 Kinga K. Hosszu1
Kinga K. Hosszu1 Alisa Valentino1 Yan Ji1 Mara Matkovic1 Lina Pednekar1 Nina Rehage1 Nithin Tumma1 Ellinor I. B. Peerschke2 Berhane Ghebrehiwet1*
Alisa Valentino1 Yan Ji1 Mara Matkovic1 Lina Pednekar1 Nina Rehage1 Nithin Tumma1 Ellinor I. B. Peerschke2 Berhane Ghebrehiwet1*- 1 The Department of Medicine, Stony Brook University, Stony Brook, NY, USA
- 2 The Department of Laboratory Medicine, Memorial Sloan–Kettering Cancer Center, New York, NY, USA
The synthesis of the subunits of the C1 complex (C1q, C1s, C1r), and its regulator C1 inhibitor (C1-Inh) by human monocytes has been previously established. However, surface expression of these molecules by monocytes has not been shown. Using flow cytometry and antigen-capture enzyme-linked immunosorbent assay, we show here for the first time that, in addition to C1q, peripheral blood monocytes, and the monocyte-derived U937 cells express C1s and C1r, as well as Factor B and C1-Inh on their surface. C1s and C1r immunoprecipitated with C1q, suggesting that at least some of the C1q on these cells is part of the C1 complex. Furthermore, the C1 complex on U937 cells was able to trigger complement activation via the classical pathway. The presence of C1-Inh may ensure that an unwarranted autoactivation of the C1 complex does not take place. Since C1-Inh closely monitors the activation of the C1 complex in a sterile or infectious inflammatory environment, further elucidation of the role of C1 complex is crucial to dissect its function in monocyte, dendritic cell, and T cell activities, and its implications in host defense and tolerance.
Introduction
C1q (460 kDa) is a collagen-like, hexameric glycoprotein and consists of similar, but distinct polypeptide chains 6A, 6B, and 6C that form 6ABC triple helices (Brodsky-Doyle et al., 1976; Reid, 1989). C1q contains immunoglobulin (Ig)G/IgM binding sequences in its globular head region, which enable it to bind to immune complexes and engage in complement-mediated microbial killing and phagocytosis (Leist-Welsh and Bjornson, 1982; Bobak et al., 1987). As a result, C1q deficiency is associated with increased susceptibility to infections including otitis media, meningitis, and pneumonia (Kuis et al., 1988; Prellner et al., 1989; Vassallo et al., 2007; Pickering et al., 2008).
C1q circulates in plasma at a concentration of 70–160 μg/ml (Hughes-Jones, 1977; Schuller and Helary, 1983; Dillon et al., 2009), but like other components of innate immunity, it is produced in higher concentrations at inflammatory sites, predominantly by macrophages and dendritic cells (DCs; Bensa et al., 1983; Schwaeble et al., 1995; Kaul and Loos, 2001; Vegh et al., 2003; Castellano et al., 2004). Approximately 80% of C1q contained in the plasma is associated with the Ca2+-dependent C1r2–C1s2 tetramer (360 kDa) to form the multimeric C1 complex, the first component of the classical complement system (Muller-Eberhard and Kunkel, 1961; Lepow et al., 1963; Calcott and Muller-Eberhard, 1972; Reid and Porter, 1976; Reid et al., 1982; Sjoholm et al., 1985; Weiss et al., 1986). However, while C1q is traditionally known as the recognition unit of the classical complement pathway, it also plays a role in the antibody-dependent adaptive and antibody-independent innate arm of immunity.
Deficiencies of any of the components of the C1 complex (C1q, C1s, or C1r) are considered to be strong susceptibility factors for systemic lupus erythematosus (SLE; Lee et al., 1978; Steinsson et al., 1983; Fremeaux-Bacchi et al., 1996; Walport et al., 1998; Cortes-Hernandez et al., 2004; Ghebrehiwet and Peerschke, 2004; Amano et al., 2008). In addition, a large portion of SLE patients have high affinity autoantibodies to C1 complex components (He and Lin, 1998; Walport et al., 1998; Walport, 2002). Although most studies suggest that it is the failure to properly clear apoptotic cells in the absence of C1q that results in autoimmunity (Hurst et al., 1984; Walport et al., 1998), recent observations challenge this idea. For example, disruption of other apoptotic uptake processes, such as those mediated by CD14 (Devitt et al., 2004), β3 or β5 integrin (Lucas et al., 2006), and mannose-binding lectin (Stuart et al., 2005), all result in accumulation of apoptotic bodies without triggering autoimmunity. Therefore, in addition to recognition and clearance of apoptotic bodies, C1q and the C1 complex play an as yet unidentified role in regulating the acquired immune response and thus maintaining tolerance. Since immature DCs (iDCs) and macrophages are the most abundant sources of C1q, we hypothesize that local production of C1q by these cells serves as an autocrine signaling mechanism, which induces cellular responses by iDCs, macrophages, and their precursors (i.e., monocytes) through interactions with surface receptors (Hosszu et al., 2010).
Biosynthesis of the multimolecular C1 complex and its regulator, C1 inhibitor, by human monocytes has been previously shown (Bensa et al., 1983; Randazzo et al., 1985; Tenner and Volkin, 1986; Drouet and Reboul, 1989; Gulati et al., 1993; Lu et al., 1996; Moosig et al., 2006). Recently, we have found that monocytes freshly isolated from peripheral blood (PB) carry C1q on their surface (Hosszu et al., 2010). Since monocytes serve a critical role in adaptive and innate immunity not only by serving as precursors of macrophages and myeloid DCs, but also by their function in phagocytosis, antigen processing, and presentation, as well as secretion of cytokines, we sought to further explore the functional significance of such extracellular C1q on these cells. The data presented here show that not only C1q, but the entire multimolecular C1 complex is present on the surface of monocytes and suggest a novel role for C1q within this complex as a molecular sensor of “danger” signals. The fact that surface expressed C1 complex on U937 cells was able to activate the classical complement cascade suggests that surface-associated C1 complex initiates complement fixation in the presence of an extracellular Ag and thus supports an immediate response to pathogens under inflammatory conditions.
Materials and Methods
Chemicals and Reagents
The following reagents and chemicals were purchased or obtained from the sources indicated: Lymphoprep (Axis-Shield, Oslo, Norway); cell lysis buffer (Cell Signaling Technology, Danvers, MA, USA); C1q (Comptech, Tyler, TX, USA); heat inactivated fetal bovine serum (FBS; Hyclone, Logan, UT, USA); 100x Penicillin/Streptomycin, RPMI 1640 (Gibco-Invitrogen, Grand Island, NY, USA); Detoxi-Gel Endotoxin Removing Gel, p-nitrophenyl phosphate (pNPP), EZ-Link Sulfo-NHS-LC-Biotin, Protein A agarose, F(ab′)2 Micro Preparation Kit (Pierce, Rockford, IL, USA); Complete protease inhibitor tablets (Roche Applied Science, Indianapolis, IN, USA); human IgG, formalin (Sigma-Aldrich, St. Louis, MO, USA). Antibodies used were against: CD14 (BioLegend, San Diego, CA, USA); CD16, CD11c (Becton–Dickinson, Mountain View, CA, USA); cC1qR (Serotec, Raleigh, NC, USA); monoclonal (mAb) anti-globular and polyclonal antibody (pAb) against C1q, C4d (Quidel, Santa Clara, CA, USA); fluorescein isothiocyanate (FITC) conjugated goat anti-mouse IgG F(ab′)2 or sheep anti-rabbit IgG F(ab′)2 (Invitrogen); and alkaline phosphatase (AP)–conjugated rabbit anti-goat IgG (Pierce).
Because of the sensitivity of monocytes to endotoxin and contaminating pathogen-associated molecular patterns (PAMPs), highly purified and endotoxin-poor reagents and proteins were purchased when possible. In addition, special efforts were made to ensure that proteins used in the cell culture were low in endotoxin by passage over Detoxi-Gel columns using pyrogen-free buffers. The endotoxin removal efficiency in one passage is ≥99% (Pierce).
Monoclonal and Polyclonal Antibodies to C1s, C1r, C1 Inhibitor, C4, and Factor B
The production, characterization, and purification of various monoclonal and polyclonal antibodies to the gC1qR protein, as well as to various peptides derived from the molecule have been previously described (Ghebrehiwet et al., 1996; Hosszu et al., 2010). Similarly, rabbit or goat pAbs to C1s, C1r, C1 inhibitor, C4, and factor B were generated in our laboratory and constitute part of our complement antibody bank. None of the antibodies cross-reacted with bovine orthologs, as verified by enzyme-linked immunosorbent assay (ELISA; data not shown). F(ab′)2 IgG fragments were prepared using a Pierce kit according to the manufacturer’s instructions.
Isolation of Peripheral Blood Monocytes
Peripheral blood monocytes were isolated according to previously published methods (Hosszu et al., 2010). Briefly, PB mononuclear cells were purified from whole blood using Lymphoprep density gradient centrifugation according to the manufacturer’s instructions. Monocytes were further purified by adhesion selection on polystyrene plates (1 h, 37°C). Apart from monocyte/DCs, all other contaminating cell constituents (lymphocytes, platelets, red blood cells, polymorphonuclear cells) were absent from the culture, as assessed by scatter profiles and microscopic observations. Monocyte subset gates were calculated on the basis of forward and side light scatter profiles and cell surface display patterns of myeloid cell/DC associated markers (CD14, CD16, and CD11c). The research was done in accordance with the NIH guidelines for human subjects and approved by the Stony Brook institutional review board (#02626-5), and all human participants donating blood gave written informed consent.
Immunofluorescence Microscopy
Cell surface staining of freshly obtained monocytes was performed as follows. The cells were isolated as described above, then washed twice in PBA staining buffer (PBS containing 1% BSA and 0.01% NaN3). Non-specific binding was blocked by incubating the cells with 1 mg/ml human IgG in 100 μl PBA/1 × 106 cells (30 min, 4°C) and primary Abs or the appropriate isotype-matched controls were then added to the cells (30 min, 4°C). The cells were washed in PBA buffer and further incubated with FITC conjugated goat anti-mouse IgG F(ab′)2 (30 min, 4°C). The cells were washed in cold PBA, fixed for 5 min in 1% PBS buffered formalin, and washed with cold PBA. The cells were finally concentrated by centrifugation, and applied to microscope slides, and allowed to air dry. Subsequently cover slips were mounted onto the slides using Immuno-mount mounting solution. The slides were viewed on a Zeiss Axiovert 200 M digital deconvolution microscope, followed by image capture at 63× (oil) magnification and analysis with AxioVision 4.5 software. The results represent the analysis of 100 cells in three independent experiments.
Flow Cytometry Assisted Analysis of Cell Surface Markers
Cells were removed from the culture daily and washed twice in PBA staining buffer. Non-specific binding was blocked by incubating the cells with 1 mg/ml human IgG in 100 μl PBA/1 × 106 cells (30 min, 4°C) and primary Abs or the appropriate isotype-matched controls were then added to the cells (30 min, 4°C). The cells were then washed in PBA buffer and further incubated with FITC conjugated goat anti-mouse IgG F(ab′)2 or sheep anti-rabbit IgG F(ab′)2 (30 min, 4°C). The cells were washed in cold PBA, fixed in 1% formalin, and assessed by flow cytometric analysis using FACSCalibur (Becton–Dickinson, Mountain View, CA, USA). For each analysis, 10,000 events were collected and the data obtained was analyzed using CellQuest Pro software (BD).
Detection of Cell Surface C1q by ELISA
For detection of surface-specific C1q, U937 cells were first surface biotinylated using sulfo-NHS-LC-biotin according to the manufacturer’s instructions, followed by cell lysis in a commercially available cell lysis buffer according to the manufacturer’s instructions. The presence of C1q was tested in each sample by ELISA. To detect surface C1q, surface biotinylated cell lysates were captured on microtiter plates coated with streptavidin or BSA (negative control) at a concentration of 5 μg/ml in coating buffer (100 mM Na2CO3/NaHCO3, pH 9.6). Non-specific binding sites were blocked using 3% heat inactivated (56°C, 90 min) BSA in PBS (1 h, 37°C). In our experience, we have found that even the highest grade BSA can contain trace amounts of C1q, therefore we routinely use heat inactivated and microfiltered BSA. Because bovine complement is unusually resistant to heat inactivation, a 90-min incubation at 56°C is necessary to ensure destruction of C1q activity. Highly purified serum C1q was used as positive control. BSA was used as a negative control. Next, a C1q-specific polyclonal Ab was added (1 h, 37°C), followed by detection of the reaction with AP–conjugated rabbit anti-goat IgG (1 h, 37°C). All these steps were performed in ELISA buffer (PBS, 0.1% BSA, 0.05% Tween 20) and each step was followed by three washes with PBS/0.05% Tween 20. Enzyme activity was assessed by the addition of the substrate pNPP. The optical density (OD) at 405 nm was measured using a microplate reader at various time points.
Western Blotting
U937 cell lysates were prepared using a non-ionic cell lysis buffer (20 mM Tris–HCL (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1% Triton) supplemented with a protease inhibitor cocktail, according to the manufacturer’s specifications. Proteins in the U937 whole cell lysates were separated by SDS-PAGE using a 12% gel, transferred to a nitrocellulose membrane, blocked in 5% milk (1 h, RT), and incubated with primary antibodies (O/N, 4°C). The membrane was washed three times in TBS containing 0.05% Tween, and incubated with secondary Abs conjugated to HRP (1 h, RT). The blot was developed using SuperSignal West Pico-ECL detection reagent according to the manufacturer’s specifications and visualized by exposing to Kodak Scientific Imaging X-OMAT film.
Detection of Complement Activation by C4 Deposition
Poly-L-Lysine-coated microtiter plate wells (50 μl/well, 10 μg/ml in PBS) were incubated with 50 μl of U937 cells (2 × 106 cells/ml in PBS; 30 min, RT). The plates were spun (5 min, 400 × g), and the cells fixed using 50 μl of 0.5% glutaraldehyde in PBS (30 min, RT). Next, the cells were washed twice in PBS and non-specific binding was blocked using 0.1% BSA + 100 mM glycine (30 min, RT). The BSA for coating was always heat inactivated (90 min, 56°C) to inactivate trace amounts of bovine C1q and microfiltered to remove large aggregates. At this point the cells can be used immediately or stored in blocking solution at −20°C until needed. On the day of the experiment the cells were thawed and the blocking solution aspirated. Then 100 μl Clq depleted serum was added (1:10 dilution in 0.1% gelatin containing veronal buffer, GVB) ±1 mg/ml aggregated IgG ±200 ng/ml C4 (1 h, 37°C). After washing the cells with GVB, complement activation was assessed by incubation with biotinylated anti-C4d to measure the C4d generated (1 h, 37°C). After incubation the cells were again washed in GVB, and incubated with AP-conjugated neutrAvidin (1 h, RT), followed by the addition of the substrate pNPP. The OD at 405 nm was measured using a microplate reader at various time points. C1q-depleted serum alone was used as the negative control.
Detection of U937 Cell Surface-Associated C1 Complex Activity
U937 cells (2 × 106 cells/sample) were washed twice in PBS. To the cells we added 1 μg purified C2 + 1 μg purified C4 + 5 μg aggregated IgG ± 1 μg C1 in 0.1% GVB (2 h, 37°C). After incubation the supernatant was collected and C4 cleavage was assessed by SDS-PAGE and Western blotting.
Detection of Complement Activation by Hemolytic Assay
The activity of U937-expressed C1q was assessed in the following manner. First U937 cells (2 × 106/sample) were washed in PBS, mixed, and incubated with human sera diluted 1:10 in GVB ± 5 μg aggregated IgG (1 h, 37°C, in a shaking water bath). After incubation, antibody-sensitized sheep erythrocytes (EAs) were added to each sample and further incubated (1 h, 37°C). The cells were then centrifuged (5 min, 800 rpm) and the released hemoglobin measured in the supernatants spectrophotometrically at 415 nm in order to assess the residual classical pathway activity. The total hemolysis was obtained using sheep erythrocytes lysed in water. C1q-depleted human sera reconstituted with increasing concentrations of C1q (0–100 ng) were used to develop a standard curve. All hemolysis data points were presented as the percentage of the complete hemolysis, calculated with the ratio between the value measured for each sample and that registered for the total hemolysis.
Statistical Analysis
Student t-tests were performed using statistical software (Excel; Microsoft, Redmond, WA, USA). A value of p ≤ 0.05 was considered to be a significant difference. (n values represent separate experiments performed using different donors.)
Results
C1q is Present on the Surface of Human Monocytes and U937 Cells
To investigate the role of C1q on monocytes, we first analyzed its surface expression on PB monocytes and on the monocyte-derived U937 cell line. PB monocytes from healthy individuals were isolated and subjected to indirect cell surface staining and microscopic analyses. Our results confirmed the presence of C1q on monocytes, and revealed a punctate pattern of C1q evenly distributed over the cellular membrane (Figure 1A; right panel), while isotype-matched non-immune Ab gave little or no signal (Figure 1A; left panel). In order to gain insight into the role of surface C1q on monocytes, we employed the commercially available monocyte-derived U937 cell line as a surrogate. As expected, the presence of C1q on the surface of these cells was verified by antigen-capture ELISA experiments. Using cell membranes isolated from U937 cells, we showed the presence of C1q (Figure 1B), while an isotype-matched non-immune rabbit Ab showed negligible staining. We further confirmed these results using flow cytometric analysis, firmly establishing the presence of surface C1q on U937 cells (Figures 1C,D). These data are consistent with previously published results showing that C1q is present on the surface of monocytes (Hosszu et al., 2010) and U937 cells (Arvieux et al., 1984).
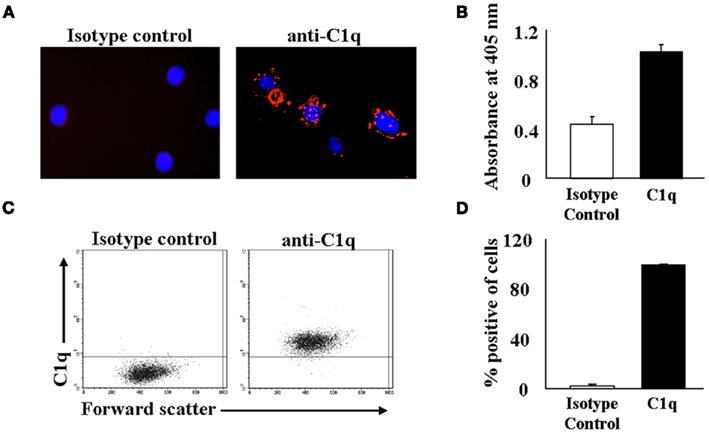
Figure 1. Monocytes display C1q on their surface. (A) C1q is present on the surface of mononuclear cells. Fresh mononuclear cells were stained with anti-C1q or an isotype control and detected using PE-conjugated secondary Ab. Cells were applied to microscope slides and viewed on a Zeiss Axiovert 200 M digital deconvolution microscope at 63× (oil). Monocytes were identified by co-staining for monocyte specific markers (not shown) in addition to anti-C1q. (B) C1q is expressed on the surface of U937 cells. Biotinylated U937 cell membrane proteins were captured on a streptavidin-coated ELISA plate and the binding of C1q was detected using pAb RaH C1q. Rabbit IgG was used as an isotype control (n = 2). (C+D) C1q is present on the surface of U937 cells as assessed by flow cytometry. U937 cells were stained with GaH C1q followed by secondary Ab conjugated to Alexa 488. One experiment is shown (C), representative of six experiments (D).
Macromolecular C1 Complex is Present on the Surface of Human Monocytes
Since most of the C1q – save approximately 20% – circulates in plasma as part of the multimeric C1 complex (C1q1C1r2–C1s2), we hypothesized that the C1q detected on the monocyte surface may be part of the C1 complex. To test this premise, monocytes were analyzed for surface expression of C1s and C1r, using flow cytometry. In addition, two other complement proteins, Factor B and C1 inhibitor, were assessed as well. Using cell surface staining and flow cytometric analysis, we found that in addition to C1q, nearly all monocytes were positive for the presence of both C1r and C1s (Figures 2A,B). Furthermore, C1-Inh was also on the cells, possibly ensuring that unwarranted classical complement activation does not take place through the surface bound C1 complex. We were also able to detect the components of the C1 complex on U937 cells by flow cytometric analysis (Figures 2C,D). Additionally, Western blotting confirmed the presence of C1s and C1r in U937 cell lysates (Figure 2E). Not surprisingly, Factor B, a complement protein synthesized by monocytes (Hogasen et al., 1995), was also found on the cells. Next we investigated whether C1s, C1r, and C1q assemble into the C1 complex on U937 cells. As Figure 2F shows anti-C1q conjugated agarose beads were able to pull down C1s and C1r from U937 whole cell lysates, confirming that the entire C1 complex is present on the cell surface.
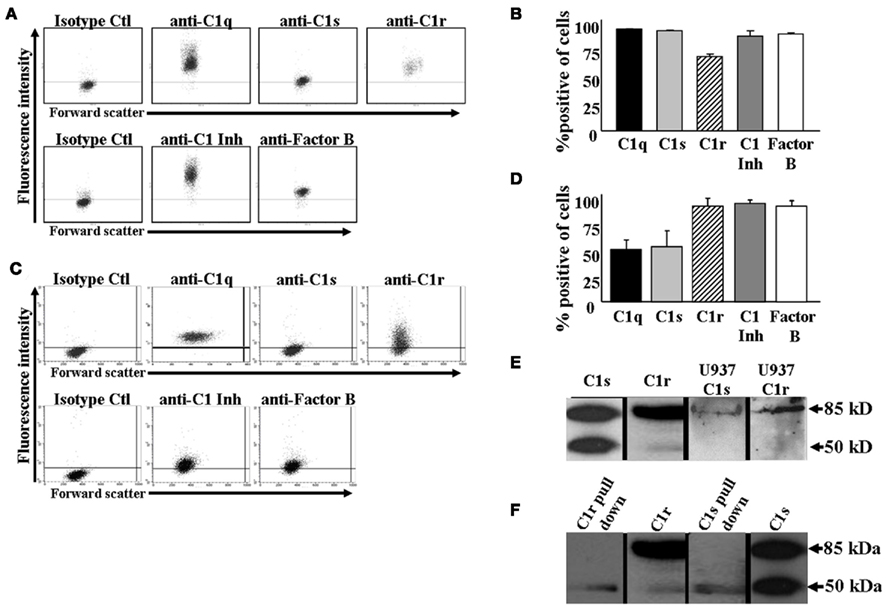
Figure 2. C1 complex is located on the surface of monocytes. (A+B) C1 complex is present on the surface of human monocytes as assessed by flow cytometry. Human monocytes were isolated from whole blood and stained with GaH C1s, C1r, C1q, or RaH C1-Inh, Factor B Ab followed by secondary Ab conjugated to Alexa 488. One experiment is shown (A), representative of three experiments (B). (C+D) C1 complex is present on the surface of U937 cells as assessed by flow cytometry. U937 cells were stained with GaH C1s, C1r, C1q, or RaH C1-Inh, Factor B Ab followed by secondary Ab conjugated to Alexa 488. One experiment is shown (C), representative of six experiments (D). (E) C1s and C1r are produced by U937 cells. U937 whole cell lysates were subjected to SDS-PAGE and subsequently visualized by Western blotting using GaH C1s and RaH C1r, followed by HRP-conjugated secondary Abs. Purified C1s and C1r were run as positive controls (n = 3). (F) C1 complex is formed on U937 cells. Anti-C1q conjugated agarose beads were incubated with U937 whole cell lysates. The bound proteins were subjected to SDS-PAGE followed by Western blotting using anti-C1s and anti-C1r and HRP-conjugated secondary Abs. Purified C1s and C1r were run as positive controls (n = 2).
To examine whether the level of these surface molecules decreases over time, we cultured U937 cells in serum-free (SF) media for 48 h, ensuring that the surface molecules could not be replenished from the media over time. Interestingly, we detected no significant change in the cell surface levels of C1q or C1r after 48 h, while C1s, Factor B, and C1 inhibitor all significantly decreased on the surface of U937 cells (Figures 3A,B). To investigate whether the surface molecule loss of C1s, Factor B, and C1 inhibitor was due to endocytosis or external release, we determined the levels of all five molecules in the SF supernatant of U937 cell cultures by Western blotting. Figure 3C shows that C1s, Factor B, and C1 inhibitor were all readily detected in the supernatants, while no detectable quantities of C1q or C1r were released over the 48-h culture period.
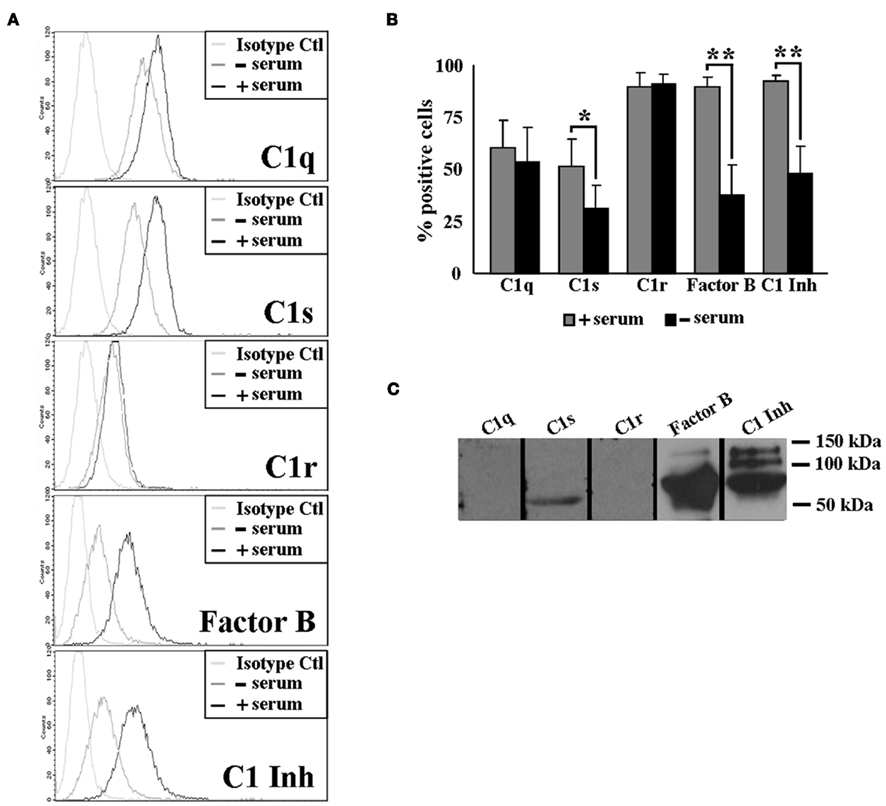
Figure 3. Surface levels of C1 complex components decrease over time on U937 cells. (A+B) C1s, Factor B, and C1 inhibitor are lost from the surface of U937 cells after 48 h. U937 cells were grown in RPMI + 10% FBS (+ serum) or in Macrophage Serum-Free Media (-serum) for 48 h and stained with GaH C1q, C1s, C1r, or RaH C1-Inh, Factor B and isotype-matched controls followed by secondary Ab conjugated to Alexa 488. The cells were analyzed by flow cytometry. One experiment is shown (A), representative of three experiments. (B) *p ≤ 0.05; **p ≤ 0.01 (C) C1 complex components are released into the supernatant of U937 cells. Supernatants of U937 cell cultures were subjected to SDS-PAGE and subsequently visualized by Western blotting using the same primary Abs as above, followed by HRP-conjugated secondary Abs (n = 3).
C1 Complex on U937 Cells Triggers Complement Activation
Next we sought to determine the functional significance of the C1q on U937 cells by assessing the ability of the surface C1q to activate the classical complement pathway. U937 cells attached to microplate wells were incubated with C1q-depleted human sera, and mixed with aggregated IgG and C4, followed by detection with an anti-C4d mAb. As expected, the C1q on the surface of U937 cells was able to initiate complement (Figure 4A) as evidenced by the formation of C4d. Aggregated IgG alone was able to produce C4d, but to a lesser degree than when additional C4 was supplemented to the reaction, indicating the rapid consumption of the C4 available in the system (Figure 4A).
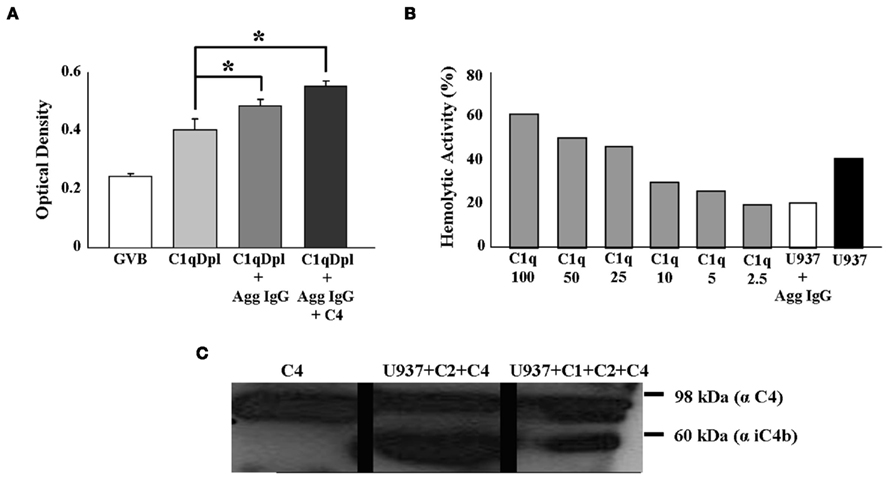
Figure 4. Surface-associated C1 complex of U937 cells can initiate complement activation. (A) C1q on the surface of U937 cells triggers the complement cascade of the classical pathway. U937 cells were attached to a microtiter plate and incubated with a 1:10 dilution of Clq depleted serum (C1qDpl) ± aggregated IgG (Agg IgG) ± C4. Activation of complement was detected by sequential addition and incubation of biotinylated anti-C4d Ab, alkaline phophatase conjugated neutrAvidin, followed by pNPP. C1q-depleted serum was used as a negative control (n = 3) *p < 0.05. (B) Surface C1q on U937 cells activates complement in a hemolytic assay. 2 × 106 U937 cells were incubated with C1q-depleted serum ± aggregated IgG (Agg IgG) + Ab-sensitized sheep erythrocytes (EAs). The residual complement activity was assessed by addition of EAs. C1q-depleted human serum reconstituted with increasing concentrations of C1q (0–100 ng) was used for the standard curve and as a positive control (n = 2). (C) C1 complex on U937 cells is able to cleave C4. U937 cells were incubated with purified C2 and C4 + Agg IgG for 2 h at 37°C. The supernatants were collected, the proteins were separated on SDS-PAGE, followed by Western blotting with anti-C4 and HRP-conjugated secondary Ab. C1 complex was added to the cell mixture as positive control (n = 2).
Assessment of in vitro hemolytic activity of the C1q on U937 cells using sheep erythrocytes was also performed (Figure 4B). Cells pre-incubated with C1q-depleted human serum induced depletion of total complement hemolytic activities (Figure 4B). The amount of hemolytically active surface C1q was quantified using the dose-dependent curve, whereby increasing concentrations of C1q were added to C1q-depleted human sera and complement pathway activity was measured. Calculated from the linear progression curve, the hemolytic activity of 2 × 106 U937 cells was equivalent to 20 ng C1q (Figure 4B). Based on this number, each U937 cell has 10 fg C1q on its surface, which represents approximately 10,000 C1q molecules/cell. These experiments indicate that U937 cell surface-associated C1q is able to contribute to the activation of the classical pathway.
Next we sought to determine whether the entire C1 complex was functional on the surface of these cells. When U937 cells were mixed with purified C2, C4, and aggregated IgG, the C1 complex on U937 cells was able to cleave C4, as evidenced by the inactivated C4b cleavage product, iC4b (Figure 4C). Taken together these experiments suggest that the C1 complex is fully assembled on the surface of U937 cells and it is able to initiate the classical complement cascade.
Discussion
The consensus that almost all of the complement proteins are synthesized in the liver has been gradually challenged over the years by findings that demonstrate extrahepatic synthesis of a wide array of complement proteins and their regulators (Laufer et al., 2001). In particular, the multimolecular C1 complex and its regulator, C1 inhibitor, have been shown to be synthesized by human monocytes and the monocyte-derived U937 cell line (Bensa et al., 1983; Randazzo et al., 1985; Tenner and Volkin, 1986; Drouet and Reboul, 1989; Gulati et al., 1993; Lu et al., 1996; Moosig et al., 2006). Our previously published data have shown that C1q is present on the surface of circulating monocytes (Hosszu et al., 2010). Since monocytes produce all components of the C1 complex, here we sought to investigate whether the cell surface C1q is part of the C1 macromolecular complex. Our data show, that contrary to the previously held postulate that macrophages and maturing DCs, but not monocytes express C1q (Kaul and Loos, 2001; Vegh et al., 2003), freshly isolated PB monocytes and U937 cells also express not only C1q, but also the other subcomponents of C1, namely C1r and C1s, as well as the regulator, C1-Inh.
The presence of C1-Inh on the surface is significant, as it intimates that C1-Inh closely monitors the role of the C1 complex in a sterile or infectious microenvironment and inflammation. Monocyte-associated C1-Inh may regulate how C1q, which binds to a plethora of microbial or non-microbial activators at inflammatory sites, can communicate with DCs or T cells to enhance or abort immune responses. Under physiological conditions complement activation is tightly regulated to prevent complement-mediated tissue damage. The presence of C1-Inh with the C1 complex probably represents a primary level of protection against autoimmunity.
Our data indicate that the C1 complex on the surface of U937 cells is able to initiate the classical complement cascade. When U937 cells – as a source of C1 complex – were incubated with purified C2 and C4, we were able to detect iC4b, an inactive cleavage product of C4b in the cell supernatants. The amount of activity initiated by 2 × 106 U937 cells was equivalent to that of approximately 20 ng of purified C1q, as measured by in vitro hemolytic activity using sheep erythrocytes. These data suggest that under pathogenic conditions, pathogen associated, or self-derived molecules can trigger complement activation in close proximity to the monocyte surface. Self-damage is controlled by the presence and quick removal of C1r2–C1s2 by C1-Inh, and in turn by the rapid capture and proteolysis of C2 and C4. Thus, C1 complex on monocytes may have implications for immediate response to pathogens under inflammatory conditions, and C1q and the C1 complex may act as an “early warning” autocrine sensor of danger and assist to instantaneously “chaperone” extracellular Ag for uptake into monocytes.
Under physiological conditions, surface-associated C1 complex – similar to surface-associated C1q (Hosszu et al., 2010) – may also function as an early signaling molecule on monocytes and may reflect an as yet unidentified regulatory mechanism that sustains innate immune functions.
Although previous reports have documented that PB monocytes, including those obtained from C1-Inh deficient patients are capable of producing C1-Inh (40% of normal; Lappin et al., 1989), and our own work has demonstrated the synthesis of this protein by the monocyte-like cell line U937 (Randazzo et al., 1985), what remains to be determined is how the C1 complex and C1-Inh are anchored on the surface. Indeed, real-time antigen-capture assay (not shown) has shown that U937 cells, grown in serum-free culture medium can produce measurable amounts of C1q, C1r, and C1s, indicating that these cells actively synthesize and secrete these proteins. Although experiments are undergoing to confirm such a scenario, we speculate that the most likely anchoring protein in this complex would be C1q, which could be anchored by a short transmembrane collagen piece similar to that described earlier (Kaul and Loos, 1995) on the surface of monocyte-derived macrophages. This transiently expressed C1 complex on the monocyte surface may be later cleaved off and released into the pericellular milieu.
Alternatively, circulating C1 complex may be captured from blood by monocytes via cell surface receptors. Two C1q receptors, gC1qR and cC1qR (calreticulin) have been well described on the monocyte surface (Malhotra, 1993; Ghebrehiwet et al., 1994). Additionally, the presence of other C1q-binding surface molecules, such as SIGN-like C-type lectins and beta-1 integrins have been shown (Tuckwell et al., 1996; Kang et al., 2006). Circulating C1 complex may be captured from blood and retained on the cell surface through these receptors.
In summary, our results show that the entire multimolecular C1 complex is present on the surface of monocytes and U937 cells, and suggests that surface-associated C1 complex is actively able to fix complement in the presence of an extracellular antigen, thus providing antigenic material for cellular uptake in the vicinity of the cell. This suggests a novel role for locally synthesized C1q, and since monocytes are critical in the adaptive and innate arms of immunity, the presence and functional activity of the C1 complex on their surface may indicate a new form of a molecular sensor of “danger” signals that could provide an immediate local response to either pathogen- or damage-associated molecular ligands.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
cC1qR/CR, receptor for the collagen tail of C1q (calreticulin); gC1q, globular heads of C1q; gC1qR, receptor for the globular heads of C1q; pAb, polyclonal antibody.
References
Amano, M. T., Ferriani, V. P., Florido, M. P., Reis, E. S., Delcolli, M. I., Azzolini, A. E., Assis-Pandochi, A. I., Sjoholm, A. G., Farah, C. S., Jensenius, J. C., and Isaac, L. (2008). Genetic analysis of complement C1s deficiency associated with systemic lupus erythematosus highlights alternative splicing of normal C1s gene. Mol. Immunol. 45, 1693–1702.
Arvieux, J., Reboul, A., Bensa, J. C., and Colomb, M. G. (1984). Characterization of the C1q receptor on a human macrophage cell line, U937. Biochem. J. 218, 547–555.
Bensa, J. C., Reboul, A., and Colomb, M. G. (1983). Biosynthesis in vitro of complement subcomponents C1q, C1s and C1 inhibitor by resting and stimulated human monocytes. Biochem. J. 216, 385–392.
Bobak, D. A., Gaither, T. A., Frank, M. M., and Tenner, A. J. (1987). Modulation of FcR function by complement: subcomponent C1q enhances the phagocytosis of IgG-opsonized targets by human monocytes and culture-derived macrophages. J. Immunol. 138, 1150–1156.
Brodsky-Doyle, B., Leonard, K. R., and Reid, K. B. (1976). Circular-dichroism and electron-microscopy studies of human subcomponent C1q before and after limited proteolysis by pepsin. Biochem. J. 159, 279–286.
Calcott, M. A., and Muller-Eberhard, H. J. (1972). C1q protein of human complement. Biochemistry 11, 3443–3450.
Castellano, G., Woltman, A. M., Nauta, A. J., Roos, A., Trouw, L. A., Seelen, M. A., Schena, F. P., Daha, M. R., and van Kooten, C. (2004). Maturation of dendritic cells abrogates C1q production in vivo and in vitro. Blood 103, 3813–3820.
Cortes-Hernandez, J., Fossati-Jimack, L., Petry, F., Loos, M., Izui, S., Walport, M. J., Cook, H. T., and Botto, M. (2004). Restoration of C1q levels by bone marrow transplantation attenuates autoimmune disease associated with C1q deficiency in mice. Eur. J. Immunol. 34, 3713–3722.
Devitt, A., Parker, K. G., Ogden, C. A., Oldreive, C., Clay, M. F., Melville, L. A., Bellamy, C. O., Lacy-Hulbert, A., Gangloff, S. C., Goyert, S. M., and Gregory, C. D. (2004). Persistence of apoptotic cells without autoimmune disease or inflammation in CD14-/- mice. J. Cell Biol. 167, 1161–1170.
Dillon, S. P., D’Souza, A., Kurien, B. T., and Scofield, R. H. (2009). Systemic lupus erythematosus and C1q: a quantitative ELISA for determining C1q levels in serum. Biotechnol. J. 4, 1210–1214.
Drouet, C., and Reboul, A. (1989). Biosynthesis of C1r and C1s subcomponents. Behring Inst. Mitt. 80–88.
Fremeaux-Bacchi, V., Weiss, L., Demouchy, C., Blouin, J., and Kazatchkine, M. D. (1996). Autoantibodies to the collagen-like region of C1q are strongly associated with classical pathway-mediated hypocomplementemia in systemic lupus erythematosus. Lupus 5, 216–220.
Ghebrehiwet, B., Lim, B. L., Peerschke, E. I., Willis, A. C., and Reid, K. B. (1994). Isolation, cDNA cloning, and overexpression of a 33-kD cell surface glycoprotein that binds to the globular “heads” of C1q. J. Exp. Med. 179, 1809–1821.
Ghebrehiwet, B., Lu, P. D., Zhang, W., Lim, B. L., Eggleton, P., Leigh, L. E., Reid, K. B., and Peerschke, E. I. (1996). Identification of functional domains on gC1Q-R, a cell surface protein that binds to the globular “heads” of C1Q, using monoclonal antibodies and synthetic peptides. Hybridoma 15, 333–342.
Ghebrehiwet, B., and Peerschke, E. I. (2004). Role of C1q and C1q receptors in the pathogenesis of systemic lupus erythematosus. Curr. Dir. Autoimmun. 7, 87–97.
Gulati, P., Lemercier, C., Guc, D., Lappin, D., and Whaley, K. (1993). Regulation of the synthesis of C1 subcomponents and C1-inhibitor. Behring Inst. Mitt. 196–203.
He, S., and Lin, Y. L. (1998). In vitro stimulation of C1s proteolytic activities by C1s-presenting autoantibodies from patients with systemic lupus erythematosus. J. Immunol. 160, 4641–4647.
Hogasen, A. K., Hestdal, K., Hogasen, K., and Abrahamsen, T. G. (1995). Transforming growth factor beta modulates C3 and factor B biosynthesis and complement receptor 3 expression in cultured human monocytes. J. Leukoc. Biol. 57, 287–296.
Hosszu, K. K., Santiago-Schwarz, F., Peerschke, E. I., and Ghebrehiwet, B. (2010). Evidence that a C1q/C1qR system regulates monocyte-derived dendritic cell differentiation at the interface of innate and acquired immunity. Innate Immun. 16, 115–127.
Hughes-Jones, N. C. (1977). Functional affinity constants of the reaction between 125I-labelled C1q and C1q binders and their use in the measurement of plasma C1q concentrations. Immunology 32, 191–198.
Hurst, N. P., Nuki, G., and Wallington, T. (1984). Evidence for intrinsic cellular defects of “complement” receptor-mediated phagocytosis in patients with systemic lupus erythematosus (SLE). Clin. Exp. Immunol. 55, 303–312.
Kang, Y. S., Do, Y., Lee, H. K., Park, S. H., Cheong, C., Lynch, R. M., Loeffler, J. M., Steinman, R. M., and Park, C. G. (2006). A dominant complement fixation pathway for pneumococcal polysaccharides initiated by SIGN-R1 interacting with C1q. Cell 125, 47–58.
Kaul, M., and Loos, M. (1995). Collagen-like complement component C1q is a membrane protein of human monocyte-derived macrophages that mediates endocytosis. J. Immunol. 155, 5795–5802.
Kaul, M., and Loos, M. (2001). Expression of membrane C1q in human monocyte-derived macrophages is developmentally regulated and enhanced by interferon-gamma. FEBS Lett. 500, 91–98.
Kuis, W., de Graeff-Meeder, E. R., Rijkers, G. T., Stoop, J. W., and Zegers, B. J. (1988). Disorders in humoral defense: clinical aspects, diagnosis and therapy. Tijdschr. Kindergeneeskd. 56, 184–192.
Lappin, D. F., McPhaden, A. R., Yap, P. L., Carter, P. E., Birnie, G. D., Fothergill, J. E., and Whaley, K. (1989). Monocyte C1-inhibitor synthesis in patients with C1-inhibitor deficiency. Eur. J. Clin. Invest. 19, 45–52.
Laufer, J., Katz, Y., and Passwell, J. H. (2001). Extrahepatic synthesis of complement proteins in inflammation. Mol. Immunol. 38, 221–229.
Lee, S. L., Wallace, S. L., Barone, R., Blum, L., and Chase, P. H. (1978). Familial deficiency of two subunits of the first component of complement. C1r and C1s associated with a lupus erythematosus-like disease. Arthritis Rheum. 21, 958–967.
Leist-Welsh, P., and Bjornson, A. B. (1982). Immunoglobulin-independent utilization of the classical complement pathway in opsonophagocytosis of Escherichia coli by human peripheral leukocytes. J. Immunol. 128, 2643–2651.
Lepow, I. H., Naff, G. B., Todd, E. W., Pensky, J., and Hinz, C. F. (1963). Chromatographic resolution of the first component of human complement into three activities. J. Exp. Med. 117, 983–1008.
Lu, J., Le, Y., Kon, O. L., Chan, J., and Lee, S. H. (1996). Biosynthesis of human ficolin, an Escherichia coli-binding protein, by monocytes: comparison with the synthesis of two macrophage-specific proteins, C1q and the mannose receptor. Immunology 89, 289–294.
Lucas, M., Stuart, L. M., Zhang, A., Hodivala-Dilke, K., Febbraio, M., Silverstein, R., Savill, J., and Lacy-Hulbert, A. (2006). Requirements for apoptotic cell contact in regulation of macrophage responses. J. Immunol. 177, 4047–4054.
Malhotra, R. (1993). Collectin receptor (C1q receptor): structure and function. Behring Inst. Mitt. 254–261.
Moosig, F., Damm, F., Knorr-Spahr, A., Ritgen, M., Zeuner, R. A., Kneba, M., Ernst, M., and Schroder, J. O. (2006). Reduced expression of C1q-mRNA in monocytes from patients with systemic lupus erythematosus. Clin. Exp. Immunol. 146, 409–416.
Muller-Eberhard, H. J., and Kunkel, H. G. (1961). Isolation of a thermolabile serum protein which precipitates gamma-globulin aggregates and participates in immune hemolysis. Proc. Soc. Exp. Biol. Med. 106, 291–295.
Pickering, M. C., Macor, P., Fish, J., Durigutto, P., Bossi, F., Petry, F., Botto, M., and Tedesco, F. (2008). Complement C1q and C8beta deficiency in an individual with recurrent bacterial meningitis and adult-onset systemic lupus erythematosus-like illness. Rheumatology (Oxford) 47, 1588–1589.
Prellner, K., Sjoholm, A. G., Harsten, G., Heldrup, J., Kalm, O., and Kornfalt, R. (1989). C1q and C1 subcomponent complexes in otitis-prone and non-otitis-prone children. A prospective study of children during their first years of life. Acta Paediatr. Scand. 78, 911–917.
Randazzo, B. P., Dattwyler, R. J., Kaplan, A. P., and Ghebrehiwet, B. (1985). Synthesis of C1 inhibitor (C1-INA) by a human monocyte-like cell line, U937. J. Immunol. 135, 1313–1319.
Reid, K. B., Gagnon, J., and Frampton, J. (1982). Completion of the amino acid sequences of the A and B chains of subcomponent C1q of the first component of human complement. Biochem. J. 203, 559–569.
Reid, K. B., and Porter, R. R. (1976). Subunit composition and structure of subcomponent C1q of the first component of human complement. Biochem. J. 155, 19–23.
Schuller, E., and Helary, M. (1983). Determination in the nanogram range of C1q in serum and unconcentrated CSF by electro-immunodiffusion. J. Immunol. Methods 56, 159–165.
Schwaeble, W., Schafer, M. K., Petry, F., Fink, T., Knebel, D., Weihe, E., and Loos, M. (1995). Follicular dendritic cells, interdigitating cells, and cells of the monocyte-macrophage lineage are the C1q-producing sources in the spleen. Identification of specific cell types by in situ hybridization and immunohistochemical analysis. J. Immunol. 155, 4971–4978.
Sjoholm, A. G., Martensson, U., and Laurell, A. B. (1985). C1 dissociation in serum: estimation of free C1q by electroimmunoassay. Acta Pathol. Microbiol. Immunol. Scand. C 93, 161–168.
Steinsson, K., McLean, R. H., Merrow, M., Rothfield, N. F., and Weinstein, A. (1983). Selective complete Clq deficiency associated with systemic lupus erythematosus. J. Rheumatol. 10, 590–594.
Stuart, L. M., Takahashi, K., Shi, L., Savill, J., and Ezekowitz, R. A. (2005). Mannose-binding lectin-deficient mice display defective apoptotic cell clearance but no autoimmune phenotype. J. Immunol. 174, 3220–3226.
Tenner, A. J., and Volkin, D. B. (1986). Complement subcomponent C1q secreted by cultured human monocytes has subunit structure identical with that of serum C1q. Biochem. J. 233, 451–458.
Tuckwell, D. S., Reid, K. B., Barnes, M. J., and Humphries, M. J. (1996). The A-domain of integrin alpha 2 binds specifically to a range of collagens but is not a general receptor for the collagenous motif. Eur. J. Biochem. 241, 732–739.
Vassallo, G., Newton, R. W., Chieng, S. E., Haeney, M. R., Shabani, A., and Arkwright, P. D. (2007). Clinical variability and characteristic autoantibody profile in primary C1q complement deficiency. Rheumatology (Oxford) 46, 1612–1614.
Vegh, Z., Goyarts, E. C., Rozengarten, K., Mazumder, A., and Ghebrehiwet, B. (2003). Maturation-dependent expression of C1q-binding proteins on the cell surface of human monocyte-derived dendritic cells. Int. Immunopharmacol. 3, 345–357.
Walport, M. J. (2002). Complement and systemic lupus erythematosus. Arthritis Res. 4(Suppl. 3), S279–S293.
Walport, M. J., Davies, K. A., and Botto, M. (1998). C1q and systemic lupus erythematosus. Immunobiology 199, 265–285.
Keywords: immunology, complement, C1 complex, monocytes, U937 cells
Citation: Hosszu KK, Valentino A, Ji Y, Matkovic M, Pednekar L, Rehage N, Tumma N, Peerschke EIB and Ghebrehiwet B (2012) Cell surface expression and function of the macromolecular C1 complex on the surface of human monocytes. Front. Immun. 3:38. doi: 10.3389/fimmu.2012.00038
Received: 31 October 2011; Paper pending published: 23 November 2011;
Accepted: 16 February 2012; Published online: 05 March 2012.
Edited by:
Claudia Kemper, King’s College London, UKReviewed by:
Peter Kraiczy, University Hospital of Frankfurt, GermanyMiki Nakao, Kyushu University, Japan
Mihaela Gadjeva, Harvard Medical School, USA
Dan Anthony Mitchell, University of Warwick, UK
Teizo Fujita, Fukushima Medical University, Japan
Copyright: © 2012 Hosszu, Valentino, Ji, Matkovic, Pednekar, Rehage, Tumma, Peerschke and Ghebrehiwet. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Berhane Ghebrehiwet, Health Sciences Center, Stony Brook University School of Medicine, T-16, Room 040, Stony Brook, NY, 11794-8161, USA. e-mail:YmVyaGFuZS5naGVicmVoaXdldEBzdG9ueWJyb29rLmVkdQ==