 Fabienne Tacchini-Cottier*
Fabienne Tacchini-Cottier* Tiffany Weinkopff and Pascal Launois†
Tiffany Weinkopff and Pascal Launois†
- Department of Biochemistry, WHO Immunology Research and Training Center, University of Lausanne, Epalinges, Switzerland
The murine model of Leishmania major infection has been an invaluable tool in understanding T helper differentiation in vivo. The initial evidence for a role of distinct CD4+ T helper subsets in the outcome of infection was first obtained with this experimental model. The development of CD4+ Th1 cells was associated with resolution of the lesion, control of parasite replication, and resistance to re-infection in most of the mouse strains investigated (i.e., C57BL/6). In contrast, differentiation of CD4+ Th2 cells correlated with the development of unhealing lesions, and failure to control parasite load in a few strains (i.e., BALB/c). Since these first reports, an incredible amount of effort has been devoted to understanding the various parameters involved in the differentiation of these, and more recently discovered T helper subsets such as Th17 and T regulatory cells. The discovery of cross-talk between T helper subsets, as well as their plasticity force us to reevaluate the events driving a protective/deleterious T helper immune response following infection with L. major in mice. In this review, we describe the individual contributions of each of these CD4+ T helper subsets following L. major inoculation, emphasizing recent advances in the field, such as the impact of different substrains of L. major on the pathogenesis of disease.
Introduction
The first description of the existence of two subsets of CD4+ T cells secreting distinct cytokines was made already 25 years ago (Mosmann et al., 1986). Two CD4+ T helper subsets named Th1 and Th2, first described in vitro, were shown to produce distinct cytokines with Th1 cells secreting IFNγ, and Th2 cells secreting IL-4, IL-13, and IL-5. The first evidence for a functional role of these CD4+ T helper subsets in vivo was obtained with the experimental model of cutaneous leishmaniasis, where Leishmania major promastigotes were inoculated subcutaneously in mice (Scott et al., 1988; Heinzel et al., 1989). Initially, resistance and susceptibility were shown to be correlated with the development of a CD4+ Th1 or Th2 response, respectively. Recently, several other T helper subsets have been identified first in vitro, and then in vivo. Among these, Th17 as well as the T regulatory (Treg) cells were shown to influence the pathogenesis of disease in L. major-infected mice.
Among the factors driving the differentiation of specific T helper subsets, cytokines present at the site of parasite inoculation and in the draining lymph node (dLN) were shown to be critical. In addition, the dose of antigen, the strength of T cell receptor signaling, and the recognition of pathogen-associated molecular patterns were also shown to influence the differentiation of T helper subsets in several experimental models of infection. Furthermore, the plasticity of some of the CD4+ T helper subsets, reviewed in O’Shea and Paul (2010) complicates our understanding of the factors involved in susceptibility or resistance to infection. Following inoculation of L. major, other parameters such as the size of the parasite inoculum, the mode (needle versus sand fly), and site of parasite inoculation, as well as the substrain of L. major further influence T helper differentiation and the outcome of the disease in this experimental model.
In this review we will focus on experimental leishmaniasis caused by L. major, one of the most extensively studied models of cutaneous leishmaniasis. We will (1) review experimental evidence revealing that the Th1/Th2 paradigm and its relevance to susceptibility or resistance to infection with L. major is not as strict as previously thought, and (2) discuss the impact that different L. major substrains (Table 1) have on the differentiation of T helper cells and susceptibility to infection.

Table 1. Origins of the L. major substrains discussed in this review.
The Factors Involved in the Differentiation of Th1 Cells and Control of Lesion Development Following L. major Inoculation
Several parameters are involved in the differentiation of naïve CD4+ T cells into CD4+ Th1 cells. Most laboratory mouse strains (C3H/He, CBA, C57BL/6, 129Sv/Ev) infected with L. major develop a small lesion that heals spontaneously, and these mice are resistant to re-infection. The healing phenotype has been correlated with the development of a CD4+ Th1 response, characterized by the generation of CD4+ T cells secreting IFNγ, a critical cytokine triggering macrophage activation that leads to parasite killing.
The critical role of IFNγ in the control of the infection was first determined in mice treated with anti-IFNγ antibodies, and later using mice deficient in IFNγ. These mice were unable to control lesion size and parasite growth, developing a CD4+ Th2 response (Belosevic et al., 1989; Scott, 1991). Mice lacking the IFNγ receptor also failed to control lesion size and parasite growth confirming that IFNγ signaling is required in the healing process (Wang et al., 1994; Swihart et al., 1995). However, despite the development of unhealing lesions, C57BL/6 IFNγR−/− mice infected with the L. major LV39 substrain developed a CD4+ Th1 response (Swihart et al., 1995), while 129/Sv/Ev IFNγR−/− mice infected with the L. major IR173 substrain developed a Th2 immune response. Different substrains of L. major have been shown to induce distinct immune responses (Table 2), suggesting that the differences measured in these two studies initially attributed to the different genetic backgrounds of the infected mice, may result from the use of distinct L. major substrains. Further support in the importance of IFNγ in the control of L. major infection, was demonstrated using mice deficient in T-bet, the master transcription factor of Th1 cells, that also renders otherwise resistant mouse strains susceptible to infection (Szabo et al., 2002).
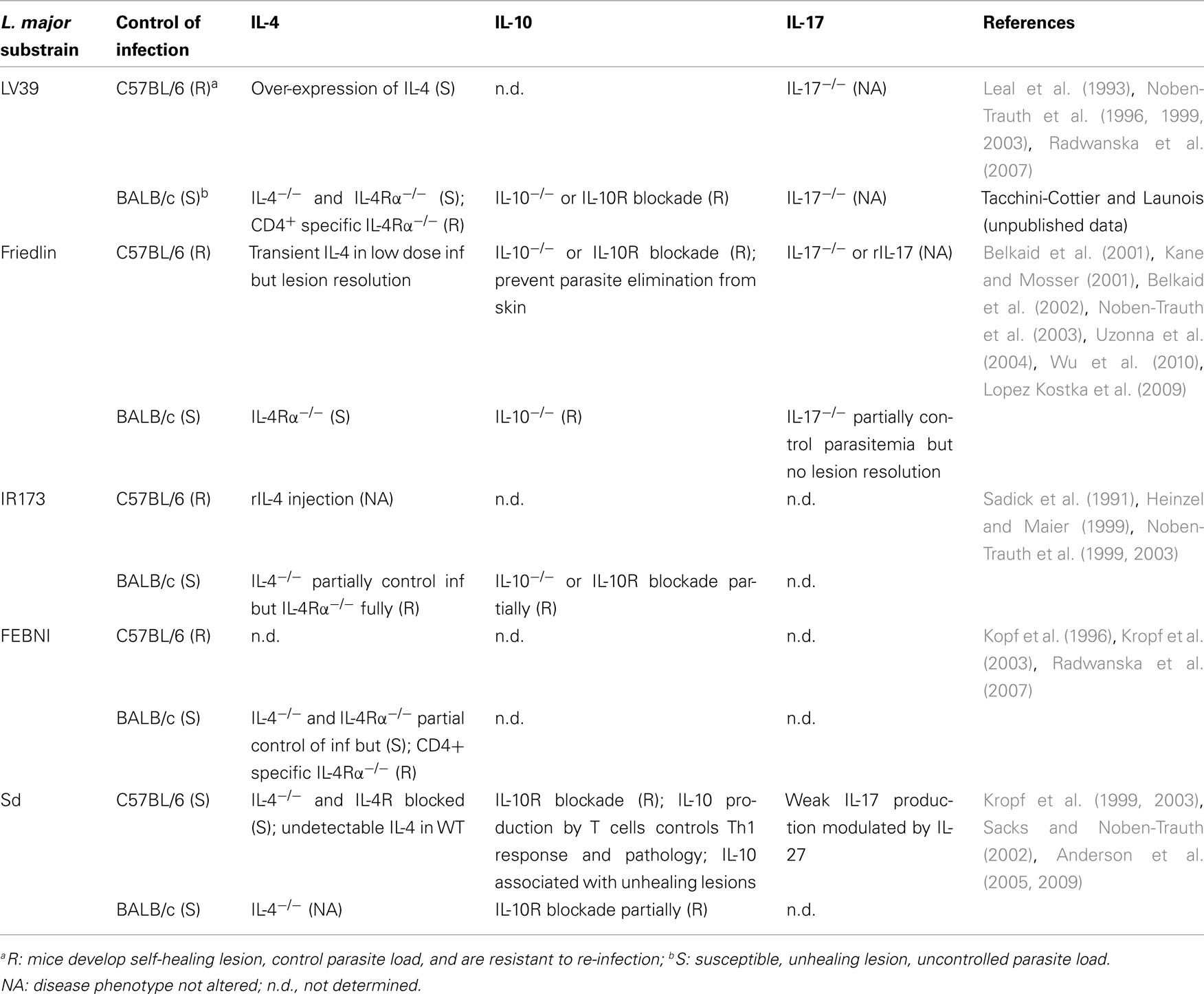
Table 2. Differential immune responses to Leishmania majo r substrains.
Following infection with L. major, IFNγ is mainly secreted by CD4+ Th1 cells and NK cells. The role of NK cell-derived IFNγ in CD4+ Th1 differentiation following L. major infection is currently controversial and may also depend on the L. major substrain. Neutralization of IL-12 in C57BL/6 mice prevents the early production of IFNγ by NK cells, abrogating the development of Th1 cells and leads to susceptibility to L. major Friedlin, Sd, and FEBNI strains (Laskay et al., 1993; Sypek et al., 1993; Scharton-Kersten and Scott, 1995). However, mice deficient in both T cells and IFNγ reconstituted with wild type (WT) CD4+ T cells as the sole source of IFNγ could differentiate functional Th1 cells upon challenge with L. major IR173 (Wakil et al., 1998), demonstrating that the IFNγ derived from NK cells is dispensable for Th1 differentiation using this L. major substrain. Furthermore, NK cells appeared to be dispensable for the development of CD4+ Th1 cells following infection with L. major LV39 substrain (Satoskar et al., 1999). It remains to be investigated if different substrains of L. major parasites induce distinct NK cell functions.
Th1 development subsequent to L. major infection has been associated with lesion healing. The IFNγ secreted by CD4+ Th1 cells synergizes with TNF to activate macrophage leishmanicidal activity. In absence of TNF or TNFR, L. major-infected mice on the C57BL/6 genetic background develop functional CD4+ Th1 cells, are able control parasite load with some variations depending on the L. major substrain inoculated (Vieira et al., 1996; Nashleanas et al., 1998; Kanaly et al., 1999; Wilhelm et al., 2001; Chakour et al., 2003; Ritter et al., 2004). However, these mice were unable to heal their lesions, revealing that TNF, and particularly the transmembrane form of TNF (Allenbach et al., 2008) is critical in the resolution of the inflammatory lesion, a process distinct from the development of CD4+ Th1 cells.
Lack of correlation between Th1 differentiation and control of lesion size in C57BL/6 mice was also observed following inoculation of the L. major Seidman (Sd) strain. C57BL/6 mice inoculated with the Sd strain developed unhealing lesions despite the development of a strong Th1 immune response (Anderson et al., 2005). In this case, a population of Th1 cells secreting both IFNγ and IL-10, as well as regulatory T cells were later shown to prevent the healing potential in this infection (Anderson et al., 2009). Collectively, these experiments reveal that the control of the inflammatory lesion involves several factors not always linked to the development of CD4+ Th1 cells.
IL-12 is a key cytokine leading to the generation of a protective CD4+ Th1 immune response following L. major infection. IL-12p70 is a heterodimeric molecule primarily secreted by antigen presenting cells (APCs), formed by the IL-12p40 and IL-12 p35 subunits (Trinchieri et al., 2003). Mice deficient in either IL-12p40 or IL-12p35 are unable to control infection (Mattner et al., 1996, 1997; Park et al., 2002) and mice deficient in the IL-12 signaling molecule STAT4 developed non-healing lesions following L. major infection (Jacobson et al., 1995; Stamm et al., 1999). Conversely, susceptible BALB/c mice treated with exogenous IL-12 developed a Th1 response leading to parasite control (Heinzel et al., 1993; Sypek et al., 1993; Sakai et al., 2000). IL-12 not only initiates the development of a Th1 response to L. major infection but it is also required to maintain Th1 cells during infection (Park et al., 2000, 2002; Stobie et al., 2000), and continued production of IL-12 during L. major infection is necessary for the development of IFNγ-producing Th1 effector cells from a plastic central memory CD4+ T cell pool (Pakpour et al., 2008).
In addition to IL-12, several other cytokines such as IL-18 and IL-27 can also contribute to CD4+ Th1 differentiation. IL-18, a member of the IL-1 cytokine family, is primarily produced by macrophages and dendritic cells (DCs) and serves as a cofactor during IL-12-mediated Th1 responses by optimizing IFNγ production from effector Th1 cells and activating NK cells. IL-12 upregulates the IL-18 receptor preferentially on Th1 cells suggesting a synergistic effect of IL-12 and IL-18 on the development of a Th1 response, reviewed in Nakanishi et al. (2001). Even though IL-18 is dispensable during L. major infection in C57BL/6 mice (Monteforte et al., 2000), the role of IL-18 in mediating protection appears to depend on the genetic background of the mouse strain (Xu et al., 2000; Wei et al., 2004). Administration of rIL-18 alone in BALB/c mice exacerbated the disease (Ohkusu et al., 2000; Li et al., 2004), revealing that IL-18 is not sufficient to induce Th1 differentiation in these Th2-prone mice. While IL-18 may not be required for the induction of a Th1 response after L. major inoculation, these data collectively suggest that this cytokine can increase IFNγ production and thus potentiate the effects of IL-12 in the development of a CD4+ Th1 response.
IL-27 is a cytokine closely related to IL-12 which is produced by phagocytes. It can also induce the differentiation of naïve T cells toward Th1 cells through the early induction of IFNγ production by T cells and NK cells (Pflanz et al., 2002). During L. major infection, C57BL/6 mice deficient in the IL-27R (WSX-1−/−) showed early impaired IFNγ response and increased IL-4 secretion and were transiently more susceptible than WT controls (Yoshida et al., 2001); however, these mice eventually developed a L. major-specific robust IFNγ response and healed their lesions in the absence of IL-4 (Artis et al., 2004). Further studies confirmed a transient role for IL-27 in Th1 differentiation in C57BL/6 mice, which is probably linked to its control of early IL-4 secretion (Anderson et al., 2009). A role for IL-27 in the control of Th17 differentiation was also described, linking Th1 and Th17 differentiation (Anderson et al., 2009).
The Factors Involved in the Differentiation of Th2 Cells and Susceptibility Following L. major Inoculation in BALB/c Mice
The murine model of infection with L. major provided the first association between expansion of CD4+ Th2 cells and progressive disease (Scott et al., 1988; Heinzel et al., 1989). Given that mRNA levels or the production of IFNγ were similar early in infection in resistant and susceptible mice (Morris et al., 1993), the strict correlation between susceptibility to infection and failure to mount a Th1 response was put into question. These experiments suggested that the development of pathology observed in BALB/c mice inoculated with L. major does not result from a defective Th1 development.
Thereafter, an important role for the IL-4 cytokine in mediating Th2 cell differentiation and susceptibility to infection with L. major was established. It was first observed that administration of neutralizing antibodies against IL-4 reversed the disease phenotype in BALB/c mice, rendering them resistant to infection (Sadick et al., 1990). Conversely, IL-4 transgenic C57BL/6 mice expressing IL-4 failed to clear the infection (Leal et al., 1993). Furthermore, several studies established that the IL-4 produced during the early phases of infection is important for the development of CD4+ Th2 cells, but the cellular source of this early IL-4 production remains a matter of debate. CD4+ T cells with the Vβ4Vα8 T cell receptor were shown to be the source of this early IL-4 secretion in response to the immunodominant epitope from Leishmania activated C kinase (LACK; Julia et al., 1996; Launois et al., 1997a,b, 2002; Himmelrich et al., 2000). However, subsequent studies questioned the role of LACK-specific CD4+ T cells as the sole source of IL-4 needed for the development of a Th2 response and thus the susceptibility of BALB/c mice. First, precursor frequency expansion and IL-4 mRNA expression of LACK-specific Vβ4Vα8 CD4+ T cells were shown to be comparable in both L. major susceptible and resistant strains of mice (Scott et al., 1996; Stetson et al., 2002). In addition, whereas inoculation of LACK-mutated L. major parasites, that are not able to activate the Vβ4Vα8 CD4+ T cells, resulted in reduced expansion of these cells in vivo, the frequency of IL-4-producing cells in infected mice was similar to those obtained following injection of WT L. major parasites (Kelly and Locksley, 2004). Collectively, these results demonstrated that the early IL-4 production by CD4+ T cells was important but not the only signal required for Th2 differentiation in BALB/c mice.
In parallel with these data, further experiments performed in IL-4 deficient mice strengthened the importance of other factors in driving Th2 responses during L. major infection. IL-4 deficient BALB/c mice infected with the L. major LV39 strain remained susceptible to infection (Noben-Trauth et al., 1996) while those infected with two other L. major substrains (FEBNI or IR173), were able to control the infection (Kopf et al., 1996; Noben-Trauth et al., 1999). Interestingly, these studies revealed not only that other factors than IL-4 contribute to susceptibility to L. major, but that different substrains of L. major can induce distinct immune responses in BALB/c mice. These data demonstrated that other Th2 cytokines such as IL-13 and IL-10 also contribute to susceptibility to infection.
A role for IL-13 in susceptibility to infection with L. major was demonstrated in IL-4Rα deficient BALB/c mice. IL-4Rα is a component of both the IL-4R and the IL-13R thus both IL-4 and IL-13 are inactive in these mice. Comparing the outcome of infection in these and IL-4−/− mice allowed the determination of the role of IL-4 and IL-13 in the development of a Th2 response. Although IL-4−/− BALB/c mice infected with L. major IR73 strain partially controlled the infection, IL-4Rα−/− mice inoculated with the same strain of L. major were fully resistant (Noben-Trauth et al., 1999), demonstrating the importance of IL-13 in the susceptibility of mice infected with L. major IR173. These results were corroborated by other studies showing that IL-13 deficient BALB/c mice became resistant to infection and that over-expression of IL-13 in resistant C57BL/6 rendered these mice susceptible to infection with L. major LV39 (Matthews et al., 2000). However, since injection of IL-13Rα2 fusion protein, which blocks the biological activity of IL-13 in IL-4−/− BALB/c mice infected with L. major LV39 did not modify the course of infection and parasite burden (Kropf et al., 1999) and since both IL-4−/− and IL-4Rα−/− BALB/c mice infected with L. major LV39 remained susceptible to infection (Noben-Trauth et al., 1999), mechanisms/cytokines other than IL-4 and IL-13 are likely to be involved in the susceptibility to infection. In contrast, a protective role for an IL-4/IL-13 responsive non-T cell in the development of a healing phenotype following infection with L. major LV39 was reported in BALB/c mice with the abrogation of IL-4 and IL-13 signaling selectively in CD4+ T cells (Radwanska et al., 2007).
In addition to IL-4 and IL-13, a crucial role for IL-10 in susceptibility was shown in BALB/c mice doubly deficient for the IL-4Rα and IL-10, as these mice were resistant to infection with L. major LV39 (Noben-Trauth et al., 2003). Interestingly, IL-10−/− BALB/c mice were more resistant to infection with L. major Friedlin than WT mice (Kane and Mosser, 2001). The cellular source of IL-10 production is another factor influencing the effect of IL-10 on susceptibility to infection with L. major. C57BL/6 mice expressing an IL-10 transgene under the IL-2 promoter, directing IL-10 secretion in T cells, remained resistant to L. major (Hagenbaugh et al., 1997). In contrast, C57BL/6 mice that express an IL-10 transgene under the control of the MHC promoter, directing IL-10 expression mainly in APCs, were susceptible to infection with L. major (Groux et al., 1999). In this context, we have recently demonstrated that a subset of L. major-specific B cells expressing the CD1B and CD5 antigens are necessary for the development of a Th2 cell response in BALB/c mice infected with the L. major LV39 strain through the production of IL-10 (Ronet et al., 2010).
Altogether these studies showed that several cytokines are involved in the development and maintenance of susceptibility to infection in response to L. major in susceptible BALB/c mice, and that the requirement of IL-4 and/or IL-13 and/or IL-10 for the development of a Th2 response is linked with several factors including the particular substrain of L. major inoculated (Table 2). Since these three cytokines have powerful inhibiting effects on macrophage activation and on IFNγ-mediated parasite killing (Bogdan et al., 1996), a small production of any of these cytokines could be sufficient to have an impact on parasite control.
In this line, different substrains of L. major were shown to influence macrophage activation and the ability to kill Leishmania parasites. For instance, killing of L. major LV39 requires greater levels of IFNγ than killing of the IR173 substrain (Noben-Trauth et al., 2003). Thus, although resistance to infection with L. major IR173 can be induced by the removal of IL-4, rendering BALB/c mice resistant to infection with L. major LV39 requires the inactivation of not only IL-4 but also of IL-13 and IL-10 in order for IFNγ to reach the threshold activity needed for parasite killing.
Altogether, the murine model has been crucial in the understanding of the multiple factors involved in susceptibility to L. major infection. Numerous evidences point to IL-4 as a critical cytokine leading to development of susceptibility to L. major in BALB/c mice, however additional factors including IL-10 and IL-13 also contribute to the pathology of the disease. It is likely that other factors yet to be described, some of them upstream of cytokines, will likely contribute to susceptibility to infection.
The Development of Th17 Cells Following L. major Inoculation
Th17 cells are a recently described T helper cell subset producing primarily IL-17A, IL-17F, and IL-22 as well as many other cytokines including TNF, IL-21, and GM-CSF. RORγt and RORα are the two master transcription factors involved in Th17 differentiation, but other transcription factors including STAT3, IRF4, and BATF also contribute to this process (reviewed in Hirota et al., 2010). The development of Th17 cells in mice and then in humans was initially described as requiring TGFβ and IL-6, and to be inhibited by IL-4 and IFNγ (reviewed in Korn et al., 2009). Recently Th17 cells were also shown to differentiate from naïve CD4+ T cells in absence of TGFβ, when exposed to IL-6, IL-23, and IL-1β (Ghoreschi et al., 2010). Subsequently, two distinct Th17 subsets were identified, conventional Th17 cells [obtained in vitro in response to TGFβ and IL-6 (Th17β)], and inflammatory Th17 cells, obtained in response to IL-6 and IL-23, Th17(23). Different function and transcriptional profiles were ascribed to these subsets, with Th17β cells expressing more IL-9, IL-10, and CCL10, while Th17(23) cells express higher levels of Tbx21, IL33, and CXCR3 (Ghoreschi et al., 2010).
Leishmania major susceptible mouse strains such as BALB/c mice develop unhealing lesions that are characterized by the presence of parasites and a persistent neutrophil infiltration at the lesion site (Beil et al., 1992; Tacchini-Cottier et al., 2000; Ribeiro-Gomes et al., 2004). One of the hallmarks of Th17 cells is the induction of the secretion of chemokines attracting neutrophils by various cell types, so it was of interest to investigate if Th17 cells played a role in the persistence of neutrophils observed in lesions of mice susceptible to L. major. The group of von Stebut measured higher levels of IL-17 and IL-23 in dLNs of BALB/c mice than in C57BL/6 mice inoculated with the L. major Friedlin substrain, suggesting the presence of inflammatory Th17 cells in dLN of BALB/c mice. DC-derived IL-23 was thought to induce and maintain Th17 cells in these mice. In addition to Th17-derived IL-17, secretion of IL-17 by recruited neutrophils appeared to also be important in mediating the persistence of an inflammatory lesion. IL-17A−/− BALB/c mice inoculated with a high dose (2 × 105) or low dose (103) of L. major Friedlin developed smaller lesion volume and parasite load (Lopez Kostka et al., 2009). However, the Th2 profile measured in dLNs of L. major-infected IL-17A−/− mice was similar to that of control mice, once more separating the Th2 response from the development of unhealing lesions. Of interest, we did not see any difference in lesion size or parasite control when IL-17−/− BALB/c mice were inoculated with a high dose of L. major LV39 (Launois and Tacchini-Cottier, unpublished data); in addition, IL-23−/− mice on a C57BL/6 genetic background infected with L. major LV39 did not show any difference in the control of inflammatory lesions nor the differentiation of Th1 cells (Launois and Tacchini-Cottier, unpublished data). Collectively, these data reveal that different substrains of L. major may also impact the levels of IL-17 and/or Th17 cells in susceptibility to infection.
Interestingly, neutrophil recruitment was different in L. major-infected IL-17−/− and control mice from 2 to 3 weeks post parasite inoculation, but did not differ significantly during the first week of infection (Lopez Kostka et al., 2009). IL-17 contributes to the early recruitment of neutrophils after L. major LV39 inoculation (Tacchini-Cottier and Launois, unpublished data), suggesting that compensatory mechanisms may induce early neutrophil recruitment in IL-17−/− mice. These data support the current hypothesis that the recruitment of neutrophils during the first days following L. major inoculation involves multiple factors including CXCL6 (GCP-2; Uyttenhove et al., 2011), and possibly CXCL1 (KC), CXCL2 (MIP-2), as well as parasite-derived factors, reviewed in Charmoy et al. (2010).
A detrimental role for Th17 cells on lesion development was also revealed following inoculation of the L. major Sd substrain which causes non-healing lesions in otherwise L. major resistant C57BL/6 mice. Following parasite injection, IL-27R-deficient mice (WSX.1−/− mice) developed more severe lesions associated with the appearance of Th17 cells, showing that IL-27 is controlling the development of Th17 cells in L. major Sd susceptible mice (Anderson et al., 2009).
In contrast to the above studies, IL-17 was shown to correlate with protection against infection in C57BL/6 mice vaccinated with live L. major Friedlin and CpG (Wu et al., 2010). First, vaccination enhanced the development of Th17 cells, and second, vaccinated IL-17R−/− mice developed higher lesion sizes and parasite burdens at early but not later time points after infection. Despite the induction of an inflammatory response, no IL-23 was detectable at the site of parasite inoculation or in the dLN of vaccinated mice. These data suggest that the type of Th17 cells measured in this study resemble classical Th17 cells (Th17β), which is different from those measured in the other studies that appear more similar to inflammatory Th17(23) cells (Lopez Kostka et al., 2009), although definitive confirmation of the existence of these Th17 subpopulations remains to be established in these experimental models. Similar vaccinations in BALB/c mice did not induce the development of Th17 cells (Wu et al., 2010).
Collectively these studies show that IL-17, whether produced by Th17 cells or another cell type, is a modulator of L. major infection. Furthermore, the impact of Th17 cells appears to differ significantly in response to distinct strains of L. major parasites and lesion development also varies depending strain, and thus the type of immune response.
The Development of Treg Cells Following L. major Inoculation
CD4+Foxp3+ Treg cell functions involve the maintenance of immune tolerance, as well as the prevention of inflammatory diseases. In addition, these cells are critical in regulating the T helper immune response generated in response to infection, reviewed in Campbell and Koch (2011). The balance of various cytokines including IL-6, IL-1, IL-23, and retinoic acid links the development of Tregs to that of Th17 cells (Lochner et al., 2008; Yang et al., 2008; Zhou, 2008).
The role of CD4+Foxp3+ Tregs in the development of Th1 cells during L. major infection was elegantly demonstrated by Belkaid and colleagues. They first described the accumulation of Tregs at the site of parasite inoculation (skin) in C57BL/6 mice. The presence of Tregs suppressed the ability of effector Th1 cells to totally eliminate L. major, allowing the maintenance of concomitant immunity after clinical cure (Belkaid et al., 2002). Depletion of Tregs in C57BL/6 mice enhanced the development of Th1 cells, rendering L. major resistant strains of mice more resistant to the primary infection (based on smaller lesion size and increased Th1 cytokine production, and decreased parasite load) but susceptible to a challenge secondary infection (Belkaid et al., 2002). It was further demonstrated that the IL-10 secreted by CD4+Foxp3+ Tregs was critical in preventing total elimination of the parasites in the skin (Suffia et al., 2005).
Unlike reports in C57BL/6 mice, the susceptibility to L. major in BALB/c IL-4Rα−/− mice was not caused by the secretion of IL-10 by Tregs (Nagase et al., 2007). Furthermore, inoculation of C57BL/6 mice with the L. major Sd substrain showed that the susceptibility is dependent on the secretion of IL-10 by IFNγ-secreting CD4+ T cells and not by Tregs (Anderson et al., 2005, 2009).
We and others have shown that CD4+Foxp3+ Tregs also modulate the extent of L. major-specific Th2 immune responses. Tregs controlled the magnitude of infection in L. major-infected BALB/c mice. Depletion of Treg cells prior to parasite inoculation enhanced parasite loads, Th2 responses, and lesion size rendering susceptible mice more susceptible to infection (Aseffa et al., 2002). Experimental reconstitution of SCID mice with T cells was further used to investigate the suppressive properties of Treg cells. Transfer of 107 naïve BALB/c spleen cells prior to infection led to resistance to L. major, associated with the development of a Th1 response, while the transfer of 108 cells rendered the mice highly susceptible to infection, associated with the development of a Th2 immune response (Mitchell et al., 1981). Transfer of 107 cells depleted of CD4+Foxp3+ Tregs suppressed the development of a protective Th1 response in reconstituted SCID mice infected with L. major (Aseffa et al., 2002; Liu et al., 2003; Xu et al., 2003), allowing the progressive development of an unhealing lesion linked to a Th2 response. The effect of CD4+Foxp3+ Tregs on the outcome of disease in these reconstituted SCID mice varied with the timing of CD4+Foxp3+ Treg reconstitution with a major effect on the inflammatory lesion appearing late in infection (Aseffa et al., 2002; Liu et al., 2003; Xu et al., 2003). Altogether, the suppressive activity of CD4+Foxp3+ Tregs modulates both Th1 and Th2 L. major-driven immune responses. However, the relative importance of the IL-10 secreted by Tregs appears to differ in mice susceptible or resistant to infection, with a critical role in resistance to infection, but no suppressive role in unhealing lesions. A role for other types of regulatory T cells such as inducible Tregs in the control of L. major infection remains to be determined.
Conclusion
The Th1/Th2 paradigm is still considered as the basis of vaccine development and is used in pre-clinical trials. Indeed antigens that induce a Th1 response are considered as potential protective antigens and those inducing a Th2 response are associated with pathology. Based on the observations (1) that the ablation of Th2 cytokines such as IL-4, IL-13, and IL-10 also confers resistance to infection and (2) that antigens such as LACK or cysteine protease antigens that strongly induce Th2 responses early after infection with Leishmania are protective when administered with appropriate adjuvants (Mougneau et al., 1995; Pollock et al., 2003); it appears that in addition to promoting a Th1 response, vaccination should also abolish the Th2 response induced by infection with Leishmania parasites. Additional work will be needed to understand if plasticity of the different T helper subsets occurs during Leishmania infection, before extrapolating results obtained in experimental cutaneous leishmaniasis for the design of new immuno-intervention tools or prophylactic approaches. Such work will also be important in defining surrogate markers of protection that may be used for diagnostics. IFNγ has been widely used as a marker of protection, however, as discussed here and shown in a recent study (Nylen et al., 2006), it may not always be the best marker for resistance.
The experimental model of L. major infection has and will continue to provide invaluable information on the mechanisms involved in T helper differentiation, with applications in leishmaniasis and other diseases where T helper subsets are contributing to pathology. It is becoming increasingly apparent that different strains of parasites causing cutaneous lesions, from the Old and New World, induce distinct types of immune responses with non-healing lesions often associated with the failure to develop a Th1 response rather than the induction of a CD4+ Th2 response, reviewed in Alexander and Bryson (2005). In order to apply the informations gained by studying the mechanisms of protection analyzed in experimental murine cutaneous leishmaniasis to humans, one should be careful to also take into account the diversity of immune responses induced by different substrains of parasites, as seen for L. major, as well as by other Leishmania species and strains causing cutaneous lesions in different parts of the world. Sequencing of the different substrains should provide important clues on the understanding of the parasite specific features leading to distinct immune responses.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank current and previous members of the WHO-IRTC for their input in some of the studies presented, and Dr. Jacques Mauël for critical reading of the manuscript. Work in Fabienne Tacchini-Cottier and Pascal Launois laboratories is funded by grants from the Swiss National Science Foundation.
References
Alexander, J., and Bryson, K. (2005). T helper (h)1/Th2 and Leishmania: paradox rather than paradigm. Immunol. Lett. 99, 17–23.
Allenbach, C., Launois, P., Mueller, C., and Tacchini-Cottier, F. (2008). An essential role for transmembrane TNF in the resolution of the inflammatory lesion induced by Leishmania major infection. Eur. J. Immunol. 38, 720–731.
Anderson, C. F., Mendez, S., and Sacks, D. L. (2005). Nonhealing infection despite Th1 polarization produced by a strain of Leishmania major in C57BL/6 mice. J. Immunol. 174, 2934–2941.
Anderson, C. F., Stumhofer, J. S., Hunter, C. A., and Sacks, D. (2009). IL-27 regulates IL-10 and IL-17 from CD4+ cells in nonhealing Leishmania major infection. J. Immunol. 183, 4619–4627.
Artis, D., Johnson, L. M., Joyce, K., Saris, C., Villarino, A., Hunter, C. A., and Scott, P. (2004). Cutting edge: early IL-4 production governs the requirement for IL-27-WSX-1 signaling in the development of protective Th1 cytokine responses following Leishmania major infection. J. Immunol. 172, 4672–4675.
Aseffa, A., Gumy, A., Launois, P., MacDonald, H. R., Louis, J. A., and Tacchini-Cottier, F. (2002). The early IL-4 response to Leishmania major and the resulting Th2 cell maturation steering progressive disease in BALB/c mice are subject to the control of regulatory CD4+CD25+ T cells. J. Immunol. 169, 3232–3241.
Beil, W. J., Meinardus-Hager, G., Neugebauer, D. C., and Sorg, C. (1992). Differences in the onset of the inflammatory response to cutaneous leishmaniasis in resistant and susceptible mice. J. Leukoc. Biol. 52, 135–142.
Belkaid, Y., Hoffmann, K. F., Mendez, S., Kamhawi, S., Udey, M. C., Wynn, T. A., and Sacks, D. L. (2001). The role of interleukin (IL)-10 in the persistence of Leishmania major in the skin after healing and the therapeutic potential of anti-IL-10 receptor antibody for sterile cure. J. Exp. Med. 194, 1497–1506.
Belkaid, Y., Piccirillo, C. A., Mendez, S., Shevach, E. M., and Sacks, D. L. (2002). CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature 420, 502–507.
Belosevic, M., Finbloom, D., Van Der Meide, P., Slayter, M., and Nacy, C. (1989). Administration of monoclonal anti-IFN-gamma antibodies in vivo abrogates natural resistance of C3H/HeN mice to infection with Leishmania major. J. Immunol. 143, 266–274.
Bogdan, C., Gessner, A., Solbach, W., and Rollinghoff, M. (1996). Invasion, control and persistence of Leishmania parasites. Curr. Opin. Immunol. 8, 517–525.
Campbell, D. J., and Koch, M. A. (2011). Treg cells: patrolling a dangerous neighborhood. Nat. Med. 17, 929–930.
Chakour, R., Guler, R., Bugnon, M., Allenbach, C., Garcia, I., Mauel, J., Louis, J., and Tacchini-Cottier, F. (2003). Both the Fas ligand and inducible nitric oxide synthase are needed for control of parasite replication within lesions in mice infected with Leishmania major whereas the contribution of tumor necrosis factor is minimal. Infect. Immun. 71, 5287–5295.
Charmoy, M., Auderset, F., Allenbach, C., and Tacchini-Cottier, F. (2010). The prominent role of neutrophils during the initial phase of infection by Leishmania parasites. J. Biomed. Biotechnol. 2010, 719361.
Ghoreschi, K., Laurence, A., Yang, X. P., Tato, C. M., McGeachy, M. J., Konkel, J. E., Ramos, H. L., Wei, L., Davidson, T. S., Bouladoux, N., Grainger, J. R., Chen, Q., Kanno, Y., Watford, W. T., Sun, H. W., Eberl, G., Shevach, E. M., Belkaid, Y., Cua, D. J., Chen, W., and O’shea, J. J. (2010). Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature 467, 967–971.
Groux, H., Cottrez, F., Rouleau, M., Mauze, S., Antonenko, S., Hurst, S., McNeil, T., Bigler, M., Roncarolo, M. G., and Coffman, R. L. (1999). A transgenic model to analyze the immunoregulatory role of IL-10 secreted by antigen-presenting cells. J. Immunol. 162, 1723–1729.
Hagenbaugh, A., Sharma, S., Dubinett, S. M., Wei, S. H., Aranda, R., Cheroutre, H., Fowell, D. J., Binder, S., Tsao, B., Locksley, R. M., Moore, K. W., and Kronenberg, M. (1997). Altered immune responses in interleukin 10 transgenic mice. J. Exp. Med. 185, 2101–2110.
Heinzel, F. P., and Maier, R. A. Jr. (1999). Interleukin-4-independent acceleration of cutaneous leishmaniasis in susceptible BALB/c mice following treatment with anti-CTLA4 antibody. Infect. Immun. 67, 6454–6460.
Heinzel, F. P., Sadick, M. D., Holaday, B. J., Coffman, R. L., and Locksley, R. M. (1989). Reciprocal expression of interferon gamma or interleukin 4 during the resolution or progression of murine leishmaniasis. Evidence for expansion of distinct helper T cell subsets. J. Exp. Med. 169, 59–72.
Heinzel, F. P., Schoenhaut, D. S., Rerko, R. M., Rosser, L. E., and Gately, M. K. (1993). Recombinant interleukin 12 cures mice infected with Leishmania major. J. Exp. Med. 177, 1505–1509.
Himmelrich, H., Launois, P., Maillard, I., Biedermann, T., Tacchini-Cottier, F., Locksley, R. M., Rocken, M., and Louis, J. A. (2000). In BALB/c mice, IL-4 production during the initial phase of infection with Leishmania major is necessary and sufficient to instruct Th2 cell development resulting in progressive disease. J. Immunol. 164, 4819–4825.
Hirota, K., Martin, B., and Veldhoen, M. (2010). Development, regulation and functional capacities of Th17 cells. Semin. Immunopathol. 32, 3–16.
Jacobson, N. G., Szabo, S. J., Weber-Nordt, R. M., Zhong, Z., Schreiber, R. D., Darnell, J. E. Jr., and Murphy, K. M. (1995). Interleukin 12 signaling in T helper type 1 (Th1) cells involves tyrosine phosphorylation of signal transducer and activator of transcription (Stat)3 and Stat4. J. Exp. Med. 181, 1755–1762.
Julia, V., Rassoulzadegan, M., and Glaichenhaus, N. (1996). Resistance to Leishmania major induced by tolerance to a single antigen. Science 274, 421–423.
Kanaly, S. T., Nashleanas, M., Hondowicz, B., and Scott, P. (1999). TNF receptor p55 is required for elimination of inflammatory cells following control of intracellular pathogens. J. Immunol. 163, 3883–3889.
Kane, M. M., and Mosser, D. M. (2001). The role of IL-10 in promoting disease progression in leishmaniasis. J. Immunol. 166, 1141–1147.
Kelly, B. L., and Locksley, R. M. (2004). The Leishmania major LACK antigen with an immunodominant epitope at amino acids 156 to 173 is not required for early Th2 development in BALB/c mice. Infect. Immun. 72, 6924–6931.
Kopf, M., Brombacher, F., Kohler, G., Kienzle, G., Widmann, K. H., Lefrang, K., Humborg, C., Ledermann, B., and Solbach, W. (1996). IL-4-deficient Balb/c mice resist infection with Leishmania major. J. Exp. Med. 184, 1127–1136.
Korn, T., Bettelli, E., Oukka, M., and Kuchroo, V. K. (2009). IL-17 and Th17 Cells. Annu. Rev. Immunol. 27, 485–517.
Kropf, P., Herath, S., Klemenz, R., and Muller, I. (2003). Signaling through the T1/ST2 molecule is not necessary for Th2 differentiation but is important for the regulation of type 1 responses in nonhealing Leishmania major infection. Infect. Immun. 71, 1961–1971.
Kropf, P., Schopf, L. R., Chung, C. L., Xu, D., Liew, F. Y., Sypek, J. P., and Muller, I. (1999). Expression of Th2 cytokines and the stable Th2 marker ST2L in the absence of IL-4 during Leishmania major infection. Eur. J. Immunol. 29, 3621–3628.
Laskay, T., Rollinghoff, M., and Solbach, W. (1993). Natural killer cells participate in the early defense against Leishmania major infection in mice. Eur. J. Immunol. 23, 2237–2241.
Launois, P., Gumy, A., Himmelrich, H., Locksley, R. M., Rocken, M., and Louis, J. A. (2002). Rapid IL-4 production by Leishmania homolog of mammalian RACK1-reactive CD4(+) T cells in resistant mice treated once with anti-IL-12 or -IFN-gamma antibodies at the onset of infection with Leishmania major instructs Th2 cell development, resulting in nonhealing lesions. J. Immunol. 168, 4628–4635.
Launois, P., Maillard, I., Pingel, S., Swihart, K. G., Xenarios, I., Acha-Orbea, H., Diggelmann, H., Locksley, R. M., MacDonald, H. R., and Louis, J. A. (1997a). IL-4 rapidly produced by V beta 4 V alpha 8 CD4+ T cells instructs Th2 development and susceptibility to Leishmania major in BALB/c mice. Immunity 6, 541–549.
Launois, P., Swihart, K. G., Milon, G., and Louis, J. A. (1997b). Early production of IL-4 in susceptible mice infected with Leishmania major rapidly induces IL-12 unresponsiveness. J. Immunol. 158, 3317–3324.
Leal, L. M., Moss, D. W., Kuhn, R., Muller, W., and Liew, F. Y. (1993). Interleukin-4 transgenic mice of resistant background are susceptible to Leishmania major infection. Eur. J. Immunol. 23, 566–569.
Li, Y., Ishii, K., Hisaeda, H., Hamano, S., Zhang, M., Nakanishi, K., Yoshimoto, T., Hemmi, H., Takeda, K., Akira, S., Iwakura, Y., and Himeno, K. (2004). IL-18 gene therapy develops Th1-type immune responses in Leishmania major-infected BALB/c mice: is the effect mediated by the CpG signaling TLR9? Gene Ther. 11, 941–948.
Liu, H., Hu, B., Xu, D., and Liew, F. Y. (2003). CD4+CD25+ regulatory T cells cure murine colitis: the role of IL-10, TGF-beta, and CTLA4. J. Immunol. 171, 5012–5017.
Lochner, M., Peduto, L., Cherrier, M., Sawa, S., Langa, F., Varona, R., Riethmacher, D., Si-Tahar, M., Di Santo, J. P., and Eberl, G. (2008). In vivo equilibrium of proinflammatory IL-17+ and regulatory IL-10+ Foxp3+ RORgamma t+ T cells. J. Exp. Med. 205, 1381–1393.
Lopez Kostka, S., Dinges, S., Griewank, K., Iwakura, Y., Udey, M. C., and Von Stebut, E. (2009). IL-17 promotes progression of cutaneous leishmaniasis in susceptible mice. J. Immunol. 182, 3039–3046.
Matthews, D. J., Emson, C. L., McKenzie, G. J., Jolin, H. E., Blackwell, J. M., and McKenzie, A. N. (2000). IL-13 is a susceptibility factor for Leishmania major infection. J. Immunol. 164, 1458–1462.
Mattner, F., Di Padova, K., and Alber, G. (1997). Interleukin-12 is indispensable for protective immunity against Leishmania major. Infect. Immun. 65, 4378–4383.
Mattner, F., Magram, J., Ferrante, J., Launois, P., Di Padova, K., Behin, R., Gately, M. K., Louis, J. A., and Alber, G. (1996). Genetically resistant mice lacking interleukin-12 are susceptible to infection with Leishmania major and mount a polarized Th2 cell response. Eur. J. Immunol. 26, 1553–1559.
Mitchell, G. F., Curtis, J. M., Scollay, R. G., and Handman, E. (1981). Resistance and abrogation of resistance to cutaneous leishmaniasis in reconstituted BALB/c nude mice. Aust. J. Exp. Biol. Med. Sci. 59, 539–554.
Monteforte, G. M., Takeda, K., Rodriguez-Sosa, M., Akira, S., David, J. R., and Satoskar, A. R. (2000). Genetically resistant mice lacking IL-18 gene develop Th1 response and control cutaneous Leishmania major infection. J. Immunol. 164, 5890–5893.
Morris, L., Aebischer, T., Handman, E., and Kelso, A. (1993). Resistance of BALB/c mice to Leishmania major infection is associated with a decrease in the precursor frequency of antigen-specific CD4+ cells secreting interleukin-4. Int. Immunol. 5, 761–767.
Mosmann, T., Cherwinski, H., Bond, M., Giedlin, M., and Coffman, R. (1986). Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J. Immunol. 136, 2348–2357.
Mougneau, E., Altare, F., Wakil, A. E., Zheng, S., Coppola, T., Wang, Z. E., Waldmann, R., Locksley, R. M., and Glaichenhaus, N. (1995). Expression cloning of a protective Leishmania antigen. Science 268, 563–566.
Nagase, H., Jones, K. M., Anderson, C. F., and Noben-Trauth, N. (2007). Despite increased CD4+Foxp3+ cells within the infection site, BALB/c IL-4 receptor-deficient mice reveal CD4+Foxp3-negative T cells as a source of IL-10 in Leishmania major susceptibility. J. Immunol. 179, 2435–2444.
Nakanishi, K., Yoshimoto, T., Tsutsui, H., and Okamura, H. (2001). Interleukin-18 regulates both Th1 and Th2 responses. Annu. Rev. Immunol. 19, 423–474.
Nashleanas, M., Kanaly, S., and Scott, P. (1998). Control of Leishmania major infection in mice lacking TNF receptors. J. Immunol. 160, 5506–5513.
Noben-Trauth, N., Kropf, P., and Muller, I. (1996). Susceptibility to Leishmania major infection in interleukin-4-deficient mice. Science 271, 987–990.
Noben-Trauth, N., Lira, R., Nagase, H., Paul, W. E., and Sacks, D. L. (2003). The relative contribution of IL-4 receptor signaling and IL-10 to susceptibility to Leishmania major. J. Immunol. 170, 5152–5158.
Noben-Trauth, N., Paul, W. E., and Sacks, D. L. (1999). IL-4- and IL-4 receptor-deficient BALB/c mice reveal differences in susceptibility to Leishmania major parasite substrains. J. Immunol. 162, 6132–6140.
Nylen, S., Khamesipour, A., Mohammadi, A., Jafari-Shakib, R., Eidsmo, L., Noazin, S., Modabber, F., and Akuffo, H. (2006). Surrogate markers of immunity to Leishmania major in leishmanin skin test negative individuals from an endemic area re-visited. Vaccine 24, 6944–6954.
Ohkusu, K., Yoshimoto, T., Takeda, K., Ogura, T., Kashiwamura, S., Iwakura, Y., Akira, S., Okamura, H., and Nakanishi, K. (2000). Potentiality of interleukin-18 as a useful reagent for treatment and prevention of Leishmania major infection. Infect. Immun. 68, 2449–2456.
O’Shea, J. J., and Paul, W. E. (2010). Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science 327, 1098–1102.
Pakpour, N., Zaph, C., and Scott, P. (2008). The central memory CD4+ T cell population generated during Leishmania major infection requires IL-12 to produce IFN-gamma. J. Immunol. 180, 8299–8305.
Park, A. Y., Hondowicz, B., Kopf, M., and Scott, P. (2002). The role of IL-12 in maintaining resistance to Leishmania major. J. Immunol. 168, 5771–5777.
Park, A. Y., Hondowicz, B. D., and Scott, P. (2000). IL-12 is required to maintain a Th1 response during Leishmania major infection. J. Immunol. 165, 896–902.
Pflanz, S., Timans, J. C., Cheung, J., Rosales, R., Kanzler, H., Gilbert, J., Hibbert, L., Churakova, T., Travis, M., Vaisberg, E., Blumenschein, W. M., Mattson, J. D., Wagner, J. L., To, W., Zurawski, S., McClanahan, T. K., Gorman, D. M., Bazan, J. F., De Waal Malefyt, R., Rennick, D., and Kastelein, R. A. (2002). IL-27, a heterodimeric cytokine composed of EBI3 and p28 protein, induces proliferation of naive CD4(+) T cells. Immunity 16, 779–790.
Pollock, K. G., McNeil, K. S., Mottram, J. C., Lyons, R. E., Brewer, J. M., Scott, P., Coombs, G. H., and Alexander, J. (2003). The Leishmania mexicana cysteine protease, CPB2.8, induces potent Th2 responses. J. Immunol. 170, 1746–1753.
Radwanska, M., Cutler, A. J., Hoving, J. C., Magez, S., Holscher, C., Bohms, A., Arendse, B., Kirsch, R., Hunig, T., Alexander, J., Kaye, P., and Brombacher, F. (2007). Deletion of IL-4Ralpha on CD4 T cells renders BALB/c mice resistant to Leishmania major infection. PLoS Pathog. 3, e68. doi: 10.1371/journal.ppat.0030068
Ribeiro-Gomes, F. L., Otero, A. C., Gomes, N. A., Moniz-De-Souza, M. C., Cysne-Finkelstein, L., Arnholdt, A. C., Calich, V. L., Coutinho, S. G., Lopes, M. F., and Dosreis, G. A. (2004). Macrophage interactions with neutrophils regulate Leishmania major infection. J. Immunol. 172, 4454–4462.
Ritter, U., Mattner, J., Rocha, J. S., Bogdan, C., and Korner, H. (2004). The control of Leishmania (Leishmania) major by TNF in vivo is dependent on the parasite strain. Microbes Infect. 6, 559–565.
Ronet, C., Hauyon-La Torre, Y., Revaz-Breton, M., Mastelic, B., Tacchini-Cottier, F., Louis, J., and Launois, P. (2010). Regulatory B cells shape the development of Th2 immune responses in BALB/c mice infected with Leishmania major through IL-10 production. J. Immunol. 184, 886–894.
Sacks, D., and Noben-Trauth, N. (2002). The immunology of susceptibility and resistance to Leishmania major in mice. Nat. Rev. Immunol. 2, 845–858.
Sadick, M. D., Heinzel, F. P., Holaday, B. J., Pu, R. T., Dawkins, R. S., and Locksley, R. M. (1990). Cure of murine leishmaniasis with anti-interleukin 4 monoclonal antibody. Evidence for a T cell-dependent, interferon gamma-independent mechanism. J. Exp. Med. 171, 115–127.
Sadick, M. D., Street, N., Mosmann, T. R., and Locksley, R. M. (1991). Cytokine regulation of murine leishmaniasis: interleukin 4 is not sufficient to mediate progressive disease in resistant C57BL/6 mice. Infect. Immun. 59, 4710–4714.
Sakai, T., Hisaeda, H., Nakano, Y., Ishikawa, H., Maekawa, Y., Ishii, K., Nitta, Y., Miyazaki, J., and Himeno, K. (2000). Gene gun-mediated delivery of an interleukin-12 expression plasmid protects against infections with the intracellular protozoan parasites Leishmania major and Trypanosoma cruzi in mice. Immunology 99, 615–624.
Satoskar, A. R., Stamm, L. M., Zhang, X., Satoskar, A. A., Okano, M., Terhorst, C., David, J. R., and Wang, B. (1999). Mice lacking NK cells develop an efficient Th1 response and control cutaneous Leishmania major infection. J. Immunol. 162, 6747–6754.
Scharton-Kersten, T., and Scott, P. (1995). The role of the innate immune response in Th1 cell development following Leishmania major infection. J. Leukoc. Biol. 57, 515–522.
Scott, P. (1991). IFN-gamma modulates the early development of Th1 and Th2 responses in a murine model of cutaneous leishmaniasis. J. Immunol. 147, 3149–3155.
Scott, P., Eaton, A., Gause, W. C., Di Zhou, X., and Hondowicz, B. (1996). Early IL-4 production does not predict susceptibility to Leishmania major. Exp. Parasitol. 84, 178–187.
Scott, P., Natovitz, P., Coffman, R. L., Pearce, E., and Sher, A. (1988). Immunoregulation of cutaneous leishmaniasis. T cell lines that transfer protective immunity or exacerbation belong to different T helper subsets and respond to distinct parasite antigens. J. Exp. Med. 168, 1675–1684.
Stamm, L. M., Satoskar, A. A., Ghosh, S. K., David, J. R., and Satoskar, A. R. (1999). STAT-4 mediated IL-12 signaling pathway is critical for the development of protective immunity in cutaneous leishmaniasis. Eur. J. Immunol. 29, 2524–2529.
Stetson, D. B., Mohrs, M., Mallet-Designe, V., Teyton, L., and Locksley, R. M. (2002). Rapid expansion and IL-4 expression by Leishmania-specific naive helper T cells in vivo. Immunity 17, 191–200.
Stobie, L., Gurunathan, S., Prussin, C., Sacks, D. L., Glaichenhaus, N., Wu, C. Y., and Seder, R. A. (2000). The role of antigen and IL-12 in sustaining Th1 memory cells in vivo: IL-12 is required to maintain memory/effector Th1 cells sufficient to mediate protection to an infectious parasite challenge. Proc. Natl. Acad. Sci. U.S.A. 97, 8427–8432.
Suffia, I., Reckling, S. K., Salay, G., and Belkaid, Y. (2005). A role for CD103 in the retention of CD4+CD25+ Treg and control of Leishmania major infection. J. Immunol. 174, 5444–5455.
Swihart, K., Fruth, U., Messmer, N., Hug, K., Behin, R., Huang, S., Del Giudice, G., Aguet, M., and Louis, J. A. (1995). Mice from a genetically resistant background lacking the interferon gamma receptor are susceptible to infection with Leishmania major but mount a polarized T helper cell 1-type CD4+ T cell response. J. Exp. Med. 181, 961–971.
Sypek, J. P., Chung, C. L., Mayor, S. E., Subramanyam, J. M., Goldman, S. J., Sieburth, D. S., Wolf, S. F., and Schaub, R. G. (1993). Resolution of cutaneous leishmaniasis: interleukin 12 initiates a protective T helper type 1 immune response. J. Exp. Med. 177, 1797–1802.
Szabo, S. J., Sullivan, B. M., Stemmann, C., Satoskar, A. R., Sleckman, B. P., and Glimcher, L. H. (2002). Distinct effects of T-bet in TH1 lineage commitment and IFN-gamma production in CD4 and CD8 T cells. Science 295, 338–342.
Tacchini-Cottier, F., Zweifel, C., Belkaid, Y., Mukankundiye, C., Vasei, M., Launois, P., Milon, G., and Louis, J. A. (2000). An immunomodulatory function for neutrophils during the induction of a CD4+ Th2 response in BALB/c mice infected with Leishmania major. J. Immunol. 165, 2628–2636.
Trinchieri, G., Pflanz, S., and Kastelein, R. A. (2003). The IL-12 family of heterodimeric cytokines: new players in the regulation of T cell responses. Immunity 19, 641–644.
Uyttenhove, C., Marillier, R. G., Tacchini-Cottier, F., Charmoy, M., Caspi, R. R., Damsker, J. M., Goriely, S., Su, D., Van Damme, J., Struyf, S., Opdenakker, G., and Van Snick, J. (2011). Amine-reactive OVA multimers for auto-vaccination against cytokines and other mediators: perspectives illustrated for GCP-2 in L. major infection. J. Leukoc. Biol. 89, 1001–1007.
Uzonna, J. E., Joyce, K. L., and Scott, P. (2004). Low dose Leishmania major promotes a transient T helper cell type 2 response that is down-regulated by interferon gamma-producing CD8+ T cells. J. Exp. Med. 199, 1559–1566.
Vieira, L. Q., Goldschmidt, M., Nashleanas, M., Pfeffer, K., Mak, T., and Scott, P. (1996). Mice lacking the TNF receptor p55 fail to resolve lesions caused by infection with Leishmania major, but control parasite replication. J. Immunol. 157, 827–835.
Wakil, A. E., Wang, Z. E., Ryan, J. C., Fowell, D. J., and Locksley, R. M. (1998). Interferon gamma derived from CD4(+) T cells is sufficient to mediate T helper cell type 1 development. J. Exp. Med. 188, 1651–1656.
Wang, Z. E., Reiner, S. L., Zheng, S., Dalton, D. K., and Locksley, R. M. (1994). CD4+ effector cells default to the Th2 pathway in interferon gamma-deficient mice infected with Leishmania major. J. Exp. Med. 179, 1367–1371.
Wei, X. Q., Niedbala, W., Xu, D., Luo, Z. X., Pollock, K. G., and Brewer, J. M. (2004). Host genetic background determines whether IL-18 deficiency results in increased susceptibility or resistance to murine Leishmania major infection. Immunol. Lett. 94, 35–37.
Wilhelm, P., Ritter, U., Labbow, S., Donhauser, N., Rollinghoff, M., Bogdan, C., and Korner, H. (2001). Rapidly fatal leishmaniasis in resistant C57BL/6 mice lacking TNF. J. Immunol. 166, 4012–4019.
Wu, W., Huang, L., and Mendez, S. (2010). A live Leishmania major vaccine containing CpG motifs induces the de novo generation of Th17 cells in C57BL/6 mice. Eur. J. Immunol. 40, 2517–2527.
Xu, D., Liu, H., Komai-Koma, M., Campbell, C., McSharry, C., Alexander, J., and Liew, F. Y. (2003). CD4+CD25+ regulatory T cells suppress differentiation and functions of Th1 and Th2 cells, Leishmania major infection, and colitis in mice. J. Immunol. 170, 394–399.
Xu, D., Trajkovic, V., Hunter, D., Leung, B. P., Schulz, K., Gracie, J. A., McInnes, I. B., and Liew, F. Y. (2000). IL-18 induces the differentiation of Th1 or Th2 cells depending upon cytokine milieu and genetic background. Eur. J. Immunol. 30, 3147–3156.
Yang, X. O., Nurieva, R., Martinez, G. J., Kang, H. S., Chung, Y., Pappu, B. P., Shah, B., Chang, S. H., Schluns, K. S., Watowich, S. S., Feng, X. H., Jetten, A. M., and Dong, C. (2008). Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity 29, 44–56.
Yoshida, H., Hamano, S., Senaldi, G., Covey, T., Faggioni, R., Mu, S., Xia, M., Wakeham, A. C., Nishina, H., Potter, J., Saris, C. J., and Mak, T. W. (2001). WSX-1 is required for the initiation of Th1 responses and resistance to L. major infection. Immunity 15, 569–578.
Keywords: Leishmania, L. major, T helper cells, Th1, Th2, Th17, Treg, IL-17
Citation: Tacchini-Cottier F, Weinkopff T and Launois P (2012) Does T helper differentiation correlate with resistance or susceptibility to infection with L. major? Some insights from the murine model. Front. Immun. 3:32. doi: 10.3389/fimmu.2012.00032
Received: 24 November 2011; Accepted: 13 February 2012;
Published online: 27 February 2012.
Edited by:
Nathan Peters, National Institute of Allergy and Infectious Diseases, USAReviewed by:
Matthew Dale Woolard, Louisiana State University Health Sciences Center at Shreveport, USANathalie Labrecque, University of Montreal, Canada
Copyright: © 2012 Tacchini-Cottier, Weinkopff and Launois. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Fabienne Tacchini-Cottier, Department of Biochemistry, WHO Immunology Research and Training Center, University of Lausanne, 155, CH-1066 Epalinges, Switzerland. e-mail:ZmFiaWVubmUudGFjY2hpbmktY290dGllckB1bmlsLmNo
†Present address: Pascal Launois, Special Programme for Research and Training in Tropical Diseases, World Health Organization, 20 Avenue Appia, 1211 Geneva 27, Switzerland.