

 Vasile I. Pavlov
Vasile I. Pavlov
- Department of Anesthesiology, Perioperative and Pain Medicine, Center for Experimental Therapeutics and Reperfusion Injury, Harvard Institutes of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, USA
Hyperglycemia, in the absence of type 1 or 2 diabetes, is an independent risk factor for cardiovascular disease. We have previously demonstrated a central role for mannose binding lectin (MBL)-mediated cardiac dysfunction in acute hyperglycemic mice. In this study, we applied whole-genome microarray data analysis to investigate MBL’s role in systematic gene expression changes. The data predict possible intracellular events taking place in multiple cellular compartments such as enhanced insulin signaling pathway sensitivity, promoted mitochondrial respiratory function, improved cellular energy expenditure and protein quality control, improved cytoskeleton structure, and facilitated intracellular trafficking, all of which may contribute to the organismal health of MBL null mice against acute hyperglycemia. Our data show a tight association between gene expression profile and tissue function which might be a very useful tool in predicting cellular targets and regulatory networks connected with in vivo observations, providing clues for further mechanistic studies.
Introduction
Diabetes mellitus has emerged as a major threat to worldwide health (Bojadzievski and Gabbay, 2011). Cardiovascular complications associated with hyperglycemia include cardiomyopathy and vasculopathy (Van Gaal et al., 2006). The pathophysiology of diabetic cardiomyopathy is likely multifactorial, with experimental evidence pointing to involvement of the rennin–angiotensin system (RAS), reactive oxygen species (ROS), and advanced glycosylation end (AGE) products and their receptors, which activate a number of secondary messenger pathways leading to activation and recruitment of inflammatory cells (Giacco and Brownlee, 2010). These molecular and cellular events culminate in the development of cardiac fibrosis, hypertrophy, and vascular dysfunction leading to cardiac dysfunction and enhanced injury. A connection between hyperglycemia-induced vasculopathy and the innate immune system, particularly mannose binding lectin (MBL), was demonstrated in diabetic patients (Hansen et al., 2004; Hovind et al., 2005; Fortpied et al., 2010).
Acute hyperglycemia, reported in many critical care settings, was originally regarded as non-problematic and potentially beneficial, by ensuring an adequate glucose supply to the brain and immune system. However, hyperglycemia, in the absence of type 1 or 2 diabetes mellitus, is an independent risk factor for cardiovascular disease (Davidson and Parkin, 2009). Hyperglycemia-induced injuries phenotypically mirror those of pressure-overloaded cardiac hypertrophy, and hyperglycemic patients have the same cardiovascular risk as patients who have had a myocardial infarction (Ceriello, 2005). Acute hyperglycemic disturbances are potent predictors of mortality in patients without previously diagnosed diabetes mellitus (Gustafsson et al., 2007).
We have established a short term (14-day) hyperglycemic mouse model that yields many of the features of hyperglycemia-induced cardiomyopathy and enhanced injury following myocardial ischemia and reperfusion (Busche et al., 2008). As compared to MBL null mice, hyperglycemic WT mice presented with significantly decreased left ventricular ejection fractions and increased serum troponin I levels. Echocardiographic measurements demonstrated signs of dilative cardiomyopathy, whereas heart:body weight ratios suggested hypertrophic cardiac remodeling after 2 weeks of hyperglycemia. Immunohistochemical analysis of cardiac progenitor cells showed significantly lower numbers in diabetic WT hearts (Busche et al., 2008). Our followup study further revealed that short term hyperglycemia (4 and 14 day), independent of oxidative stress or insulin levels, led to dilated cardiomyopathy with significantly increased short and long axis area measurements during systole and diastole in hyperglycemic WT mice compared to MBL null mice. The EC50 for acetylcholine-induced relaxation of mesenteric arterioles in WT mice following 4 days of hyperglycemia demonstrated a significant loss of nitric oxide-mediated relaxation. The hyperglycemic WT vasculopathy phenotype could be restored in the MBL null mice by addition of recombinant MBL during the hyperglycemic period. Myocardial histochemistry and western blots revealed a significant influx of macrophages, altered morphology, and increased elastin and collagen deposition in hyperglycemic WT hearts. Serum TGF-β1 levels were significantly lower in hyperglycemic MBL null compared to WT mice, suggesting decreased pro-fibrotic signaling (Pavlov et al., 2012).
It is likely that the molecular mechanisms involved in the hyperglycemia-induced cardiomyopathy and vasculopathy differences observed in the WT and MBL null genotypes are multifactorial. In the present study, we explored the whole-genome gene expression profiles of left ventricular transcripts from WT and MBL null mice following acute hyperglycemia in order to provide a basis for future study and investigation on the molecular mechanisms involved in this model. We used high-throughput whole-genome microarray scans as well as the database for annotation, visualization, and integrated discovery (DAVID) analysis tool to compare the gene transcriptional variances from 12 murine left ventricle samples (File Sa0 in Supplementary Material). The purpose of this study was to establish the intrinsic relationship between gene transcriptional changes and animal organ functional outcomes. The predicted cellular targets and regulatory networks connecting MBL with hyperglycemia-induced vasculopathy and cardiomyopathy presented in this study will facilitate future mechanistic studies in this model. We have already used these data to investigate successfully several molecular mechanisms involved and several additional manuscripts are in preparation. We believe that this study demonstrates a successful way to use microarray data to predict preclinical endpoints (MAQC-Consortium, 2010).
Materials and Methods
Agents and Chemicals
Streptozotocin (STZ) was purchased (Sigma-Aldrich; St. Louis, MO, USA). Citrated saline was purchased from ALEXIS (Lausen, Switzerland).
Animals
Mannose binding lectin null mice were developed by our collaborators and now have been deposited to The Jackson Laboratory (Takahashi et al., 2002; Shi et al., 2004). Male C57BL/6 (WT) mice (8–12 weeks old, Taconic Farms) were used as background controls for MBL null mice as directed by the online instruction1. The MBL null line was backcrossed to greater than 96% C57BL/6 (Moller-Kristensen et al., 2007). The following groups were studied: (1) WT and MBL null mice following 4-days of hyperglycemia and (2) WT and MBL null mice following 14-days of hyperglycemia (n = 3/group). Mice were housed four per cage and had unlimited access to water and standard mouse chow. All procedures were reviewed and conducted according to the Institute’s Animal Care and Use Committee. All experiments were performed under the standards and principles set forth in the Guide for Care and Use of Laboratory Animals, published by National Institute of Health (NIH Publication No. 85-23, Revised 1996).
Hyperglycemia Induction
Hyperglycemia was induced by a single injection of freshly prepared STZ solution (200 mg/kg body weight, i.p. in citrated saline, pH 4.2). Urinary glucose was tested on the fourth day following STZ injection and once more before the mice euthanized for tissue sampling and freezing. Mice with urinary glucose levels greater than 500 mg/dl (measured with Glucostix by Diastix, Bayer, Elkhart, IN, USA) in both tests were considered hyperglycemic. Control mice were injected with citrated saline. Sera insulin and glucose levels in this model have been published previously (Pavlov et al., 2012).
Genome-Wide Expression Profiling
Left ventricle free walls of mice were sent to GenUs BioSystems2 for microarray processing. Briefly, RNA was extracted and purified using Ribopure (Ambion) RNA isolation. The quality of total RNA was assessed using an Agilent Bioanalyzer. After first and second strand cDNA preparation, cRNA target was acquired from the DNA template and verified on the Bioanalyzer. cRNA was then fragmented to uniform size and hybridized to Agilent Mouse GE 4 K × 44 K v2 arrays. Slides were washed and scanned on an Agilent G2565 Microarray Scanner. Data were analyzed with Agilent Feature Extraction and GeneSpring GX v7.3.1 software packages.
Microarray Data Analysis
The original microarray probe intensity data are listed (File Sa0 in Supplementary Material). Only genes differently expressed (MBL null vs. WT, t-test, p < 0.05) were uploaded with their Entrez Gene ID to DAVID3. Four or fourteen-day hyperglycemic groups, up-regulated (MBL null/WT > 1) or down-regulated (MBL null/WT < 1) genes were analyzed separately following the published protocol for DAVID (Huang et al., 2009). Gene functional classification was employed to explore and view functionally related genes together, as a unit, to concentrate on the larger biological network rather than at the level of individual genes. The mouse whole-genome was set as the population background in enrichment analysis. We used the highest classification stringency to generate less functional gene groups with more tightly associated genes in each group. Only groups with an enrichment score greater than 2 (which means statistical significance p < 0.01 for the gene enrichment) were counted. The physiological meaning of clustered genes was extracted by analyzing the biological functions of those gene families. The gene summary of individual genes (by clicking the gene name) as well as gene ontology terms (by clicking the red “T” on the right of enrichment score) was also explored to assist in understanding these highly organized biologically meaningful modules.
Statistical Analysis
Microarray probe intensity comparisons between WT and MBL null groups were made using Student’s t-test. The differences were considered significant at p < 0.05.
Results
Differentially expressed genes (File Sa1–4 in Supplementary Material) were functionally classified into groups by DAVID (File Sb1–4 in Supplementary Material). Those gene clusters as well as affiliated gene families were also categorized in Tables 1– 4.
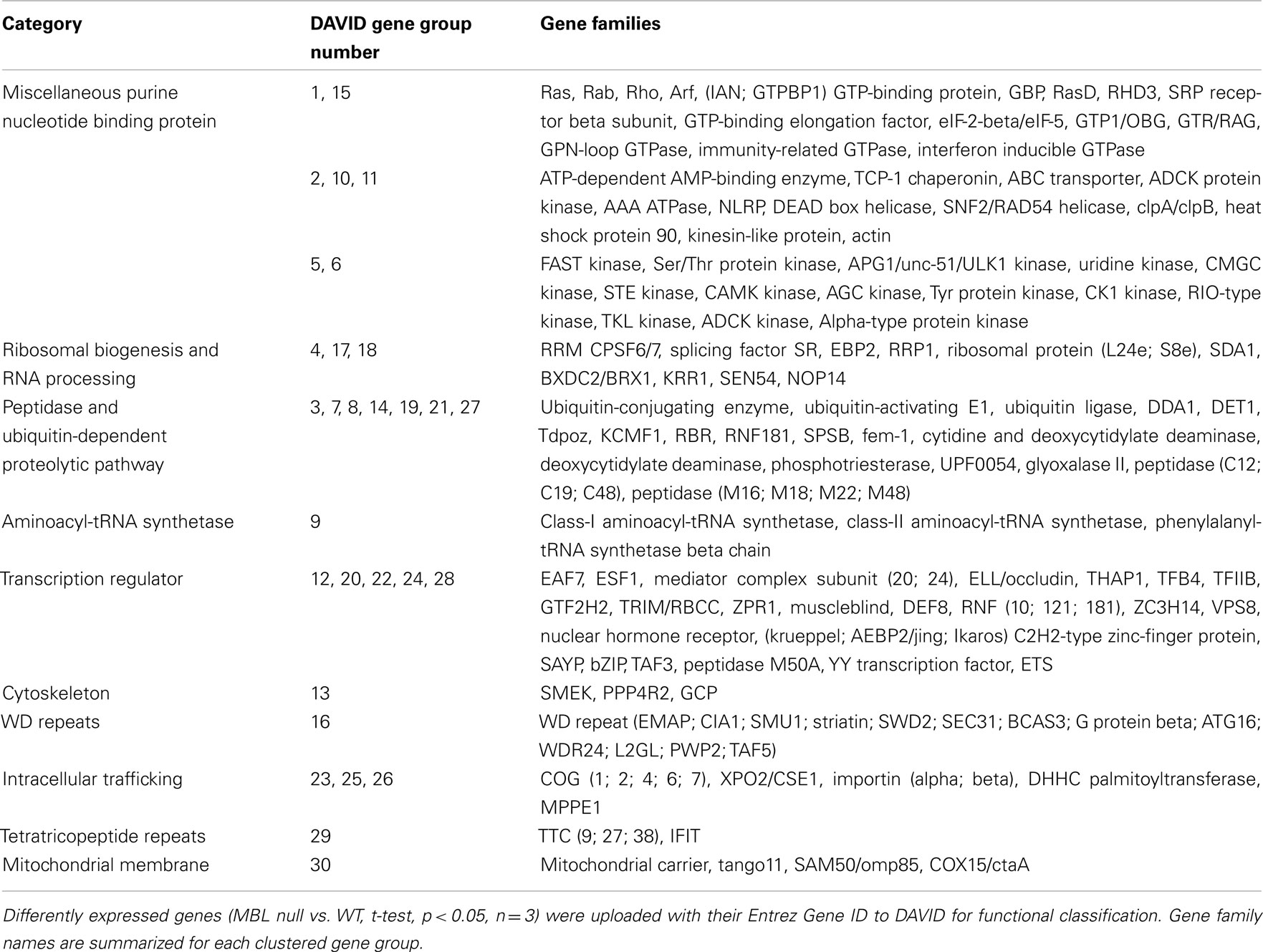
Table 1. Gene families of up-regulated genes in 4-day hyperglycemic MBL null mice compared to WT control.
Higher Expression of Downstream Effectors in Hyperglycemic MBL Null Mice May Sensitize Insulin Signaling Pathways
STZ selectively kills insulin-secreting β cells and the subsequent lack of insulin leads to increased blood and urinary glucose. Insulin has important cardiac actions. Diminished or resistant insulin signaling results in cardiomyopathy and vasculopathy, with complications including non-ischemic heart failure, which is a consequence of lipotoxicity, sympathetic up-regulation, inflammation, oxidative stress, and fibrosis (Witteles and Fowler, 2008); as well as hypertension, coronary heart disease, and atherosclerosis which are all characterized by endothelial cell dysfunction (Muniyappa et al., 2007).
Insulin signaling transduction is initiated by phosphorylation of the insulin receptor (Tyr protein kinase family; White, 2002). The insulin-growth factor-1 receptor (Tyr protein kinase family) can also be activated by insulin. As a consequence, two main signaling pathways are activated: phosphatidylinositol 3-kinase (PI3K)–AKT/protein kinase B (AGC Ser/Thr protein kinase family) pathway, which is responsible for a majority of the metabolic actions of insulin, and the Ras–mitogen-activated protein kinase (MAPK, CMGC Ser/Thr protein kinase family) pathway which regulates cell growth and differentiation (Taniguchi et al., 2006).
PI3K activates critical regulators of insulin signaling by catalyzing the formation of the lipid second messenger, PIP3, in the cell. Proteins with pleckstrin homology (PH) domains bind PIP3, which allows for their activation (Taniguchi et al., 2006). These proteins belong to the AGC Ser/Thr protein kinase, Tyr kinase, and Rho family. Glycogen synthase kinase-3 (CMGC Ser/Thr protein kinase family) is a target of AKT which regulates glycogen synthesis. AKT also regulates glucose uptake by triggering the activation of Rab small GTPases (Rab family) which involves cytoskeletal re-organization that is required for translocation of the glucose transporter (Taniguchi et al., 2006). AKT also effectively activates the mTOR pathway and regulates protein synthesis by phosphorylating the p70 ribosomal protein S6 kinase (AGC Ser/Thr protein kinase family). Atypical protein kinase C (aPKC, AGC Ser/Thr protein kinase family) is another downstream effector of PI3K and plays a role in glucose homeostasis (Taniguchi et al., 2006).
Activation of MAPK triggers the activation of the small GTPase Ras (Ras family) and a kinase cascade including MEK1 and 2 (STE Ser/Thr protein kinase family), ERKs (CMGC Ser/Thr protein kinase family), p90RSK (AGC Ser/Thr protein kinase family), and a transcription factor ELK1 (ETS family). Small GTPase Rac and CDC42 (Rho family) have also been implicated in insulin’s action by triggering downstream effectors involved in the rearrangement of the actin cytoskeleton (Taniguchi et al., 2006).
All gene families mentioned above can be found in Tables 1 and 3, and are significantly up-regulated in 4 and 14-day hyperglycemic MBL null mice compared to the respective WT group. Robustly expressed members of these gene families may act synergistically to sensitize the insulin signaling pathway in MBL null mice.
Cardiac Mitochondrial Functions are Preserved in Hyperglycemic MBL Null Mice
Mitochondria are the primary energy source fueling the contractile apparatus of the heart and are therefore key players in the progression of heart failure (Rosca and Hoppel, 2010). Damage to the cardiovascular endothelium is one of the first steps leading to heart disease, detectable even before structural changes to the vessel wall are apparent, as we recently demonstrated (Pavlov et al., 2012). Disruption of mitochondrial activities affects endothelial function and conceivably contributes to heart disease (Davidson, 2010).
We found scores of mitochondrial functional genes up-regulated in 4 and 14-day hyperglycemic MBL null mice (Tables 1 and 3) compared to their WT counterparts in response to hyperglycemic stress.
A down-regulation of the entire pathway of mitochondrial biogenesis appears in heart failure (Rosca and Hoppel, 2010). Mitochondrial biogenesis is a complex and dynamic process that regulates and determines the content and composition of mitochondria including mtDNA replication, nuclear-encoded protein synthesis, and incorporation into mitochondrial membranes and matrix (Bugger and Abel, 2010). Division of pre-existent organelles is dependent on mitochondrial fission, which also requires protein biosynthesis. In hyperglycemic MBL null mice, dozens of up-regulated mitochondrial ribosomal protein families were observed, including those of the Tango11 family, which may represent partially preserved mitochondrial biogenesis capability. Members of SAM50/omp85, Tom, Tim, small Tim, and metaxin families work as chaperones in guiding protein incorporation into mitochondrial membranes.
The myocardium depends on continuous oxidation of energy metabolic substrates for sufficient ATP generation to maintain contractile function. The myocardium uses even more energy under hyperglycemic conditions. The mitochondrial respiratory chain is the principal source of cellular ROS, which can lead to cellular damage and may ultimately result in reduced ATP production and impair contractility (Bugger and Abel, 2010). The primary factor governing mitochondrial ROS generation is the redox state of the respiratory chain. Abundant mitochondrial respiratory chain complexes help overcome energy-generating deficiencies, thereby maintaining a positive redox state with diminished ROS production. Up-regulated gene families in hyperglycemic MBL null mice which are components of the mitochondria respiratory chain include: complex I NDUFB5 subunit, complex I LYR, methyltransferase, AAA ATPase, cytochrome c oxidase VIIa, COX15/ctaA, and SURF1. The mitochondrial carrier and porin families are groups of proteins which import or export specific molecules across mitochondrial membranes. Facilitated solute trafficking/metabolites exchange plays a vital role in maintaining normal mitochondrial function (Nury et al., 2006).
Advantageous Transcriptional and Translational Adaption in Hyperglycemic MBL Null Mice
The myocardium is a high energy-consuming organ requiring a constant energy supply to maintain intracellular ATP levels essential for the uninterrupted myocardial contraction/relaxation cycle (Bertrand et al., 2008). It is reasonable that protein synthesis, which also requires high energy supply, would be suppressed in the insulin deficient state in favor of normal cardiac physiological function (Bertrand et al., 2008).
We found evidence that overall left ventricular protein synthesis is decreased more in hyperglycemic MBL null mice compared WT counterparts, likely to further lower energy usage during hyperglycemia. Furthermore, synthesis of specific proteins might be differentially regulated by transcriptional regulators in order to maintain a more effective defense against hyperglycemia. The ribosome is responsible for protein synthesis in all cells, whose large and small subunits consist of ribosomal RNA and proteins (Schmeing and Ramakrishnan, 2009). We observed an overwhelming down-regulation of ribosomal protein families in both 4 and 14-day hyperglycemic MBL null mice (Table 2, gene group 1 and Table 4, gene group 2), indicating ribosomal biogenesis may be greatly suppressed, leading to decreased protein synthesis. However, synthesis of specific proteins may remain active, as members of aminoacyl-tRNA synthetase families were increased (Table 1, gene group 9 and Table 3, gene group 3) as well as some mRNA translation activators, the GTP-binding elongation factor family and eIF-2-beta/eIF-5 family (Table 1, gene group 1). In addition, elevated DNA/RNA helicase (Table 1, gene group 10) and reduced histone expression (Table 2, gene group 7) were suggestive of an active transcription/translation process.

Table 2. Gene families of down-regulated genes in 4-day hyperglycemic MBL null mice compared to WT control.
It is likely that the specificity of those proteins still being synthesized is determined by transcription regulators. Unfortunately, we were not able to fully elucidate these regulators because of the complex interactions of these gene families. Therefore, we only extracted partial functional features of those genes and analyzed their physiological implications.
Selectively enhanced gene families include: promoting transcription activities (Table 1, gene groups 12 and 20), cell growth, division and differentiation (Table 1, gene groups 22, 24, and 28), and hormone receptors (Table 1, gene group 24). These gene families were only regulated in early (4-day) but not later (14-day) hyperglycemic stages. Therefore, they are likely offering initial emergent protection to cells against acute hyperglycemia.
Selectively inhibited gene families in 4 and 14-day samples include certain transcription suppressors (Table 2, gene group 2; Table 4, gene groups 1, 10, and 13); cell growth inhibition, terminal differentiation proteins and DNA damage sensors (Table 2, gene group 5; Table 4, gene groups 3 and 10), and hormone receptors (Table 4, gene groups 5 and 13). Generally, these changes reflect sustained gene transcription, cell growth, and prevention of apoptosis. However, in 14-day hyperglycemia samples, down-regulated hormone receptors, and DNA helicase expression (Table 4, gene group 12) plus restored histone expression (Table 3, gene group 18) indicate negative feedback loops may be observed.
Induced Ubiquitin-Dependent Proteolytic Cascade Helps Cardiac Cells Survive Initial Hyperglycemic Stress in MBL Null Mice
Gene families associated with the ubiquitin-dependent proteolytic pathway were overwhelmingly expressed at higher levels in 4-day hyperglycemic MBL null murine left ventricles (Table 1, gene groups 3, 7, 8, 14, 19, 21, and 27). Members of these gene families are involved in various steps of ubiquitin-dependent proteolytic cascades. This increased expression, however, was not obvious in the hearts of 14-day hyperglycemic mice. Therefore, this may be another rescue measure for cardiac cells to survive initial acute hyperglycemia in MBL null mice.
Hyperglycemia induces protein misfolding and results in altered organ function or failure (Herczenik and Gebbink, 2008). Cell protein quality control functions to minimize the production of abnormal proteins, remove abnormal proteins, or prevent abnormal proteins from damaging the cell. The myocardium is arguably one of the most stressed organs in the body, therefore, protein quality control is a vital cellular process for cardiomyocyte and cardiovascular endothelial cell functions (Stangl and Stangl, 2010; Su and Wang, 2010).
It is noteworthy that the role of the ubiquitin proteasome system in protein quality control has expanded with new studies delineating its cross-talk with the process of autophagy. Autophagy also works as a cellular housekeeper and plays a vital role to eliminate defective proteins and organelles, prevent abnormal protein aggregate accumulation, and remove intracellular pathogens (Levine and Kroemer, 2008; Willis et al., 2010).
Cardiac Contractile Dysfunction May Be Reversed by Advantageous Cytoskeleton Alterations in MBL Null Mice
The cytoskeleton is a complex network of filaments and tubules which transmit mechanical and chemical stimuli within and between cells. The cytoskeleton contributes to cell stability by anchoring subcellular structures such as mitochondria, Golgi apparatus, nuclei, and myofibrils (Hein et al., 2000).
Vascular endothelial cells are vulnerable to glucose-derived oxoaldehydes through the formation of AGEs, which induce cytoskeleton rearrangement and endothelial barrier dysfunction (Sliman et al., 2010). Heart failure is also characterized by cytoskeletal alterations resulting in reduced ventricular function. Cytoskeletal abnormalities rather than myofilament changes are responsible for cellular contractile dysfunction (Hein et al., 2000).
The absence of changes in mRNA levels of actin (actin family) but the abundance of tubulin (tubulin family) mRNA and microtubules were observed in pigs with heart failure (Hein et al., 2000). Meanwhile, contractile dysfunction and increased microtubules/cytoskeletal stiffness in failing hearts can be reversed by reducing the rate of tubulin polymerization (Hein et al., 2000). A progressive increase of desmin (intermediate filament family) was observed also during the transition from hypertrophy to failure in guinea pig hearts after aortic banding (Hein et al., 2000).
The expression of PPP4R2 and SMEK (PPP4R2 and SMEK families) leads to microtubule disruption and dysfunction (Liu et al., 2007; Martin-Granados et al., 2008). SFI1 (SFI1 family) is an essential gene required for spindle pole body duplication and bipolar spindle assembly (Anderson et al., 2007).
We observed in the early stage of hyperglycemia (4-day), actin gene family (with a main role in intracellular trafficking) expression levels are increased (Table 1) while intermediate filament and tubulin gene family expression levels are suppressed (Table 2). Therefore, microtubule depolymerization would be enhanced in the hyperglycemic MBL null mouse. In the later stages of hyperglycemia (14-day), despite the restored tubulin expression (Table 3), a down-regulation of SFI1 family genes (Table 4), which assists in anchoring tubulin, still puts microtubule polymerization under strict control. All the changes mentioned above would contribute to beneficial cytoskeleton rearrangements in MBL null mouse hearts in response to acute hyperglycemic stress compared to the WT genotype.

Table 3. Gene families of up-regulated genes in 14-day hyperglycemic MBL null mice compared to WT control.
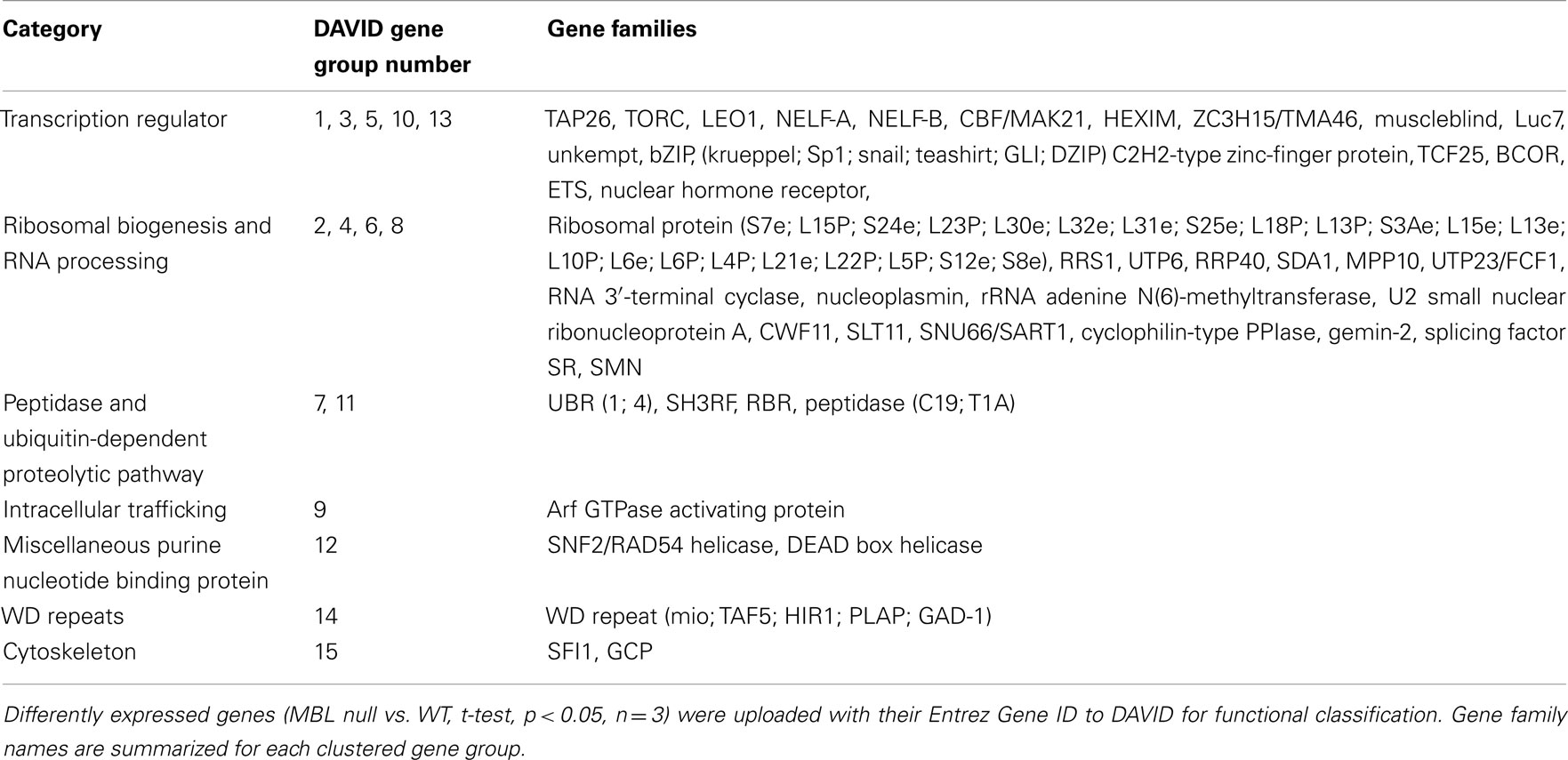
Table 4. Gene families of down-regulated genes in 14-day hyperglycemic MBL null mice compared to WT control.
Possible Facilitated Intracellular Trafficking Helps Maintain Normal Physiological Myocardial Function in Hyperglycemic MBL Null Mice
The Golgi apparatus is a hub for membrane-trafficking and plays a key role in processing and secretion of glycoproteins, glycolipids, and proteoglycans. COG families (Table 1) are among the list of up-regulated gene families. The COG complex is preferentially localized to the cis/medial cisternae of the Golgi apparatus and is involved in Golgi membrane traffic (Shestakova et al., 2006). The DHHC palmitoyltransferase family (Table 1) is responsible for adding palmitates to cytoplasmic proteins participating in the process of protein trafficking between organelles and in the segregation/clustering of proteins in membrane compartments (Linder and Deschenes, 2007). The MPPE1 family (Table 1) is required for transport of GPI-anchor proteins from the endoplasmic reticulum to the Golgi (Fujita et al., 2009). The endosomal system functions to sort and deliver internalized proteins to appropriate subcellular destinations. The Sorting nexin family (Table 3) regulates endosomal sorting of internalized proteins through modulating endosomal-to-lysosomal sorting (Carlton et al., 2004). Both elevated Arf GTPase (Tables 1 and 3) or attenuated Arf GTPase activating protein family (Table 4) expression may help maintain Arf GTPase activity which is essential for the recruitment of coat protein complex I and clathrin coat complexes needed for the generation of transport vesicles (Benjamin et al., 2011).
During interphase, thousands of nuclear proteins and RNAs cross the nuclear envelope to move from the cellular compartment in which they are synthesized to their final destination. Transport occurs through nuclear pore complexes and is mediated by nucleocytoplasmic transport receptors. Cse1 (XPO2/CSE1 family, Table 1) is the exportin that mediates the recycling of importin α (importin families, Table 1; Cook et al., 2005). Increased expression of these gene family members may indicate a facilitated nuclear transport in the MBL null genotype during hyperglycemia.
The ABC transporter gene family is among those up-regulated genes in both 4 and 14-day hyperglycemic MBL null mice (Tables 1 and 3). ABC transporters are transmembrane proteins that utilize ATP hydrolysis to translocate a wide variety of substrates across extra- and intracellular membranes, including amino acids, sugars, ions, glycans, peptides, proteins, lipids, and sterols (Dean et al., 2001; Couture et al., 2006). Expression of ABC transporters in the heart is associated with regulation of normal physiological function as well as protection and detoxification of the organism (Couture et al., 2006, 2007).
Discussion
Poor glycemic control is associated with microvascular and macrovascular disease, yet a link with cardiovascular events is less clear. Previous studies from our group support an important role for MBL and the lectin complement pathway in mediating the cardiomyopathy and vasculopathy associated with acute hyperglycemia (Busche et al., 2008; Pavlov et al., 2012). Hyperglycemic MBL null mice are protected from loss of nitric oxide-mediated relaxation of arterioles, displayed less myocardial inflammation, and less ventricular remodeling compared to hyperglycemic WT controls (Pavlov et al., 2012). The purpose of the present study was to broadly investigate the early and later genetic changes in the myocardium of hyperglycemic MBL null and WT mice so that potential molecular mechanisms involved could be investigated in subsequent studies. This preliminary study will allow us and others to advance the field so that the findings can be ultimately translated to the clinic (Collard et al., 2000).
Mouse models using STZ on the C57BL/6 background, which closely mimic the human disease, are recommended for the studies of hyperglycemia-induced cardiovascular complications (Shen and Bornfeldt, 2007). STZ results in acute hyperglycemia and is cleared from the body within 3 days. Therefore, the short term hyperglycemia (4–14 days) models rule out any potential cardiotoxic effects of STZ and enable us to focus on the assessment of molecular mechanisms in early stage development of cardiovascular pathology as well as further test human treatment solutions for the prevention of such diseases in the future. We have already developed an acute hyperglycemia model to investigate the role of complement and myocardial injury (Busche et al., 2008). In this study, we were able to provide more evidence that MBL deficiency leads to systematic gene expression changes compared to WT mice following hyperglycemia. We speculate that possible intracellular events taking place in multiple cellular compartments include enhanced insulin signaling pathway sensitivity, promoted mitochondrial respiratory function, better cell energy expenditure and protein quality control, improved cytoskeletal structure, and facilitated intracellular trafficking. All of these events may contribute to the organismal health of MBL null mice during acute hyperglycemia. Our data demonstrate an association between gene expression profile and tissue function which could be a very useful tool in predicting cellular targets and regulatory networks connected with in vivo observations and generate novel hypotheses for further mechanistic studies.
High-throughput genomic, proteomic, and bioinformatics scanning approaches provide significant capabilities to study a large variety of biological mechanisms associated with diseases (Huang et al., 2009). The DAVID gene functional classification tool classifies large gene lists into functionally related gene groups, highlights the importance of the discovered gene groups with an enrichment score, and summarizes the major biological importance of these gene groups. Our data reveal that all genes in each gene group were highly arrayed and belonged to one or multiple gene families. A gene family is a set of several similar genes, formed by duplication of a single original gene and likely to have highly similar functions (Demuth et al., 2006). Grouping genes based on gene families (functional similarity) can systematically enhance biological interpretation of large lists of genes derived from high-throughput studies. Therefore, it provides a rapid means to organize large lists of genes into functionally related groups to help unravel the biological content captured by high-throughput technologies.
The redundancy existing within biological networks means that modulating a single target might not be sufficient to produce the desired efficacy in treating complex diseases while, at the same time, minimizing adverse effects. Therefore, modulating multiple protein targets simultaneously might be a conceivable solution for the design of therapeutic protocols (Morphy and Rankovic, 2007). The combination of high-throughput microarray scans and DAVID analysis may have potential application in the multi-target therapeutic intervention evaluation as well as the early stage forecast of potential detrimental consequences of treatment. As we advance our current studies examining the role of MBL in various hyperglycemic models, we believe these data will drive our studies to testable hypotheses that could not have been anticipated, if not for this genomic screen.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Supported in part by a NIH fellowship HL099043 (Laura R. La Bonte) and NIH grants HL056086, HL099130, and AI089781 (Gregory L. Stahl).
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/molecular_innate_immunity/10.3389/fimmu.2012.00015/abstract
Footnotes
References
Anderson, V. E., Prudden, J., Prochnik, S., Giddings, T. H. Jr., and Hardwick, K. G. (2007). Novel sfi1 alleles uncover additional functions for Sfi1p in bipolar spindle assembly and function. Mol. Biol. Cell 18, 2047–2056.
Benjamin, J. J. R., Poon, P. P., Lewis, S. M., Auger, A. A., Wong, T. A., Singer, R. A., and Johnston, G. C. (2011). The yeast Arf GTPase-activating protein Age1 is regulated by phospholipase D for post-Golgi vesicular transport. J. Biol. Chem. 286, 5187–5196.
Bertrand, L., Horman, S., Beauloye, C., and Vanoverschelde, J.-L. (2008). Insulin signalling in the heart. Cardiovasc. Res. 79, 238–248.
Bojadzievski, T., and Gabbay, R. A. (2011). Patient-centered medical home and diabetes. Diabetes Care 34, 1047–1053.
Bugger, H., and Abel, E. D. (2010). Mitochondria in the diabetic heart. Cardiovasc. Res. 88, 229–240.
Busche, M., Walsh, M., McMullen, M., Guikema, B., and Stahl, G. (2008). Mannose-binding lectin plays a critical role in myocardial ischaemia and reperfusion injury in a mouse model of diabetes. Diabetologia 51, 1544–1551.
Carlton, J., Bujny, M., Peter, B. J., Oorschot, V. M. J., Rutherford, A., Mellor, H., Klumperman, J., McMahon, H. T., and Cullen, P. J. (2004). Sorting nexin-1 mediates tubular endosome-to-TGN transport through coincidence sensing of high- curvature membranes and 3-phosphoinositides. Curr. Biol. 14, 1791–1800.
Ceriello, A. (2005). Acute hyperglycaemia: a ‘new’ risk factor during myocardial infarction. Eur. Heart J. 26, 328–331.
Collard, C. D., Vakeva, A., Morrissey, M. A., Agah, A., Rollins, S. A., Reenstra, W. R., Buras, J. A., Meri, S., and Stahl, G. L. (2000). Complement activation after oxidative stress: role of the lectin complement pathway. Am. J. Pathol. 156, 1549–1556.
Cook, A., Fernandez, E., Lindner, D., Ebert, J., Schlenstedt, G., and Conti, E. (2005). The structure of the nuclear export receptor Cse1 in its cytosolic state reveals a closed conformation incompatible with cargo binding. Mol. Cell 18, 355–367.
Couture, L., Nash, J. A., and Turgeon, J. (2006). The ATP-binding cassette transporters and their implication in drug disposition: a special look at the heart. Pharmacol. Rev. 58, 244–258.
Couture, L., Nash, J. A., and Turgeon, J. (2007). Role of ATP-binding cassette transporters in drug distribution to the heart and protection from toxic compounds. Heart Metab. 35, 1–6.
Davidson, J. A., and Parkin, C. G. (2009). Is hyperglycemia a causal factor in cardiovascular disease? Diabetes Care 32, S331–S333.
Dean, M., Hamon, Y., and Chimini, G. (2001). The human ATP-binding cassette (ABC) transporter superfamily. J. Lipid Res. 42, 1007–1017.
Demuth, J. P., Bie, T. D., Stajich, J. E., Cristianini, N., and Hahn, M. W. (2006). The evolution of mammalian gene families. PLoS ONE 1, e85. doi:10.1371/journal.pone.0000085
Fortpied, J., Vertommen, D., and Van Schaftingen, E. (2010). Binding of mannose-binding lectin to fructosamines: a potential link between hyperglycaemia and complement activation in diabetes. Diabetes Metab. Res. Rev. 26, 254–260.
Fujita, M., Maeda, Y., Ra, M., Yamaguchi, Y., Taguchi, R., and Kinoshita, T. (2009). GPI glycan remodeling by PGAP5 regulates transport of GPI-anchored proteins from the ER to the Golgi. Cell 139, 352–365.
Giacco, F., and Brownlee, M. (2010). Oxidative stress and diabetic complications. Circ. Res. 107, 1058–1070.
Gustafsson, I., Kistorp, C. N., James, M. K., Faber, J. O., Dickstein, K., and Hildebrandt, P. R. (2007). Unrecognized glycometabolic disturbance as measured by hemoglobin A1c is associated with a poor outcome after acute myocardial infarction. Am. Heart J. 154, 470–476.
Hansen, T. K., Tarnow, L., Thiel, S., Steffensen, R., Stehouwer, C. D., Schalkwijk, C. G., Parving, H.-H., and Flyvbjerg, A. (2004). Association between mannose-binding lectin and vascular complications in type 1 diabetes. Diabetes 53, 1570–1576.
Hein, S., Kostin, S., Heling, A., Maeno, Y., and Schaper, J. (2000). The role of the cytoskeleton in heart failure. Cardiovasc. Res. 45, 273–278.
Herczenik, E., and Gebbink, M. F. B. G. (2008). Molecular and cellular aspects of protein misfolding and disease. FASEB J. 22, 2115–2133.
Hovind, P., Hansen, T. K., Tarnow, L., Thiel, S., Steffensen, R., Flyvbjerg, A., and Parving, H.-H. (2005). Mannose-binding lectin as a predictor of microalbuminuria in type 1 diabetes. Diabetes 54, 1523–1527.
Huang, D. W., Sherman, B. T., and Lempicki, R. A. (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57.
Linder, M. E., and Deschenes, R. J. (2007). Palmitoylation: policing protein stability and traffic. Nat. Rev. Mol. Cell Biol. 8, 74–84.
Liu, Y., Lagowski, J., Sundholm, A., Sundberg, A., and Kulesz-Martin, M. (2007). Microtubule disruption and tumor suppression by mitogen-activated protein kinase phosphatase 4. Cancer Res. 67, 10711–10719.
MAQC-Consortium. (2010). The microarray quality control (MAQC)-II study of common practices for the development and validation of microarray-based predictive models. Nat. Biotechnol. 28, 827–838.
Martin-Granados, C., Philp, A., Oxenham, S. K., Prescott, A. R., and Cohen, P. T. W. (2008). Depletion of protein phosphatase 4 in human cells reveals essential roles in centrosome maturation, cell migration and the regulation of Rho GTPases. Int. J. Biochem. Cell Biol. 40, 2315–2332.
Moller-Kristensen, M., Hamblin, M. R., Thiel, S., Jensenius, J. C., and Takahashi, K. (2007). Burn injury reveals altered phenotype in mannan-binding lectin-deficient mice. J. Invest. Dermatol. 127, 1524–1531.
Morphy, R., and Rankovic, Z. (2007). Fragments, network biology and designing multiple ligands. Drug Discov. Today 12, 156–160.
Muniyappa, R., Montagnani, M., Koh, K. K., and Quon, M. J. (2007). Cardiovascular actions of insulin. Endocr. Rev. 28, 463–491.
Nury, H., Dahout-Gonzalez, C., Trezeguet, V., Lauquin, G. J. M., Brandolin, G., and Pebay-Peyroula, E. (2006). Relations between structure and function of the mitochondrial ADP/ATP carrier. Annu. Rev. Biochem. 75, 713–741.
Pavlov, V. I., La Bonte, L. R., Baldwin, W. M., Markiewski, M. M., Lambris, J. D., and Stahl, G. L. (2012). Absence of mannose-binding lectin prevents hyperglycemic cardiovascular complications. Am. J. Pathol. 180, 104–112.
Schmeing, T. M., and Ramakrishnan, V. (2009). What recent ribosome structures have revealed about the mechanism of translation. Nature 461, 1234–1242.
Shen, X. I. A., and Bornfeldt, K. E. (2007). Mouse models for studies of cardiovascular complications of type 1 diabetes. Ann. N. Y. Acad. Sci. 1103, 202–217.
Shestakova, A., Zolov, S., and Lupashin, V. (2006). COG complex-mediated recycling of golgi glycosyltransferases is essential for normal protein glycosylation. Traffic 7, 191–204.
Shi, L., Takahashi, K., Dundee, J., Shahroor-Karni, S., Thiel, S., Jensenius, J. C., Gad, F., Hamblin, M. R., Sastry, K. N., and Ezekowitz, R. A. B. (2004). Mannose-binding lectin-deficient mice are susceptible to infection with Staphylococcus aureus. J. Exp. Med. 199, 1379–1390.
Sliman, S., Eubank, T., Kotha, S., Kuppusamy, M., Sherwani, S., Butler, E., Kuppusamy, P., Roy, S., Marsh, C., Stern, D., and Parinandi, N. (2010). Hyperglycemic oxoaldehyde, glyoxal, causes barrier dysfunction, cytoskeletal alterations, and inhibition of angiogenesis in vascular endothelial cells: aminoguanidine protection. Mol. Cell. Biochem. 333, 9–26.
Stangl, K., and Stangl, V. (2010). The ubiquitin-proteasome pathway and endothelial (dys)function. Cardiovasc. Res. 85, 281–290.
Su, H., and Wang, X. (2010). The ubiquitin-proteasome system in cardiac proteinopathy: a quality control perspective. Cardiovasc. Res. 85, 253–262.
Takahashi, K., Gordon, J., Liu, H., Sastry, K. N., Epstein, J. E., Motwani, M., Laursen, I., Thiel, S., Jensenius, J. C., Carroll, M., and Ezekowitz, R. A. B. (2002). Lack of mannose-binding lectin-A enhances survival in a mouse model of acute septic peritonitis. Microbes Infect. 4, 773–784.
Taniguchi, C. M., Emanuelli, B., and Kahn, C. R. (2006). Critical nodes in signalling pathways: insights into insulin action. Nat. Rev. Mol. Cell Biol. 7, 85–96.
Van Gaal, L. F., Mertens, I. L., and De Block, C. E. (2006). Mechanisms linking obesity with cardiovascular disease. Nature 444, 875–880.
White, M. F. (2002). IRS proteins and the common path to diabetes. Am. J. Physiol. Endocrinol. Metab. 283, E413–E422.
Willis, M. S., Townley-Tilson, W. H. D., Kang, E. Y., Homeister, J. W., and Patterson, C. (2010). Sent to destroy: the ubiquitin proteasome system regulates cell signaling and protein quality control in cardiovascular development and disease. Circ. Res. 106, 463–478.
Keywords: hyperglycemia, cardiomyopathy, vasculopathy, microarray, mannose binding lectin
Citation: Zou C, La Bonte LR, Pavlov VI and Stahl GL (2012) Murine hyperglycemic vasculopathy and cardiomyopathy: whole-genome gene expression analysis predicts cellular targets and regulatory networks influenced by mannose binding lectin. Front. Immun. 3:15. doi: 10.3389/fimmu.2012.00015
Received: 12 October 2011;
Accepted: 29 January 2012;
Published online: 10 February 2012.
Edited by:
Zvi Fishelson, Tel Aviv University, IsraelReviewed by:
Cordula M. Stover, University of Leicester, UKWilhelm Johannes Schwaeble, University of Leicester, UK
Copyright: © 2012 Zou, La Bonte, Pavlov and Stahl. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Gregory L. Stahl, Center for Experimental Therapeutics and Reperfusion Injury, HIM 845A, 77 Avenue Louis Pasteur, Boston, MA 02115, USA. e-mail:Z3N0YWhsQHpldXMuYndoLmhhcnZhcmQuZWR1