 Verónica I. Dumit1,2
Verónica I. Dumit1,2 Jörn Dengjel1,2,3*
Jörn Dengjel1,2,3*- 1 School of Life Sciences – LifeNet, Freiburg Institute for Advanced Studies, University of Freiburg, Freiburg, Germany
- 2 ZBSA Center for Biological Systems Analysis, University of Freiburg, Freiburg, Germany
- 3 BIOSS Center for Biological Signalling Studies, University of Freiburg, Freiburg, Germany
Autophagy is a constitutive, catabolic process leading to the lysosomal degradation of cytosolic proteins and organelles. However, it is also induced under stress conditions, remodeling the eukaryotic cell by regulating energy, protein, and lipid homeostasis. It is likely that the autophagosomal/lysosomal pathway evolved primordially to recycle cell components, but further functionally developed as to become part of the immune system to defend against invading pathogens. Likewise, pathogenic, foreign agents developed strategies to fight back and even to employ the autophagy machinery to their own benefit. Hence, the regulation of autophagy has many implications on human health and disease. This review summarizes the molecular dynamics of autophagosome formation, maturation, and target selection. Membrane dynamics, as well as protein–protein and protein–membrane interactions are particularly addressed. In addition, it recapitulates current knowledge of the influences of influenza virus infection on the process.
So It Begins
Autophagy comprises several diverse lysosomal degradation pathways. Chaperone-mediated autophagy involves the direct translocation of cytosolic proteins across the lysosomal membrane (Dice, 2007). During microautophagy the lysosomal membrane invaginates or protrudes to sequester and deliver portions of cytoplasm directly into lysosomes (Klionsky et al., 2007). Macroautophagy, hereafter referred to as autophagy, is a lysosomal degradation pathway mediated by specialized organelles, autophagosomes, and will be the focus of this review. Autophagosomes enclose part of the cytoplasm destined for recycling. The exact mechanism of autophagosome formation remains still unknown. However, growing evidence suggests that a subdomain of the endoplasmic reticulum (ER) is crucial for autophagosome biogenesis (Hayashi-Nishino et al., 2009, 2010; Ylä-Anttila et al., 2009). Particularly, it has been observed by electron microscopy that ER cisternae often associate with early autophagic structures (Hayashi-Nishino et al., 2010). Nevertheless, the origin of the autophagosomal membrane and how it is formed is still under debate (Chen and Klionsky, 2011). Next to the ER, mitochondria (Hailey et al., 2010), and the plasma membrane (Ravikumar et al., 2010) have been discussed as membrane sources. Almost two decades ago, complementation screening of yeast genes allowed identification of minimally 15 genes responsible for autophagosome formation (Tsukada and Ohsumi, 1993). Lately this list has grown to 33 entries of which 17 are required for all autophagy subtypes (Inoue and Klionsky, 2010). These genes are named ATG (autophagy-related) and their orthologs are essentially conserved in all eukaryotes (Noda et al., 2009). In this work, Atg refers to autophagy genes in yeast and ATG is reserved for their mammalian orthologs.
Although autophagy is a constitutive process, it can also be induced by different stress conditions, e.g., amino acid starvation or growth factor deprivation (Figure 1A). These treatments induce autophagy through the inhibition of the mammalian target of rapamycin (mTOR), a serine–threonine kinase central in autophagy regulation. mTOR exists as part of at least two complexes: complex 1 (mTORC1) is sensitive to nutrient abundance and is made-up of mTOR along with the subunits Raptor, mLST8 (also known as G protein beta subunit-like) and PRAS40. When activated, mTORC1 stimulates cell growth by promoting protein translation and ribosome synthesis, while it inhibits cellular degradation by autophagy (Chan, 2009). mTORC2, containing Rictor, mSin1, mLST8, and Protor next to mTOR, is discussed as acting on the cytoskeleton through other kinases, such as the serine–threonine kinase Akt and SGK1 kinase (Chan, 2009; Kim and Guan, 2011; Zoncu et al., 2011). In Drosophila melanogaster a third TOR complex was identified, dTTT (Drosophila TOR, TELO2, TTI1) which is required for dTORC1/dTORC2 activity and cell growth (Glatter et al., 2011).
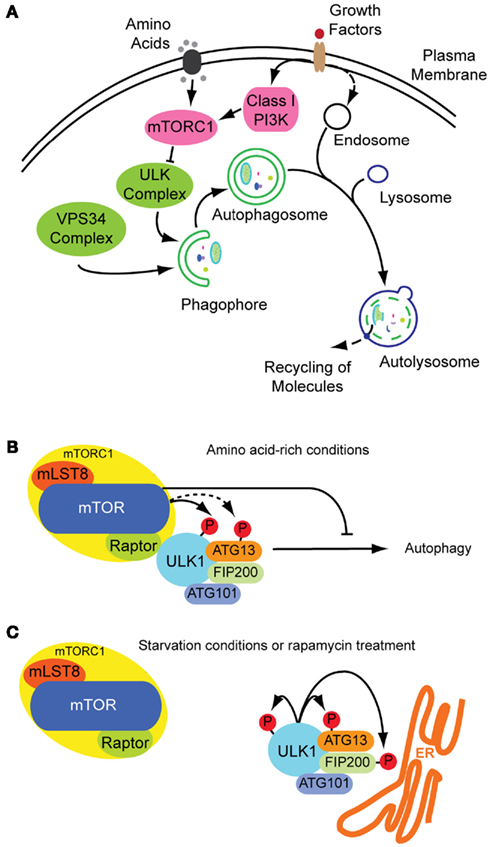
Figure 1. Autophagosome formation and underlying signaling events. (A) Autophagy is a cellular stress response that can be induced inter alia by nutrition and growth factor deprivation. Active mTORC1 inhibits autophagosome generation, whereas active ULK- and VPS34 class III PI3K-complexes are prerequisites for autophagosome formation. The phagophore enraptures cytoplasm and matures to an autophagosome, also fusing with endosomes. Acidic hydrolases degrade the constituents in autolysosomes enabling a recycling of bio-molecules (green: stimulatory signals; pink: inhibitory signals; figure not complete). (B) Under growth conditions mTOR is active inhibiting the ULK-complex and by this autophagy. (C) Under starvation conditions mTOR is inactive and ULK active, phosphorylating itself and its binding partners and translocating to pre-autophagosomal structures.
It is known from yeasts, that inhibition of TORC1 by rapamycin, starvation, or other stresses induces formation of an activated Atg1 complex along with the cofactors Atg13 and Atg17, both needed for maximal Atg1 catalytic activity (Mizushima, 2010). The Atg1–Atg13–Atg17 complex has serine–threonine kinase activity and its formation leads to autophagy induction. In contrast, when TORC1 is active, it leads to Atg13 phosphorylation and subsequent destabilization of the complex and effective Atg1 inactivation (Chan, 2009). It remains unclear, however, whether TOR directly phosphorylates Atg13 (Mizushima, 2010).
The functional counterparts to this complex in mammals are ULKs, ATG13 and FIP200 (orthologs of Atg1, Atg13, and Atg17, respectively). ULK1 is the best characterized Atg1 homolog. The role of the other isoforms ULK2 and ULK3 in autophagy are yet less clear. Although it seems likely that ULK2 is partially redundant to ULK1 (Chan, 2009), ULK3 may not have equivalent functions (Mizushima, 2010). Under non-stress conditions, mTORC1 associates with the ULK1–ATG13–FIP200–ATG101 complex by a direct interaction between Raptor and ULK1 (Chan, 2009; Mizushima, 2010), and phosphorylates ULK1 and ATG13, inhibiting their activity (Figure 1B). On the other hand, when mTORC1 is inactive, it dissociates from the ULK1 complex, leading to ULK1 activation. In its active state, ULK1 undergoes autophosphorylation and phosphorylates ATG13 and FIP200 (Figure 1C). ULK1, ATG13, FIP200, and ATG101 accomplish their function by translocating from the cytosol to subdomains of the ER, and are thus essential for initiation of autophagosome formation (Mizushima, 2010). These proteins lead to the isolation of membrane subdomains by recruitment of a class III phosphatidylinositol-3-OH kinase (PI3K) complex to the ER. The PI3K complex includes VPS34 (also known as PIK3C3), VPS15 (PIK3R4 and p150), Beclin-1 (ATG6), ATG14, and AMBRA1 (Levine et al., 2011). At this point of autophagy induction, not only protein components have decisive functions in autophagosome formation, also the role of lipids is crucial in its regulation. In the following section, we focus on the hinge role of lipids within protein dynamics in autophagy.
The Lipid Connection
Phosphatidylinositols (PI) are negatively charged phospholipids present as minor component at the cytosolic side of eukaryotic cell membranes (Leevers et al., 1999). PI can be phosphorylated on its inositol ring to form PI-phosphate (PIP), PI-bisphosphate (PIP2), and PI-trisphosphate (PIP3; Burman and Ktistakis, 2010). PIP, PIP2, and PIP3 are collectively called phosphoinositides (Leevers et al., 1999).
In general, PI3Ks are responsible for phosphorylating the 3′OH-position of the inositol ring of PI, yielding PI3P (Burman and Ktistakis, 2010). Synthesis of PI3P is a strictly necessary requirement for all organisms undergoing autophagy (Burman and Ktistakis, 2010). The function of PI3P is to gather signaling proteins containing specific lipid-binding domains to the membrane. Particularly in autophagy, such effectors are the double FYVE-containing protein 1 (DFCP1) and WD40-repeat domain phosphoinositide-interacting (WIPI, homolog to Atg18 in yeast) family proteins (Levine et al., 2011). DFCP1, in contrast to most FYVE domain proteins that localize to endosomes, is located mainly at the ER, where PI3P is usually absent until autophagy is induced (Noda et al., 2010). Then, DFCP1 translocates to the autophagosome formation site, drawn by PI3P, to produce ER-associated Ω-like structures called omegasomes (Axe et al., 2008). The other effector of PI3P during autophagy, WIPI/Atg18, functions downstream of DFCP1 and was suggested to help the development of omegasomes into autophagosomes. WIPI2 is the major isoform among the four WIPI isoforms in most mammalian cells (Polson et al., 2010).
The FYVE domain is named after the four proteins in which it has been found: Fab1p (yeast ortholog of PIKfyve), YOTB, Vac1p (vesicle transport protein), and EEA1 (early endosome antigen 1). It is characterized by having two zinc ions and eight potential zinc coordinating cysteine residues. Additionally, several basic amino acids are localized around the cysteines. FYVE domains are part of cysteine-rich proteins, which bind PI3P in a way dependent on their metal ion coordination and interaction of their basic amino acids with the negative charged PI3P head group (Krauss and Haucke, 2007). Figure 2A shows the tertiary structure of a FYVE domain and the coordination mode of the two metal ions to the eight cysteine residues. Figure 2B shows EEA1 and its binding mode to inositol-1,3-diphosphate, as a representative example of their interaction. It has been suggested that the binding causes conformational changes regulating protein–protein or lipid–protein interactions (Leevers et al., 1999). Most pathways regulated by PI, including autophagy, depend on their generation and likewise on their consumption. A situation where PI persist longer than the lipid signal is needed will result in loss of homeostasis. Jumpy is a PI3P phosphatase, which inhibits recruitment of WIPI to the autophagic membranes, and thus is in charge of PI3P signal termination (Vergne et al., 2009).
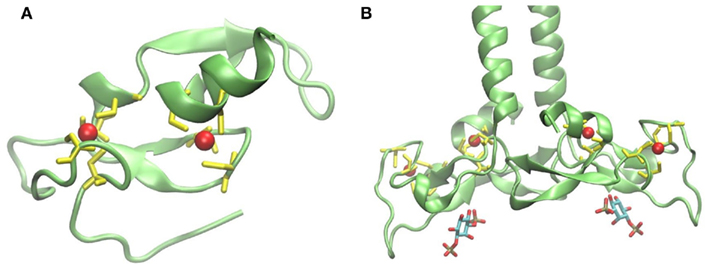
Figure 2. (A) Structure of protein FYVE domain of the RUN and FYVE domain containing protein 1; Zn atoms are shown in red and coordinating cysteins in yellow (PDB code 2yw8). (B) Homodimer of EEA1’s C-terminal FYVE domain bound to inositol-1,3-diphosphate (PDB code 1joc). The picture was prepared using the program VMD (Humphrey et al., 1996).
Independently of the conditions triggering the catabolic pathway, autophagy begins with activation of the class III PI3K Beclin-1-complex in mammals, necessary to target membranes for autophagosome generation and posterior maturation. Beclin-1, homolog of yeast Atg6, will be treated in detail below as target of various viruses to abort autophagy and to use the autophagy machinery for their own infectious purposes.
One Way Ticket to the Autophagosome
Two ubiquitin-like conjugation systems (UBL), ATG12, and LC3, have been implicated in biogenesis and membrane expansion of autophagosomes (Münz, 2011a; Weidberg et al., 2011a). Modification of proteins with ubiquitin-like proteins follows a similar mechanism like modification with ubiquitin itself. For this reason, it is worthwhile to briefly summarize the general process (Figure 3A): Ubiquitin is a 76 amino acid-residue polypeptide, whose role is to direct proteins to the proteasome for degradation, among other regulatory functions. Ubiquitin is activated by an E1 enzyme and, subsequently, E2 enzymes pick up activated ubiquitin by transthiolation and together with E3 enzymes catalyze ubiquitination of substrates. E3 enzymes function to recognize substrates and are also capable of interacting with E2, allowing conjugation of ubiquitin to target proteins. There are two major types of E3 enzymes in eukaryotes, defined by the presence of either a HECT or a RING domain (Deshaies and Joazeiro, 2009). HECT and RING E3s catalyze ubiquitin transfer by different mechanisms (Figure 3A). HECT-domain containing E3s bind themselves ubiquitin from an E2 protein before transferring it to its target protein. RING E3s function as a bridge between an activated E2 and a target protein. Ubiquitination can be a repetitive process leading to the generation of polyubiquitin chains or multiple mono-ubiquitinations. It is also a dynamic process. Deubiquitinating enzymes are able to trim polyubiquitin chains or to remove single moieties allowing their recycling. Although eukaryotic cells have only one or few E1 enzymes, they encode more than 40 isoforms of E2 and more than 600 E3 enzymes (Grabbe et al., 2011). As it is to be expected, the number of E2 and E3 isoforms show a higher degree of complexity of the ubiquitin conjugation systems in human cells relative to yeast (Hicke et al., 2005), enabling to recognize diverse proteins in a highly specific manner (Hochstrasser, 2009).
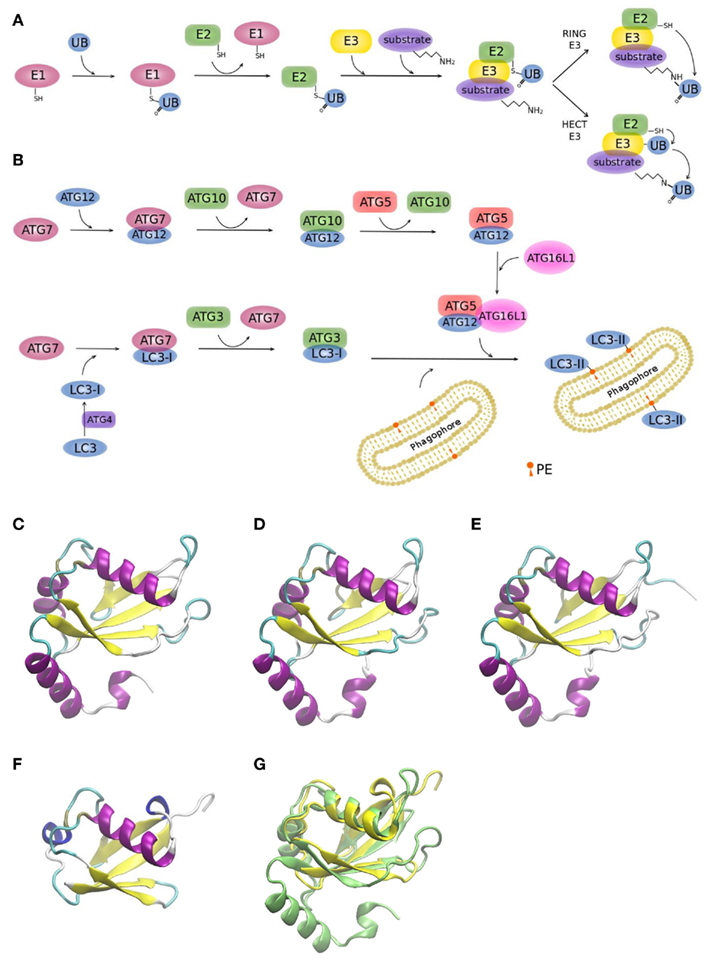
Figure 3. Ubiquitin and ubiquitin-like conjugation systems. (A) The ubiquitination system. E1–E2–E3 enzymatic cascades are depicted. (B) The whole set of ubiquitin-like reactions taking part in autophagy involves the E1-, E2-, and E3-like enzymes ATG7, ATG3/ATG10, and ATG12–ATG5/ATG16L1, respectively. Crystallographic structure of (C) LC3 (PDB code 1ugm), (D) GABARAP (PDB code 1kjt), (E) GABARAPL2 (PDB code 1eo6), (F) ubiquitin (PDB code 1aar), and (G) superimposition of ubiquitin (yellow) and LC3 (green) crystallographic structures. MultiProt Server was employed for protein alignment based on their structures (Shatsky et al., 2004). The picture was prepared using the program VMD (Humphrey et al., 1996).
Figure 3B shows the two UBL systems operating in autophagy. Firstly, the ATG12–ATG5 conjugation is generated by ATG7 (E1-like) and ATG10 (E2-like). ATG12–ATG5 bind to ATG16L1 and promote autophagosome formation (Fujita et al., 2008a; Weidberg et al., 2011a). It has been shown in yeast, that it is the Atg12–Atg5 complex itself that catalyzes the transfer of Atg8 from Atg3 to the substrate, phosphatidylethanolamine (PE), thus, behaving as a ubiquitin–protein ligase E3-like enzyme (Hanada et al., 2007). Secondly, LC3s, mammalian Atg8 homologs, are synthesized as precursors with an extra sequence at the C-terminus, which must be cleaved by the protease ATG4, resulting in the LC3 form I (LC3-I; Mizushima et al., 2011). LC3-I is then readily conjugated to PE (forming LC3-II). PE is a lipid found in biological membranes and lipidation of LC3/Atg8 during autophagy anchors this protein to the autophagosomal membrane. LC3/Atg8 may serve different purposes. Yeast Atg8 has been shown to be important for phagophore membrane elongation (Abeliovich et al., 2000). In addition, LC3/Atg8 functions as a membrane anchor enabling the targeting of substrates to the autophagosome (Münz, 2011a). It was also shown that it is important for membrane tethering and fusion (Nakatogawa et al., 2007; Weidberg et al., 2011b). However, these studies were performed in vitro and in a recent in vivo study it was suggested that soluble NSF attachment protein receptor (SNARE) proteins are required for membrane fusion, given that Atg8 is not able to mediate membrane fusion under physiological PE levels (Nair et al., 2011).
Yeast has a single Atg8 protein while mammals have several paralogs: three MAP1 light chain three (LC3A, LC3B, and LC3C) and four gamma-aminobutyrate receptor associated protein (GABARAP) and GABARAP-like proteins (GABARAPL1-3), collectively referred to as LC3s. The roles of LC3s remained unclear for a long time, since the other isoforms partially compensated the loss of function of a specific LC3 form in knockout studies (Noda et al., 2009). Figures 3C–E displays the remarkable structural similarities of the different LC3 forms among themselves and to ubiquitin (Figure 3F). Superimposition of LC3 and ubiquitin structures clearly shows the high resemblance between these two proteins (Figure 3G).
Anchoring of autophagosomal substrates by LC3/Atg8 is accomplished by direct interaction with cargo proteins or by adaptor proteins. NIX is a mitochondrial membrane protein capable of interacting with LC3s during mitophagy, the selective removal of mitochondria by autophagy (Novak et al., 2010). Examples of adaptor proteins are p62/sequestosome 1 (Bjorkoy et al., 2005; Pankiv et al., 2007), optineurin (Wild et al., 2011), NBR1 (Kirkin et al., 2009), or NDP52 (Thurston et al., 2009), which favor interaction between LC3/Atg8 and polyubiquitinated substrates (Figure 4A; Münz, 2011a). p62 and NBR1 differ in size but their structures share three domains: an N-terminal PB1 domain, a LIR motif capable of interacting with LC3 proteins, and a C-terminal UBA domain interacting with ubiquitin. Both cargo proteins, p62 and NBR1, cooperate in the targeting of ubiquitinated proteins to the autophagosome, and are both required for their degradation by autophagy (Lamark et al., 2009). The binding is mediated by the 22 amino acid-residue sequence forming the conserved LIR motif with the core W/YxxL/I, which is indispensable for LC3 recruitment into p62-positive inclusion bodies (Figures 4B,C; Pankiv et al., 2007; Shpilka et al., 2011). It could be shown that phosphorylation of p62’s UBA domain enhances degradation of polyubiquitinated proteins (Matsumoto et al., 2011).
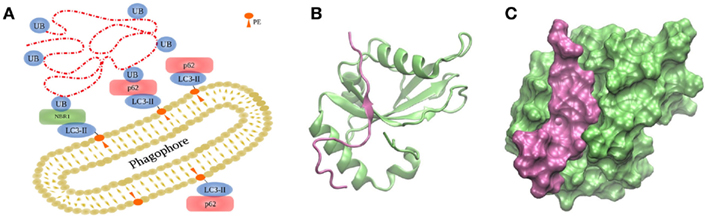
Figure 4. Targeting of substrates to the autophagosome. (A) During autophagy, the autophagosome engulfs the cargo via LC3/Atg8, covalently bound to PE on both sides of the membranes by two ubiquitin-like conjugation systems depicted in Figure 3. (B,C) Interaction between human GABARAPL1 and the LIR motiv of NBR1, displayed in two different representation forms (PDB code 2l8j).
The next step in the maturation of an autophagosome is its closure. It is known that a defect in LC3 function leads to unclosed autophagosomes (Fujita et al., 2008b). The current model suggests that when the autophagosome double membrane is about to close forming a narrow pore, LC3 molecules, especially GABARAP proteins, meet and catalyze the mixture of lipids of the pore, forming a stalk. Disintegration of the stalk completes autophagosome closure (Noda et al., 2009). However, also here a role of SNARE proteins cannot be ruled out. The completed autophagosome is then ready to fuse with late endosomes, forming amphisomes (Gordon and Seglen, 1988), followed by fusion with lysosomes, resulting in autolysosomes, and its inner membrane and cargo get degraded by lysosomal hydrolysis (Figure 1A; Münz, 2011a).
Traditionally, autophagy has been described as unselective bulk degradation (Seglen et al., 1990), however, the discovery of autophagy receptors like p62 and NBR1 which also shuttle polyubiquitinated protein aggregates to autophagosomes during classical autophagy indicates the existence of target recognition and possibly directed transport. We could show by a global proteomics approach that the subcellular localization of proteins influences their degradation dynamics by autophagy (Kristensen et al., 2008). During 36 h of amino acid starvation cytosolic proteins were initially degraded, only followed at later time points by organellar proteins, such as ribosomal and mitochondrial proteins. In a follow-up study we compared the proteomic composition of autophagosomes after different autophagy-inducing stimuli (Dengjel et al., 2012). The proteomic composition of stress-induced autophagosomes during amino acid starvation differed clearly from autophagosomes induced by rapamycin treatment or by block of basal autophagy by concanamycin A, an inhibitor of the lysosomal H+-ATPase. Comparing the proteome of autophagosomes over time also highlighted quantitative abundance differences of autophagosomal proteins. Hence, the inducing stimuli, as well as the time frame of stimulation, seem to influence the composition of autophagosomes. If (macro)autophagy is per definition unspecific, we might have to consider the possibility that only basal macroautophagy is truly unspecific and that all variants of stress-induced (macro)autophagy should instead be regarded as specific, similar to organelle-specific autophagy subtypes. Thus, we might want to discriminate, e.g., between growth factor- and amino acid-starvation induced (macro)autophagy. Another possibility might be that (macro)autophagy per se is not unspecific and we just did not succeed to elucidate the underlying signaling events until now. Modern high-throughput “omics” approaches in combination with systems biology allow for generation of large datasets and construction of mathematical models which may help in shedding more light on these complex biological processes (Zimmermann et al., 2010; Engelke et al., 2012).
Autophagy and Antigen Processing
Even though autophagy mainly serves as protein degrading mechanism, it is likely that the same machinery has been adopted to participate in adaptive immunity (Levine et al., 2011). Thus, it could be shown that autophagy can be induced by activation of innate immune receptors in antigen presenting cells. Conveniently, fragments of proteins from infecting pathogens, which are degraded through autophagy, result in foreign peptides which can be complexed to MHC molecules and presented to T cells (for detailed review see Münz, 2011b). Classically, MHC class I molecules were regarded to present peptides from intracellular antigens to CD8+ T cells. Peptides are generated by proteasomal processing and translocate to the ER via the transporter for antigenic peptides where they are loaded to nascent MHC class I complexes with the help of chaperones. In the cytosol and the ER peptides may be further processed by additional proteases before binding to MHC class I chains. Finally, MHC-I-peptide complexes transit to the cell surface where they can be recognized by CD8+ T cells. An alternative pathway, called cross-presentation, allows presentation of peptides from exogenous antigens on MHC class I molecules (Crotzer and Blum, 2010). Whereas the classical MHC class I presentation pathway seems not to be influenced by autophagy, it could be shown that autophagy modulates MHC class I presentation during late stage herpes simplex virus (HSV) infection (English et al., 2009).
Under normal conditions, MHC class II molecules present peptides on antigen presenting cells to CD4+ T cells, but during infection or inflammation MHC-II expression can be induced in non-immune cells as well. MHC class II molecules were viewed to present mainly peptides from extracellular antigens. However, MHC-II peptide analyses revealed that also peptides from intracellular source proteins are presented on MHC class II molecules. MHC class II α and β chains are synthesized into the ER and the chaperone invariant chain (Ii) prevents the binding of antigenic peptides to the class II binding groove. In acidic vesicular compartments Ii is cleaved and antigenic peptides can bind to MHC class II heterodimers with the help of chaperones. In contrast to MHC-I presentation, the role of autophagy in MHC-II presentation is more clear. Thus, it could be shown by several groups that macroautophagy and chaperone-mediated autopahgy play major roles in promoting presentation of peptides derived from cytoplasmic and nuclear proteins on MHC-II (Nimmerjahn et al., 2003; Dengjel et al., 2005; Zhou et al., 2005). This is also true for the presentation of some virus derived peptides, like from Epstein Barr virus nuclear antigen 1 (Paludan et al., 2005) and from influenza virus matrix protein (Schmid et al., 2007). Interestingly, for MHC class I as well as for MHC class II presentation a dual dependency on proteasome- and autophagy-activity could be observed highlighting a crosstalk between the two degradation pathways (Dörfel et al., 2005; English et al., 2009). Along this line, we could show that the proteasome is one of the “favorite substrates” of autophagosomes and that proteasome activity is modulated by functional autophagy (Dengjel et al., 2012). However, the exact molecular mechanisms underlying autophagy–proteasome crosstalk are still not fully unveiled and more work has to be done. E.g., it is not clear if proteasomes are active inside autophagosomes and if autophagosomes may thus be regarded as scaffolds bringing together the proteasome with its substrates.
Influenza Virus versus Autophagy. Who Takes Control?
Many pathogens compromise peptide presentation on MHC molecules by blocking the induction of autophagy or the maturation of autophagosomes. Moreover, it is known that some viruses induce autophagy but inhibit autophagosome–lysosome fusion (Deretic and Levine, 2009). Hence, several viruses inhibit autophagy at the level of autophagosome initiation by antagonizing Beclin-1. Examples of them are the α-herpervirus (HSV-1; Orvedahl et al., 2007), and γ-herpesviruses, which include human pathogens such as Epstein Barr virus, Kaposi’s sarcoma associated herpesvirus (KSHV) and murine γ-HV68 (Liang et al., 2008). In contrast to DNA viruses, RNA viruses, including HIV (Kyei et al., 2009; Blanchet et al., 2010), hepatitis C (Ait-Goughoulte et al., 2008; Dreux et al., 2009), and poliovirus (Dales et al., 1965; Jackson et al., 2005), block autophagosome maturation and consequently degradation, possibly to benefit from vesicular organelles for their replication (Rossman and Lamb, 2009; Münz, 2011a). Non-maturing autophagosomes offer a propitious environment for virus replication, due to the fact that high concentrations of viral proteins can be accumulated while being unnoticed by the adaptive immune system (Rossman and Lamb, 2009). Importantly, viruses inhibiting autophagosome maturation target Beclin-1 as well, as it is also involved in the maturation process as binding partner of UVRAG.
Although it is well accepted that influenza virus infection affects autophagy, controversy still remains in many aspects of the underlying mechanisms and functional strategies employed by the virus to succeed in its infective purpose. Various independent studies suggest that an increasingly sophisticated connection exists between autophagy, apoptosis and viral replication. Moreover, interconnection between these three processes appears to be cell-line dependent, further complicating interpretation of the gathered data on the effects of influenza virus infection.
Autophagy is induced by reactive oxygen species (Huang et al., 2011) which are also produced after influenza infection (Vlahos et al., 2012) highlighting a potential point of crosstalk. Oxidizing molecules are suggested to modulate ATG4 activity leading to LC3-II accumulation (Scherz-Shouval et al., 2007). Influenza virus has been also proposed to up-regulate the expression of ATG7, ATG5, and ATG12 (Dai et al., 2012). Hence, all of these actions lead to autophagosome accumulation and may be tracked back to influenza virus infection. Significantly, it has been suggested that autophagy is involved in virus-dependent cytokine induction, which is thought to be the main cause of death of infected patients (Law et al., 2010).
A closer look at the association between influenza virus infection and autophagy raises many questions. It is well documented that influenza virus inhibits autophagy at the stage of autophagosome fusion with lysosomes, and thus leads to an accumulation of autophagosomes in human lung carcinoma-derived cells (Gannagé et al., 2009). On the other hand, it was shown that different strains of influenza virus induce functional autophagy, as detected by degradation of the autophagy receptor p62 in infected primary human blood macrophages (Law et al., 2010). A third report stated as well that influenza virus infection does induce functional autophagy in several different cell lines, with no detectable block in the pathway, as concluded from both GFP-LC3 and p62 degradation measurements (Comber et al., 2011). In an attempt to conciliate all apparently contradicting results, the authors of the latter work suggested that discrepancies may be due to the cell types or the influenza virus strains used for the experiments (Comber et al., 2011).
Another polemic aspect is the functional association between autophagy and viral replication. The purpose of compromising a key homeostatic pathway of the cell by influenza virus is still under debate. It was observed that influenza virus infection decreases cell survival by inducing apoptosis and inhibiting autophagy. The induction of apoptosis was suggested to circumvent an anti-viral immune response (Gannagé et al., 2009). However, autophagy had apparently a negligible influence on viral yields, given that loss of the degradation process did not affect virus replication. Therefore it was concluded that viral replication does not require the autophagosome environment to take its course (Gannagé et al., 2009). Hence, it has been suggested that influenza virus seems to remain in the cytoplasm and nucleus for its replication (Rossman and Lamb, 2009). But why does influenza virus compromise autophagy? In contrast to the mentioned study, another investigation showed that inhibition of autophagy reduces replication of influenza virus (Zhou et al., 2009). On top, it was suggested that influenza virus induces autophagy only when apoptosis is first inhibited (McLean et al., 2009). This evidence was proposed to be the reason for the apparently opposing results attained before (Rossman and Lamb, 2009).
On the molecular level, there is solid evidence that binding of influenza virus M2 protein to Beclin-1 compromises autophagy at the step of lysosome fusion to autophagosomes (Gannagé et al., 2009). Beclin-1 contains a conserved BH3 domain (Oberstein et al., 2007). Such domains were first discovered in the context of apoptosis, but then could also be related to regulation of autophagy (Sinha and Levine, 2008). Figures 5A,B show the amphipathic BH3 helix of Beclin-1 interacting with a conserved hydrophobic groove of Bcl-XL and M11, respectively. Bcl-XL belongs to the Bcl-2 family of proteins, known to regulate apoptotic and autophagic processes in the cell (Sinha and Levine, 2008). M11 is a Bcl-2 homolog present in the human pathogen γ-herpesvirus 68, able to regulate autophagy through interaction with Beclin-1 (Sinha et al., 2008).
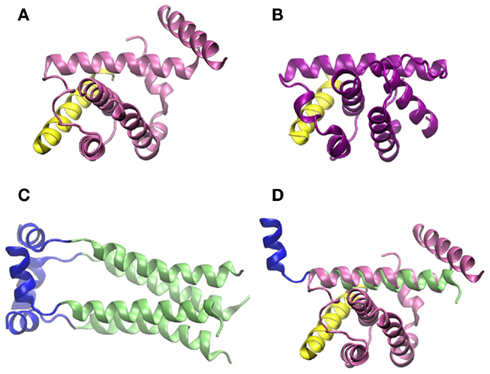
Figure 5. Crystallographic structure of the BH3 helix of Beclin-1, shown in yellow, complexed to (A) Bcl-XL (PDB code 2p1l) and (B) M11 (PDB code 3bl2). (C) Crystallographic structure of M2 transmembrane protein from influenza virus (PDB code 2kih); residues 23–49 including the transmembrane domain are shown in green and the cytoplasmic helices (residues 50–60) in blue. (D) Superimposition of Beclin-1 complexed to Bcl-XL and influenza virus M2 protein. The picture was prepared using the program VMD (Humphrey et al., 1996), and MultiProt Server was employed for protein alignment (Shatsky et al., 2004).
It could be shown that influenza virus M2 integral membrane protein is necessary and sufficient to block autophagosome–lysosome fusion (Gannagé et al., 2009). Transient expression of influenza A virus M2 protein reproduced the same phenotype as viral infection, i.e., autophagosomes accumulation due to a block in autophagosome maturation and not to an increase in autophagy. Silencing M2 expression during influenza A virus infection, or infecting cells with a M2 knockout influenza A virus, reverted the phenotype of classical infection, allowing autophagosome–lysosome fusion (Gannagé et al., 2009, 2010). M2 is a proton-selective ion channel responsible for acidification of the viral core once the virus reaches endosomes, causing dissociation of the viral particles and release of the genome into the cytoplasm (Wang et al., 2011). Surprisingly, it has been observed that autophagosome maturation is not inhibited by the M2 ion channel activity itself and that the first 60 residues of the protein are sufficient to inhibit autophagy by binding to Beclin-1 (Gannagé et al., 2009, 2010). The precise mechanism of inhibition has not been clearly determined, but it has been proposed that M2 is likely to interact with Beclin-1 through either the ectodomain (residues 1-24) or the cytoplasmic amphipatic helix (residues 46–62), but not through the transmembrane domain which should be shielded from access (Rossman and Lamb, 2009).
As shown in Figure 5, the secondary structures of both Bcl-XL and M11 contain mostly α-helices, as do the first 70 amino acid-residues of influenza virus M2 protein, which are needed to inhibit Beclin-1. If crystallographic structures of the complex Beclin-1–Bcl-XL and M2 are aligned, one of the transmembrane α-helices of M2 superimposes with one of the α-helices of Bcl-XL which interacts with Beclin-1 (Figures 5C,D). In our opinion, it cannot be excluded that this ion channel transmembrane domain of M2 binds to Beclin-1. This may happen before the M2 homotetramer complex is fully assembled in the membrane indicating a second, non-membrane-bound, function/role of this protein and possibly explaining the observation that the M2 ion channel activity seems not to be involved in its functions in autophagy regulation. To fully understand the modulations of autophagy by influenza virus and to outline the underlying molecular mechanisms more work has to be done, e.g., specifically addressing protein dynamics and protein–protein interactions under various conditions.
Conclusion
Autophagy is a highly complex process and only the identification of autophagy-related genes in the genetically tractable organism yeast and the fact that the process is conserved in humans have allowed the elucidation of underlying molecular mechanisms leading to the generation of autophagosomes in mammalian cells. Although we have gained a tremendous amount of knowledge in the last decade there are still many unanswered questions, especially related to human diseases. Viruses employ autophagy for their own goods and the studying of autophagy modulation by viral infection and viral proteins will allow a deeper insight into underlying molecular mechanisms shedding more light onto this basal cell biological process. We are confident that the newly generated knowledge will not only allow the design of new anti-viral therapies but will also help in targeting autophagy in other disease settings.
Regarding macroautophagy, a lot of work has to be done to fully understand target selection. Can a cell actually allow a completely unspecific bulk degradation process to happen? Large-scale “omics” approaches should help in generating enough data to comprehensively tackle this problem on a global scale.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to Victoria Küttner for carefully reading the manuscript. The research leading to these results has received funding from the Excellence Initiative of the German Federal and State Governments through FRIAS and BIOSS, from the Deutsche Forschungsgemeinschaft (GZ DE1757/2-1), and from the Federal Ministry of Education and Research through GerontoSys II – NephAge (031 5896 A). We thank all Protein Dynamics group members, as well as M. Boerries, H. Busch, A. Schlosser, and colleagues from the ZBSA, for helpful discussions and support. We apologize to colleagues whose work was not covered in this review due to space limitations.
References
Abeliovich, H., Dunn, W. A., Kim, J., and Klionsky, D. J. (2000). Dissection of autophagosome biogenesis into distinct nucleation and expansion steps. J. Cell Biol. 151, 1025–1034.
Ait-Goughoulte, M., Kanda, T., Meyer, K., Ryerse, J. S., Ray, R. B., and Ray, R. (2008). Hepatitis C virus genotype 1a growth and induction of autophagy. J. Virol. 82, 2241–2249.
Axe, E. L., Walker, S. A., Manifava, M., Chandra, P., Roderick, H. L., Habermann, A., Griffiths, G., and Ktistakis, N. T. (2008). Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 182, 685–701.
Bjorkoy, G., Lamark, T., Brech, A., Outzen, H., Perander, M., Overvatn, A., Stenmark, H., and Johansen, T. (2005). p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 171, 603–614.
Blanchet, F. P., Moris, A., Nikolic, D. S., Lehmann, M., Cardinaud, S., Stalder, R., Garcia, E., Dinkins, C., Leuba, F., Wu, L., Schwartz, O., Deretic, V., and Piguet, V. (2010). Human immunodeficiency virus-1 inhibition of immunoamphisomes in dendritic cells impairs early innate and adaptive immune responses. Immunity 32, 654–669.
Burman, C., and Ktistakis, N. T. (2010). Regulation of autophagy by phosphatidylinositol 3-phosphate. FEBS Lett. 584, 1302–1312.
Chan, E. Y. (2009). mTORC1 phosphorylates the ULK1-mAtg13-FIP200 autophagy regulatory complex. Sci. Signal. 2, pe51.
Chen, Y., and Klionsky, D. J. (2011). The regulation of autophagy – unanswered questions. J. Cell Sci. 124, 161–170.
Comber, J. D., Robinson, T. M., Siciliano, N. A., Snook, A. E., and Eisenlohr, L. C. (2011). Functional macroautophagy induction by influenza A virus without a contribution to major histocompatibility complex class II-restricted presentation. J. Virol. 85, 6453–6463.
Dai, J., Wang, G., Li, W., Zhang, L., Yang, J., Zhao, X., Chen, X., Xu, Y., and Li, K. (2012). High-throughput screening for anti-influenza a virus drugs and study of the mechanism of procyanidin on influenza a virus induced autophagy. J. Biomol. Screen. doi: 10.1177/1087057111435236
Dales, S., Eggers, H. J., Tamm, I., and Palade, G. E. (1965). Electron microscopic study of the formation of poliovirus. Virology 26, 379–389.
Dengjel, J., Hoyer-Hansen, M., Nielsen, M. O., Eisenberg, T., Harder, L. M., Schandorff, S., Farkas, T., Kirkegaard, T., Becker, A. C., Schroeder, S., Vanselow, K., Lundberg, E., Nielsen, M. M., Kristensen, A. R., Akimov, V., Bunkenborg, J., Madeo, F., Jäättelä, M., and Andersen, J. S. (2012). Identification of autophagosome-associated proteins and regulators by quantitative proteomic analysis and genetic screens. Mol. Cell. Proteomics. doi: 10.1074/mcp.M111.014035
Dengjel, J., Schoor, O., Fischer, R., Reich, M., Kraus, M., Müller, M., Kreymborg, K., Altenberend, F., Brandenburg, J., Kalbacher, H., Brock, R., Driessen, C., Rammensee, H. G., and Stevanovic, S. (2005). Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc. Natl. Acad. Sci. U.S.A. 102, 7922–7927.
Deretic, V., and Levine, B. (2009). Autophagy, immunity, and microbial adaptations. Cell Host Microbe 5, 527–549.
Deshaies, R. J., and Joazeiro, C. A. (2009). RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 78, 399–434.
Dörfel, D., Appel, S., Grünebach, F., Weck, M. M., Müller, M. R., Heine, A., and Brossart, P. (2005). Processing and presentation of HLA class I and II epitopes by dendritic cells after transfection with in vitro-transcribed MUC1 RNA. Blood 105, 3199–3205.
Dreux, M., Gastaminza, P., Wieland, S. F., and Chisari, F. V. (2009). The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl. Acad. Sci. U.S.A. 106, 14046–14051.
Engelke, R., Becker, A. C., and Dengjel, J. (2012). The degradative inventory of the cell: proteomic insights. Antioxid Redox Signal. doi: 10.1089/ars.2011.4393
English, L., Chemali, M., Duron, J., Rondeau, C., Laplante, A., Gingras, D., Alexander, D., Leib, D., Norbury, C., Lippé, R., and Desjardins, M. (2009). Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat. Immunol. 10, 480–487.
Fujita, N., Itoh, T., Omori, H., Fukuda, M., Noda, T., and Yoshimori, T. (2008a). The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell 19, 2092–2100.
Fujita, N., Hayashi-Nishino, M., Fukumoto, H., Omori, H., Yamamoto, A., Noda, T., and Yoshimori, T. (2008b). An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol. Biol. Cell 19, 4651–4659.
Gannagé, M., Dormann, D., Albrecht, R., Dengjel, J., Torossi, T., Rämer, P. C., Lee, M., Strowig, T., Arrey, F., Conenello, G., Pypaert, M., Andersen, J., García-Sastre, A., and Münz, C. (2009). Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 6, 367–380.
Gannagé, M., Rämer, P. C., and Münz, C. (2010). Targeting Beclin 1 for viral subversion of macroautophagy. Autophagy 6, 166–167.
Glatter, T., Schittenhelm, R. B., Rinner, O., Roguska, K., Wepf, A., Jünger, M. A., Köhler, K., Jevtov, I., Choi, H., Schmidt, A., Nesvizhskii, A. I., Stocker, H., Hafen, E., Aebersold, R., and Gstaiger, M. (2011). Modularity and hormone sensitivity of the Drosophila melanogaster insulin receptor/target of rapamycin interaction proteome. Mol. Syst. Biol. 7, 547–547.
Gordon, P. B., and Seglen, P. O. (1988). Prelysosomal convergence of autophagic and endocytic pathways. Biochem. Biophys. Res. Commun. 151, 40–47.
Grabbe, C., Husnjak, K., and Dikic, I. (2011). The spatial and temporal organization of ubiquitin networks. Nat. Rev. Mol. Cell Biol. 12, 295–307.
Hailey, D. W., Rambold, A. S., Satpute-Krishnan, P., Mitra, K., Sougrat, R., Kim, P. K., and Lippincott-Schwartz, J. (2010). Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 141, 656–667.
Hanada, T., Noda, N. N., Satomi, Y., Ichimura, Y., Fujioka, Y., Takao, T., Inagaki, F., and Ohsumi, Y. (2007). The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J. Biol. Chem. 282, 37298–37302.
Hayashi-Nishino, M., Fujita, N., Noda, T., Yamaguchi, A., Yoshimori, T., and Yamamoto, A. (2009). A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol. 11, 1433–1437.
Hayashi-Nishino, M., Fujita, N., Noda, T., Yamaguchi, A., Yoshimori, T., and Yamamoto, A. (2010). Electron tomography reveals the endoplasmic reticulum as a membrane source for autophagosome formation. Autophagy 6, 301–303.
Hicke, L., Schubert, H. L., and Hill, C. P. (2005). Ubiquitin-binding domains. Nat. Rev. Mol. Cell Biol. 6, 610–621.
Huang, J., Lam, G. Y., and Brumell, J. H. (2011). Autophagy signaling through reactive oxygen species. Antioxid Redox Signal. 14, 2215–2231.
Humphrey, W., Dalke, A., and Schulten, K. (1996). VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38.
Inoue, Y., and Klionsky, D. J. (2010). Regulation of macroautophagy in Saccharomyces cerevisiae. Semin. Cell Dev. Biol. 21, 664–670.
Jackson, W. T., Giddings, T. H., Taylor, M. P., Mulinyawe, S., Rabinovitch, M., Kopito, R. R., and Kirkegaard, K. (2005). Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 3, 861–871. doi: 10.1371/journal.pbio.0030156
Kim, J., and Guan, K. L. (2011). Amino acid signaling in TOR activation. Annu. Rev. Biochem. 80, 1001–1032.
Kirkin, V., Lamark, T., Sou, Y. S., Bjorkoy, G., Nunn, J. L., Bruun, J. A., Shvets, E., McEwan, D. G., Clausen, T. H., Wild, P., Bilusic, I., Theurillat, J. P., Øvervatn, A., Ishii, T., Elazar, Z., Komatsu, M., Dikic, I., and Johansen, T. (2009). A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol. Cell 33, 505–516.
Klionsky, D. J., Cuervo, A. M., and Seglen, P. O. (2007). Methods for monitoring autophagy from yeast to human. Autophagy 3, 181–206.
Krauss, M., and Haucke, V. (2007). Phosphoinositide-metabolizing enzymes at the interface between membrane traffic and cell signalling. EMBO Rep. 8, 241–246.
Kristensen, A. R., Schandorff, S., Hoyer-Hansen, M., Nielsen, M. O., Jäättelä, M., Dengjel, J., and Andersen, J. S. (2008). Ordered organelle degradation during starvation-induced autophagy. Mol. Cell. Proteomics 7, 2419–2428.
Kyei, G. B., Dinkins, C., Davis, A. S., Roberts, E., Singh, S. B., Dong, C., Wu, L., Kominami, E., Ueno, T., Yamamoto, A., Federico, M., Panganiban, A., Vergne, I., and Deretic, V. (2009). Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J. Cell Biol. 186, 255–268.
Lamark, T., Kirkin, V., Dikic, I., and Johansen, T. (2009). NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle 8, 1986–1990.
Law, A. H., Lee, D. C., Yuen, K. Y., Peiris, M., and Lau, A. S. (2010). Cellular response to influenza virus infection: a potential role for autophagy in CXCL10 and interferon-alpha induction. Cell. Mol. Immunol. 7, 263–270.
Leevers, S. J., Vanhaesebroeck, B., and Waterfield, M. D. (1999). Signalling through phosphoinositide 3-kinases: the lipids take centre stage. Curr. Opin. Cell Biol. 11, 219–225.
Levine, B., Mizushima, N., and Virgin, H. W. (2011). Autophagy in immunity and inflammation. Nature 469, 323–335.
Liang, C., E, X., and Jung, J. U. (2008). Downregulation of autophagy by herpesvirus Bcl-2 homologs. Autophagy 4, 268–272.
Matsumoto, G., Wada, K., Okuno, M., Kurosawa, M., and Nukina, N. (2011). Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol. Cell 44, 279–289.
McLean, J. E., Datan, E., Matassov, D., and Zakeri, Z. F. (2009). Lack of Bax prevents influenza A virus-induced apoptosis and causes diminished viral replication. J. Virol. 83, 8233–8246.
Mizushima, N. (2010). The role of the Atg1/ULK1 complex in autophagy regulation. Curr. Opin. Cell Biol. 22, 132–139.
Mizushima, N., Yoshimori, T., and Ohsumi, Y. (2011). The role of atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 27, 107–132.
Münz, C. (2011b). Antigen processing by macroautophagy for MHC presentation. Front. Immunol. 2:1–7. doi: 10.3389/fimmu.2011.00042
Nair, U., Jotwani, A., Geng, J., Gammoh, N., Richerson, D., Yen, W. L., Griffith, J., Nag, S., Wang, K., Moss, T., Baba, M., McNew, J. A., Jiang, X., Reggiori, F., Melia, T. J., and Klionsky, D. J. (2011). SNARE proteins are required for macroautophagy. Cell 146, 290–302.
Nakatogawa, H., Ichimura, Y., and Ohsumi, Y. (2007). Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 130, 165–178.
Nimmerjahn, F., Milosevic, S., Behrends, U., Jaffee, E. M., Pardoll, D. M., Bornkamm, G. W., and Mautner, J. (2003). Major histocompatibility complex class II-restricted presentation of a cytosolic antigen by autophagy. Eur. J. Immunol. 33, 1250–1259.
Noda, T., Fujita, N., and Yoshimori, T. (2009). The late stages of autophagy: how does the end begin? Cell Death Differ. 16, 984–990.
Noda, T., Matsunaga, K., Taguchi-Atarashi, N., and Yoshimori, T. (2010). Regulation of membrane biogenesis in autophagy via PI3P dynamics. Semin. Cell Dev. Biol. 21, 671–676.
Novak, I., Kirkin, V., McEwan, D. G., Zhang, J., Wild, P., Rozenknop, A., Rogov, V., Löhr, F., Popovic, D., Occhipinti, A., Reichert, A. S., Terzic, J., Dötsch, V., Ney, P. A., and Dikic, I. (2010). Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 11, 45–51.
Oberstein, A., Jeffrey, P. D., and Shi, Y. (2007). Crystal structure of the Bcl-XL-Beclin 1 peptide complex: Beclin 1 is a novel BH3-only protein. J. Biol. Chem. 282, 13123–13132.
Orvedahl, A., Alexander, D., Tallóczy, Z., Sun, Q., Wei, Y., Zhang, W., Burns, D., Leib, D. A., and Levine, B. (2007). HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 1, 23–35.
Paludan, C., Schmid, D., Landthaler, M., Vockerodt, M., Kube, D., Tuschl, T., and Münz, C. (2005). Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 307, 593–596.
Pankiv, S., Clausen, T. H., Lamark, T., Brech, A., Bruun, J. A., Outzen, H., Øvervatn, A., Bjorkoy, G., and Johansen, T. (2007). p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 282, 24131–24145.
Polson, H. E., de Lartigue, J., Rigden, D. J., Reedijk, M., Urbé, S., Clague, M. J., and Tooze, S. A. (2010). Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 6, 506–522.
Ravikumar, B., Moreau, K., Jahreiss, L., Puri, C., and Rubinsztein, D. C. (2010). Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat. Cell Biol. 12, 747–757.
Rossman, J. S., and Lamb, R. A. (2009). Autophagy, apoptosis, and the influenza virus M2 protein. Cell Host Microbe 6, 299–300.
Scherz-Shouval, R., Shvets, E., Fass, E., Shorer, H., Gil, L., and Elazar, Z. (2007). Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 26, 1749–1760.
Schmid, D., Pypaert, M., and Münz, C. (2007). Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity 26, 79–92.
Seglen, P. O., Gordon, P. B., and Holen, I. (1990). Non-selective autophagy. Semin. Cell Biol. 1, 441–448.
Shatsky, M., Nussinov, R., and Wolfson, H. J. (2004). A method for simultaneous alignment of multiple protein structures. Proteins 56, 143–156.
Shpilka, T., Weidberg, H., Pietrokovski, S., and Elazar, Z. (2011). Atg8: an autophagy-related ubiquitin-like protein family. Genome Biol. 12, 226–226.
Sinha, S., Colbert, C. L., Becker, N., Wei, Y., and Levine, B. (2008). Molecular basis of the regulation of Beclin 1-dependent autophagy by the gamma-herpesvirus 68 Bcl-2 homolog M11. Autophagy 4, 989–997.
Sinha, S., and Levine, B. (2008). The autophagy effector Beclin 1: a novel BH3-only protein. Oncogene 27(Suppl. 1), 137–148.
Thurston, T. L., Ryzhakov, G., Bloor, S., von Muhlinen, N., and Randow, F. (2009). The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat. Immunol. 10, 1215–1221.
Tsukada, M., and Ohsumi, Y. (1993). Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 333, 169–174.
Vergne, I., Roberts, E., Elmaoued, R. A., Tosch, V., Delgado, M. A., Proikas-Cezanne, T., Laporte, J., and Deretic, V. (2009). Control of autophagy initiation by phosphoinositide 3-phosphatase Jumpy. EMBO J. 28, 2244–2258.
Vlahos, R., Stambas, J., and Selemidis, S. (2012). Suppressing production of reactive oxygen species (ROS) for influenza A virus therapy. Trends Pharmacol. Sci. 33, 3–8.
Wang, J., Qiu, J. X., Soto, C., and DeGrado, W. F. (2011). Structural and dynamic mechanisms for the function and inhibition of the M2 proton channel from influenza A virus. Curr. Opin. Struct. Biol. 21, 68–80.
Weidberg, H., Shvets, E., and Elazar, Z. (2011a). Biogenesis and cargo selectivity of autophagosomes. Annu. Rev. Biochem. 80, 125–156.
Weidberg, H., Shpilka, T., Shvets, E., Abada, A., Shimron, F., and Elazar, Z. (2011b). LC3 and GATE-16 N termini mediate membrane fusion processes required for autophagosome biogenesis. Dev. Cell 20, 444–454.
Wild, P., Farhan, H., McEwan, D. G., Wagner, S., Rogov, V. V., Brady, N. R., Richter, B., Korac, J., Waidmann, O., Choudhary, C., Dötsch, V., Bumann, D., and Dikic, I. (2011). Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 333, 228–233.
Ylä-Anttila, P., Vihinen, H., Jokitalo, E., and Eskelinen, E. L. (2009). 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy 5, 1180–1185.
Zhou, D., Li, P., Lin, Y., Lott, J. M., Hislop, A. D., Canaday, D. H., Brutkiewicz, R. R., and Blum, J. S. (2005). Lamp-2a facilitates MHC class II presentation of cytoplasmic antigens. Immunity 22, 571–581.
Zhou, Z., Jiang, X., Liu, D., Fan, Z., Hu, X., Yan, J., Wang, M., and Gao, G. F. (2009). Autophagy is involved in influenza A virus replication. Autophagy 5, 321–328.
Zimmermann, A. C., Zarei, M., Eiselein, S., and Dengjel, J. (2010). Quantitative proteomics for the analysis of spatio-temporal protein dynamics during autophagy. Autophagy 6, 1009–1016.
Keywords: autophagy, influenza, organelle, autophagosome, protein interaction, ubiquitin
Citation: Dumit VI and Dengjel J (2012) Autophagosomal protein dynamics and influenza virus infection. Front. Immun. 3:43. doi: 10.3389/fimmu.2012.00043
Received: 09 December 2011; Paper pending published: 11 January 2012;
Accepted: 23 February 2012; Published online: 14 March 2012.
Edited by:
Christian Munz, University of Zurich, SwitzerlandReviewed by:
Laura Santambrogio, Albert Einstein College of Medicine, USALoredana Saveanu, Institut National de la Sante et de la Recherche Medicale, France
Copyright: © 2012 Dumit and Dengjel. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Jörn Dengjel, School of Life Sciences – LifeNet, Freiburg Institute for Advanced Studies, University of Freiburg, Albertstr. 19, 79104 Freiburg, Germany. e-mail:am9lcm4uZGVuZ2plbEBmcmlhcy51bmktZnJlaWJ1cmcuZGU=