
 Roberto Tinoco
Roberto Tinoco
- Infectious and Inflammatory Disease Center, Sanford-Burnham Medical Research Institute, La Jolla, CA, USA
Despite the widespread use of the cell-surface receptor CD44 as a marker for antigen (Ag)-experienced, effector and memory T cells, surprisingly little is known regarding its function on these cells. The best-established function of CD44 is the regulation of cell adhesion and migration. As such, the interactions of CD44, primarily with its major ligand, the extracellular matrix (ECM) component hyaluronic acid (HA), can be crucial for the recruitment and function of effector and memory T cells into/within inflamed tissues. However, little is known about the signaling events following engagement of CD44 on T cells and how cooperative interactions of CD44 with other surface receptors affect T cell responses. Recent evidence suggests that the CD44 signaling pathway(s) may be shared with those of other adhesion receptors, and that these provide contextual signals at different anatomical sites to ensure the correct T cell effector responses. Furthermore, CD44 ligation may augment T cell activation after Ag encounter and promote T cell survival, as well as contribute to regulation of the contraction phase of an immune response and the maintenance of tolerance. Once the memory phase is established, CD44 may have a role in ensuring the functional fitness of memory T cells. Thus, the summation of potential signals after CD44 ligation on T cells highlights that migration and adhesion to the ECM can critically impact the development and homeostasis of memory T cells, and may differentially affect subsets of T cells. These aspects of CD44 biology on T cells and how they might be modulated for translational purposes are discussed.
Introduction
CD44 is a carbohydrate-binding, type 1 transmembrane protein that is the product of a single gene, and is represented by a family of six isoforms that arise by mRNA splicing of 10 variably expressed exons (Ponta et al., 2003). Further molecular heterogeneity on various cell types is introduced by glycosylation, primarily on the CD44 variants (English et al., 1998). CD44 is found constitutively on the surface of many cell types, including resting T cells, which exclusively express the invariant or standard form. CD44 localizes in lipid rafts with the T cell receptor (TCR) complex and is upregulated early after TCR engagement (DeGrendele et al., 1997). Expression of CD44 variant isoforms may also be induced, although the functional implications remain unclear and this aspect of CD44 regulation has not been widely studied in T cells (Konig et al., 1998; Forster-Horvath et al., 2001). Most biological and signaling functions of CD44 are associated with binding of the primary ligand, HA, a flexible copolymer of N-acetylglucosamine and glucuronic acid that occurs naturally and is an integral part of the extracellular matrix (ECM). Whereas there have been reports that isoform usage may affect HA binding, the N-terminal site, present in all isoforms, is sufficient for recognition of HA (He et al., 1992; Liao et al., 1993). Once CD44 is upregulated on responding T cells, its expression is sustained on effector cells as well as those cells that survive to become memory cells after an immune response subsides. As such, CD44 is the most widely used indicator of prior exposure to Ag.
CD44 is best known as a critical regulator of biological processes involving migrating cells, including the recruitment of effector T cells and other leukocytes to sites of infection (Ponta et al., 2003). It has long been recognized that T cells are intrinsically reprogrammed to migrate after their encounter with Ag (Sallusto and Lanzavecchia, 2009). However, much less attention has been devoted to the roles of adhesion receptors as vital signaling molecules, which can have a profound impact not only on differentiation of effector T cells and their responses, but also on the development and homeostasis of memory T cells. Both CD4+ and CD8+ memory T cells are heterogeneous with respect to adhesion receptor expression and the potential cues they may receive from the microenvironments in which they localize and migrate can further influence their effector function as well as their transition from effector to memory cells. However, both populations of memory T cells maintain increased levels of CD44 after immune activation. Importantly, CD44 may contribute to the functional specialization as well as the regulation of memory in T cells through the engagement of specific signaling cascades. Current evidence predicts that CD44 becomes repeatedly engaged during an immune response and periodically thereafter during the homeostatic regulation of memory T cells. Here, we discuss the multi-functional roles of CD44 in the migration, activation, differentiation, and homeostasis of T cells in the context of signals received based on anatomical location, ligand availability, and “co”-receptor interaction. The multiplicity of potential signaling outcomes after CD44 ligation on T cells highlights that migration and adhesion to the ECM, or its components displayed on other cells, can make critical contributions to T cell function and homeostasis, as well as the regulation of memory.
Regulation of T Cell Recruitment to Tissues by CD44
CD44-dependent adhesion mechanisms are important for the mobilization of effector T cells to sites of infection and inflammation in many systems (Camp et al., 1993; Wittig et al., 2000; Savinov and Strongin, 2007; discussed in more detail below). The mechanisms underlying lymphocyte migration into tissues have common attributes that have led to the general cascade of events that is elicited by initial engagement, termed rolling/tethering, firm adhesion, and transmigration (for review see Ley et al., 2007; Figures 1 and 2). At sites of infection, endothelial cells become activated in response to inflammatory cytokines and express endothelial selectins (E- and P-selectin) that mediate rolling (Ley and Kansas, 2004; Bonder et al., 2006). Rolling can also occur as a result of CD44 presentation of HA on the luminal surface of vascular endothelial cells to CD44 on the surface of effector cells (Figure 1, left panel), which is enhanced by pro-inflammatory cytokines, including IL-1 and tumor necrosis factor (TNF)-α (Jiang et al., 2011). Chemokines presented or secreted at the endothelial cell-surface induce signaling via G-protein coupled receptors (GPCRs), which results in increased integrin affinity/adhesiveness (“inside-out” signaling). Subsequently, integrin signaling itself secures adhesion stability and may contribute to transmigration (Ley et al., 2007). In general, the integrins LFA-1 (αLβ2), VLA-4 (α4β1), and α4β7 and their co-receptors ICAM-1, vascular cell adhesion molecule (VCAM)-1, and mucosal addressin cell adhesion molecule-1, respectively, regulate trafficking into sites of inflammation. The VLA-4-ligand VCAM-1 is induced on endothelial cells in response to inflammatory cytokines and is thought to cooperate with CD44 in cell migration (Alon et al., 1995; Siegelman et al., 2000). For example, in human mesenchymal stem cells, engagement of CD44 by HA triggered GPCR-dependent “inside-out” upregulation of VLA-4–VCAM binding, which enhanced adhesion and subsequent transendothelial migration without a requirement for chemokines (Thankamony and Sackstein, 2011). Adhesion receptors exhibit considerable functional redundancy, which is thought to enable the use of specific combinations for controlled entry into particular tissues. In addition, their function may be dependent on, or augmented by, other adhesion receptors and specific combinations of receptor expression patterns could result in a variety of potential outcomes following interaction with the endothelium during migration.
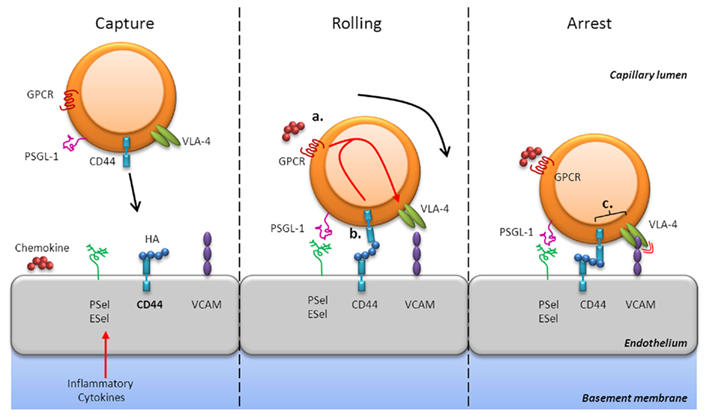
Figure 1. CD44-mediated rolling and arrest. CD44-dependent adhesion mechanisms are important for the mobilization of effector T cells in sites of infection and inflammation. At these sites, endothelial cells secrete chemokines and express selectins (E- and P-selectin), HA, and integrin ligands (e.g., VCAM-1) in response to inflammatory cytokines (left panel). CD44 can mediate rolling interactions with vascular endothelial cells that express HA, its natural ligand, or even E-selectin (center panel). Chemokine signaling via GPCRs results in increased integrin affinity, such as VLA-4, which enhances adhesiveness [“inside-out” signaling (a)]. Engagement of CD44 with HA may also trigger GPCR-dependent “inside-out” upregulation of VLA-4–VCAM binding (b). Furthermore, CD44 may associate with VLA-4 in the membrane of activated T cells and augment signaling [(c) right panel]. Thus, CD44 contributes to mechanisms that allow T cells to successfully interpret environmental signals to migrate into sites of inflammation.
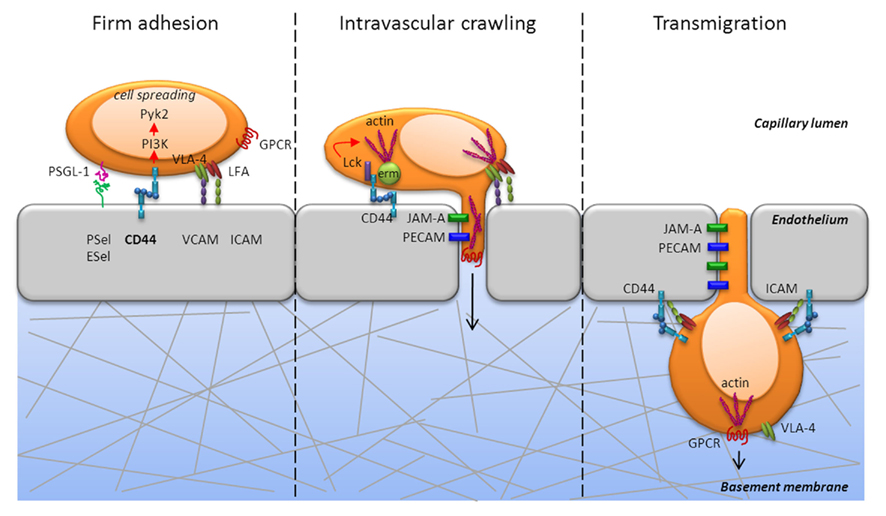
Figure 2. CD44 is critically involved during firm adhesion and intravascular crawling to mediate transmigration of T cells. CD44 engagement activates the non-receptor tyrosine kinase, Pyk2, in a PI3K dependent fashion and mediates cell spreading during firm adhesion (left panel). In addition to Pyk2, CD44 engagement activates the Src kinase, Lck, which is required for actin polymerization (center panel). Further, cytoskeletal rearrangement is mediated by the ERM proteins, which are also induced following PI3K signaling. CD44 can directly associate with the ERM proteins, which crosslink CD44 and other proteins to the actin cytoskeleton. The development of cell polarity and the diapedesis occurs through PECAM and JAM that enable extrusion of filopodia into the junctions of endothelial cells and transmigration into tissue (center and right panel). Thus, signaling via CD44 can promote the cytoskeletal changes necessary for T cell extravasation from the blood into tissues.
In addition to the individual role of CD44 during migration and its effect on the signaling outcome of other receptor–ligand interactions, it can directly share signaling pathways with other adhesion receptors. CD44 can associate with VLA-4 in the membrane of activated T cells through its cytoplasmic tail and thereby provide access to its signaling pathways that would have otherwise been unavailable (Marhaba et al., 2006). For example, focal adhesion kinase is associated with VLA-4, and auto-phosphorylation creates a docking site for the Src family kinase Lck, which is associated with CD44 (Lefebvre et al., 2010). The formation of this complex promoted resistance of primary T cells to apoptosis via the Akt/PI3K pathway (Marhaba et al., 2006) and was crucial for T cell recruitment in a peritonitis model (Nandi et al., 2004). CD44 can also cooperate with LFA-1 during T cell recruitment (Fanning et al., 2005). Interestingly, in leukocytes, CD44 is a ligand for E-selectin, as is P-selectin glycoprotein-1 (PSGL-1), and both molecules elicit LFA-1-dependent slow rolling via ICAM-1 by activating Src family kinases (Yago et al., 2010). Furthermore, both CD44 and PSGL-1 engage the ERM proteins ezrin, radixin, and moesin, which regulate actin cytoskeleton rearrangement and thereby convert signals from membrane proteins into cell movement. There are many similarities between the effects of cross-linking CD44 or LFA-1 on human T cells, both of which can elicit in vitro migration that is protein kinase C dependent (Fanning et al., 2005). It seems therefore, that engagement of common signaling pathways by multiple adhesion receptors provides an additional mechanism whereby T cells may successfully interpret environmental signals to achieve the goal of ensuring efficient recruitment into sites of inflammation and/or tissue damage.
Firm adhesion is typically associated with integrin function and is essential for arrest and cell spreading, which are in turn necessary for successful transmigration from blood into tissue. CD44 contributes to these processes by eliciting signaling via PI3K and Src family kinases (Figure 2). Engagement of the PI3K signaling pathway through CD44 also activates the non-receptor proline-rich tyrosine kinase 2 (Pyk2) that is related to focal adhesion kinase, which mediates signals via integrins. A recent study indicated that Pyk2 activation was required for CD44-mediated elongated cell spreading (Wong et al., 2011). In addition to activating Pyk2, CD44 engagement activated Lck, which was required for actin polymerization in T cells (Nandi et al., 2004). Furthermore, our studies showed that ligation of CD44 elicited PI3K signaling in Th1 cells (Baaten et al., 2010), which also induced cytoskeletal rearrangement through the ERM proteins (Ponta et al., 2003). CD44 directly associated with the ERM proteins (Mori et al., 2008) and co-localization correlated with HA binding (Brown et al., 2005). CD44 can thereby provide a direct link from the plasma membrane to the cellular machinery and enable directed movement. The ERM–CD44 complex can also regulate signaling by Rho-ROCK family GTPases to re-organize the actin cytoskeleton. Rho GTPases have important roles in integrin-mediated adhesion, cell division, and cell survival, and CD44 signaling may affect other receptor signaling pathways via cross-talk (Tybulewicz and Henderson, 2009). The development of cell polarity and diapedesis occurs through the engagement of multiple receptors that include platelet/endothelial adhesion molecule-1 (PECAM-1) and junctional adhesion molecule A (JAM-A; Ley et al., 2007) that enable extrusion of filopodia into the junctions of endothelial cells and transmigration into the target tissue (Figure 3). Thus, signaling via CD44 can promote the cytoskeletal changes necessary for T cell recruitment from the blood into tissue and may participate in the signaling events that affect T cell differentiation programs and/or responses in the local microenvironment.
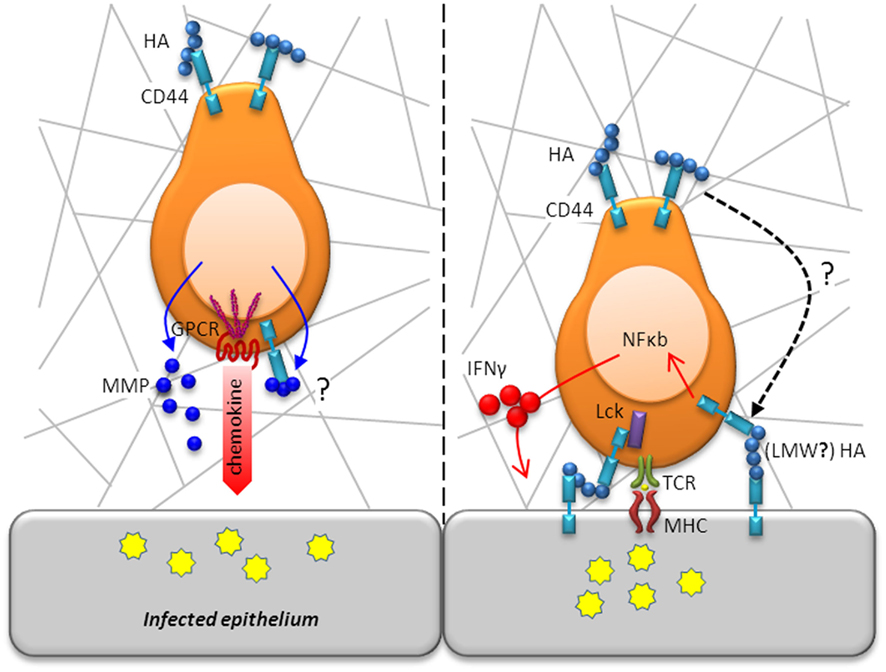
Figure 3. T cell motility and activation in situ is modulated by CD44. Following diapedesis, T cells adopt an amoeboid shape and migrate in a polarized fashion along collagen fibrils in response to a chemotactic gradient toward the site of infection/inflammation (left panel). Whereas chemotactic receptors are located in the leading edge, CD44 localizes in the uropod, which is in contact with the ECM, to maintain a polarized shape. As movement takes place, CD44 anchorage of the cell’s uropod to the ECM is disengaged. Migration through the parenchymal basement membrane is facilitated by enzymatic digestions mediated by secretion of MMPs, which may even be bound to CD44 for directed proteolytic activity. Following efficient migration to areas of infection, T cells will encounter, recognize, and respond to antigen (right panel). CD44 augments TCR signaling by increasing the availability of Lck for downstream engagement of ZAP-70. CD44 ligation activates the transcription factor, NF-κB, and induces expression of IFN-γ highlighting its contribution to T cell activation.
A Role for CD44 in T Cell Motility within Tissues
Little is known regarding the movement of T cells in non-lymphoid tissues. To enter the tissue parenchyma, cells must move through the underlying ECM, including the basement membrane, which is primarily comprised of laminins, collagen type IV, nidogens, heparan sulfate, and proteoglycans (Sorokin, 2010). HA is distributed throughout the ECM in soluble form where it contributes to the scaffolds that maintain tissue integrity (Girish and Kemparaju, 2007). In the absence of inflammation, the majority of HA is found as large, high molecular weight (HMW) polymers (>1000 kDa) that are integral to the ECM. The degradation of the ECM into fragments, particularly of collagen and low molecular weight (LMW) HA, is a characteristic feature of inflammation that stimulates tissue remodeling, but may also modify the activity of T cells via signaling through CD44 (see below).
T cell traversal of the endothelial basement membrane is regulated by integrins that include VLA-4 and α6β1. Migration through the parenchymal basement membrane is mediated by secretion of matrix metalloproteinases (MMPs), most notably the gelatinases MMP-2 and MMP-9 whose expression is regulated by proinflammatory cytokines (Elkington et al., 2005). CD44 has been identified to bind MMP-9 on the cell surface of tumor cells, which was shown to be required for tumor invasion (Yu and Stamenkovic, 1999). A similar mechanism may assist in the directed degradation of the ECM to allow for T cell motility in the basement membrane (Figure 3). After penetrating into the parenchyma, the cells adopt an amoeboid shape and migrate in a polarized fashion along collagen fibrils in response to a chemotactic gradient toward the site of infection/inflammation. To achieve directed movement within tissues, T cells need to convert microenvironmental cues into intracellular signals that result in the development of a polarized shape with the formation of a lamellipodium at the leading edge of the migrating cell and with the uropod at the rear (Figure 3). For T cells, chemotactic receptors are located on the leading edge, enabling them to sense chemokine gradients (del Pozo et al., 1995). Chemotaxis results in the localization of CD44 and other adhesion receptors in the uropod, which is in contact with the ECM (Rosenman et al., 1993; del Pozo et al., 1995). As movement takes place, CD44’s anchorage of the cell’s uropod to the ECM is disengaged by proteolytic activity. Cleavage of the extracellular domain of CD44 is mediated by membrane type metalloproteinases on responding T cells (Savinov et al., 2005). The development of cell polarity depends upon remodeling of the actin cytoskeleton that is regulated by the Rho family of GTPases (Tybulewicz and Henderson, 2009). Actin polymerization at the leading edge regulates forward movement and the formation of the uropod. During this process, the microtubule organizing center, Golgi apparatus, and other organelles are redistributed to the rear of the migrating cell. There is increasing recognition that signals transmitted during movement can be essential for cell function. It is important to bear in mind that the signaling pathways induced by adhesion receptors can converge at the level of cyoskeletal rearrangement. In vitro, cross-linking of CD44 on activated T cells by ligation with antibody or with HA-containing ECM lattices was sufficient to induce cytoskeletal rearrangement and polarized morphology as well as motility (Fanning et al., 2005). A recent study using intravital imaging of CD8+ T cells found impaired motility of Cd44−/− cells within tumor explants, which could be reconstituted by reintroduction of full length CD44 (Mrass et al., 2008). While CD44 was not required for homing, it was needed to maintain a polarized shape and movement, which were essential for effective cytotoxic activity and tumor eradication. Thus, in addition to being involved in the recruitment of T cells to the tissue, CD44 also plays a crucial role in the motility of T cells within the tissue, allowing them to gain access to areas of inflammation or infection and function appropriately.
CD44 Signaling in T Cells in Response to Antigen Encounter
Following efficient migration to areas of infection, T cells will encounter, recognize, and respond to Ag. During this phase, CD44 ligation could augment T cell activation, maintain polarization, and ensure survival and further differentiation. Most of the biological responses that are mediated by CD44 are due to its binding of HA in the N-terminal region of the molecule (Banerji et al., 2007). Whereas the interaction between HA and CD44 is of low affinity (Kd = 10–100 μM), the repetitive structure of HA is such that a single large polymer can consist of up to 10,000 disaccharides and can bind to a large number of CD44 molecules (>1000; Wolny et al., 2010). In response to inflammation, LMW fragments of HA (<500 kDa, typically 8–40 mers) are generated by degradation of HMW HA by hyaluronidases. An octomer of HA appears to represent the smallest polymer capable of mediating maximal binding to CD44 (Banerji et al., 2007). Whereas LMW HA induces reversible CD44 binding, larger polymers seem to bind irreversibly (Wolny et al., 2010). However, little is known regarding the effects of LMW versus HMW HA binding by T cells (see below). In general, LMW HA has agonist functions, whereas HMW HA has antagonist activities in a variety of cell types (Jiang et al., 2011). The size of HA could influence the extent of CD44 aggregation and association with other receptors in the cell membrane, thereby modulating the downstream signaling pathways that are engaged.
CD44 itself can interconvert between low and high affinity HA binding states (Ogino et al., 2010), which in T cells are influenced by the activation status with respect to TCR signaling (Lesley et al., 1994; Maeshima et al., 2011). Although the basis for this transition is incompletely understood, the main mechanism involves enzymatic cleavage of an inhibitory terminal sialic acid residue from N-linked sugar chains on the HA binding domain of CD44 by an inducible sialidase (Katoh et al., 1999; Gee et al., 2003). Additional conformational changes occur upon HA binding (Takeda et al., 2006), which may support multimerization of CD44 within the cell membrane and thereby increase the strength of signaling. The development of HA binding is also required for proteolytic cleavage of extracellular and intracellular domains of CD44, which supports feed-back regulation of CD44 synthesis (Nagano and Saya, 2004). Although it has been reported that CD44 binds to the ECM components fibronectin and collagen in vitro, these interactions have not been corroborated in vivo (Ponta et al., 2003).
CD44 is contained in detergent-insoluble lipid rafts in the cell membrane and localizes with the TCR–CD3 signaling complex in the immunological synapse (Hegde et al., 2008). After signaling, the TCR–CD3 complex moves to the center of the immunological synapse, where interactions with MHC molecules on antigen presenting cells (APC) are facilitated. During synapse formation, CD44, as well as CD28, LFA-1, and probably VLA-4, relocate to the outside edge of the complex known as the supra-molecular activation complex (Huppa and Davis, 2003). Thus, CD44 is located in proximity to other molecules known to participate in augmenting adhesion to APC during T cell signaling. Presentation of HA by CD44 expressed on dendritic cells (DCs) has been shown to facilitate T cell–DC conjugate formation (Hegde et al., 2008), as does LFA-1 through its interactions with ICAM-1 on APC (Fooksman et al., 2010; Figure 4A). The finding that cross-linking CD44 activates Lck suggests an interconnection with TCR signaling (Taher et al., 1996; Lefebvre et al., 2010). As observed with CD44 ligation, TCR signaling also engages ERM proteins through activation of Lck (Autero et al., 2003), which may stabilize interactions with APC (Roumier et al., 2001). It is thought that CD44 may contribute to and enhance signal transduction in the context of TCR engagement by increasing the availability of Lck for downstream engagement of ZAP-70, which binds to the CD3ζ chain of the TCR and transmits the signals necessary for T cell activation. In support of this conclusion, ligation of CD44 leads to tyrosine phosphorylation of Zap-70 (Taher et al., 1996). Activated Lck also contributes to signaling by CD28 and could play a similar role in regulating signaling by CD44 (Rozsnyay, 1999) at later stages in an immune response.
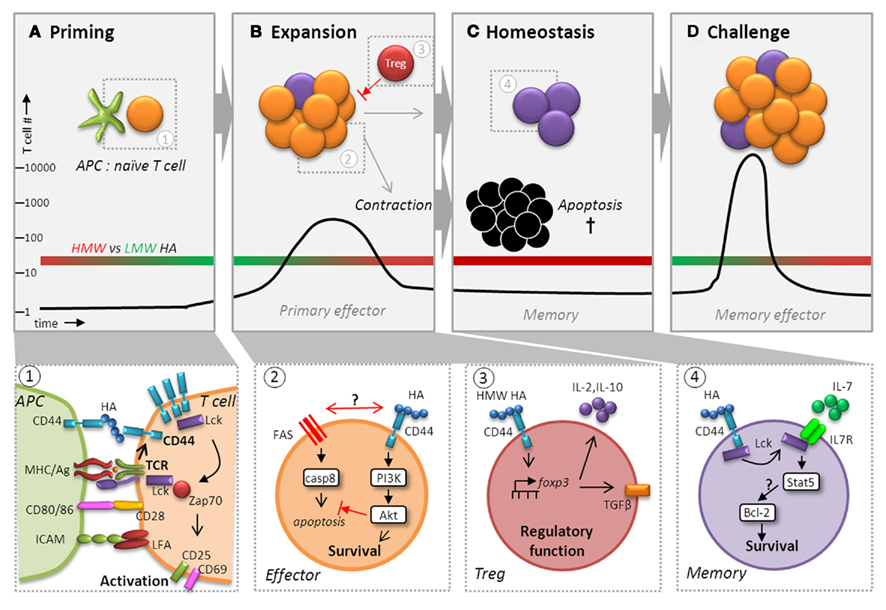
Figure 4. CD44 potentiates T cell activation, expansion, contraction, and maintenance of memory cells. Besides playing a pivotal role during recruitment, CD44 ligation affects T cell memory function by augmenting activation (A), ensuring survival (B), and regulating homeostasis (C) to facilitate maximum expansion following challenge (D). Following recognition of Ag via TCR, CD44 affinity and expression are upregulated and it localizes with the TCR signaling complex in the immunological synapse where it receives signals from HA presented by CD44 expressed on DCs (panel 1). CD44 ligation contributes to and enhances signal transduction in the context of TCR engagement by increasing the availability of Lck for downstream engagement of ZAP-70, which transmits the signals necessary for T cell activation. Signaling events in the primary response to infections can profoundly affect the generation and maintenance of T cell memory (panel 2). HA binding to CD44 can induce a survival signal via the PI3K/Akt pathway, which may inhibit apoptosis during contraction by interfering with cell death either through direct obstruction of Fas to prevent DISC assembly and caspase 8 activation or by affecting Fas signaling itself. Although differences in the capacity of LMW versus HMW HA to elicit CD44-mediated signaling in T cells has not been carefully investigated, recent studies suggest that HMW (red bar), but not LMW (green bar), HA (A–D) can upregulate the transcription factor FoxP3 in Tregs cells via CD44 signaling (panel 3). CD44-regulated FoxP3 expression was linked to a greater capacity for IL-10 production and the development of surface TGF-β1 expression, which are important for their regulatory function. We hypothesize that effector T cells are more likely to respond to LMW HA generated during inflammation. Furthermore, during homeostasis, we envision that HA binding by CD44 elicits enhanced survival due to engagement of Lck (panel 4). CD44 signaling may increase the responsiveness of IL-7Rα by making more Lck available leading to IL-7-mediated upregulation of Bcl-2 via Stat5 signaling. Thus, CD44 could have a global function in regulating the transition of effector cells to memory cells, as well as in maintaining memory T cells that exceeds its roles in migration and motility.
A role for CD44 as a potential co-stimulatory molecule is suggested by the finding that low doses of anti-CD3, that did not support T cell proliferation or IL-2 induction in vitro, were rendered stimulatory after antibody-mediated cross-linking of CD44 (Foger et al., 2000). Furthermore, in the context of low TCR signaling, CD44 ligation induced expression of CD69 and CD25, supporting the potential of CD44 to contribute to T cell activation. In addition, HA engagement during T cell–DC interactions promoted the induction of IL-2 in vitro (Mummert et al., 2002). Thus, for memory T cells that already express higher levels of CD44, ligation could provide an immediate co-stimulatory signal leading to more rapid T cell activation compared to naïve T cells. One caveat for the studies of CD44 signaling in T cells is that most work has focused on cell lines and in vitro analyses with monoclonal antibodies. Signals transmitted by cross-linking CD44 with various monoclonal antibodies are likely to differ since they can have widely disparate effects on the induction of HA binding, as well as CD44 external domain shedding (Zheng et al., 1995). Efforts to recapitulate CD44 interactions with HA and/or ECM lattices in vitro may also not faithfully reveal the signaling pathways that are engaged physiologically. Nevertheless, it is likely that in the context of HA binding, CD44 can augment TCR signaling and impact the T cell response in a context-dependent manner. With the perspective of the potential outcomes of CD44 signaling described above, HA availability on DCs and other APCs in sites of inflammation is likely to be a significant factor. The extent of co-stimulation can greatly impact the differentiation of Th subsets and is essential for the development of memory. It is notable that CD4+ T cells from Cd44−/− mice appear to be defective in Th1 cell responses. They exhibit enhanced expression of the Th2 transcription factor GATA3 and diminished expression of the Th1 transcription factor T-bet after in vivo immunization (Guan et al., 2009). Furthermore, naive CD4+ T cells from Cd44−/− mice more readily differentiate into Th2 than Th1 cells in vitro, which is seemingly regulated on an epigenetic level (Guan et al., 2011). Consistent with these findings, our studies indicated that CD44 regulates the survival of Th1 cells, but not Th2 cells, which could account for Th2 bias in Cd44−/− mice (Baaten et al., 2010).
At sites of inflammation, tissue damage causes the accumulation of LMW HA fragments and the release of multiple proinflammatory mediators from DCs and macrophages, including IL-12, TNF-α, and several chemokines (Konig et al., 1998). Whether T cells can be induced to produce cytokines in response to LMW HA binding in vivo is unknown, but in vitro induction of interferon (IFN)-γ has been observed (Blass et al., 2001). A recent study using the murine multiple sclerosis model experimental autoimmune encephalitis (EAE), demonstrated that Cd44−/− mice had greatly reduced severity of disease with fewer CD4+ T cells in CNS infiltrates, which produced lower levels of IFN-γ and IL-17 compared to cells from WT mice (Guan et al., 2011), suggesting a potential role for CD44 in either regulating the development of effector T cells or their production of cytokines. Another mechanism that could account for regulation of activated T cells by CD44 is through the cytokine, osteopontin (OPN), which is widely produced in sites of inflammation and binds to v7-containing isoforms of CD44. OPN upregulated T-bet in CD4+ T cells and enhanced Th1 cell differentiation in vitro (Shinohara et al., 2005). Furthermore, OPN has been previously shown to promote the survival of activated T cells in EAE in part by inhibiting the transcription factor Foxo3a and activating the transcription factor NF-κB (Hur et al., 2007), as well as modulating the levels of proapoptotic Bcl-2 family members. Whether this occurs in response to direct-binding of CD44 by OPN was not addressed, but both could occur via modulation of Akt signaling (Lin and Yang-Yen, 2001).
Although differences in the capacity of LMW versus HMW HA to elicit CD44-mediated signaling in T cells has not been carefully investigated, recent studies suggest that there are likely to be important distinctions. Interestingly, CD44–HA interactions have been demonstrated to control the activity of naturally occurring FoxP3+, CD25+ regulatory T cells (Tregs). HMW, but not LMW, HA can upregulate the transcription factor FoxP3 in Tregs cells via CD44 signaling (Figure 4). The development of FoxP3 expression was associated with the induction of regulatory function (Bollyky et al., 2007). Surprisingly, CD44 regulated FoxP3 expression in part by inducing IL-2 production, which was linked to a greater capacity for IL-10 production and the development of surface TGF-β1 expression (Bollyky et al., 2009). Consistent with these findings, Tregs from Cd44−/− mice exhibited impaired regulatory function. Expression levels of CD44 were upregulated on Tregs after activation in vitro, and a significant proportion bound exogenous HA, which was linked to a greater capacity to inhibit allogeneic and graft versus host responses in vivo (Firan et al., 2006). Together, these results reveal that T cells can engage HMW HA and suggest that CD44–HA binding could have an unexpected role in the contraction of immune responses, in the resolution of inflammation, and in maintaining tolerance to self-Ags. Although not yet studied, presumably effector T cells that become activated in the context of an immune response and inflammation will have the opportunity to bind LMW HA.
Potential Roles of CD44 in the Regulation of T Cell Memory
Signaling events in the primary response to infections can profoundly affect the generation and maintenance of T cell memory. Our recent study demonstrated a previously unexpected role for signaling via CD44 during a primary response to influenza virus infection in the survival and generation of memory in Th1 cells, but not Th2 or Th17 cells (Baaten et al., 2010). Thus, as with CD28 and OX40, signaling via CD44 could contribute to maintaining effector responses through engagement of survival mechanisms, particularly in Th1 cells, and thereby contribute to the development of memory. However, ligation of CD28 (Boise et al., 1995) or OX40 (Rogers et al., 2001) induced an increase in the anti-apoptotic Bcl-2 family member Bcl-xL that was not observed with CD44. Instead, we showed that CD44 can signal via the PI3K/Akt pathway selectively in Th1 cells (Baaten et al., 2010), a mechanism by which HA binding to CD44 promotes survival and growth of some tumor cells (Bourguignon et al., 2008; Figure 4). This pathway can also be engaged by CD28 in CD4+ T cells, and inhibit apoptosis by interfering with Fas-mediated cell death by preventing assembly of the death-inducing signaling complex (DISC) that is necessary to activate caspase 8 (Jones et al., 2002). Indeed, other studies in cancer cells showed that CD44 may directly interfere with Fas to prevent DISC assembly and thereby control resistance versus susceptibility to Fas-mediated cell death (Hauptschein et al., 2005) via interaction with the ECM (Lee et al., 2008). Although studies of Jurkat T cells, which lack CD44, showed that transfection with CD44 variant isoforms, rather than the standard form, could inhibit Fas-mediated apoptosis (Mielgo et al., 2006), we could not discern any differences in CD44 variant usage between primary Th1 and Th2 cells (Baaten et al., 2010). Nevertheless, Th1 cells have been previously demonstrated to be more susceptible to apoptosis than Th2 cells (Varadhachary et al., 1997; Zhang et al., 1997) and we showed that Th1 cells express higher levels of Fas than do Th2 or Th17 cells, irrespective of the presence of CD44 (Baaten et al., 2010). Thus, control of survival in Th1 cells may be more dependent on regulation by CD44 because of higher expression of Fas, and lower inherent expression of Akt/PI3K (Varadhachary et al., 1999). Alternatively, CD44 signaling might have an indirect effect on T cell survival. Given its role in trafficking, CD44 may affect T cell localization to a niche where memory T cells receive survival signals via other mechanisms. However, in our hands, T cell localization was not dramatically affected in the absence of CD44 (Baaten et al., 2010). Nevertheless, the deleterious effects of Cd44 deficiency on memory CD4 T cell survival highlight the vital role CD44 plays in memory T cell development.
Although the molecular basis of the regulated changes in central trafficking molecules during effector T cell development is beginning to be understood, much less is known regarding how changes in adhesion receptor expression impact memory T cells. There has been considerable effort devoted to investigating the functional regulation of memory T cells by compartmentalizing them on the basis of their trafficking molecules (Sallusto et al., 2004). Central memory cells (TCM) remain in the lymphoid compartment and express the lymph node homing receptor, CD62L, and/or the chemokine receptor, CCR7. More differentiated effector memory cells (TEM) lack these molecules and more readily localize in non-lymphoid tissues in addition to being found in the circulation. The sustained high level of CD44 expression on all Ag-experienced T cells suggests that this molecule has the potential to regulate memory T cells during migration, adhesion, and motility as detailed above. Whereas both CD4+ and CD8+ T cells can undergo initial activation and expansion in the absence of CD44 (Mrass et al., 2008; Baaten et al., 2010), it is possible that CD44 is not generally needed for T cell priming when Ag levels are high and TCR signaling is strong during the initial phases of the immune response. Because CD44 signals can enhance T cell responses when TCR signals are low (Foger et al., 2000), they may contribute to the formation of memory as Ag levels diminish during the course of an immune response. The paucity of Ag also favors the generation of CD62L+ TCM cells that are less differentiated than TEM cells, which retain the capacity for immediate effector function (Catron et al., 2006). We have demonstrated that Cd44-deficient Th1 cells that are withdrawn from overt TCR stimulation fail to survive as memory cells over time when transferred into naïve recipients (Baaten et al., 2010). We did not find differences in CD44 expression on CD4+ effector T cells that were sorted on the basis of CD62L expression to distinguish precursors of TCM and TEM memory cells. Moreover, in the absence of CD44 expression, both subsets were equally susceptible to decay in vivo in the absence of overt stimulation by Ag. These results imply that, at least for CD4+ T cells, CD44 could play a role in the survival of those effectors cells that persist through the contraction phase to become memory cells.
In addition to the roles in effector and memory generation, CD44 may also contribute to memory T cell homeostasis. A recent study shows that a subset of memory CD8+ T cells expresses CD44-mediated HA binding (Maeshima et al., 2011). These T cells are enriched in TCM phenotype memory cells, and exhibit an enhanced capacity to proliferate in response to the common γ-chain cytokines IL-7 and IL-15, which are required for the maintenance of memory T cells. In some models of infection, memory CD8+ T cell precursors have been subdivided based on the expression of two surface markers, IL-7 receptor α (IL-7Rα) and killer cell lectin-like receptor subfamily G, member 1 (KLRG1; Kaech et al., 2003; Joshi et al., 2007). Whereas short-lived effector cells are characterized by low IL-7Rα and high KLRG1 expression, memory precursor effector cells (MPECs) are characterized by high IL-7Rα and low KLRG1 expression. It is not known whether levels of cell-surface CD44 differ among these T cell subsets or whether they might be differentially regulated by CD44-mediated signaling. Previous studies indicate that Lck associates with the IL-7Rα and is activated in response to IL-7 engagement, which itself is dependent on the activation status of the T cell (Page et al., 1995, 1997). We hypothesize that HA binding by CD44 elicits a greater homeostatic response to IL-7 by increasing the availability of Lck (Figure 4). Thus, increased responsiveness of IL-7Rα to IL-7 in response to HA binding could promote enhanced survival in response to IL-7-mediated upregulation of Bcl-2, a key target of IL-7R signaling. Since HA binding by CD44 is upregulated in response to TCR signaling, it is possible that memory cells expressing this activity represent the population undergoing homeostatic turnover driven by peptide-MHC recognition on DCs that express surface HA (Boyman et al., 2009), which contributes to the maintenance of memory T cell function (Kassiotis et al., 2002). In this scenario, we envision that HA binding is acquired transiently with periodic cycles of TCR engagement. However, it is possible that the frequency of HA-binding T cells has been underestimated in the contexts in which it has been studied, particularly since it is often detected only by the ability to take up exogenous HA, which does not take into account that HA is potentially already bound and can be detected on the surface with HA binding protein (Cichy et al., 2002). Furthermore, the cycling between HA binding and non-binding states of CD44 may permit engagement and disengagement of memory cells from HA during migration and interaction with DCs. Thus, consistent with the sustained expression of CD44 on memory T cells, emerging evidence suggests that, beyond roles in migration and motility, CD44 could have a global function in possibly regulating the transition of effector cells to memory cells, as well as in maintaining memory T cells. Therefore, on T cells, engagement of CD44 could impact the outcome of memory responses at multiple points after an initial Ag encounter.
CD44 and its Importance for Optimal T Cell Immunity during Infection
CD44 has been shown to be important in mediating innate immune cell function (Matsumoto et al., 1998; Larkin et al., 2006; Shirali and Goldstein, 2008) and innate immunity following bacterial infections. For example, lung infection with Mycobacterium tuberculosis in Cd44-deficient mice led to increased neutrophil infiltration with no differences in leukocyte entry or bacterial clearance (Kipnis et al., 2003). In addition, pulmonary infection with Escherichia coli promoted increased neutrophil infiltration and inflammation in the lung, which was not observed when mice were infected with Streptococcus (Wang et al., 2002). However, little progress has been made in examining the function of CD44 on T cells in response to infection with microbial pathogens. We recently showed a critical function for CD44 in optimal Th1 cell memory development against influenza virus infection (Baaten et al., 2010). It is possible that inappropriate CD4+ T cell help during the immune response against pathogens could impact optimal development of effector and/or memory CD8+ T cells after influenza virus infection, as has been shown in other acute (Bourgeois et al., 2002; Janssen et al., 2003; Shedlock and Shen, 2003; Sun and Bevan, 2003) and chronic infections (Matloubian et al., 1994). Indeed, in the absence of CD4+ T cell help, maintenance and recall of CD8+ memory to influenza virus challenge is compromised and the clearance of residual virus delayed (Belz et al., 2002; Johansen et al., 2004). Additionally, in the absence of Th1 cells, B cell responses and the generation of long-lived plasma cells may be compromised. Profound immune defects from the absence of Th1 cells can result in enhanced pathogen spread or delayed pathogen clearance following infection.
In another study, the intrinsic effect of CD44 on T cell expansion following acute lymphocytic choriomeningitis virus infection was examined, and despite finding similar numbers of IFN-γ-producing CD4+ T cells, virus-specific CD8+ T cell expansion was decreased in the Cd44-deficient compartment (Graham et al., 2007). Antibody blockade of CD44 in mice infected with Toxoplasma gondii resulted in a lack of IFN-γ production by CD4+ T cells, which protected treated mice from exuberant inflammation (Blass et al., 2001). Interestingly, IFN-γ production in CD4+ T cells was restored upon addition of LMW HA to T cell cultures. Together, these findings indicate that CD44-mediated signals are important in T cell function and survival and are necessary for both CD4+ and CD8+ T cell responses after infection. However, more studies are necessary to investigate the role of CD44 during infections, as the few that have been done highlight the complexity of CD44 function. It may play specific contrasting roles on different cell types and the outcome for the host may depend on the pathogen encountered. It will be important to determine whether there are extrinsic roles for CD44, e.g. on APCs or Tregs, which can impact and/or influence an ongoing T cell response during infection. Along with TCR–MHC and co-stimulatory signals, the contextual signals through CD44 and its ligand may also impact effector and/or memory T cell fates after microbial infections.
Targeting CD44 on T Cells to Ameliorate Disease: The Example of Type 1 Diabetes
CD44 has long been considered to have important potential as a therapeutic target to control inflammation. Early studies on the effects of blocking CD44 showed that skin contact sensitivity responses were diminished because of reduced lymphocyte and leukocyte infiltration (Camp et al., 1993), a result also seen in atopic dermatitis in Cd44-deficient mice (Gonda et al., 2005). In addition, pulmonary injury and allergic responses were reduced by anti-CD44 antibody treatment (Katoh et al., 2003; Li et al., 2011). In contrast, Cd44-deficient animals cannot control acute induced pulmonary inflammation due to the inability to clear soluble HA (Teder et al., 2002). Likewise, in different models of hepatic inflammation, targeting CD44 can have protective (McDonald et al., 2008) or deleterious effects (Chen et al., 2001). Despite the potential for such diverse outcomes, consistent therapeutic effects are observed after interfering with CD44 function in models of chronic inflammation, particularly autoimmune diseases. These include induced models of arthritis, multiple sclerosis, colitis, and type 1 diabetes (T1D; reviewed in Johnson and Ruffell, 2009). Overall, these studies reveal that amelioration of disease is due to reduced inflammatory infiltrates in the target tissues. However, studies of T1D suggested that control of inflammation can be achieved by altering the migration and survival of effector/memory T cells and Tregs in addition to migration as summarized below.
Type 1 diabetes represents an important example of a chronic disease that can be prevented by treatment with anti-CD44 during the prediabetic phase in the NOD mouse model (Weiss et al., 2000). Although the development of this disease is multifactorial, T cells orchestrate the inflammation that leads to destruction of insulin-producing islet β-cells. Blocking CD44 activation by enzymatic cleavage abolished the migration of islet-Ag-specific CD8+ T cells into the pancreas and prevented development of diabetes (Savinov and Strongin, 2007). We observe that anti-CD44 treatment prevents diabetogenic CD4+ and CD8+ T cells from accumulating in the pancreas (unpublished observations). Many additional studies as well as our own have provided evidence that blocking HA–CD44 binding can promote the death of activated T cells (Ruffell and Johnson, 2008; Baaten et al., 2010), which is associated with the amelioration of inflammation in a colitis model (Wittig et al., 1998). Thus, the development and survival of memory in Th1 CD4+ T cells may also be controlled by targeting CD44 in T1D. Importantly, the activity of regulatory T cells can be augmented after CD44 ligation due to a direct effect on their inhibitory function (Bollyky et al., 2009), revealing an unexpected role of CD44 in the maintenance of tolerance. Together, these findings suggest that the combined effect of modulating CD44-signaling on T cells could control the inflammatory response in T1D through multiple mechanisms.
Another area of investigation where anti-CD44 treatment may provide a benefit for T1D is in islet-transplantation to restore insulin production after diabetes onset. A recent study in a non-autoimmune model demonstrated that administration of anti-CD44 prolonged survival of allogeneic islets after transplantation (Peng et al., 2011). This improvement was associated with greatly reduced accumulation of CD4+ T cells within the grafts and diminished levels of IFN-γ in the sera of treated mice. These results support the concept that CD44 on T cells represents an important target for the treatment of T1D in particular, but possibly other T cell-dependent autoimmune diseases as well. However, before the clinical translation of biologics or small molecules that target CD44 can be considered, a better understanding of the consequences are needed in light of CD44’s multiple distinct roles in several cell types.
Conclusion
Modulation of signaling via adhesion receptors can contribute to multiple, often overlooked aspects of T cell regulation. While increased expression of several adhesion receptors on activated and memory T cells signifies greater migratory capacity, the example of CD44 demonstrates the underappreciated roles that a single constitutively expressed adhesion receptor can play in integrating signals within the microenvironment, which can impact T cell activation, proliferation, differentiation, survival, locomotion, and function. By these processes, CD44 can influence not only the fates of responding T cells, but also the generation and maintenance of memory and control of autoimmunity.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank M. Douglass for review of the manuscript. This work was supported by NIH grants AI079584 to Bas J. G. Baaten and AI061615, AI046530, and AI081238 to Linda M. Bradley. The authors have no conflicting financial interests.
References
Alon, R., Kassner, P. D., Carr, M. W., Finger, E. B., Hemler, M. E., and Springer, T. A. (1995). The integrin VLA-4 supports tethering and rolling in flow on VCAM-1. J. Cell Biol. 128, 1243–1253.
Autero, M., Heiska, L., Ronnstrand, L., Vaheri, A., Gahmberg, C. G., and Carpen, O. (2003). Ezrin is a substrate for Lck in T cells. FEBS Lett. 535, 82–86.
Baaten, B. J., Li, C. R., Deiro, M. F., Lin, M. M., Linton, P. J., and Bradley, L. M. (2010). CD44 regulates survival and memory development in Th1 cells. Immunity 32, 104–115.
Banerji, S., Wright, A. J., Noble, M., Mahoney, D. J., Campbell, I. D., Day, A. J., and Jackson, D. G. (2007). Structures of the Cd44-hyaluronan complex provide insight into a fundamental carbohydrate-protein interaction. Nat. Struct. Mol. Biol. 14, 234–239.
Belz, G. T., Wodarz, D., Diaz, G., Nowak, M. A., and Doherty, P. C. (2002). Compromised influenza virus-specific CD8(+)-T-cell memory in CD4(+)-T-cell-deficient mice. J. Virol. 76, 12388–12393.
Blass, S. L., Pure, E., and Hunter, C. A. (2001). A role for CD44 in the production of IFN-gamma and immunopathology during infection with Toxoplasma gondii. J. Immunol. 166, 5726–5732.
Boise, L. H., Minn, A. J., Noel, P. J., June, C. H., Accavitti, M. A., Lindsten, T., and Thompson, C. B. (1995). CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-XL. Immunity 3, 87–98.
Bollyky, P. L., Falk, B. A., Long, S. A., Preisinger, A., Braun, K. R., Wu, R. P., Evanko, S. P., Buckner, J. H., Wight, T. N., and Nepom, G. T. (2009). CD44 costimulation promotes FoxP3+ regulatory T cell persistence and function via production of IL-2, IL-10, and TGF-beta. J. Immunol. 183, 2232–2241.
Bollyky, P. L., Lord, J. D., Masewicz, S. A., Evanko, S. P., Buckner, J. H., Wight, T. N., and Nepom, G. T. (2007). Cutting edge: high molecular weight hyaluronan promotes the suppressive effects of CD4+CD25+ regulatory T cells. J. Immunol. 179, 744–747.
Bonder, C. S., Clark, S. R., Norman, M. U., Johnson, P., and Kubes, P. (2006). Use of CD44 by CD4+ Th1 and Th2 lymphocytes to roll and adhere. Blood 107, 4798–4806.
Bourgeois, C., Rocha, B., and Tanchot, C. (2002). A role for CD40 expression on CD8+ T cells in the generation of CD8+ T cell memory. Science 297, 2060–2063.
Bourguignon, L. Y., Peyrollier, K., Xia, W., and Gilad, E. (2008). Hyaluronan-CD44 interaction activates stem cell marker Nanog, Stat-3-mediated MDR1 gene expression, and ankyrin-regulated multidrug efflux in breast and ovarian tumor cells. J. Biol. Chem. 283, 17635–17651.
Boyman, O., Letourneau, S., Krieg, C., and Sprent, J. (2009). Homeostatic proliferation and survival of naive and memory T cells. Eur. J. Immunol. 39, 2088–2094.
Brown, K. L., Birkenhead, D., Lai, J. C., Li, L., Li, R., and Johnson, P. (2005). Regulation of hyaluronan binding by F-actin and colocalization of CD44 and phosphorylated ezrin/radixin/moesin (ERM) proteins in myeloid cells. Exp. Cell Res. 303, 400–414.
Camp, R. L., Scheynius, A., Johansson, C., and Pure, E. (1993). CD44 is necessary for optimal contact allergic responses but is not required for normal leukocyte extravasation. J. Exp. Med. 178, 497–507.
Catron, D. M., Rusch, L. K., Hataye, J., Itano, A. A., and Jenkins, M. K. (2006). CD4+ T cells that enter the draining lymph nodes after antigen injection participate in the primary response and become central-memory cells. J. Exp. Med. 203, 1045–1054.
Chen, D., Mckallip, R. J., Zeytun, A., Do, Y., Lombard, C., Robertson, J. L., Mak, T. W., Nagarkatti, P. S., and Nagarkatti, M. (2001). CD44-deficient mice exhibit enhanced hepatitis after concanavalin A injection: evidence for involvement of CD44 in activation-induced cell death. J. Immunol. 166, 5889–5897.
Cichy, J., Bals, R., Potempa, J., Mani, A., and Pure, E. (2002). Proteinase-mediated release of epithelial cell-associated CD44. Extracellular CD44 complexes with components of cellular matrices. J. Biol. Chem. 277, 44440–44447.
DeGrendele, H. C., Kosfiszer, M., Estess, P., and Siegelman, M. H. (1997). CD44 activation and associated primary adhesion is inducible via T cell receptor stimulation. J. Immunol. 159, 2549–2553.
del Pozo, M. A., Sanchez-Mateos, P., Nieto, M., and Sanchez-Madrid, F. (1995). Chemokines regulate cellular polarization and adhesion receptor redistribution during lymphocyte interaction with endothelium and extracellular matrix. Involvement of cAMP signaling pathway. J. Cell Biol. 131, 495–508.
Elkington, P. T., O’Kane, C. M., and Friedland, J. S. (2005). The paradox of matrix metalloproteinases in infectious disease. Clin. Exp. Immunol. 142, 12–20.
English, N. M., Lesley, J. F., and Hyman, R. (1998). Site-specific de-N-glycosylation of CD44 can activate hyaluronan binding, and CD44 activation states show distinct threshold densities for hyaluronan binding. Cancer Res. 58, 3736–3742.
Fanning, A., Volkov, Y., Freeley, M., Kelleher, D., and Long, A. (2005). CD44 cross-linking induces protein kinase C-regulated migration of human T lymphocytes. Int. Immunol. 17, 449–458.
Firan, M., Dhillon, S., Estess, P., and Siegelman, M. H. (2006). Suppressor activity and potency among regulatory T cells is discriminated by functionally active CD44. Blood 107, 619–627.
Foger, N., Marhaba, R., and Zoller, M. (2000). CD44 supports T cell proliferation and apoptosis by apposition of protein kinases. Eur. J. Immunol. 30, 2888–2899.
Fooksman, D. R., Vardhana, S., Vasiliver-Shamis, G., Liese, J., Blair, D. A., Waite, J., Sacristan, C., Victora, G. D., Zanin-Zhorov, A., and Dustin, M. L. (2010). Functional anatomy of T cell activation and synapse formation. Annu. Rev. Immunol. 28, 79–105.
Forster-Horvath, C., Bocsi, J., Raso, E., Orban, T. I., Olah, E., Timar, J., and Ladanyi, A. (2001). Constitutive intracellular expression and activation-induced cell surface up-regulation of CD44v3 in human T lymphocytes. Eur. J. Immunol. 31, 600–608.
Gee, K., Kozlowski, M., and Kumar, A. (2003). Tumor necrosis factor-alpha induces functionally active hyaluronan-adhesive CD44 by activating sialidase through p38 mitogen-activated protein kinase in lipopolysaccharide-stimulated human monocytic cells. J. Biol. Chem. 278, 37275–37287.
Girish, K. S., and Kemparaju, K. (2007). The magic glue hyaluronan and its eraser hyaluronidase: a biological overview. Life Sci. 80, 1921–1943.
Gonda, A., Gal, I., Szanto, S., Sarraj, B., Glant, T. T., Hunyadi, J., and Mikecz, K. (2005). CD44, but not L-selectin, is critically involved in leucocyte migration into the skin in a murine model of allergic dermatitis. Exp. Dermatol. 14, 700–708.
Graham, V. A., Marzo, A. L., and Tough, D. F. (2007). A role for CD44 in T cell development and function during direct competition between CD44+ and CD44- cells. Eur. J. Immunol. 37, 925–934.
Guan, H., Nagarkatti, P. S., and Nagarkatti, M. (2009). Role of CD44 in the differentiation of Th1 and Th2 cells: CD44-deficiency enhances the development of Th2 effectors in response to sheep RBC and chicken ovalbumin. J. Immunol. 183, 172–180.
Guan, H., Nagarkatti, P. S., and Nagarkatti, M. (2011). CD44 reciprocally regulates the differentiation of encephalitogenic Th1/Th17 and Th2/regulatory T cells through epigenetic modulation involving DNA methylation of cytokine gene promoters, thereby controlling the development of experimental autoimmune encephalomyelitis. J. Immunol. 186, 6955–6964.
Hauptschein, R. S., Sloan, K. E., Torella, C., Moezzifard, R., Giel-Moloney, M., Zehetmeier, C., Unger, C., Ilag, L. L., and Jay, D. G. (2005). Functional proteomic screen identifies a modulating role for CD44 in death receptor-mediated apoptosis. Cancer Res. 65, 1887–1896.
He, Q., Lesley, J., Hyman, R., Ishihara, K., and Kincade, P. W. (1992). Molecular isoforms of murine CD44 and evidence that the membrane proximal domain is not critical for hyaluronate recognition. J. Cell Biol. 119, 1711–1719.
Hegde, V. L., Singh, N. P., Nagarkatti, P. S., and Nagarkatti, M. (2008). CD44 mobilization in allogeneic dendritic cell-T cell immunological synapse plays a key role in T cell activation. J. Leukoc. Biol. 84, 134–142.
Huppa, J. B., and Davis, M. M. (2003). T-cell-antigen recognition and the immunological synapse. Nat. Rev. Immunol. 3, 973–983.
Hur, E. M., Youssef, S., Haws, M. E., Zhang, S. Y., Sobel, R. A., and Steinman, L. (2007). Osteopontin-induced relapse and progression of autoimmune brain disease through enhanced survival of activated T cells. Nat. Immunol. 8, 74–83.
Janssen, E. M., Lemmens, E. E., Wolfe, T., Christen, U., Von Herrath, M. G., and Schoenberger, S. P. (2003). CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature 421, 852–856.
Jiang, D., Liang, J., and Noble, P. W. (2011). Hyaluronan as an immune regulator in human diseases. Physiol. Rev. 91, 221–264.
Johansen, P., Stamou, P., Tascon, R. E., Lowrie, D. B., and Stockinger, B. (2004). CD4 T cells guarantee optimal competitive fitness of CD8 memory T cells. Eur. J. Immunol. 34, 91–97.
Johnson, P., and Ruffell, B. (2009). CD44 and its role in inflammation and inflammatory diseases. Inflamm. Allergy Drug Targets 8, 208–220.
Jones, R. G., Elford, A. R., Parsons, M. J., Wu, L., Krawczyk, C. M., Yeh, W. C., Hakem, R., Rottapel, R., Woodgett, J. R., and Ohashi, P. S. (2002). CD28-dependent activation of protein kinase B/Akt blocks Fas-mediated apoptosis by preventing death-inducing signaling complex assembly. J. Exp. Med. 196, 335–348.
Joshi, N. S., Cui, W., Chandele, A., Lee, H. K., Urso, D. R., Hagman, J., Gapin, L., and Kaech, S. M. (2007). Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity 27, 281–295.
Kaech, S. M., Tan, J. T., Wherry, E. J., Konieczny, B. T., Surh, C. D., and Ahmed, R. (2003). Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat. Immunol. 4, 1191–1198.
Kassiotis, G., Garcia, S., Simpson, E., and Stockinger, B. (2002). Impairment of immunological memory in the absence of MHC despite survival of memory cells. Nat. Immunol. 3, 244–250.
Katoh, S., Matsumoto, N., Kawakita, K., Tominaga, A., Kincade, P. W., and Matsukura, S. (2003). A role for CD44 in an antigen-induced murine model of pulmonary eosinophilia. J. Clin. Invest. 111, 1563–1570.
Katoh, S., Miyagi, T., Taniguchi, H., Matsubara, Y., Kadota, J., Tominaga, A., Kincade, P. W., Matsukura, S., and Kohno, S. (1999). Cutting edge: an inducible sialidase regulates the hyaluronic acid binding ability of CD44-bearing human monocytes. J. Immunol. 162, 5058–5061.
Kipnis, A., Basaraba, R. J., Turner, J., and Orme, I. M. (2003). Increased neutrophil influx but no impairment of protective immunity to tuberculosis in mice lacking the CD44 molecule. J. Leukoc. Biol. 74, 992–997.
Konig, H., Ponta, H., and Herrlich, P. (1998). Coupling of signal transduction to alternative pre-mRNA splicing by a composite splice regulator. EMBO J. 17, 2904–2913.
Larkin, J., Renukaradhya, G. J., Sriram, V., Du, W., Gervay-Hague, J., and Brutkiewicz, R. R. (2006). CD44 differentially activates mouse NK T cells and conventional T cells. J. Immunol. 177, 268–279.
Lee, J. L., Wang, M. J., Sudhir, P. R., and Chen, J. Y. (2008). CD44 engagement promotes matrix-derived survival through the CD44-SRC-integrin axis in lipid rafts. Mol. Cell. Biol. 28, 5710–5723.
Lefebvre, D. C., Lai, J. C., Maeshima, N., Ford, J. L., Wong, A. S., Cross, J. L., and Johnson, P. (2010). CD44 interacts directly with Lck in a zinc-dependent manner. Mol. Immunol. 47, 1882–1889.
Lesley, J., Howes, N., Perschl, A., and Hyman, R. (1994). Hyaluronan binding function of CD44 is transiently activated on T cells during an in vivo immune response. J. Exp. Med. 180, 383–387.
Ley, K., and Kansas, G. S. (2004). Selectins in T-cell recruitment to non-lymphoid tissues and sites of inflammation. Nat. Rev. Immunol. 4, 325–335.
Ley, K., Laudanna, C., Cybulsky, M. I., and Nourshargh, S. (2007). Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat. Rev. Immunol. 7, 678–689.
Li, Y., Jiang, D., Liang, J., Meltzer, E. B., Gray, A., Miura, R., Wogensen, L., Yamaguchi, Y., and Noble, P. W. (2011). Severe lung fibrosis requires an invasive fibroblast phenotype regulated by hyaluronan and CD44. J. Exp. Med. 208, 1459–1471.
Liao, H. X., Levesque, M. C., Patton, K., Bergamo, B., Jones, D., Moody, M. A., Telen, M. J., and Haynes, B. F. (1993). Regulation of human CD44H and CD44E isoform binding to hyaluronan by phorbol myristate acetate and anti-CD44 monoclonal and polyclonal antibodies. J. Immunol. 151, 6490–6499.
Lin, Y. H., and Yang-Yen, H. F. (2001). The osteopontin-CD44 survival signal involves activation of the phosphatidylinositol 3-kinase/Akt signaling pathway. J. Biol. Chem. 276, 46024–46030.
Maeshima, N., Poon, G. F., Dosanjh, M., Felberg, J., Lee, S. S., Cross, J. L., Birkenhead, D., and Johnson, P. (2011). Hyaluronan binding identifies the most proliferative activated and memory T cells. Eur. J. Immunol. 41, 1108–1119.
Marhaba, R., Freyschmidt-Paul, P., and Zoller, M. (2006). In vivo CD44-CD49d complex formation in autoimmune disease has consequences on T cell activation and apoptosis resistance. Eur. J. Immunol. 36, 3017–3032.
Matloubian, M., Concepcion, R. J., and Ahmed, R. (1994). CD4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection. J. Virol. 68, 8056–8063.
Matsumoto, M., Nghiem, M. P., Nozaki, N., Schmits, R., and Penninger, J. M. (1998). Cooperation between CD44 and LFA-1/CD11a adhesion receptors in lymphokine-activated killer cell cytotoxicity. J. Immunol. 160, 5781–5789.
McDonald, B., Mcavoy, E. F., Lam, F., Gill, V., De La Motte, C., Savani, R. C., and Kubes, P. (2008). Interaction of CD44 and hyaluronan is the dominant mechanism for neutrophil sequestration in inflamed liver sinusoids. J. Exp. Med. 205, 915–927.
Mielgo, A., Van Driel, M., Bloem, A., Landmann, L., and Gunthert, U. (2006). A novel antiapoptotic mechanism based on interference of Fas signaling by CD44 variant isoforms. Cell Death Differ. 13, 465–477.
Mori, T., Kitano, K., Terawaki, S., Maesaki, R., Fukami, Y., and Hakoshima, T. (2008). Structural basis for CD44 recognition by ERM proteins. J. Biol. Chem. 283, 29602–29612.
Mrass, P., Kinjyo, I., Ng, L. G., Reiner, S. L., Pure, E., and Weninger, W. (2008). CD44 mediates successful interstitial navigation by killer T cells and enables efficient antitumor immunity. Immunity 29, 971–985.
Mummert, M. E., Mummert, D., Edelbaum, D., Hui, F., Matsue, H., and Takashima, A. (2002). Synthesis and surface expression of hyaluronan by dendritic cells and its potential role in antigen presentation. J. Immunol. 169, 4322–4331.
Nagano, O., and Saya, H. (2004). Mechanism and biological significance of CD44 cleavage. Cancer Sci. 95, 930–935.
Nandi, A., Estess, P., and Siegelman, M. (2004). Bimolecular complex between rolling and firm adhesion receptors required for cell arrest; CD44 association with VLA-4 in T cell extravasation. Immunity 20, 455–465.
Ogino, S., Nishida, N., Umemoto, R., Suzuki, M., Takeda, M., Terasawa, H., Kitayama, J., Matsumoto, M., Hayasaka, H., Miyasaka, M., and Shimada, I. (2010). Two-state conformations in the hyaluronan-binding domain regulate CD44 adhesiveness under flow condition. Structure 18, 649–656.
Page, T. H., Lali, F. V., and Foxwell, B. M. (1995). Interleukin-7 activates p56lck and p59fyn, two tyrosine kinases associated with the p90 interleukin-7 receptor in primary human T cells. Eur. J. Immunol. 25, 2956–2960.
Page, T. H., Lali, F. V., Groome, N., and Foxwell, B. M. (1997). Association of the common gamma-chain with the human IL-7 receptor is modulated by T cell activation. J. Immunol. 158, 5727–5735.
Peng, Y., Chen, J., Shao, W., Wang, F., Dai, H., Cheng, P., Xia, J., Huang, R., Zhu, Q., and Qi, Z. (2011). Xenoreactive CD4(+) memory T cells resist inhibition by anti-CD44 mAb and reject islet grafts via a Th2-dependent pathway. Xenotransplantation 18, 252–261.
Ponta, H., Sherman, L., and Herrlich, P. (2003). CD44: from adhesion molecules to signaling regulators. Nat. Rev. Mol. Cell Biol. 4, 33–45.
Rogers, P. R., Song, J., Gramaglia, I., Killeen, N., and Croft, M. (2001). OX40 promotes Bcl-xL and Bcl-2 expression and is essential for long-term survival of CD4 T cells. Immunity 15, 445–455.
Rosenman, S. J., Ganji, A. A., Tedder, T. F., and Gallatin, W. M. (1993). Syn-capping of human T lymphocyte adhesion/activation molecules and their redistribution during interaction with endothelial cells. J. Leukoc. Biol. 53, 1–10.
Roumier, A., Olivo-Marin, J. C., Arpin, M., Michel, F., Martin, M., Mangeat, P., Acuto, O., Dautry-Varsat, A., and Alcover, A. (2001). The membrane-microfilament linker ezrin is involved in the formation of the immunological synapse and in T cell activation. Immunity 15, 715–728.
Rozsnyay, Z. (1999). Signaling complex formation of CD44 with src-related kinases. Immunol. Lett. 68, 101–108.
Ruffell, B., and Johnson, P. (2008). Hyaluronan induces cell death in activated T cells through CD44. J. Immunol. 181, 7044–7054.
Sallusto, F., Geginat, J., and Lanzavecchia, A. (2004). Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu. Rev. Immunol. 22, 745–763.
Sallusto, F., and Lanzavecchia, A. (2009). Heterogeneity of CD4+ memory T cells: functional modules for tailored immunity. Eur. J. Immunol. 39, 2076–2082.
Savinov, A. Y., Rozanov, D. V., Golubkov, V. S., Wong, F. S., and Strongin, A. Y. (2005). Inhibition of membrane type-1 matrix metalloproteinase by cancer drugs interferes with the homing of diabetogenic T cells into the pancreas. J. Biol. Chem. 280, 27755–27758.
Savinov, A. Y., and Strongin, A. Y. (2007). Defining the roles of T cell membrane proteinase and CD44 in type 1 diabetes. IUBMB Life 59, 6–13.
Shedlock, D. J., and Shen, H. (2003). Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science 300, 337–339.
Shinohara, M. L., Jansson, M., Hwang, E. S., Werneck, M. B., Glimcher, L. H., and Cantor, H. (2005). T-bet-dependent expression of osteopontin contributes to T cell polarization. Proc. Natl. Acad. Sci. U.S.A. 102, 17101–17106.
Shirali, A. C., and Goldstein, D. R. (2008). Activation of the innate immune system by the endogenous ligand hyaluronan. Curr. Opin. Organ Transplant 13, 20–25.
Siegelman, M. H., Stanescu, D., and Estess, P. (2000). The CD44-initiated pathway of T-cell extravasation uses VLA-4 but not LFA-1 for firm adhesion. J. Clin. Invest. 105, 683–691.
Sorokin, L. (2010). The impact of the extracellular matrix on inflammation. Nat. Rev. Immunol. 10, 712–723.
Sun, J. C., and Bevan, M. J. (2003). Defective CD8 T cell memory following acute infection without CD4 T cell help. Science 300, 339–342.
Taher, T. E., Smit, L., Griffioen, A. W., Schilder-Tol, E. J., Borst, J., and Pals, S. T. (1996). Signaling through CD44 is mediated by tyrosine kinases. Association with p56lck in T lymphocytes. J. Biol. Chem. 271, 2863–2867.
Takeda, M., Ogino, S., Umemoto, R., Sakakura, M., Kajiwara, M., Sugahara, K. N., Hayasaka, H., Miyasaka, M., Terasawa, H., and Shimada, I. (2006). Ligand-induced structural changes of the CD44 hyaluronan-binding domain revealed by NMR. J. Biol. Chem. 281, 40089–40095.
Teder, P., Vandivier, R. W., Jiang, D., Liang, J., Cohn, L., Pure, E., Henson, P. M., and Noble, P. W. (2002). Resolution of lung inflammation by CD44. Science 296, 155–158.
Thankamony, S. P., and Sackstein, R. (2011). Enforced hematopoietic cell E- and L-selectin ligand (HCELL) expression primes transendothelial migration of human mesenchymal stem cells. Proc. Natl. Acad. Sci. U.S.A. 108, 2258–2263.
Tybulewicz, V. L., and Henderson, R. B. (2009). Rho family GTPases and their regulators in lymphocytes. Nat. Rev. Immunol. 9, 630–644.
Varadhachary, A. S., Perdow, S. N., Hu, C., Ramanarayanan, M., and Salgame, P. (1997). Differential ability of T cell subsets to undergo activation-induced cell death. Proc. Natl. Acad. Sci. U.S.A. 94, 5778–5783.
Varadhachary, A. S., Peter, M. E., Perdow, S. N., Krammer, P. H., and Salgame, P. (1999). Selective up-regulation of phosphatidylinositol 3’-kinase activity in Th2 cells inhibits caspase-8 cleavage at the death-inducing complex: a mechanism for Th2 resistance from Fas-mediated apoptosis. J. Immunol. 163, 4772–4779.
Wang, Q., Teder, P., Jeudd, N. P., Noble, P. W., and Doerschuk, C. M. (2002). CD44 deficiency leads to enhanced neutrophil migration and lung injury in Escherichia coli pneumonia in mice. Am. J. Pathol. 161, 2219–2228.
Weiss, L., Slavin, S., Reich, S., Cohen, P., Shuster, S., Stern, R., Kaganovsky, E., Okon, E., Rubinstein, A. M., and Naor, D. (2000). Induction of resistance to diabetes in non-obese diabetic mice by targeting CD44 with a specific monoclonal antibody. Proc. Natl. Acad. Sci. U.S.A. 97, 285–290.
Wittig, B., Schwarzler, C., Fohr, N., Gunthert, U., and Zoller, M. (1998). Curative treatment of an experimentally induced colitis by a CD44 variant V7-specific antibody. J. Immunol. 161, 1069–1073.
Wittig, B. M., Johansson, B., Zoller, M., Schwarzler, C., and Gunthert, U. (2000). Abrogation of experimental colitis correlates with increased apoptosis in mice deficient for CD44 variant exon 7 (CD44v7). J. Exp. Med. 191, 2053–2064.
Wolny, P. M., Banerji, S., Gounou, C., Brisson, A. R., Day, A. J., Jackson, D. G., and Richter, R. P. (2010). Analysis of CD44-hyaluronan interactions in an artificial membrane system: insights into the distinct binding properties of high and low molecular weight hyaluronan. J. Biol. Chem. 285, 30170–30180.
Wong, N. K., Lai, J. C., Maeshima, N., and Johnson, P. (2011). CD44-mediated elongated T cell spreading requires Pyk2 activation by Src family kinases, extracellular calcium, phospholipase C and phosphatidylinositol-3 kinase. Cell. Signal. 23, 812–819.
Yago, T., Shao, B., Miner, J. J., Yao, L., Klopocki, A. G., Maeda, K., Coggeshall, K. M., and Mcever, R. P. (2010). E-selectin engages PSGL-1 and CD44 through a common signaling pathway to induce integrin alphaLbeta2-mediated slow leukocyte rolling. Blood 116, 485–494.
Yu, Q., and Stamenkovic, I. (1999). Localization of matrix metalloproteinase 9 to the cell surface provides a mechanism for CD44-mediated tumor invasion. Genes Dev. 13, 35–48.
Zhang, X., Brunner, T., Carter, L., Dutton, R. W., Rogers, P., Bradley, L., Sato, T., Reed, J. C., Green, D., and Swain, S. L. (1997). Unequal death in T helper cell (Th)1 and Th2 effectors: Th1, but not Th2, effectors undergo rapid Fas/FasL-mediated apoptosis. J. Exp. Med. 185, 1837–1849.
Keywords: CD44, extracellular matrix, T cell, memory, migration
Citation: Baaten BJG, Tinoco R, Chen AT and Bradley LM (2012) Regulation of antigen-experienced T cells: lessons from the quintessential memory marker CD44. Front. Immun. 3:23. doi: 10.3389/fimmu.2012.00023
Received: 13 September 2011; Paper pending published: 03 October 2011;
Accepted: 08 February 2012; Published online: 27 February 2012.
Edited by:
Donna L. Farber, Columbia University Medical Center, USAReviewed by:
Gabrielle Belz, Walter and Eliza Hall Institute of Medical Research, AustraliaTania H. Watts, University of Toronto, Canada
Copyright: © 2012 Baaten, Tinoco, Chen and Bradley. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Linda M. Bradley, Infectious and Inflammatory Disease Center, Sanford-Burnham Medical Research Institute, 10901 North Torrey Pines Road, La Jolla, CA, USA. e-mail:bGJyYWRsZXlAc2FuZm9yZGJ1cm5oYW0ub3Jn