


- Department of Academic Rheumatology, Centre for Molecular and Cellular Biology of Inflammation, King’s College London, London, UK
Heat shock proteins (HSPs) and other members of the much broader stress protein family have been shown to play important roles in coordinating multiple phases of immunological reactions; from facilitating immunological recognition, to promoting and regulating immunological responses and finally augmenting the resolution of inflammation and return to immunological homeostasis. In this review, we consider the challenges facing the stress protein field as we enter 2012; in particular we consider the role that HSPs and stress proteins may play in the initiation and termination of immunological responses. Special attention is afforded to the resolution-associated molecular pattern, binding immunoglobulin protein (BiP, also known as glucose regulated protein-78). We review the evidence that resolution-promoting proteins such as BiP may herald a new generation of biologics for inflammatory disease and reflect on the challenges of achieving clinical remission in rheumatoid arthritis with novel therapeutics and correlating clinical remission with immunological parameters of resolution of inflammation.
Introduction
Heat shock proteins (HSP) and stress proteins are a collection of highly evolutionarily conserved proteins, grouped by molecular weight, with intracellular functions involving protein chaperoning and folding and protection of the cell during physiologically stressful conditions, including heat-shock, hypoxia, hypoglycemia, and intracellular electrolyte abnormalities. The term “stress protein” provides an umbrella for all those proteins upregulated in response to physiological stress, while HSP refers to the sub-family of proteins that have specific promoters allowing them to respond to heat shock. Stress proteins may be intracellular, cell surface expressed, and/or extracellular proteins and, therefore, have access to multiple subcellular and extracellular compartments. Consequently, there has been much discussion around potential roles that stress proteins may play in innate and adaptive immune responses.
Immunological responses may be grouped into four overlapping and interdependent phases: recognition, response, regulation, and resolution. Much data suggest that stress proteins play important roles in all of the phases of the immunological response. In this review, we briefly discuss how stress proteins can influence the resolution of inflammation, in particular, with respect to binding immunoglobulin protein (BiP, also known as glucose regulated protein-78). We also explore the concept of third generation biologic therapies for immunological diseases such as rheumatoid arthritis (RA). The third generation of biologics for immune-mediated diseases will be therapeutic agents that significantly modulate the dysregulated immune system, restoring immune homeostasis for prolonged periods without further biologic administration. We believe that BiP and other members of the recently defined resolution associated molecular pattern (RAMP) family (Shields et al., 2011) may herald a new generation of such novel biologics.
Elephants in the Laboratory; A 2011 View of the Stress Protein Field
Extensive research has now shown that stress proteins are involved in multiple stages of the immune response (Figure 1): stress proteins participate in the recognition of immunological danger, facilitate peptide carriage between cells and subsequent cross presentation of antigen, can modulate the immunological profile of myeloid lineage cells in both a pro- and anti-inflammatory direction and promote the regulation and resolution of inflammation. However, research in the stress protein field has been dogged by two controversies for many years. Firstly, there remains suspicion that stress protein family members possess no intrinsic immunological activity and all observed activity is the consequence of bacterial contaminants, particularly endotoxin, within the protein preparation (Wallin et al., 2002; Gao and Tsan, 2003a,b; Ye and Gan, 2007). Secondly, compounding this problem, is the ever-increasing variety of cell surface receptors identified for HSP. Using HSP70 as an example, the broad array of putative receptors include, TLR2, TLR4, CD14, CD40, CD91, CCR5, LOX-1, SREC-1 (Calderwood et al., 2007). However, there remain inconsistencies when trying to replicate HSP binding to null cells transfected with some of these specific molecules, such as CD14, TLR2, and TLR4, thus throwing doubt on whether they are true receptors (Theriault et al., 2005).
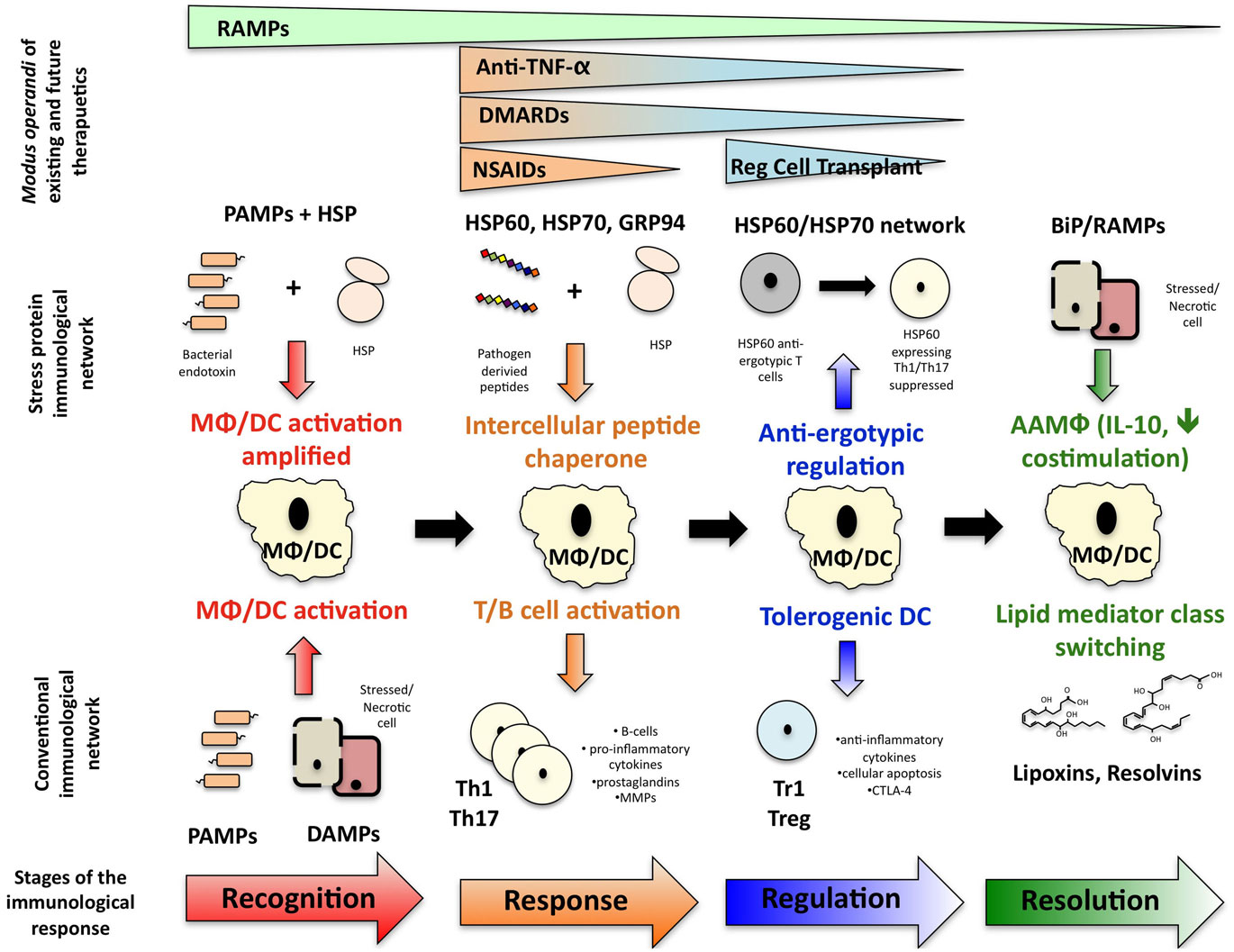
Figure 1. How stress protein networks influences immunological reactions. Immunological events can be broken down into a series of interdependent phases: immunological recognition, an immunological response following by regulation of that response and resolution of inflammation leading to a return to homeostasis. The stress protein network influences each phase of the immunological event and ultimately, resolution-associated molecular patterns may help facilitate the return to immunological homeostasis. The different phases at which existing and future therapeutics act are also illustrated.
The major challenge facing the stress protein field is consolidating and refining the results of the extensive body of research conducted to date into a more comprehensive theory of how stress protein networks facilitate the initiation, progression, and resolution of an immunological event. To achieve this, careful distinctions should be made between the immunological activity of mycobacterial HSP and mammalian HSP and, more pertinently, between the immunological effects derived from whole extracellular stress protein molecules and those effects caused by peptides derived from stress protein molecules coordinating the generation or expansion of self-reactive HSP T cells.
Moreover, stress protein researchers must address the elephants in their experimental data. It is increasingly clear that stress proteins bind bacterial products; however, the potential immunological implications of stress protein–bacterial product interactions have been over-shadowed by heated debate as to the immunological activity of ultra-pure stress protein preparations and their role as damage-associated molecular patterns (DAMP; Gao and Tsan, 2003a,b; Osterloh et al., 2004; Henderson et al., 2010). More recently, further discussion has arisen surrounding subtle differences between danger-associated and DAMP (van Eden et al., 2011), with van Eden et al. convincingly arguing that HSP should be excluded from the DAMP family given the paucity of evidence that they alone can activate an immunological response. Yet, the very fact that HSP are upregulated in response to cellular stress and can associate with pathogen associated molecular patterns (PAMP) is of great immunological significance in it’s own right, particularly with respect to the danger hypothesis, for the following reasons:
(1) Cellular stress is synonymous with potential threats to tissue viability and is therefore synonymous with immunological danger, in accordance with the danger hypothesis (Matzinger, 2002). Cellular stress can arise from infectious and non-infectious sources (Macario and Conway de Macario, 2005; Rath and Haller, 2011) and results in the upregulation and redistribution of stress proteins, including members of the HSP family, to the cell surface and beyond, into the extracellular fluid (Multhoff and Hightower, 1996; Delpino and Castelli, 2002; Mambula and Calderwood, 2006; Corrao et al., 2010; Merendino et al., 2010; Sreekumar et al., 2010).
(2) Cell surface HSP and other stress proteins (e.g., gp96) signal immunological danger by inducing and/or enhancing inflammatory cytokine production, dendritic cell (DC) maturation, and NK cell activity (Chen et al., 2002; Liu et al., 2003; Osterloh et al., 2004). While this does not qualify the molecules as DAMPs, as laid out in the criteria defined by Kono and Rock (2008), it emphasizes their importance in signaling immunological danger. Furthermore, while on the cell surface, it has been hypothesized that stress proteins associate with PAMPs and thus serve as a primitive antigen presentation system (Li et al., 2002).
(3) There is considerable evidence to suggest that extracellular HSP60, HSP70, HSP90, and gp96 bind to LPS and potentiate its immunostimulatory effects (Triantafilou et al., 2001; Warger et al., 2006; Osterloh et al., 2007). High-mobility group box protein-1 has shown similar properties (Pisetsky et al., 2008). This suggests that stress proteins may facilitate the recognition of a pathogenic infection, particularly if that infection has caused direct cellular insult. This may be of significance in maintaining immunological homeostasis at epithelial surfaces.
(4) Beyond the cell, stress protein family members appear to bind multiple extracellular protein and receptor targets (Calderwood et al., 2007). The immunological significance of these protein–protein interactions remain unclear. However, HSP do appear to be “sticky” proteins. Given their interaction with PAMPs, this promiscuity may facilitate the containment of shed bacterial products, preventing their dissemination, while providing a localized cache of PAMPs permitting a more anatomically targeted immunological response.
In summary, the specific properties of an extracellular stress protein are likely to depend both on the protein itself, the surrounding extracellular environment and the nature of the tissue from which the stress protein was released. Furthermore, the possibility that the extracellular environment modulates the activity of stress proteins during the course of an immunological response requires further attention; one could envisage a paradigm where extracellular stress proteins evolve from poachers, inciting inflammation, and tissue destruction, into gamekeepers, promoting regulation, and resolution during the course of an immunological event. Indeed a post-translational modification of high-mobility G protein-1 (HMGB1) caused by oxidation attenuates pro-inflammatory functions and makes the protein anti-inflammatory (Urbonaviciute et al., 2009). However, since stress proteins exhibit such radically different extracellular functions it should not be surprising that they show different affinities for a variety of receptors and may even bind different receptors depending on cell type. Such activity would enhance their flexibility and diversity of function (Calderwood et al., 2007). Hence, a greater understanding of how the stress protein network interacts with wider immunological networks would be extremely beneficial, however, a systems biology approach will likely be necessary to extend our knowledge further.
The Third Generation of Biologics: Promoting Resolution
At present all therapies for immune-mediated diseases – autoimmune or allotransplant – require either continuous administration of immunosuppressive drugs or intermittent dosing at frequent intervals. For example, despite the effectiveness of anti-TNF-α therapy at suppressing inflammation in RA, cessation of therapy is associated with clinical relapse and radiological progression (Quinn et al., 2005) and although there is some in vitro evidence that anti-TNF-α therapy may modulate adaptive immune responses, particularly with respect to regulatory T cells (Ehrenstein et al., 2004), there is little evidence that long-term therapy alters the underlying immunological mechanisms that contribute to the chronic autoimmune state. Indeed, even after achieving low disease activity following long-term infliximab treatment, 45% of patients displayed further radiological disease progression within just a year of cessation of therapy (Tanaka et al., 2010).
Nevertheless, the transplantation field has offered hope that physicians can induce a state of tolerance to alloantigen in the absence of long-term immunosuppression. The best-characterized example of this phenomenon is liver transplantation: 20% of liver transplant recipients are capable of achieving a state of clinical operational tolerance (defined as a well functioning graft, without histological signs of rejection following the complete cessation of immunosuppression for over 1 year; Orlando et al., 2009). Tolerance is rarer with other solid-organ transplants, but has been reported in renal transplantation (Orlando et al., 2010); immunologists are beginning to describe biomarkers and molecular signatures that characterize and can be used to monitor clinical operational tolerance which include reduced co-stimulatory molecule expression, immune quiescence, apoptosis, and memory T cell responses are important in the maintenance of operational tolerance (Brouard et al., 2007; Hernandez-Fuentes and Lechler, 2010).
With respect to RA, the clinical goal is not a tolerogenic state per se, but the induction of drug-free remission. The ACR–EULAR agreed definition of disease remission in RA is a simplified disease activity index (SDAI) of ≤13.3 at any one time, or a total joint count of ≤1, and a swollen joint count ≤1, and a CRP ≤1 mg/dl and a patient global assessment score of ≤1 (Bykerk, 2011). Remission of RA, may indeed involve the re-establishment of immunological tolerance to cognate autoantigens. However, extensive cell–cell interactions between immune and stromal cells within the synovial architecture add a layer a complexity beyond the extracelluar cytokine and stress protein networks that drive the pathogenesis and maintain chronic inflammation within the rheumatoid joint (McInnes and Schett, 2011).
Currently, rheumatologists do not possess any immunological biomarkers to predict which patients will achieve or maintain a state of disease-free remission or indeed the mechanisms by which clinical remission is achieved (Isaacs, 2010). Thus, the attainment of drug-free remission in RA is the premier challenge facing rheumatologists and immunologist in the twenty-first century.
Early, aggressive intervention in the disease process is now the gold-standard for achieving maximum clinical response and potential remission in the RA patient (Quinn et al., 2005). B-cell depletion therapy has also offered a glimpse that more permanent changes to the immunological phenotype are possible. For example, rituximab, a B-cell depleting anti-CD20 monoclonal antibody, induces enduring clinical responses that do not correlate with peripheral blood levels of CD20+ B cells after treatment (Breedveld et al., 2007). Multiple potential therapies and possible mechanisms for the re-establishment of immune tolerance and subsequent resolution of chronic inflammation have been discussed at length elsewhere (Albani et al., 2011). Needless to say that, like the conditions necessary for operational tolerance of renal grafts, tolerance in the rheumatoid patient involves modification of DC function such that T regulatory cells, of various phenotypes and mode of action are generated.
Since stress proteins have pleiotropic functions, and have been called “moonlighting” proteins (Huberts and van der Klei, 2010), it may be that the immune down regulating properties of some of them are due to an effect on DCs. Hence when used for the treatment of human disease they may be able to produce prolonged drug-free disease remissions or even tolerance. What is the evidence for this dramatic claim? Examination of the multiple extracellular functions of BiP may provide an answer.
BiP – A Brief History
Binding immunoglobulin protein is a member of the HSP70 family and an ubiquitously expressed, endogenous protein. BiP is constitutively expressed in the endoplasmic reticulum (ER), and essential for the correct folding of many nascent peptides (Gething, 1999). BiP is also the master regulator of the unfolded protein response (UPR), a transcriptional program designed to relieve ER stress by promoting the correct folding of ER luminal proteins (Hendershot, 2004). BiP, therefore, is regulated at two levels, constitutive and stress induced. Stress induced upregulation occurs in environments of high cellular activity in inadequately vascularized tissue, for example, during inflammation or neoplasia, pathologies characterized by relative hypoglycemia and hypoxia (Lee, 2007). Principally, during perturbation of the ER, BiP protects the cell from the accumulation of misfolded and denatured proteins and, thus, prevents apoptosis. Testament to the fundamental importance of BiP is the fact that BiP knock-out mice and mice where BiP is constitutively targeted to the incorrect subcellular compartment are not viable (Luo et al., 2006; Mimura et al., 2007). Many previous reviews have covered the intracellular chaperone function and protective role of BiP, during health and disease, so these aspects of BiP biology will not be reviewed here.
In contrast, our work for the last decade has focused exclusively on the extracellular properties of cell-free human BiP and its immunoregulatory role in inflammation. Like many other stress proteins, BiP is now known to be cell surface expressed and detectable at relatively high concentrations in serum, synovial fluid and oviductal fluid (Delpino and Castelli, 2002; Corrigall et al., 2004; Marin-Briggiler et al., 2010). As such, BiP has several physiological properties. Although BiP is a member of the HSP70 family we have previously hypothesized that, unlike HSP70 itself, BiP acts as a RAMP (Shields et al., 2011). RAMPs are protein molecules released alongside DAMPs from stressed or necrotic cells, which provide negative inputs into immunological networks, antagonizing pro-inflammatory mediators, and helping restore the immune system to homeostasis. Unlike the “DAMPing” or regulatory effect that HSPs exert on the immune system which appear to act via HSP derived peptides expanding sets of HSP-specific regulatory T cells (van Eden et al., 2005; Quintana and Cohen, 2011), the RAMP family members act predominantly on the myeloid lineage, setting the scene for the resolution of inflammation. Furthermore, what makes these molecules unique, is that unlike the increasing number of resolution inducing molecules being described (e.g., lipoxins and resolvins), the protein members of the RAMP family are constitutively expressed and thus able to affect the course of inflammation from the outset (Shields et al., 2011).
BiP: Driving Resolution of Inflammation
What sets BiP apart from other potential biologic therapeutics appears to be its mode of action. Our research provides evidence that BiP offers long-lasting prophylactic, and therapeutic protection from disease in the murine model of collagen induced arthritis (CIA; Corrigall et al., 2001; Brownlie et al., 2006). Adoptive transfer studies confirm that the end-point of the mode of action involves immunological changes to cell function and suggests that regulatory T cells are induced rapidly by BiP either in naïve animals, following intravenous injection of BiP, or in vitro, when their splenocytes and lymph nodes are cultured in the presence of BiP (Corrigall et al., 2001; Brownlie et al., 2006). Importantly, the message from these studies is that BiP-treated cells, when adoptively transferred into arthritic mice are therapeutic and give long-term relief in the absence of repeated administration.
Another indicator that BiP is a potentially successful therapeutic in RA arises from our experimental pre-clinical investigation into a xenogeneic model where small pieces of inflamed synovium from RA patients were transplanted subcutaneously into severe combined immunodeficient (SCID) mice (Yoshida et al., 2011). Following successful vascular anastomosis of the graft, a single intravenous dose of BiP significantly reduced histological features of inflammation in the synovial explants. In addition, the histological expression of pro-inflammatory cytokines (TNF-α and IL-6) and the co-stimulatory molecules HLA-DR and CD86 was significantly down regulated in the grafts from the BiP-treated animals (Yoshida et al., 2011). The down-regulation of co-stimulatory molecules would significantly reduce the efficiency of antigen presentation. This SCID/RA synovial membrane chimera has been used to validate other biologic therapies, anti-TNF-α neutralizing antibody inhibitor and anti-soluble IL-6 receptor, that are now in the clinic.
Binding immunoglobulin protein treatment induces key changes in T cell and monocyte development. Firstly, T cell development is skewed to a Th2 profile with the production of IL-4, IL-5, and IL-10 (Bodman-Smith et al., 2003). Human BiP-specific T cell clones derived from healthy PBMC are predominantly CD8+ and produce little or no interferon γ while splenocyte and lymph node cell suspensions from BiP-treated CIA mice also show a Th2 profile of cytokine release, IL-4, IL-5, and IL-10, on re-stimulation with BiP in vitro (Brownlie et al., 2006). Serum samples from these mice suggest that BiP suppresses the production of the pathogenic anti-collagen type II antibodies, which drive CIA, while stimulating the production of non-pathogenic IgG1 isotype antibodies indicative of a Th2 environment (Brownlie et al., 2006). Secondly, BiP is capable of binding to a receptor expressed by >95% human peripheral blood monocytes, up to 50% B cells and 10% T cells (Corrigall et al., 2003). Currently, the identity of this receptor(s) remains elusive. The immediate result of BiP stimulation, in human peripheral blood mononuclear cells, is the attenuation of TNF-α production after 7 h and increased IL-10 production to a plateau lasting over 96 h (Corrigall et al., 2004). On being stimulated by BiP, monocytes show phenotypic changes similar to deactivated macrophages. Macrophage deactivation is achieved via the stimulation of macrophages with IL-10, TGF-β, steroids, or interactions between CD200 and CD200R and CD47 and CD172a; the consequences of deactivation include increased production of IL-10, TGF-β, PGE-2, and reduced expression of MHC-II molecules (Gordon and Taylor, 2005). The various differentiation pathways and activation states of the monocyte–macrophage lineage remain under intense investigation, however, BiP induced changes are temporally different from those induced exclusively by IL-10. The major effect is the complete inhibition of TNF-α production with increased production of IL-10, soluble TNF receptors, and IL-1 receptor antagonist (Corrigall et al., 2004). Ultimately, the consequence of BiP stimulation is non-phlogistic activation of the monocyte–macrophage. Monocytes cultured in the presence of IL-4 and GM-CSF and BiP fail to differentiate into mature DC (Corrigall et al., 2009). Remarkably these BiP-treated cells were highly positive for indoleamine 2,3-dioxygenase (IDO), a characteristic of tolerogenic DC. When these DCs were co-cultured with autologous T cells they induced regulatory CTLA-4+ T cells. This process was attributable to IDO up regulation because CTLA-4 up regulation was reversible in the presence of 1 methyl tryptophan, the IDO inhibitor. On separation from the DC the CTLA-4+ T cells showed regulatory T cell function capable of suppressing T cell stimulation by anti-CD3. Although BiP was essential for the induction of the DCs, no additional BiP was required in the secondary cultures in which regulatory T cell function was assessed (Corrigall et al., 2009). The mechanism underlying the induction of regulatory T cells by BiP is under investigation by our laboratory. There are three possible scenarios: firstly, that there is direct action on the T cell via a receptor mediated process, but this is unlikely given the relative lack of extracellular BiP protein in the culture system during DC–T cell col-culture; secondly, that BiP drives the induction of DC of a tolerogenic phenotype, including increasing IDO expression, which we have shown directly leads to upregulated CTLA-4+ regulatory T cells, either in the presence or absence of peptide presentation. Finally BiP-specific peptide presentation may expand existing regulatory T cells in a peptide dependent process. Cross-reactivity with peptides from other HSP70 family members is a possibility in this case as 68% amino acid homology exists between HSP70 and BiP. If cross-reactivity between HSP70 and BiP occurs and is responsible for any of the T-cell dependent regulatory effects of either protein, it lends further weight to the hypothesis that an intricate network of extracellular stress proteins exists which facilitates the maintenance of immunological homeostasis (Panayi et al., 2004).
These studies are all in immunological models. In the TNF-α transgenic mouse a spontaneous arthritis develops that resembles RA in many of its features (Li and Schwarz, 2003) although the joint inflammation and destruction is independent on immune mechanisms. However, since BiP deactivates monocytes both at the transcriptional and the translational level a prediction would be that BiP would also have a therapeutic effect in this model. Indeed a single intraperitoneal dose of BiP was able to significantly suppress joint inflammation and systemic bone damage for several weeks (Corrigall et al., manuscript submitted).
The spontaneous arthritis observed in the transgenic mice is almost certainly induced by TNF-α activation of transcription factors including NF-κB, which is responsible for the induction of many of the major inflammatory cytokines. Intracellularly, BiP and NF-κB are counter regulated (Pahl and Baeuerle, 1995). A recent review by Kitamura (2011) has reported that although early UPR activity may drive NF-κB activation later UPR involvement tends to attenuate NF-κB function. As an extracellular protein we have shown that BiP acts to inhibit MAPK phosphorylation (Corrigall et al., manuscript submitted) and downregulates protein levels of these signaling molecules. As a stress protein, BiP is upregulated intracellularly in response to ER stress prior to its release into the extracellular environment. Thus, both the intracellular and extracellular actions of BiP may operate to diminish the pro-inflammatory effects of NF-κB thus helping the resolution of acute inflammation.
The Other Resolution-Associated Molecular Patterns
Binding immunoglobulin protein is just one member of a family of proteins we have recently defined as the RAMPs. We have extensively reviewed the properties of the RAMP family elsewhere (Shields et al., 2011). Needless to say, we believe this family of proteins, whose founder members include HSP10, HSP27, and α-B-crystallin may have great potential as resolution-promoting therapeutics. Resolution-promoting regulatory signals are subtly different from immunoregulatory immunological signals; resolution-promoting signals are those that specifically promote the non-phlogistic activation of macrophages, the phagocytosis of apoptotic neutrophils, the prevention of influx of inflammatory cells and the restoration of parenchymal cells to the non-inflammatory state (Serhan et al., 2007). The crux of resolution is, therefore, centered around myeloid lineage cells and the parenchyma. This is why we believe the RAMP family, which principally exert their immunological effects via the myeloid lineage (thus affecting antigen presentation, co-stimulatory molecule expression, myeloid cell differentiation, and anti-inflammatory cytokine secretion (De et al., 2000; Corrigall et al., 2004, 2009; Banerjee et al., 2011; Shields et al., 2011)), have the potential to exert resolution-promoting effects, rather than simply immunoregulatory effects.
Clinical trials of HSP10 have already yielded promising results (Vanags et al., 2006; Williams et al., 2008) and pre-clinical models have shown α-B-crystallin to be an extremely effective therapeutic in a variety of different inflammatory disease models including experimental autoimmune encephalomyelitis, ischemic optic neuropathy and stroke (Ousman et al., 2007; Arac et al., 2011; Pangratz-Fuehrer et al., 2011). Further investigations are necessary to fully understand how the RAMP family exert their immunological effects and under what circumstance they might be used in clinical practice.
Conclusion
To develop the third generation of biologics, a great deal of investment will need to be made in correlating the clinical parameters of remission from chronic inflammatory disease and the immunological parameters of resolution from inflammation. Experience from the rheumatological field has shown us that aggressive immunosuppressive and biological therapy can induce remission in early RA. However, the longer the disease progresses, the more unlikely this strategy is to succeed because the mechanisms controlling the regulation and resolution of inflammation fail in chronic inflammation. Therapy in chronic inflammation can reduce ongoing inflammation but does not resolve the underlying immunological defect.
Evidence from our laboratory suggests that a single administration of BiP in models of inflammatory arthritis is sufficient to regulate and resolve chronic inflammation. The planned Stage I/II clinical trial of BiP in RA patients will teach us more regarding the properties of BiP as a therapy in human disease. However, it may be that resolution-promoting biologic therapies will have to be administered in conjunction with other biologics, which can control ongoing inflammation and set-the-scene for immunological resolution and restoration of homeostasis to occur.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Prof. Gabriel Panayi is a shareholder in Immune Regulation Ltd., which holds the patent rights for BiP for the treatment of rheumatoid arthritis. Dr. Valerie Corrigall is a shareholder in Immune Regulation Ltd. Funding Statement: As a review, this manuscript has not received direct funding although Dr. Adrian Shields received an Oliver Bird Rheumatism Foundation PhD studentship for the duration of his doctoral studies (completed July 2010) and the Corrigall laboratory receives funding from Arthritis Research UK.
References
Albani, S., Koffeman, E. C., and Prakken, B. (2011). Induction of immune tolerance in the treatment of rheumatoid arthritis. Nat. Rev. Rheumatol. 7, 272–281.
Arac, A., Brownell, S. E., Rothebard, J. B., Chen, C., Ko, R. M., Pereira, M. P., Albers, G. W., Steinman, L., and Steinberg, G. K. (2011). Systemic augmentation of αB-crystallin provides therapeutic benefit twelve hours post-stroke onset via immune modulation. Proc. Natl. Acad. Sci. U.S.A. [Epub ahead of print].
Banerjee, S., Lin, C. F., Skinner, K. A., Schiffhauer, L. M., Peacock, J., Hicks, D. G., Redmond, E. M., Morrow, D., Huston, A., Shayne, M., Langstein, H. N., Miller-Graziano, C. L., Strickland, J., O’Donoghue, L., and De, A. K. (2011). Heat shock protein 27 differentiates tolerogenic macrophages that may support human breast cancer progression. Cancer Res. 71, 318–327.
Bodman-Smith, M. D., Corrigall, V. M., Kemeny, D. M., and Panayi, G. S. (2003). BiP, a putative autoantigen in rheumatoid arthritis, stimulates IL-10-producing CD8-positive T cells from normal individuals. Rheumatology (Oxford) 42, 637–644.
Breedveld, F., Agarwal, S., Yin, M., Ren, S., Li, N. F., Shaw, T. M., and Davies, B. E. (2007). Rituximab pharmacokinetics in patients with rheumatoid arthritis: B-cell levels do not correlate with clinical response. J. Clin. Pharmacol. 47, 1119–1128.
Brouard, S., Mansfield, E., Braud, C., Li, L., Giral, M., Hsieh, S. C., Baeten, D., Zhang, M., Ashton-Chess, J., Braudeau, C., Hsieh, F., Dupont, A., Pallier, A., Moreau, A., Louis, S., Ruiz, C., Salvatierra, O., Soulillou, J. P., and Sarwal, M. (2007). Identification of a peripheral blood transcriptional biomarker panel associated with operational renal allograft tolerance. Proc. Natl. Acad. Sci. U.S.A. 104, 15448–15453.
Brownlie, R. J., Myers, L. K., Wooley, P. H., Corrigall, V. M., Bodman-Smith, M. D., Panayi, G. S., and Thompson, S. J. (2006). Treatment of murine collagen-induced arthritis by the stress protein BiP via interleukin-4-producing regulatory T cells: a novel function for an ancient protein. Arthritis Rheum. 54, 854–863.
Bykerk, V. P. (2011). Rheumatoid arthritis: 2011 remission criteria are a new benchmark for RA therapy. Nat. Rev. Rheumatol. 7, 317–318.
Calderwood, S. K., Theriault, J., Gray, P. J., and Gong, J. (2007). Cell surface receptors for molecular chaperones. Methods 43, 199–206.
Chen, X., Tao, Q., Yu, H., Zhang, L., and Cao, X. (2002). Tumor cell membrane-bound heat shock protein 70 elicits antitumor immunity. Immunol. Lett. 84, 81–87.
Corrao, S., Campanella, C., Anzalone, R., Farina, F., Zummo, G., Conway De Macario, E., Macario, A. J., Cappello, F., and La Rocca, G. (2010). Human Hsp10 and Early Pregnancy Factor (EPF) and their relationship and involvement in cancer and immunity: current knowledge and perspectives. Life Sci. 86, 145–152.
Corrigall, V. M., Bodman-Smith, M. D., Brunst, M., Cornell, H., and Panayi, G. S. (2004). Inhibition of antigen-presenting cell function and stimulation of human peripheral blood mononuclear cells to express an antiinflammatory cytokine profile by the stress protein BiP: relevance to the treatment of inflammatory arthritis. Arthritis Rheum. 50, 1164–1171.
Corrigall, V. M., Bodman-Smith, M. D., Fife, M. S., Canas, B., Myers, L. K., Wooley, P., Soh, C., Staines, N. A., Pappin, D. J., Berlo, S. E., Van Eden, W., Van Der Zee, R., Lanchbury, J. S., and Panayi, G. S. (2001). The human endoplasmic reticulum molecular chaperone BiP is an autoantigen for rheumatoid arthritis and prevents the induction of experimental arthritis. J. Immunol. 166, 1492–1498.
Corrigall, V. M., Bodman-Smith, M. D., and Panayi, G. S. (2003). The stress protein, immunoglobulin binding protein (BiP), has a specific receptor (abstract). Rheumatology (Oxford) 42, 43.
Corrigall, V. M., Vittecoq, O., and Panayi, G. S. (2009). Binding immunoglobulin protein-treated peripheral blood monocyte-derived dendritic cells are refractory to maturation and induce regulatory T-cell development. Immunology 128, 218–226.
De, A. K., Kodys, K. M., Yeh, B. S., and Miller-Graziano, C. (2000). Exaggerated human monocyte IL-10 concomitant to minimal TNF-alpha induction by heat-shock protein 27 (Hsp27) suggests Hsp27 is primarily an antiinflammatory stimulus. J. Immunol. 165, 3951–3958.
Delpino, A., and Castelli, M. (2002). The 78 kDa glucose-regulated protein (GRP78/BIP) is expressed on the cell membrane, is released into cell culture medium and is also present in human peripheral circulation. Biosci. Rep. 22, 407–420.
Ehrenstein, M. R., Evans, J. G., Singh, A., Moore, S., Warnes, G., Isenberg, D. A., and Mauri, C. (2004). Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFalpha therapy. J. Exp. Med. 200, 277–285.
Gao, B., and Tsan, M. F. (2003a). Endotoxin contamination in recombinant human heat shock protein 70 (Hsp70) preparation is responsible for the induction of tumor necrosis factor alpha release by murine macrophages. J. Biol. Chem. 278, 174–179.
Gao, B., and Tsan, M. F. (2003b). Recombinant human heat shock protein 60 does not induce the release of tumor necrosis factor alpha from murine macrophages. J. Biol. Chem. 278, 22523–22529.
Gething, M. J. (1999). Role and regulation of the ER chaperone BiP. Semin. Cell Dev. Biol. 10, 465–472.
Gordon, S., and Taylor, P. R. (2005). Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 5, 953–964.
Hendershot, L. M. (2004). The ER function BiP is a master regulator of ER function. Mt. Sinai J. Med. 71, 289–297.
Henderson, B., Calderwood, S. K., Coates, A. R., Cohen, I., Van Eden, W., Lehner, T., and Pockley, A. G. (2010). Caught with their PAMPs down? The extracellular signalling actions of molecular chaperones are not due to microbial contaminants. Cell Stress Chaperones 15, 123–141.
Hernandez-Fuentes, M. P., and Lechler, R. I. (2010). A “biomarker signature” for tolerance in transplantation. Nat. Rev. Nephrol. 6, 606–613.
Huberts, D. H., and van der Klei, I. J. (2010). Moonlighting proteins: an intriguing mode of multitasking. Biochim. Biophys. Acta 1803, 520–525.
Isaacs, J. D. (2010). The changing face of rheumatoid arthritis: sustained remission for all? Nat. Rev. Immunol. 10, 605–611.
Kitamura, M. (2011). Control of NF-kappaB and inflammation by the unfolded protein response. Int. Rev. Immunol. 30, 4–15.
Kono, H., and Rock, K. L. (2008). How dying cells alert the immune system to danger. Nat. Rev. Immunol. 8, 279–289.
Lee, A. S. (2007). GRP78 induction in cancer: therapeutic and prognostic implications. Cancer Res. 67, 3496–3499.
Li, P., and Schwarz, E. M. (2003). The TNF-alpha transgenic mouse model of inflammatory arthritis. Springer Semin. Immunopathol. 25, 19–33.
Li, Z., Menoret, A., and Srivastava, P. (2002). Roles of heat-shock proteins in antigen presentation and cross-presentation. Curr. Opin. Immunol. 14, 45–51.
Liu, B., Dai, J., Zheng, H., Stoilova, D., Sun, S., and Li, Z. (2003). Cell surface expression of an endoplasmic reticulum resident heat shock protein gp96 triggers MyD88-dependent systemic autoimmune diseases. Proc. Natl. Acad. Sci. U.S.A. 100, 15824–15829.
Luo, S., Mao, C., Lee, B., and Lee, A. S. (2006). GRP78/BiP is required for cell proliferation and protecting the inner cell mass from apoptosis during early mouse embryonic development. Mol. Cell. Biol. 26, 5688–5697.
Macario, A. J., and Conway de Macario, E. (2005). Sick chaperones, cellular stress, and disease. N. Engl. J. Med. 353, 1489–1501.
Mambula, S. S., and Calderwood, S. K. (2006). Heat shock protein 70 is secreted from tumor cells by a nonclassical pathway involving lysosomal endosomes. J. Immunol. 177, 7849–7857.
Marin-Briggiler, C. I., Gonzalez-Echeverria, M. F., Munuce, M. J., Ghersevich, S., Caille, A. M., Hellman, U., Corrigall, V. M., and Vazquez-Levin, M. H. (2010). Glucose-regulated protein 78 (Grp78/BiP) is secreted by human oviduct epithelial cells and the recombinant protein modulates sperm-zona pellucida binding. Fertil. Steril. 93, 1574–1578.
McInnes, I. B., and Schett, G. (2011). The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 365, 2205–2219.
Merendino, A. M., Bucchieri, F., Campanella, C., Marciano, V., Ribbene, A., David, S., Zummo, G., Burgio, G., Corona, D. F., Conway De Macario, E., Macario, A. J., and Cappello, F. (2010). Hsp60 is actively secreted by human tumor cells. PLoS ONE 5, e9247. doi:10.1371/journal.pone.0009247
Mimura, N., Hamada, H., Kashio, M., Jin, H., Toyama, Y., Kimura, K., Iida, M., Goto, S., Saisho, H., Toshimori, K., Koseki, H., and Aoe, T. (2007). Aberrant quality control in the endoplasmic reticulum impairs the biosynthesis of pulmonary surfactant in mice expressing mutant BiP. Cell Death Differ. 14, 1475–1485.
Multhoff, G., and Hightower, L. E. (1996). Cell surface expression of heat shock proteins and the immune response. Cell Stress Chaperones 1, 167–176.
Orlando, G., Hematti, P., Stratta, R. J., Burke, G. W. III, Di Cocco, P., Pisani, F., Soker, S., and Wood, K. (2010). Clinical operational tolerance after renal transplantation: current status and future challenges. Ann. Surg. 252, 915–928.
Orlando, G., Soker, S., and Wood, K. (2009). Operational tolerance after liver transplantation. J. Hepatol. 50, 1247–1257.
Osterloh, A., Kalinke, U., Weiss, S., Fleischer, B., and Breloer, M. (2007). Synergistic and differential modulation of immune responses by Hsp60 and lipopolysaccharide. J. Biol. Chem. 282, 4669–4680.
Osterloh, A., Meier-Stiegen, F., Veit, A., Fleischer, B., Von Bonin, A., and Breloer, M. (2004). Lipopolysaccharide-free heat shock protein 60 activates T cells. J. Biol. Chem. 279, 47906–47911.
Ousman, S. S., Tomooka, B. H., Van Noort, J. M., Wawrousek, E. F., O’Connor, K. C., Hafler, D. A., Sobel, R. A., Robinson, W. H., and Steinman, L. (2007). Protective and therapeutic role for alphaB-crystallin in autoimmune demyelination. Nature 448, 474–479.
Pahl, H. L., and Baeuerle, P. A. (1995). A novel signal transduction pathway from the endoplasmic reticulum to the nucleus is mediated by transcription factor NF-kappa B. EMBO J. 14, 2580–2588.
Panayi, G. S., Corrigall, V. M., and Henderson, B. (2004). Stress cytokines: pivotal proteins in immune regulatory networks; opinion. Curr. Opin. Immunol. 16, 531–534.
Pangratz-Fuehrer, S., Kaur, K., Ousman, S. S., Steinman, L., and Liao, Y. J. (2011). Functional rescue of experimental ischemic optic neuropathy with alphaB-crystallin. Eye (Lond.) 25, 809–817.
Pisetsky, D. S., Erlandsson-Harris, H., and Andersson, U. (2008). High-mobility group box protein 1 (HMGB1): an alarmin mediating the pathogenesis of rheumatic disease. Arthritis Res. Ther. 10, 209.
Quinn, M. A., Conaghan, P. G., O’Connor, P. J., Karim, Z., Greenstein, A., Brown, A., Brown, C., Fraser, A., Jarret, S., and Emery, P. (2005). Very early treatment with infliximab in addition to methotrexate in early, poor-prognosis rheumatoid arthritis reduces magnetic resonance imaging evidence of synovitis and damage, with sustained benefit after infliximab withdrawal: results from a twelve-month randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 52, 27–35.
Quintana, F. J., and Cohen, I. R. (2011). The HSP60 immune system network. Trends Immunol. 32, 89–95.
Rath, E., and Haller, D. (2011). Inflammation and cellular stress: a mechanistic link between immune-mediated and metabolically driven pathologies. Eur. J. Nutr. 50, 219–233.
Serhan, C. N., Brain, S. D., Buckley, C. D., Gilroy, D. W., Haslett, C., O’Neill, L. A., Perretti, M., Rossi, A. G., and Wallace, J. L. (2007). Resolution of inflammation: state of the art, definitions and terms. FASEB J. 21, 325–332.
Shields, A. M., Panayi, G. S., and Corrigall, V. M. (2011). Resolution-associated molecular patterns (RAMP): RAMParts defending immunological homeostasis? Clin. Exp. Immunol. 165, 292–300.
Sreekumar, P. G., Kannan, R., Kitamura, M., Spee, C., Barron, E., Ryan, S. J., and Hinton, D. R. (2010). AlphaB crystallin is apically secreted within exosomes by polarized human retinal pigment epithelium and provides neuroprotection to adjacent cells. PLoS ONE 5, e12578. doi:10.1371/journal.pone.0012578
Tanaka, Y., Takeuchi, T., Mimori, T., Saito, K., Nawata, M., Kameda, H., Nojima, T., Miyasaka, N., and Koike, T. (2010). Discontinuation of infliximab after attaining low disease activity in patients with rheumatoid arthritis: RRR (remission induction by Remicade in RA) study. Ann. Rheum. Dis. 69, 1286–1291.
Theriault, J. R., Mambula, S. S., Sawamura, T., Stevenson, M. A., and Calderwood, S. K. (2005). Extracellular HSP70 binding to surface receptors present on antigen presenting cells and endothelial/epithelial cells. FEBS Lett. 579, 1951–1960.
Triantafilou, K., Triantafilou, M., and Dedrick, R. L. (2001). A CD14-independent LPS receptor cluster. Nat. Immunol. 2, 338–345.
Urbonaviciute, V., Meister, S., Furnrohr, B. G., Frey, B., Guckel, E., Schett, G., Herrmann, M., and Voll, R. E. (2009). Oxidation of the alarmin high-mobility group box 1 protein (HMGB1) during apoptosis. Autoimmunity 42, 305–307.
van Eden, W., Spiering, R., Broere, F., and Van Der Zee, R. (2011). A case of mistaken identity: HSP are no DAMPs but DAMPERs. Cell Stress Chaperones. [Epub ahead of print].
van Eden, W., van Der Zee, R., and Prakken, B. (2005). Heat-shock proteins induce T-cell regulation of chronic inflammation. Nat. Rev. Immunol. 5, 318–330.
Vanags, D., Williams, B., Johnson, B., Hall, S., Nash, P., Taylor, A., Weiss, J., and Feeney, D. (2006). Therapeutic efficacy and safety of chaperonin 10 in patients with rheumatoid arthritis: a double-blind randomised trial. Lancet 368, 855–863.
Wallin, R. P., Lundqvist, A., More, S. H., Von Bonin, A., Kiessling, R., and Ljunggren, H. G. (2002). Heat-shock proteins as activators of the innate immune system. Trends Immunol. 23, 130–135.
Warger, T., Hilf, N., Rechtsteiner, G., Haselmayer, P., Carrick, D. M., Jonuleit, H., Von Landenberg, P., Rammensee, H. G., Nicchitta, C. V., Radsak, M. P., and Schild, H. (2006). Interaction of TLR2 and TLR4 ligands with the N-terminal domain of Gp96 amplifies innate and adaptive immune responses. J. Biol. Chem. 281, 22545–22553.
Williams, B., Vanags, D., Hall, S., McCormack, C., Foley, P., Weiss, J., Johnson, B., Latz, E., and Feeney, D. (2008). Efficacy and safety of chaperonin 10 in patients with moderate to severe plaque psoriasis: evidence of utility beyond a single indication. Arch. Dermatol. 144, 683–685.
Ye, Z., and Gan, Y. H. (2007). Flagellin contamination of recombinant heat shock protein 70 is responsible for its activity on T cells. J. Biol. Chem. 282, 4479–4484.
Keywords: binding immunoglobulin protein, resolution-associated molecular patterns, inflammation, resolution of inflammation, immunotherapy, rheumatoid arthritis, immune networks
Citation: Shields AM, Panayi GS and Corrigall VM (2012) A new-age for biologic therapies: long-term drug-free therapy with BiP? Front. Immun. 3:17. doi: 10.3389/fimmu.2012.00017
Received: 10 December 2011;
Paper pending published: 11 January 2012;
Accepted: 01 February 2012;
Published online: 17 February 2012.
Edited by:
Willem Van Eden, Utrecht University, NetherlandsReviewed by:
Miriam Wittmann, University of Leeds, UKBernard R. Lauwerys, Université Catholique de Louvain, Belgium
Ruurd Van Der Zee, Universiteit Utrech, Netherlands
Copyright: © 2012 Shields, Panayi and Corrigall. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Valerie M. Corrigall, Department of Academic Rheumatology, Centre for Molecular and Cellular Biology of Inflammation, 1st Floor New Hunts House, Guy’s Campus, King’s College London, SE1 1UL London, UK. e-mail:dmFsZXJpZS5jb3JyaWdhbGxAa2NsLmFjLnVr