

- Laboratory of Host Defenses, Graduate School of Medical Science and Engineering, Korea Advanced Institute of Science and Technology, Daejeon, Korea
Autophagy is an ancient biological process for maintaining cellular homeostasis by degradation of long-lived cytosolic proteins and organelles. Recent studies demonstrated that autophagy is availed by immune cells to regulate innate immunity. On the one hand, cells exert direct effector function by degrading intracellular pathogens; on the other hand, autophagy modulates pathogen recognition and downstream signaling for innate immune responses. Pathogen recognition via pattern recognition receptors induces autophagy. The function of phagocytic cells is enhanced by recruitment of autophagy-related proteins. Moreover, autophagy acts as a delivery system for viral replication complexes to migrate to the endosomal compartments where virus sensing occurs. In another case, key molecules of the autophagic pathway have been found to negatively regulate immune signaling, thus preventing aberrant activation of cytokine production and consequent immune responses. In this review, we focus on the recent advances in the role of autophagy in pathogen recognition and modulation of innate immune responses.
Introduction
Eukaryotic cells contain two major protein degradation systems for maintaining cellular homeostasis: the ubiquitin-proteasome system, responsible for degradation of soluble short-lived proteins (Kloetzel, 2001), and autophagy, a conserved system that degrades long-lived proteins and organelles (Klionsky and Emr, 2000). Autophagy was originally described as a starvation-induced response that provides nutrients by degrading long-lived proteins and recycling intracellular organelles (Kuma et al., 2004; Komatsu et al., 2005). There are three types of autophagy: microautophagy, chaperone-mediated autophagy, and macroautophagy (Figure 1A; Mizushima and Klionsky, 2007). Macroautophagy is the main route of degradation. It involves the formation of a double-membrane vesicle called autophagosome, which is formed by elongation of a cup-shaped membrane, followed by wrapping of the cellular constituents and fusion with lysosomes for degradation (Mizushima et al., 1998). The molecular mechanism underlying autophagy has been thoroughly covered in excellent reviews elsewhere (Klionsky and Emr, 2000; Ohsumi, 2001; Mizushima et al., 2002). Here, we briefly describe the process relevant to innate pathogen recognition by macroautophagy. Autophagy-related gene (Atg) 6 (Beclin-1) and type III phosphatidylinositol 3-kinase (PI3K) are required for the initiation of the isolation membrane. The elongation and termination of the autophagosome are regulated by at least two ubiquitin-like systems: the microtubule-associated protein 1 light chain 3 (LC3; mammalian homolog of the yeast autophagic protein Atg8) and the Atg12 conjugation pathways. The C-terminal amino acids of LC3 are cleaved by Atg4, and this C-terminal residue then gets transferred to phosphatidylethanolamine (PE) in the newly formed isolation membrane by the E1- and E2-like enzymes Atg7 and Atg3, respectively. Although LC3 gets recycled from the outer autophagosomal membrane by deconjugation from its phospholipids, it remains attached to the inner autophagosomal membrane, and this portion is degraded along with the inner autophagosomal membrane in lysosomes and late endosomes after fusion with these vesicles (Ohsumi, 2001). Autophagosome-associated LC3 (LC3-II) and free cytosolic LC3 (LC3-I) can be distinguished by their apparent molecular weight, and autophagosomes can be visualized by using green fluorescent protein (GFP)-conjugated LC3 molecules. In the other ubiquitin-like system, Atg12 gets coupled through its C-terminal glycine residue to a lysine residue of Atg5 by the E1- and E2-like enzymes Atg7 and Atg10, respectively. The Atg12–Atg5 complex associates with Atg16 and then binds to the outer surface of the isolation membrane. Upon completion of the autophagosome, the Atg5–Atg12–Atg16 complex dissociates from the outer autophagosomal membrane and only LC3 remains associated with the completed autophagosome. Autophagosomes then fuse with late endosomes and lysosomes for degradation of their contents and membrane (Figure 1B; Levine and Deretic, 2007).
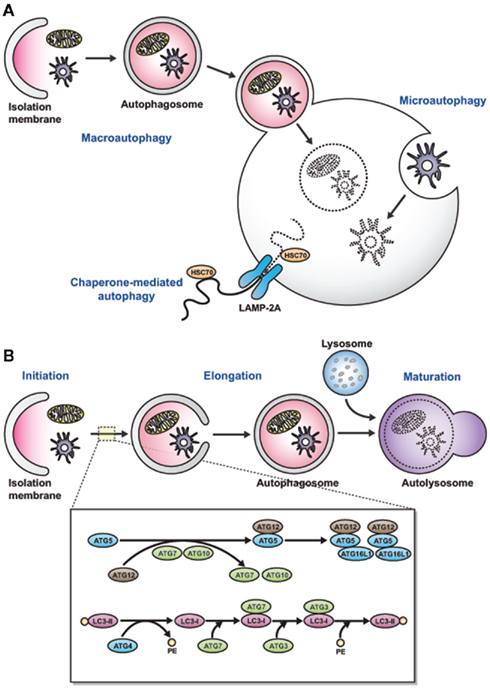
Figure 1. Three types of autophagy and cellular and molecular events in an autophagic pathway. (A) There are three types of autophagy. In macroautophagy, which is the main route of cellular degradation, an autophagosome (a double-layered membrane vesicle) is formed, which fuses with lysosomes to degrade the cellular constituents or pathogens. Microautophagy is characterized by the removal of cellular constituents via the budding of an autophagic body at the lysosomal membrane. In chaperone-mediated autophagy, a signaling motif that contains molecules is transported with the chaperone HSC70 via the LAMP-2A protein into lysosomes. (B) The autophagic pathway consists of three distinct stages. For process initiation, nucleation of the autophagic vesicle is needed. Atg6 (Beclin-1) and type III PI3-kinase are required for the initiation of isolation-membrane formation. Elongation and closure of the autophagosome membrane are regulated by two ubiquitin-like conjugation systems: LC3 and Atg12 conjugation systems. The autophagosome fuses with lysosomes and subsequently matures into an autolysosome to degrade the materials present inside the cell.
Besides maintaining cellular homeostasis, autophagy plays important roles in multiple biological processes including development, aging, and degeneration (Levine and Klionsky, 2004). Not surprisingly, aberrant regulation of autophagy induces many diseases such as cancer, neurodegenerative diseases, and myopathies (Shintani and Klionsky, 2004; Levine and Kroemer, 2008). Recently, autophagy was found to be involved in immunity. It can act as a direct effector by eliminating invading pathogens (Gutierrez et al., 2004; Nakagawa et al., 2004; Deretic, 2005; Ogawa et al., 2005), regulating innate pathogen recognition (Sanjuan et al., 2007; Xu et al., 2007; Delgado et al., 2008), contributing to antigen presentation via major histocompatibility complex (MHC) class II molecules (Dengjel et al., 2005; Paludan et al., 2005; Schmid and Munz, 2007; Schmid et al., 2007), and controlling B- and T-cell development (Li et al., 2006; Pua and He, 2007; Pua et al., 2007; Miller et al., 2008). In this review, we focus on the role of autophagy in innate pathogen recognition and its regulation.
Autophagy in Pathogen Recognition
PAMPs Stimulating Certain TLRs Induce Autophagy Induction
The innate immune system recognizes only a limited number of microbial molecular structures, so-called pathogen-associated molecular patterns (PAMPs; Iwasaki and Medzhitov, 2004; Akira et al., 2006), which are conserved within the same class of microbes but differ across classes (e.g., viruses, Gram-negative bacteria, Gram-positive bacteria, and fungi). Pattern recognition receptors (PRRs) bind to these conserved structures and initiate downstream signaling pathways. PRRs are located in various sites such as plasma membranes, endosomal vesicles, and cytoplasm, thereby enabling the recognition of various types of microbes in any of these locations. Toll-like receptors (TLRs) are the most well-known PRRs in innate pathogen recognition. TLRs can be divided into two groups based on their locations: cell surface TLRs, TLRs 1, 2, 4, 5, and 6, that mainly recognize bacterial components, and endosomal TLRs, TLRs 3, 7, 8, and 9, that generally recognize viral nucleic acids. All these receptors contain leucine-rich repeats (LRRs) in their extracellular domain for ligand binding, and a cytoplasmic Toll/IL-1 receptor (TIR) homology domain, which is essential for signaling. After recognition of PAMPs, TLRs initiate common or distinct signaling pathways via different kinds of adaptor molecules. All TLRs except TLR3 activate the transcription factors, nuclear factor (NF)-κB and activator protein-1 (AP-1), via MyD88, leading to the production of proinflammatory cytokines. TLR3 and TLR4 activate the transcription factor interferon regulatory factor 3 (IRF3) via TIR domain-containing, adapter-inducing interferon-β (TRIF), leading to the production of type I IFNs (Lee and Kim, 2007; Delgado and Deretic, 2009).
Recently, some studies have shown that activation of TLR can lead to induction of autophagy (Xu et al., 2007; Delgado et al., 2008; Shi and Kehrl, 2008). After stimulation with lipopolysaccharide (LPS), TLR4 can induce autophagy in primary human macrophages and in the murine macrophage cell line RAW264.7 (Xu et al., 2007). It was shown that LPS stimulation induces redistribution of LC3 protein from a diffuse to a punctate pattern and increases the levels of the lipidated form of LC3 (LC3-II), both of which are reliable markers of autophagy induction. Interestingly, this LPS-induced autophagy occurs via a TRIF-dependent, MyD88-independent TLR signaling pathway, for which receptor-interacting protein 1 (RIP1) and p38 mitogen-activated protein kinase (MAPK) are required (Figure 2). Moreover, Xu et al. (2007) also reported that LPS-induced autophagy results in mycobacterial colocalization with the autophagosomes, suggesting that autophagy could enhance the elimination of mycobacteria. In another study, mycobacteria elimination has been demonstrated using starvation and rapamycin for autophagy induction(Gutierrez et al., 2004).
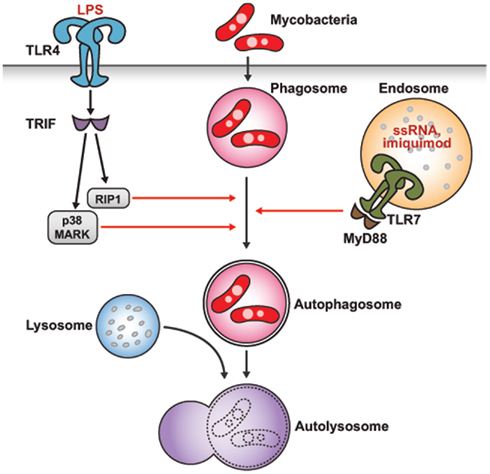
Figure 2. Autophagy induction by TLR stimulation. Activation of TLR can induce autophagy for pathogen elimination. Stimulation of TLR4 with LPS in a TRIF-dependent, MyD88-independent signaling pathway, and stimulation of endosomal TLR7 with two different TLR7 ligands via the MyD88-dependent pathway induces autophagosome formation and eliminates Mycobacteria bacilli, even though mycobacteria are normally not associated with TLR7 signaling. Atg proteins such as Atg5, Beclin-1, and PI3K are required for the formation of an autophagosome.
A study reporting the effect of TLR agonists on autophagy induction in RAW264.7 macrophages (Delgado et al., 2008) showed that ligands of TLR3, TLR4, and TLR7 could induce autophagy, and those of TLR7 generated the most potent effects (Figure 2). Two different ligands of mouse TLR7, single-stranded RNA (ssRNA) and imiquimod, induced formation of autophagosomes characterized by LC3 puncta formation in murine primary macrophages (Delgado et al., 2008). Induction of autophagy via TLR7 signaling was dependent on MyD88 and required Beclin-1. Moreover, autophagy activation by TLR7 agonists induced killing of intracellular mycobacteria, even though mycobacteria are normally not associated with TLR7 signaling. This ability of pathogen elimination was diminished by siRNA knockdown of Beclin 1, thus depending on autophagy.
Interplay between Autophagy and Phagocytosis
Unlike TLR4- and TLR7-mediated induction of autophagy, which enhances autophagosome fusion with the pathogen-containing phagosomes and promotes elimination of intracellular pathogens, certain TLR signaling pathways enhance the maturation of phagosomes by the autophagic machinery (Sanjuan et al., 2007). Upon phagocytosis of zymosan (a component of the fungal cell wall), phagosomes rapidly recruit LC3 and fuse with lysosomes for maturation (Figure 3). Pam3CSK4 coating of latex beads also induces the rapid recruitment of LC3 to phagosomes in RAW264.7 macrophages. This zymosan-mediated translocation of LC3 to phagosomes was found to be independent of MyD88 but dependent on Atg5 and Atg7. Live Saccharomyces cerevisiae engulfed by macrophages survived more frequently in Atg7 knockout macrophages than in wild-type macrophages. Interestingly, LC3 recruitment to phagosomal membranes was not associated with the double-membrane structures characteristic of autophagosomes. Instead, it was associated with phagosome fusion with lysosomes, which enhanced killing of the engulfed pathogens. Collectively, this study has shown a new way of utilizing the autophagic machinery to promote conventional functions of phagocytes after TLR activation, in the absence of autophagosome formation.
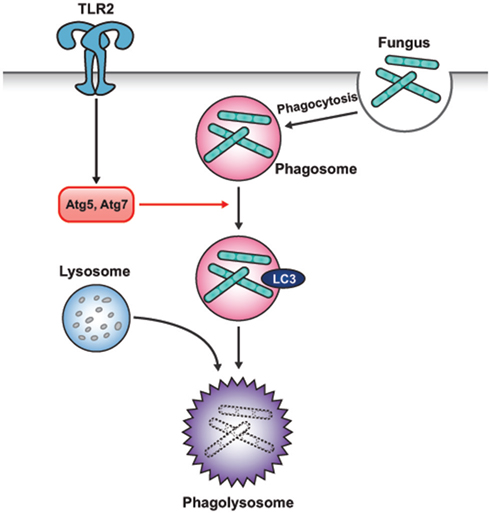
Figure 3. Interplay between autophagy and phagocytosis. TLR signaling can enhance the maturation of phagosomes with the autophagic machinery. LC3 recruitment to phagosomal membranes promotes the fusion of phagosome with lysosome for maturation. However, LC3 recruitment to phagosomal membranes is not associated with the characteristic double-membrane structure of the autophagosomes. The Atg5 and Atg7 proteins are involved in the development of this structure.
NOD-Like Receptor Signaling and Autophagy
To the best of our knowledge, there are only two reports on the NOD-like receptor (NLR) family in autophagy (Cooney et al., 2010; Travassos et al., 2010). NLRs recognize bacterial cell wall components (specifically, peptidoglycans) in the eukaryotic cell’s cytosol. NLRs are composed of three distinct domains: a C-terminal LRR, a NACHT domain, and an N-terminal effector domain, which mediate ligand sensing, activation of the NLRs, and initiation of downstream signaling, respectively. When NLRs recognize bacterial peptidoglycans, they initiate signaling transduction by recruiting the protein kinases, which, in turn, activate NF-κB and AP-1 leading to production of cytokines and other molecules involved in innate immunity (Lee and Kim, 2007).
Recent studies have shown that activation of NOD2 by muramyl dipeptide (MDP) induces autophagosome formation, which in turn enhances bacterial clearance (Figure 4; Kuma et al., 2004; Travassos et al., 2010). In human DCs, NOD2 stimulated with MDP induces autophagosome formation, which promotes MHC class II-associated antigen presentation. Atg5, Atg7, Atg16L1, and receptor-interacting serine-threonine kinase-2 (RIPK2), the latter being one of the downstream regulators of the NOD2 signaling pathway, are required for autophagosome formation and antigen presentation by MDP (Cooney et al., 2010). Another study also showed that stimulation of NOD1 and NOD2 by bacterial peptidoglycans activates the autophagy pathway in mouse embryonic fibroblasts (MEFs; Travassos et al., 2010). Upon bacterial invasion, NOD2 recruits Atg16L1 to the bacterial entry sites, facilitating bacterial trafficking to the autophagosomes. This, in turn, induces the fusion of the autophagosomes with the lysosomes to form the autophagolysosomes and promotes antigen presentation via MHC class II molecules. In MEFs, this process does not require the adaptor RIP2 and the transcription factor NF-κB. Interestingly, NOD2 mutation and single-nucleotide polymorphism in the Atg16L1 gene have been known to be associated with the development of Crohn’s disease (Cho and Weaver, 2007; Hampe et al., 2007; Rioux et al., 2007). DCs isolated from patients with Crohn’s disease and risk alleles for NOD2 or Atg16L1 showed impaired function in autophagy induction and antigen presentation (Cooney et al., 2010). Collectively, these reports revealed a close relationship between two of the most important Crohn’s disease-associated susceptibility genes. Furthermore, they functionally link bacterial sensing by NOD proteins to the autophagy pathway.
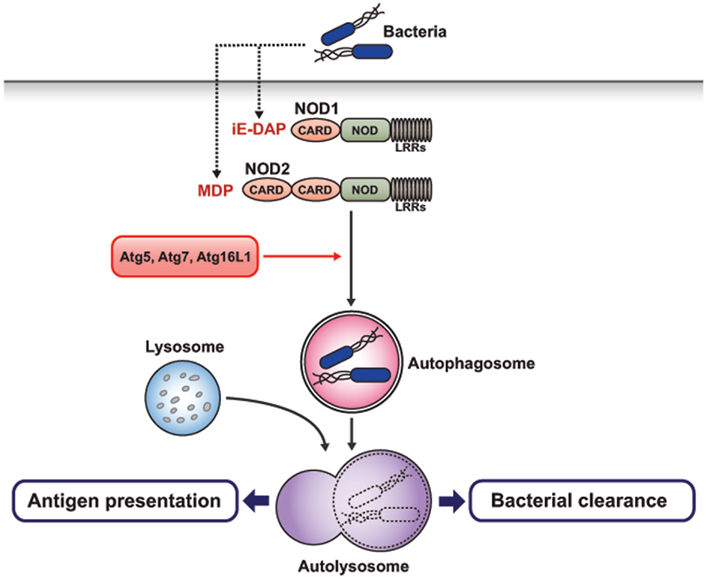
Figure 4. NOD-like receptor-mediated autophagy induction. NOD-like receptors recognize bacterial peptidoglycan in the cytosol. C-terminal leucine-rich repeats (LRRs) of NOD1 and NOD2 detect γ-D-glutamyl-meso-diaminopimelic acid (iE-DAP) and muramyl dipeptide(MDP), respectively. NOD1 and NOD2 can induce the autophagosome formation after stimulation with bacterial peptidoglycan, which promotes bacterial clearance and MHC class II-associated antigen presentation. This process depends on Atg5, Atg7, and Atg16L1.
Role of Autophagy in Viral Sensing
In addition to the aforementioned direct effector function, autophagy also works in TLR activation by delivering cytosolic PAMPs to endosomal TLRs (Lee et al., 2007). In plasmacytoid dendritic cells (pDCs), viral RNA or DNA recognized by TLR7 or TLR9, respectively, induces type I IFN and proinflammatory cytokine production. These TLRs are located in the endosomal compartment and sense the viral nucleic acids endocytosed by the host cells (Barton, 2007). In addition to NF-κB and MAPK activation, MyD88, an adaptor molecule for these TLRs, activates IRF7, leading to type I IFN production.
In the case of vesicular stomatitis virus (VSV), the replicating virus rather than the viral genome is required to initiate TLR7 signaling and produce IFN-α in pDCs. However, how these cytosolic replication intermediates gain access to the endosomal compartment where TLR7 resides is not completely known. A recent study demonstrated that cytosolic PAMP is delivered by autophagy to the lysosomes for TLR7 recognition (Figure 5; Lee et al., 2007). Thus, Atg5-deficient pDCs fail to sense VSV via TLR7, and are unable to secrete IFN-α and IL-12p40. Consequently, mice lacking Atg5 fail to defend themselves from systemic VSV infection in vivo. Moreover, IFN-α production is impaired in Atg5-deficient pDCs in response to herpes simplex virus-1 (HSV-1), which is recognized by TLR9, while IL-12 response remained intact in these cells. Thus, autophagy plays a critical role in the induction of innate immune responses by delivering viral replication intermediates from the cytosol to the endosome for recognition after ssRNA virus infection (Lee and Iwasaki, 2008; Tal and Iwasaki, 2009; Yordy and Iwasaki, 2011).
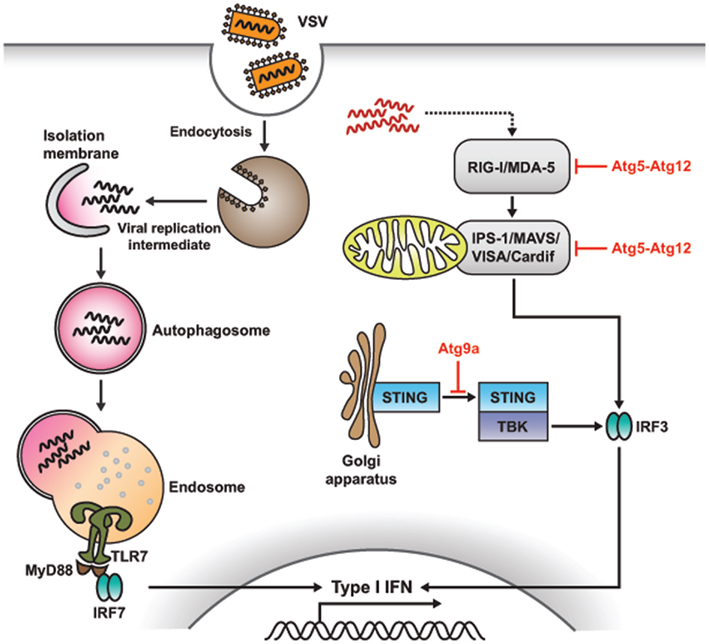
Figure 5. Role of autophagy in antiviral immune response. Autophagy and Atg proteins are required for viral sensing and regulation of antiviral immune responses. In pDCs, autophagy mediates the recognition of viral infection by delivering the viral replication intermediates in the cytosol to lysosomes, where TLR recognition occurs, which, in turn, enhances type I IFN production. However, in non-pDCs, such as mouse embryonic fibroblasts (MEFs), autophagy negatively regulates type I IFN production in response to viral infection. Atg5-Atg12 conjugates block RLR signaling by direct CARD-mediated association with RIG-I and IPS-1, resulting in the suppression of type I IFN production. In the case of dsDNA recognition, STING, a multispanning membrane protein, is translocated from the ER to the Golgi apparatus and assembled with TBK1, which phosphorylates the transcription factor IRF3, resulting in the production of type I IFN. During this process, Atg9a, an essential component of autophagy, colocalizes with STING in the Golgi apparatus, where it controls the assembly of STING.
Negative Regulation of Innate Immune Responses via Autophagy
The autophagic machinery plays key roles other than activating PRR signaling. In contrast to viral recognition in pDCs, which is mediated by endosomal TLRs, most of the other cell types utilize cytosolic sensors such as those encoded by retinoic acid-inducible gene I (RIG-I) and melanoma differentiation associated gene 5 (MDA-5) to detect the virus invasion (Yoneyama et al., 2004, 2005; Foy et al., 2005). RIG-I and MDA-5, both of which are RIG-I-like receptors (RLRs), contain a DExD/H box RNA helicase domain for ligand recognition and two caspase-recruiting domains (CARDs) for initiating downstream signaling. When these CARD-containing RNA helicases recognize double-stranded RNA (dsRNA), which is synthesized during active viral replication in the cytosol, signals through IFN-β promoter stimulator-1 [IPS-1; also known as mitochondrial antiviral signaling (MAVS), virus-induced signaling adaptor (VISA), or Cardif] activate the transcription factors IRF-3 and NF-κB, and subsequently lead to the production of type I IFN. IPS-1 is an essential adaptor molecule consisting of an N-terminal CARD domain (through which it associates with RIG-I and MDA-5) and a C-terminal domain (for mitochondrial localization; Lee and Kim, 2007).
A recent study revealed that Atg5–Atg12 conjugates, essential components of the autophagic process, regulate innate viral recognition by RIG-I and MDA-5 in MEFs (Figure 5; Jounai et al., 2007). Atg5- and Atg7-deficient MEFs, lacking Atg5–Atg12 conjugates, overproduce type I IFN in response to VSV. Conversely, biochemical analysis indicated that Atg5–Agt12 conjugates block RLR signaling by direct CARD-mediated association with RIG-I and IPS-1; additionally, they suppress type I IFN production. Thus, autophagy-related proteins involved in RLR-mediated viral sensing repress type I IFN response, acting as negative regulators of antiviral responses.
Similarly, in another report, Atg5-deficient cells showed overproduction of type I IFN through enhanced RLR signaling pathway (Tal et al., 2009). That study also showed that dysfunctional mitochondria and mitochondria-associated IPS-1 were accumulated in the absence of autophagy. In Atg5-deficient cells, reactive oxygen species associated with dysfunctional mitochondria may potentiate RLR signaling, which can be blocked by antioxidant treatment. Thus, autophagy plays important roles in the balanced regulation of innate antiviral response by acting as a scavenger of dysfunctional mitochondria.
Another recent study showed that Atg16L1, a candidate gene for Crohn’s disease, is involved in endotoxin-induced inflammasome activation in mice (Figure 6A; Saitoh et al., 2008). Atg16L1 is an essential component of the autophagosome. It forms a complex with Atg5–Atg12 conjugates and induces LC3-PE conjugation (Fujita et al., 2008). Atg16L1-deficient macrophages exhibit enhanced IL-1β and IL-18 production in response to LPS. This process is mediated by TRIF-dependent activation of caspase-1, which, in turn, activates IL-1β production. Moreover, mice lacking Atg16L1 in hematopoietic cells are highly susceptible to dextran sulfate sodium-induced acute colitis, which is alleviated by the injection of anti-IL-1β and IL-18 antibodies (Saitoh et al., 2008). Hence, Atg16L1 plays an important role in negatively regulating endotoxin-induced inflammatory immune responses.
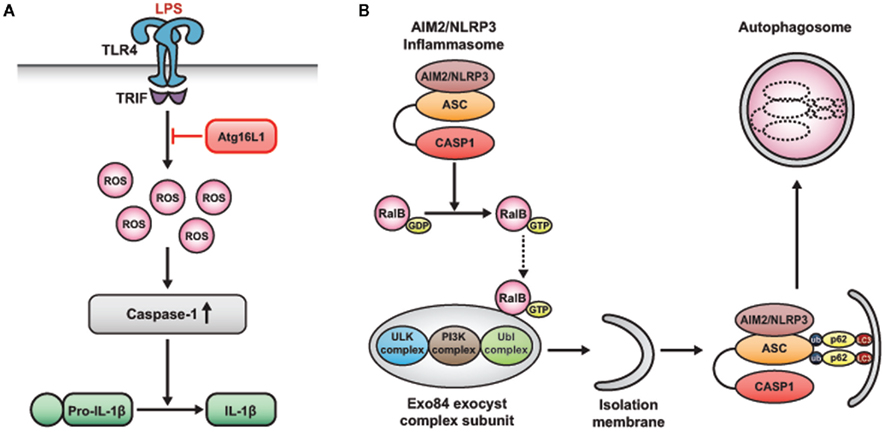
Figure 6. Suppression of inflammasome activity by autophagy. (A) Atg16L1, a candidate gene for Crohn’s disease, regulates endotoxin-induced inflammasome activation. Atg16L1-deficient macrophages exhibit enhanced IL-1β and IL-18 production in response to LPS. This process is mediated by excessive ROS, which activates caspase-1, leading to the processing of IL-1β. (B) Autophagy limits IL-1β production by eliminating active inflammasomes. Induction of AIM2 or NLRP3 inflammasomes triggers the nucleotide exchange of RalB, which promotes autophagosome formation by the binding activated RalB to the exocyst complex subunit Exo84. This binding induces the assembly of active kinase complexes on the exocyst, which are required for the formation and maturation of isolation membranes into autophagosomes. Activation of inflammasomes leads to the ubiquitination of ASC, and ubiquitinated inflammasomes are engulfed by autophagosomes via the autophagic adaptor protein, p62.
Very recently, an interesting study showed the relationship between autophagy and inflammasome activity. This study suggested that autophagy induced by inflammatory signals targets ubiquitinated inflammasomes for destruction, thereby limiting IL-1β production (Figure 6B; Shi et al., 2012). The activation of AIM2 or NLRP3 inflammasomes in macrophages triggers nucleotide exchange on RalB, thereby effecting autophagosome assembly (Bodemann et al., 2011; Shi et al., 2012). Autophagic adaptors such as p62 (sequestosome 1) and neighbor of BRCA1 gene (NBR1), which have ubiquitin-associated (UBA) domains and LC3-interacting regions (LIR), recognize ubiquitinated molecules and facilitate their elimination by autophagy (Johansen and Lamark, 2011; Deretic, 2012). Various kinds of intracellular pathogens are recognized by different kinds of autophagic adaptors and thereby eliminated by autophagy (Dupont et al., 2009; Zheng et al., 2009; Orvedahl et al., 2010; Mostowy et al., 2011). Similarly, in the present study, assembled inflammasomes were ubiquitinated, and these ubiquitinated complexes were recruited by the autophagic adaptor p62, which assisted in their entry into the autophagy pathway. Thus, autophagy limits inflammasome activity by eliminating active inflammasomes, and this tempers inflammation.
The exact mechanisms underlying recognition of dsDNA derived from bacteria or DNA viruses are still unclear, and so are the subsequent immune responses. Nonetheless, it becomes apparent that stimulation with dsDNA induces the production of type I IFNs and other inflammatory cytokines (Stetson and Medzhitov, 2006; Charrel-Dennis et al., 2008). Recent studies indicated that translocation and assembly of stimulator of IFN genes (STING) and TANK-binding kinase 1 (TBK1) are required for the induction of type I IFN responses (Ishikawa and Barber, 2008; Jin et al., 2008; Zhong et al., 2008; Sun et al., 2009). STING is a multispanning membrane protein, which is translocated from the endoplasmic reticulum to the Golgi apparatus after sensing dsDNA, followed by assembly with TBK1, which phosphorylates the transcription factor IRF3, leading to the production of type I IFNs. During this process, Atg9a colocalizes with STING in the Golgi apparatus, where it controls the assembly of STING (Saitoh et al., 2009). In Atg9a-deficient MEFs, but not in Atg7- and Atg16L1-deficient MEFs, the translocation of STING from the Golgi apparatus to the cytoplasmic punctate structures, and its assembly with TBK1, are greatly enhanced. This, in turn, induces aberrant activation of type I IFN responses (Saitoh and Akira, 2010). Overall, these findings underline a role for Atg9a in the regulation of innate immune responses.
Conclusions
Recent studies have demonstrated that autophagy acts as an important regulator of immune responses. In addition to elimination of intracellular pathogens by its original function of degradation (xenophagy), autophagy can be involved in innate pathogen recognition and modulate the downstream signaling pathway. Autophagy promotes the clearance of cytosolic pathogens via autophagosome formation, which is elicited by TLR sensing. Autophagic machinery is utilized to enhance phagosome–lysosome fusion and efficiently eliminate extracellular phagocytosed pathogens. Moreover, autophagy can promote adaptive immune responses such as MHC class II-restricted antigen presentation after bacterial sensing via NLRs. In case of viral recognition, autophagy delivers the cytosolic replication intermediates to the lysosomes, where recognition by the endosomal TLRs occurs, thus enhancing the production of type I IFN essential for antiviral response. However, autophagy does not play a role in enhancing pathogen elimination alone. It can also negatively regulate the signaling pathway mediating pathogen recognition and elimination. In case of viral recognition by cytosolic viral sensors such as RLRs, autophagy represses the signaling downstream of the innate immune response, such as that mediating the production of type I IFN. Autophagy-related proteins, including Atg16L1, are also involved in the regulation of endotoxin-induced inflammasome activation, which has been associated with Crohn’s disease. Recent advances in the study of autophagy have largely helped understanding the mode of function of the autophagic machinery in PRR-mediated innate pathogen recognition and its regulation. Considering the complexity of autophagy function in immunity, it is still unclear whether activation or suppression of autophagy could have therapeutic benefits in the treatment of infectious diseases or inflammatory disorders such as Crohn’s disease. A better understanding of the modulation of the immune system by autophagy is essential to unveil new therapeutic avenues in the future.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Jeongsu Park for his help with the figure. This study was supported by the National Research Foundation (2010-0012891) and the Converging Research Center Program (2011K000864) funded by the Ministry of Education, Science and Technology of Korea. This study was also supported by the Korean Health Technology R&D Project (A100920) and the National R&D Program for Cancer Control (1020230) funded by the Ministry of Health and Welfare, Republic of Korea.
References
Akira, S., Uematsu, S., and Takeuchi, O. (2006). Pathogen recognition and innate immunity. Cell 124, 783–801.
Bodemann, B. O., Orvedahl, A., Cheng, T., Ram, R. R., Ou, Y. H., Formstecher, E., Maiti, M., Hazelett, C. C., Wauson, E. M., Balakireva, M., Camonis, J. H., Yeaman, C., Levine, B., and White, M. A. (2011). RalB and the exocyst mediate the cellular starvation response by direct activation of autophagosome assembly. Cell 144, 253–267.
Charrel-Dennis, M., Latz, E., Halmen, K. A., Trieu-Cuot, P., Fitzgerald, K. A., Kasper, D. L., and Golenbock, D. T. (2008). TLR-independent type I interferon induction in response to an extracellular bacterial pathogen via intracellular recognition of its DNA. Cell Host Microbe 4, 543–554.
Cho, J. H., and Weaver, C. T. (2007). The genetics of inflammatory bowel disease. Gastroenterology 133, 1327–1339.
Cooney, R., Baker, J., Brain, O., Danis, B., Pichulik, T., Allan, P., Ferguson, D. J., Campbell, B. J., Jewell, D., and Simmons, A. (2010). NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat. Med. 16, 90–97.
Delgado, M. A., and Deretic, V. (2009). Toll-like receptors in control of immunological autophagy. Cell Death Differ. 16, 976–983.
Delgado, M. A., Elmaoued, R. A., Davis, A. S., Kyei, G., and Deretic, V. (2008). Toll-like receptors control autophagy. EMBO J. 27, 1110–1121.
Dengjel, J., Schoor, O., Fischer, R., Reich, M., Kraus, M., Muller, M., Kreymborg, K., Altenberend, F., Brandenburg, J., Kalbacher, H., Brock, R., Driessen, C., Rammensee, H. G., and Stevanovic, S. (2005). Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc. Natl. Acad. Sci. U.S.A. 102, 7922–7927.
Deretic, V. (2012). Autophagy as an innate immunity paradigm: expanding the scope and repertoire of pattern recognition receptors. Curr. Opin. Immunol. 24, 21–31.
Dupont, N., Lacas-Gervais, S., Bertout, J., Paz, I., Freche, B., Van Nhieu, G. T., Van Der Goot, F. G., Sansonetti, P. J., and Lafont, F. (2009). Shigella phagocytic vacuolar membrane remnants participate in the cellular response to pathogen invasion and are regulated by autophagy. Cell Host Microbe 6, 137–149.
Foy, E., Li, K., Sumpter, R., Loo, Y. M. Jr., Johnson, C. L., Wang, C., Fish, P. M., Yoneyama, M., Fujita, T., Lemon, S. M., and Gale, M. Jr. (2005). Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. Proc. Natl. Acad. Sci. U.S.A. 102, 2986–2991.
Fujita, N., Itoh, T., Omori, H., Fukuda, M., Noda, T., and Yoshimori, T. (2008). The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell 19, 2092–2100.
Gutierrez, M. G., Master, S. S., Singh, S. B., Taylor, G. A., Colombo, M. I., and Deretic, V. (2004). Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119, 753–766.
Hampe, J., Franke, A., Rosenstiel, P., Till, A., Teuber, M., Huse, K., Albrecht, M., Mayr, G., De La Vega, F. M., Briggs, J., Gunther, S., Prescott, N. J., Onnie, C. M., Hasler, R., Sipos, B., Folsch, U. R., Lengauer, T., Platzer, M., Mathew, C. G., Krawczak, M., and Schreiber, S. (2007). A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat. Genet. 39, 207–211.
Ishikawa, H., and Barber, G. N. (2008). STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678.
Iwasaki, A., and Medzhitov, R. (2004). Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 5, 987–995.
Jin, L., Waterman, P. M., Jonscher, K. R., Short, C. M., Reisdorph, N. A., and Cambier, J. C. (2008). MPYS, a novel membrane tetraspanner, is associated with major histocompatibility complex class II and mediates transduction of apoptotic signals. Mol. Cell. Biol. 28, 5014–5026.
Johansen, T., and Lamark, T. (2011). Selective autophagy mediated by autophagic adapter proteins. Autophagy 7, 279–296.
Jounai, N., Takeshita, F., Kobiyama, K., Sawano, A., Miyawaki, A., Xin, K. Q., Ishii, K. J., Kawai, T., Akira, S., Suzuki, K., and Okuda, K. (2007). The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc. Natl. Acad. Sci. U.S.A. 104, 14050–14055.
Klionsky, D. J., and Emr, S. D. (2000). Autophagy as a regulated pathway of cellular degradation. Science 290, 1717–1721.
Komatsu, M., Waguri, S., Ueno, T., Iwata, J., Murata, S., Tanida, I., Ezaki, J., Mizushima, N., Ohsumi, Y., Uchiyama, Y., Kominami, E., Tanaka, K., and Chiba, T. (2005). Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 169, 425–434.
Kuma, A., Hatano, M., Matsui, M., Yamamoto, A., Nakaya, H., Yoshimori, T., Ohsumi, Y., Tokuhisa, T., and Mizushima, N. (2004). The role of autophagy during the early neonatal starvation period. Nature 432, 1032–1036.
Lee, H. K., and Iwasaki, A. (2008). Autophagy and antiviral immunity. Curr. Opin. Immunol. 20, 23–29.
Lee, H. K., Lund, J. M., Ramanathan, B., Mizushima, N., and Iwasaki, A. (2007). Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 315, 1398–1401.
Lee, M. S., and Kim, Y. J. (2007). Signaling pathways downstream of pattern-recognition receptors and their cross talk. Annu. Rev. Biochem. 76, 447–480.
Levine, B., and Deretic, V. (2007). Unveiling the roles of autophagy in innate and adaptive immunity. Nat. Rev. Immunol. 7, 767–777.
Levine, B., and Klionsky, D. J. (2004). Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev. Cell 6, 463–477.
Li, C., Capan, E., Zhao, Y., Zhao, J., Stolz, D., Watkins, S. C., Jin, S., and Lu, B. (2006). Autophagy is induced in CD4 (T cells and important for the growth factor-withdrawal cell death. J. Immunol. 177, 5163–5168.
Miller, B. C., Zhao, Z., Stephenson, L. M., Cadwell, K., Pua, H. H., Lee, H. K., Mizushima, N. N., Iwasaki, A., He, Y. W., Swat, W., and Virgin, H. W. T. (2008). The autophagy gene ATG5 plays an essential role in B lymphocyte development. Autophagy 4, 309–314.
Mizushima, N., and Klionsky, D. J. (2007). Protein turnover via autophagy: implications for metabolism. Annu. Rev. Nutr. 27, 19–40.
Mizushima, N., Noda, T., Yoshimori, T., Tanaka, Y., Ishii, T., George, M. D., Klionsky, D. J., Ohsumi, M., and Ohsumi, Y. (1998). A protein conjugation system essential for autophagy. Nature 395, 395–398.
Mizushima, N., Ohsumi, Y., and Yoshimori, T. (2002). Autophagosome formation in mammalian cells. Cell Struct. Funct. 27, 421–429.
Mostowy, S., Sancho-Shimizu, V., Hamon, M. A., Simeone, R., Brosch, R., Johansen, T., and Cossart, P. (2011). p62 and NDP52 proteins target intracytosolic Shigella and Listeria to different autophagy pathways. J. Biol. Chem. 286, 26987–26995.
Nakagawa, I., Amano, A., Mizushima, N., Yamamoto, A., Yamaguchi, H., Kamimoto, T., Nara, A., Funao, J., Nakata, M., Tsuda, K., Hamada, S., and Yoshimori, T. (2004). Autophagy defends cells against invading group A Streptococcus. Science 306, 1037–1040.
Ogawa, M., Yoshimori, T., Suzuki, T., Sagara, H., Mizushima, N., and Sasakawa, C. (2005). Escape of intracellular Shigella from autophagy. Science 307, 727–731.
Ohsumi, Y. (2001). Molecular dissection of autophagy: two ubiquitin-like systems. Nat. Rev. Mol. Cell Biol. 2, 211–216.
Orvedahl, A., Macpherson, S., Sumpter, R., Talloczy, Z. Jr., Zou, Z., and Levine, B. (2010). Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe 7, 115–127.
Paludan, C., Schmid, D., Landthaler, M., Vockerodt, M., Kube, D., Tuschl, T., and Munz, C. (2005). Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 307, 593–596.
Pua, H. H., Dzhagalov, I., Chuck, M., Mizushima, N., and He, Y. W. (2007). A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J. Exp. Med. 204, 25–31.
Pua, H. H., and He, Y. W. (2007). Maintaining T lymphocyte homeostasis: another duty of autophagy. Autophagy 3, 266–267.
Rioux, J. D., Xavier, R. J., Taylor, K. D., Silverberg, M. S., Goyette, P., Huett, A., Green, T., Kuballa, P., Barmada, M. M., Datta, L. W., Shugart, Y. Y., Griffiths, A. M., Targan, S. R., Ippoliti, A. F., Bernard, E. J., Mei, L., Nicolae, D. L., Regueiro, M., Schumm, L. P., Steinhart, A. H., Rotter, J. I., Duerr, R. H., Cho, J. H., Daly, M. J., and Brant, S. R. (2007). Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet. 39, 596–604.
Saitoh, T., and Akira, S. (2010). Regulation of innate immune responses by autophagy-related proteins. J. Cell Biol. 189, 925–935.
Saitoh, T., Fujita, N., Hayashi, T., Takahara, K., Satoh, T., Lee, H., Matsunaga, K., Kageyama, S., Omori, H., Noda, T., Yamamoto, N., Kawai, T., Ishii, K., Takeuchi, O., Yoshimori, T., and Akira, S. (2009). Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc. Natl. Acad. Sci. U.S.A. 106, 20842–20846.
Saitoh, T., Fujita, N., Jang, M. H., Uematsu, S., Yang, B. G., Satoh, T., Omori, H., Noda, T., Yamamoto, N., Komatsu, M., Tanaka, K., Kawai, T., Tsujimura, T., Takeuchi, O., Yoshimori, T., and Akira, S. (2008). Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 456, 264–268.
Sanjuan, M. A., Dillon, C. P., Tait, S. W., Moshiach, S., Dorsey, F., Connell, S., Komatsu, M., Tanaka, K., Cleveland, J. L., Withoff, S., and Green, D. R. (2007). Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 450, 1253–1257.
Schmid, D., and Munz, C. (2007). Innate and adaptive immunity through autophagy. Immunity 27, 11–21.
Schmid, D., Pypaert, M., and Munz, C. (2007). Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity 26, 79–92.
Shi, C. S., and Kehrl, J. H. (2008). MyD88 and Trif target Beclin 1 to trigger autophagy in macrophages. J. Biol. Chem. 283, 33175–33182.
Shi, C. S., Shenderov, K., Huang, N. N., Kabat, J., Abu-Asab, M., Fitzgerald, K. A., Sher, A., and Kehrl, J. H. (2012). Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 13, 255–263.
Shintani, T., and Klionsky, D. J. (2004). Autophagy in health and disease: a double-edged sword. Science 306, 990–995.
Stetson, D. B., and Medzhitov, R. (2006). Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity 24, 93–103.
Sun, W., Li, Y., Chen, L., Chen, H., You, F., Zhou, X., Zhou, Y., Zhai, Z., Chen, D., and Jiang, Z. (2009). ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc. Natl. Acad. Sci. U.S.A. 106, 8653–8658.
Tal, M. C., and Iwasaki, A. (2009). Autophagy and innate recognition systems. Curr. Top. Microbiol. Immunol. 335, 107–121.
Tal, M. C., Sasai, M., Lee, H. K., Yordy, B., Shadel, G. S., and Iwasaki, A. (2009). Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc. Natl. Acad. Sci. U.S.A. 106, 2770–2775.
Travassos, L. H., Carneiro, L. A., Ramjeet, M., Hussey, S., Kim, Y. G., Magalhaes, J. G., Yuan, L., Soares, F., Chea, E., Le Bourhis, L., Boneca, I. G., Allaoui, A., Jones, N. L., Nunez, G., Girardin, S. E., and Philpott, D. J. (2010). Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 11, 55–62.
Xu, Y., Jagannath, C., Liu, X. D., Sharafkhaneh, A., Kolodziejska, K. E., and Eissa, N. T. (2007). Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 27, 135–144.
Yoneyama, M., Kikuchi, M., Matsumoto, K., Imaizumi, T., Miyagishi, M., Taira, K., Foy, E., Loo, Y. M., Gale, M., Akira, S. Jr., Yonehara, S., Kato, A., and Fujita, T. (2005). Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol. 175, 2851–2858.
Yoneyama, M., Kikuchi, M., Natsukawa, T., Shinobu, N., Imaizumi, T., Miyagishi, M., Taira, K., Akira, S., and Fujita, T. (2004). The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 5, 730–737.
Yordy, B., and Iwasaki, A. (2011). Autophagy in the control and pathogenesis of viral infection. Curr. Opin. Virol. 1, 196–203.
Zheng, Y. T., Shahnazari, S., Brech, A., Lamark, T., Johansen, T., and Brumell, J. H. (2009). The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J. Immunol. 183, 5909–5916.
Keywords: autophagy, Toll-like receptors, RIG-I-like receptors, NOD-like receptors
Citation: Oh JE and Lee HK (2012) Modulation of pathogen recognition by autophagy. Front. Immun. 3:44. doi: 10.3389/fimmu.2012.00044
Received: 15 December 2011; Paper pending published: 02 January 2012;
Accepted: 23 February 2012; Published online: 12 March 2012.
Edited by:
Christian Munz, University of Zurich, SwitzerlandReviewed by:
Laura Santambrogio, Albert Einstein College of Medicine, USAJürgen Wittmann, University of Erlangen-Nürnberg, Germany
Copyright: © 2012 Oh and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Heung Kyu Lee, Laboratory of Host Defenses, Graduate School of Medical Science and Engineering, Korea Advanced Institute of Science and Technology, 291 Daehak-ro, Daejeon 305-701, Korea. e-mail:aGV1bmdreXUubGVlQGthaXN0LmFjLmty