Shimizu, Y., Nakatsuru, Y., Ichinose, M., Takahashi, Y., Kume, H., Mimura, J., Fujii-Kuriyama, Y., and Ishikawa, T. (2000). Benzo[a]pyrene carcinogenicity is lost in mice lacking the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. U.S.A. 97, 779–782.
Song, J., Clagett-Dame, M., Peterson, R. E., Hahn, M. E., Westler, W. M., Sicinski, R. R., and DeLuca, H. F. (2002). A ligand for the aryl hydrocarbon receptor isolated from lung. Proc. Natl. Acad. Sci. U.S.A. 99, 14694–14699.
Sonnenberg, G. F., Fouser, L. A., and Artis, D. (2011). Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat. Immunol. 12, 383–390.
Spits, H., and Di Santo, J. P. (2011). The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nat. Immunol. 12, 21–27.
Stevens, E. A., Mezrich, J. D., and Bradfield, C. A. (2009). The aryl hydrocarbon receptor: a perspective on potential roles in the immune system. Immunology 127, 299–311.
Stockinger, B., Hirota, K., Duarte, J., and Veldhoen, M. (2011). External influences on the immune system via activation of the aryl hydrocarbon receptor. Semin. Immunol. 23, 99–105.
Sutton, C. E., Lalor, S. J., Sweeney, C. M., Brereton, C. F., Lavelle, E. C., and Mills, K. H. G. (2009). Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity 31, 331–341.
Takatori, H., Kanno, Y., Watford, W. T., Tato, C. M., Weiss, G., Ivanov, I. I., Littman, D. R., and O’Shea, J. J. (2009). Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. J. Exp. Med. 206, 35–41.
Takeda, K., Tsutsui, H., Yoshimoto, T., Adachi, O., Yoshida, N., Kishimoto, T., Okamura, H., Nakanishi, K., and Akira, S. (1998). Defective NK cell activity and Th1 response in IL-18-deficient mice. Immunity 8, 383–390.
Tanigaki, K., and Honjo, T. (2010). Two opposing roles of RBP-J in Notch signaling. Curr. Top. Dev. Biol. 92, 231–252.
Tumanov, A. V., Koroleva, E. P., Guo, X., Wang, Y., Kruglov, A., Nedospasov, S., and Fu, Y.-X. (2011). Lymphotoxin controls the IL-22 protection pathway in gut innate lymphoid cells during mucosal pathogen challenge. Cell Host Microbe 10, 44–53.
van de Pavert, S. A., and Mebius, R. E. (2010). New insights into the development of lymphoid tissues. Nat. Rev. Immunol. 10, 664–674.
Veldhoen, M., Hirota, K., Christensen, J., O’Garra, A., and Stockinger, B. (2009). Natural agonists for aryl hydrocarbon receptor in culture medium are essential for optimal differentiation of Th17 T cells. J. Exp. Med. 206, 43–49.
Wang, Y., Koroleva, E. P., Kruglov, A. A., Kuprash, D. V., Nedospasov, S. A., Fu, Y.-X., and Tumanov, A. V. (2010). Lymphotoxin beta receptor signaling in intestinal epithelial cells orchestrates innate immune responses against mucosal bacterial infection. Immunity 32, 403–413.
Wilson, C. L., and Safe, S. (1998). Mechanisms of ligand-induced aryl hydrocarbon receptor-mediated biochemical and toxic responses. Toxicol. Pathol. 26, 657–671.
Wu, H. Y., Quintana, F. J., da Cunha, A. P., Dake, B. T., Koeglsperger, T., Starossom, S. C., and Weiner, H. L. (2011). In vivo induction of Tr1 cells via mucosal dendritic cells and AHR signaling. PLoS ONE 6, e23618. doi:10.1371/journal.pone.0023618
Yang, Q., Saenz, S. A., Zlotoff, D. A., Artis, D., and Bhandoola, A. (2011). Cutting edge: natural helper cells derive from lymphoid progenitors. J. Immunol. 187, 5505–5509.
Yokota, Y., Mansouri, A., Mori, S., Sugawara, S., Adachi, S., Nishikawa, S. I., and Gruss, P. (1999). Development of peripheral lymphoid organs and natural killer cells depends on the helix–loop–helix inhibitor Id2. Nature 397, 702–706.
Yoshida, H. (1999). IL-7 receptor alpha+ CD3 cells in the embryonic intestine induces the organizing center of Peyer’s patches. Int. Immunol. 11, 643–655.
Keywords: innate lymphoid cell, IL-22, AHR, isolated lymphoid follicle, kynurenine, NKp46
Citation: Lee JS, Cella M and Colonna M (2012) AHR and the transcriptional regulation of Type-17/22 ILC. Front. Immun. 3:10. doi: 10.3389/fimmu.2012.00010
Received: 07 December 2011;
Paper pending published: 02 January 2012;
Accepted: 16 January 2012;
Published online: 06 February 2012.
Edited by:
Brigitta Stockinger, MRC National Institute for Medical Research, UKReviewed by:
Jean-Christophe Renauld, Ludwig Institute for Cancer Research, BelgiumEric Vivier, Centre d’Immunologie de Marseille-Luminy, France
Copyright: © 2012 Lee, Cella and Colonna. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Marco Colonna, Department of Pathology and Immunology, BJC Institute of Health, Washington University School of Medicine, 8th Floor Room 8107, 425 S. Euclid, St. Louis, MO 63110, USA. e-mail:bWNvbG9ubmFAcGF0aG9sb2d5Lnd1c3RsLmVkdQ==