 Sarah Bruneau1,2 Craig Bryan Woda1,2 Kevin Patrick Daly1,2 Leonard Boneschansker1,2 Namrata Gargee Jain1,2
Sarah Bruneau1,2 Craig Bryan Woda1,2 Kevin Patrick Daly1,2 Leonard Boneschansker1,2 Namrata Gargee Jain1,2 Nora Kochupurakkal1,2 Alan Gabriel Contreras1,2 Tatsuichiro Seto1,2
Nora Kochupurakkal1,2 Alan Gabriel Contreras1,2 Tatsuichiro Seto1,2 David Michael Briscoe1,2*
David Michael Briscoe1,2*- 1 The Division of Nephrology, Transplantation Research Center, Children’s Hospital Boston, Boston, MA, USA
- 2 Department of Pediatrics, Harvard Medical School, Boston, MA, USA
In this review, we discuss how changes in the intragraft microenvironment serve to promote or sustain the development of chronic allograft rejection. We propose two key elements within the microenvironment that contribute to the rejection process. The first is endothelial cell proliferation and angiogenesis that serve to create abnormal microvascular blood flow patterns as well as local tissue hypoxia, and precedes endothelial-to-mesenchymal transition. The second is the overexpression of local cytokines and growth factors that serve to sustain inflammation and, in turn, function to promote a leukocyte-induced angiogenesis reaction. Central to both events is overexpression of vascular endothelial growth factor (VEGF), which is both pro-inflammatory and pro-angiogenic, and thus drives progression of the chronic rejection microenvironment. In our discussion, we focus on how inflammation results in angiogenesis and how leukocyte-induced angiogenesis is pathological. We also discuss how VEGF is a master control factor that fosters the development of the chronic rejection microenvironment. Overall, this review provides insight into the intragraft microenvironment as an important paradigm for future direction in the field.
General Overview
In an endothelial cell (EC)-based model, the initiation of inflammation within an allograft results from the activation of donor ECs responding to pro-inflammatory cytokines released from resident macrophages in response to hypoxia (Cotran, 1994; Briscoe et al., 1998; Denton et al., 2000; Pober and Sessa, 2007; Ingulli et al., 2009). Graft EC also respond to cytokines and growth factors produced in association with alloimmune cellular and humoral targeting of the graft (Pober et al., 1996; Valujskikh and Heeger, 2003; Zhang and Reed, 2009; Halloran et al., 2010; Sis and Halloran, 2010), as well as by factors produced by infiltrating mononuclear cells that are characteristic of chronic rejection (Libby and Pober, 2001). The induced expression of adhesion molecules and chemokines by donor EC results in the recruitment of leukocytes into the graft, whereas the expression of MHC class I and II molecules on donor EC is critical for the local presentation of alloantigen to infiltrating effector/memory lymphocytes (Briscoe and Sayegh, 2002; Kreisel et al., 2002; Pober and Sessa, 2007). These events set the stage for an intragraft microenvironment that sustains donor-directed alloimmune inflammation and the development of chronic rejection.
In this review, we focus on how the integrity of the vascular endothelium is critical for a microenvironment that sustains allograft function. In vascularized solid organ allografts, such as the kidney, early ischemia–reperfusion results in profound injury to the microvasculature (Bishop et al., 1989; Vos and Briscoe, 2002; Woywodt et al., 2003; Reinders et al., 2006; Aydin et al., 2007; Contreras and Briscoe, 2007; Rabelink et al., 2007; Mayer, 2011). Furthermore, the degree of injury and microvascular EC loss at early times post transplantation can be predictive of long-term graft survival (Bishop et al., 1989; Choi et al., 2000; Fine and Norman, 2008; Mayer, 2011; Steegh et al., 2011). Indeed, it is reported that the sequential loss of peritubular capillaries, starting as early as 3 months post renal transplantation predicts the development of interstitial fibrosis and tubular atrophy (IFTA) and later chronic rejection (Steegh et al., 2011). It is proposed that the loss of the intrarenal microvasculature results in impaired delivery of oxygen and nutrients to the renal tubules, which in turn contributes to local tissue ischemia, tubular dropout, and cell death (Kang et al., 2002; Reinders et al., 2006; Contreras and Briscoe, 2007; Rabelink et al., 2007; Fine and Norman, 2008; Mayer, 2011). Thus, the initial loss of microvessels/peritubular capillaries may be a primary factor in the development of fibrosis and chronic renal disease (Kang et al., 2002; Reinders and Briscoe, 2002; Contreras and Briscoe, 2007; Mayer, 2011). Pharmacologic therapy which can protect microvascular integrity at early times post transplantation has potential to improve long-term graft survival (Johnson et al., 2006; Nakao et al., 2006; Aydin et al., 2007; Rabelink et al., 2007; Briscoe and Pal, 2008; Leonard et al., 2008; Hanto et al., 2010). If early protection and repair is not accomplished, then ongoing local ischemia will result in cellular atrophy, and chronic allograft disease will be inevitable.
However, it is underappreciated that inflammatory infiltrates also cause EC proliferation, a process called leukocyte-induced angiogenesis (Auerbach and Sidky, 1979; Cotran, 1994). In addition, the binding of alloantibodies to the graft vascular endothelium can result in EC activation and a proliferative response (Zhang and Reed, 2009). As will be discussed below, this abnormal or pathological angiogenesis response may be associated with local tissue hypoxia, and thus precedes later hypoxic tissue injury (Babu et al., 2007; Contreras and Briscoe, 2007; Goel et al., 2011). If EC proliferation occurs at later times post transplantation in association with pericyte loss, microvessels become disorganized and endothelial-to-mesenchymal transition (EndMT) may occur. This results in collagen deposition and tissue fibrosis (Schor et al., 1995; Humphreys et al., 2010; Medici et al., 2010). Central to all these events is the expression of vascular endothelial growth factor (VEGF), which is both pro-angiogenic and pro-inflammatory, and thus drives progression of the chronic rejection microenvironment (Reinders et al., 2006). Here, we discuss a new paradigm, whereby local tissue hypoxia and overexpressed intragraft VEGF are key features of a microenvironment that determine the development of chronic rejection. Cartoons illustrating this paradigm are shown in Figures 1 and 2.
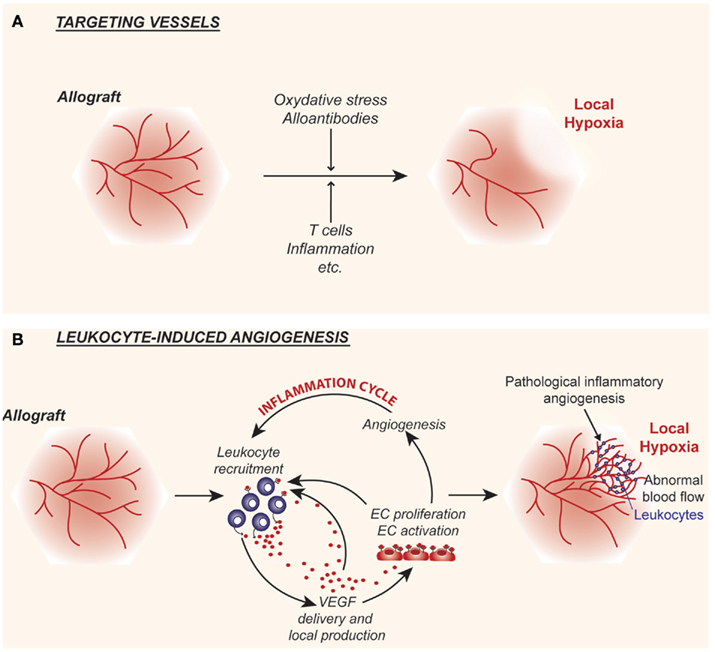
Figure 1. Cartoon illustrating the interplay between alloimmunity and the intragraft microvasculature. (A) Following transplantation, alloimmune inflammatory responses target the graft vascular endothelium resulting in the destruction of microvessels, which in turn leads to local hypoxia and tissue injury. (B) Alloimmune inflammatory responses may also stimulate endothelial cell proliferation and promote leukocyte-induced angiogenesis within allografts. The local delivery of the pro-inflammatory and pro-angiogenic factor vascular endothelial growth factor (VEGF) is central to this response. Pathological leukocyte-induced angiogenesis results in the formation of abnormal networks of capillaries that lead to chaotic blood flow patterns and paradoxically results in local hypoxia. Thus, local tissue hypoxia is the end result of acute events and direct targeting of the graft endothelial cells (A) as well as inflammation and the associated leukocyte-induced angiogenesis (B).
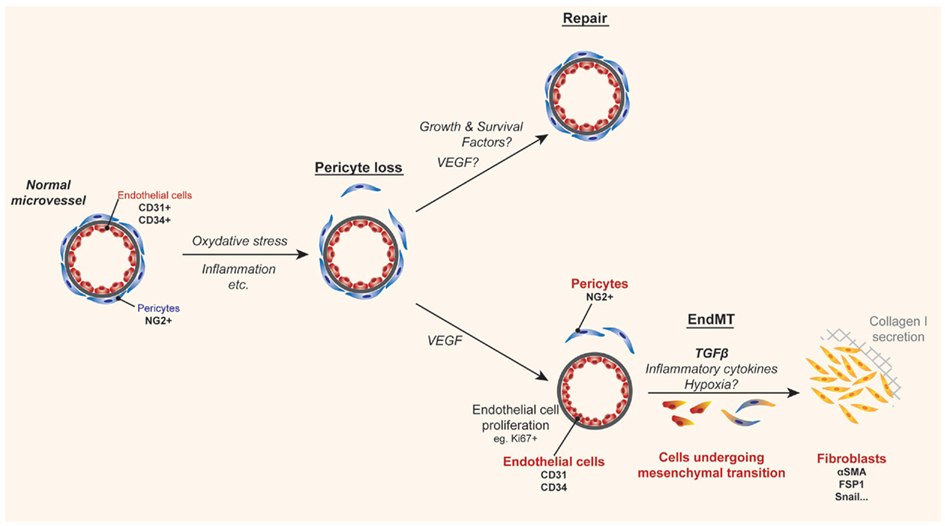
Figure 2. Cartoon illustrating a mechanism of tissue fibrosis associated with allograft rejection. During inflammation, pathological angiogenesis, and/or local hypoxia can lead to pericyte loss. Under normal conditions homeostatic repair occurs under the influence of protective growth and survival factors. In contrast, when the inflammatory microenvironment is sustained, the loss of pericytes serves as a precedent for endothelial-to-mesenchymal transition (EndMT), where endothelial cells become denuded from their basement membrane and migrate along with pericytes into the surrounding tissue. Although still under debate, it is reported that the presence of TGFβ, inflammatory cytokines, and hypoxia enables dissociated pericytes and/or endothelial cells to dedifferentiate into collagen-secreting fibroblasts, which in turn results in fibrosis and scarring.
Microvascular Patterning and the Intragraft Microenvironment
Angiogenesis, the generation of new blood vessels from pre-existing ones, is a complex process involving the degradation of the vascular basement membrane and surrounding extracellular matrix as well as EC proliferation and migration (Cotran, 1994; Folkman, 1995a,b; Brown et al., 1997; Carmeliet and Jain, 2000; Ferrara and Kerbel, 2005; Goel et al., 2011). The creation of new blood vessels is critical for normal organ growth and development and it is a requirement for normal wound healing and tissue repair (Cotran, 1994; Majno, 1998). In all of these biological conditions, angiogenesis is tightly regulated in a tissue specific manner, such that the microvascular bed provides tissues with their nutritive and oxygen demands in a manner that is sufficient for normal physiological processes. However, angiogenesis is also characteristic of many disease states, and is well-established to occur in association with cell-mediated immune responses (Cotran, 1994) and chronic inflammatory diseases (Folkman and Brem, 1992; Ferrara and Alitalo, 1999; Ezaki et al., 2001), notably inflammatory bowel disease (Kanazawa et al., 2001), arthritis (Walsh and Pearson, 2001), chronic asthma (Detoraki et al., 2010), and chronic allograft rejection (Tanaka et al., 1994; Moulton et al., 1999; Reinders and Briscoe, 2002; Denton et al., 2004; Babu et al., 2007). In disease processes, the neoangiogenesis response occurs in an abnormal and disorganized manner. In some chronic disease conditions, such as arthritis and chronic asthma, it can be uncontrolled (Walsh and Pearson, 2001; Detoraki et al., 2010). Solid tumors are a prototype example where angiogenesis can be abnormal and pathological (Fukumura and Jain, 2007; Jain, 2008; Goel et al., 2011). In this disease, local tissue hypoxia drives the production of angiogenesis factors, notably VEGF. This in turn elicits a powerful neovascular response (Brown et al., 1997; Goel et al., 2011). Since newly formed vessels are irregular in size with random branching patterns, the associated blood flow within the entire microvascular tree becomes abnormal and shunting occurs throughout the tissue (Jain, 2008; Goel et al., 2011). In this manner, some areas of the tissue have potential for increased blood flow and have adequate oxygenation. In contrast, other areas have decreased or aberrant blood flow which results in local tissue hypoxia. This latter event further drives the expression of hypoxia-inducible angiogenesis factors, including VEGF, such that the cyclical process is sustained. It is proposed that these events may also occur in association with chronic kidney disease as well as within kidney allografts in association with chronic rejection (Choi et al., 2000; Reinders et al., 2006; Contreras and Briscoe, 2007; Mayer, 2011; Figure 1). Therefore, local tissue hypoxia may occur as a result of both initial targeting and loss of microvessels (Figure 1A), or as a result of leukocyte-induced angiogenesis (Figure 1B). We propose that once an abnormal pattern of blood vessels develop within the intragraft microenvironment, it likely serves to elicit local tissue hypoxia as well as to induce VEGF expression, analogous to that described in tumors (Goel et al., 2011). We hypothesize that these events sustain inflammation and the progression of chronic rejection.
Leukocyte-Induced Angiogenesis: A Pathological Response that Sustains Intragraft Injury
Leukocyte-induced angiogenesis was initially described following the local injection of spleen cells into the skin of nude mice (Sidky and Auerbach, 1975; Auerbach and Sidky, 1979). In these original studies, it was noted that the reaction did not occur following the injection of syngeneic spleen cells, but it was dose-dependent and reproducible following the intradermal injection of allogeneic cells (Auerbach and Sidky, 1979). Subsequently, it was demonstrated that this leukocyte-induced angiogenesis reaction was mediated by CD4+ T lymphocytes (Kaminski and Auerbach, 1988), which we now understand to be critical for the initiation of the alloimmune inflammatory response (Ingulli et al., 2009). Thus, while not the intention of these studies, this model clearly indicates that alloactivated leukocytes are potent for the production of angiogenesis factors. Indeed, it is now known that both monocyte/macrophages (Koch et al., 1986; Leibovich and Wiseman, 1988; Polverini, 1997) and activated T cells (Freeman et al., 1995; Melter et al., 2000; Mor et al., 2004) secrete angiogenesis factors, including VEGF, which is a central mediator of the leukocyte-induced reaction (Leibovich et al., 1987; Giraudo et al., 1998; Reinders et al., 2006). Other factors including TNF-α (63), TGF-β, and nitric oxide (Wiseman et al., 1988; Leibovich et al., 1994) may also elicit the response. Therefore, angiogenesis may result from the local production of distinct factors, or via cytokine- and cell-mediated responses that increase local concentrations of VEGF. This interplay between cell-mediated immune reactions and the local delivery of TGF-β and VEGF by monocyte/macrophages have resulted in the development of paradigms to explain how angiogenesis and fibrosis are characteristic of chronic inflammatory disease states (Cotran, 1994; Brown et al., 1995; Freeman et al., 1995; Sharma et al., 1996; Inoue et al., 1998; Majno, 1998; Pilmore et al., 1999; Jain et al., 2000, 2002; Ezaki et al., 2001; Kanazawa et al., 2001; Mannon, 2006; Booth and Bishop, 2010).
In our own studies, we evaluated recipient angiogenesis using a humanized SCID mouse (huSCID) model of rejection (Moulton et al., 1999). Human foreskin was transplanted onto SCID mice and was found to engraft after 4–6 weeks. Functional vessels and vascular networks within the healed human skin were derived from both human and mouse EC (Briscoe et al., 1999). The subsequent adoptive transfer of human peripheral blood leukocytes into the mouse resulted in human leukocytic infiltrates within the engrafted human skin but not within the adjacent mouse skin, as evaluated by videomicroscopy and by immunohistochemistry (Briscoe et al., 1999; Moulton et al., 1999). After 2–3 days, infiltrates were present within grafts by videomicroscopy, and by 7 days, infiltrates were notable by immunohistochemistry; by day 14, cellular infiltrates were profound within grafts. In our analyses, we also found a notable angiogenesis response that was spatially associated with leukocytic infiltrates within the human skins. The angiogenesis response occurred at early time points, typically on day 3–5 by videomicroscopy and on day 7 by immunohistochemistry (Moulton et al., 1999). Notably, the response preceded the development of marked infiltrates, which were ultimately associated with microvascular destruction. Therefore, we propose that local tissue hypoxia was not the primary stimulus for the initiation of the angiogenesis response within these allografts. Rather, we believe that the angiogenesis reaction was initiated by factors produced by infiltrating leukocytes in a similar manner as previously noted by Auerbach and Sidky (1979) in their model of leukocyte-induced angiogenesis.
More recently, Babu et al. (2007) found a similar neovascularization reaction in their analysis of rejecting tracheal allografts. Although they did not characterize this response in great detail, their studies illustrated prominent angiogenesis in grafts on days 4-, 6-, and 8-post transplantation. Similar to our studies (Moulton et al., 1999; Contreras and Briscoe, 2007), they noted that it did not persist and completely disappeared by day 10–12 following transplantation in association with fulminant rejection. However, these authors also evaluated tissue oxygenation within the graft. Surprisingly, rather than finding that oxygenation was normal at sites of neovascularization, they observed that tissue pO2 decreased in day 4–6 allografts when the leukocyte-induced angiogenesis reaction was prominent (Babu et al., 2007). Their observations indicate that, contrary to expectations, hypoxia occurs within allografts prior to microvascular destruction, and it is associated with the presence of leukocyte-induced angiogenesis. This observation is consistent with extensive studies by Rakesh Jains group demonstrating that pathological neoangiogenesis and its association with abnormal blood flow patterns within a tumor is ineffective to support tissue oxygenation (Goel et al., 2011).
Angiogenesis has been demonstrated to occur in allografts with evidence of chronic rejection in association with allograft vasculopathy (Atkinson et al., 2005). Similar to our studies in the huSCID (discussed above), as well as those reported by Babu et al. (2007), increased capillary density within the parenchyma of cardiac allografts has been found to be associated with T cell and monocyte infiltrates (Tanaka et al., 1994). Also, neovessels within the intima of large vessels with vasculopathy lesions have been found to be spatially associated with inflammatory infiltrates (Tanaka et al., 1994; Denton et al., 2004). These neovessels within allografts are activated, in as much as they express cell surface adhesion molecules and MHC class II. Thus, the angiogenesis reaction itself may be pro-inflammatory in as much as it has potential to mediate the recruitment and the activation of local infiltrates (Atkinson et al., 2005).
Collectively, these findings illustrate that leukocyte-induced EC proliferation/angiogenesis occurs at different sites within allografts. We propose that the abnormal angiogenic microvasculature may be causative of disease, and that local tissue hypoxia is the result, rather than the primary stimulus of the response (Jain, 2005; Figure 1). This paradigm explains in part how the intragraft microenvironment functions to initiate and sustain the development of chronic allograft rejection. Other major issues relate to the production of cytokines and growth factors that initiate EndMT, illustrated in Figure 2. This will be discussed in more detail below.
Overlapping Nature of Inflammation and Angiogenesis and the Development of Chronic Rejection
During inflammation, the leukocyte-induced angiogenesis reaction may be associated with local areas of tissue hypoxia and tissue injury (illustrated in Figure 1). To this end, it is important to note that angiogenesis factors such as VEGF have been reported to be overexpressed in all models of chronic inflammation, including models of chronic rejection, and their expression has been found to be associated with disease progression (Leibovich et al., 1987; Folkman and Brem, 1992; Cotran, 1994; Majno, 1998; Ezaki et al., 2001; Ferrara, 2005). Consistent with this possibility, blockade of individual angiogenesis factors, including VEGF–VEGFR interactions, in animal models has been found to attenuate the progression of the chronic rejection disease process (Lemstrom et al., 2002; Nykanen et al., 2003; Reinders et al., 2003a; Denton et al., 2004; Sho et al., 2005; Malmstrom et al., 2008).
On the other hand, immune inflammation and the angiogenesis response can be antagonistic. For instance, some angiogenesis factors such as fibroblast growth factor (FGF) may inhibit adhesion molecule expression and have anti-inflammatory effects (Jain et al., 1996; Melder et al., 1996). Some inflammatory mediators such as IFNγ or the IFNγ-inducible chemokine CXCL10/IP-10 can be anti-angiogenic (Strieter et al., 1995a; Boulday et al., 2006). Moreover, the competitive binding of chemokines to their receptors (and vice versa) results in competition for angiogenesis and inflammation (Strieter et al., 1995b). So, how is it possible that the evolution of the leukocyte-induced angiogenesis response can be associated with pro-inflammation? Neovessels at sites of angiogenesis express adhesion molecules and chemokines and can facilitate the recruitment of leukocytes in part via enhanced leukocyte–endothelial adhesion events (Melder et al., 1996; Detmar et al., 1998; Kim et al., 2001; Reinders et al., 2003a). In addition, mediators of the leukocyte-induced angiogenesis reaction, such as VEGF, induce the expression of EC adhesion molecules [including E-selectin, ICAM-1, and VCAM-1 (Melder et al., 1996; Kim et al., 2001)] and pro-inflammatory chemokines [such as CXCL10/IP-10 and MCP-1 (Marumo et al., 1999; Reinders et al., 2003a; Boulday et al., 2006)]. Also, as will be discussed below, VEGF can serve as a potent leukocyte chemoattractant via direct interactions with its receptors expressed on subsets of monocyte/macrophages and T cells (Barleon et al., 1996; Shin et al., 2009; Basu et al., 2010; Suzuki et al., 2010). Therefore, once established within allografts, the initial EC activation response that results in proliferation and angiogenesis also facilitates inflammation. In contrast, inflammatory mediators can both stimulate and inhibit angiogenesis. Thus, the balance between the relative production of pro- versus anti-angiogenic factors in the course of the immune response will determine the inducible neovascular response, resulting in vascular repair or injury, and this process may be a key determinant of the outcome of rejection.
Collectively, these observations suggest that the overlapping nature of inflammation and angiogenesis create an environment that is critical to shape the rejection process. They also imply that pharmacologic manipulation of the EC response to injury or leukocyte-induced angiogenesis will target the pathological intragraft microenvironment and interrupt chronic rejection. Indeed, in animal models, several angiogenesis inhibitors have been reported to slow the progression of chronic rejection (Lemstrom et al., 2002; Denton et al., 2004; Reinders et al., 2006; Malmstrom et al., 2008). PTK787, a selective VEGFR protein tyrosine kinase angiogenesis inhibitor was found to attenuate the development of interstitial fibrosis and allograft vasculopathy in well-established rat cardiac and renal transplantation models (Lemstrom et al., 2002; Malmstrom et al., 2008). Also, TNP-470, a synthetic fumagillin derivative and a well-established angiogenesis inhibitor was found to interrupt the progression of inflammation, intragraft fibrosis, and the degree of allograft vasculopathy in the Fischer 344 into Lewis rat cardiac allograft model (Denton et al., 2004). Furthermore, we find that endostatin, another well-established angiogenesis inhibitor prevents the progression of allograft vasculopathy in the MHC class II mismatched B6.C-H2bm12 into C57BL/6 mouse model of chronic rejection (Contreras and Briscoe, unpublished observations). Therefore, it appears that transient interruption therapy with angiogenesis inhibitors has potential to normalize the vasculature and inhibit the progression of chronic rejection.
To this end, it is important to note that the mTOR kinase and its associated signaling pathway has profound effects on EC proliferation in vitro and in vivo (Dormond et al., 2007; Contreras et al., 2008). We believe that mTOR inhibitors represent the first-in-kind angiogenesis inhibitor agents that are currently being used therapeutically in humans following transplantation. Furthermore, we suggest that this biological effect may account for some of their ability to retard the progression of chronic rejection (Contreras et al., 2008). Several reports have indicated that a switch from calcineurin inhibitor therapy to an mTOR inhibitor based regimen (interruption protocol) in humans slows the progression of chronic rejection (Oberbauer et al., 2005; Schena et al., 2009; Arora et al., 2011). These observations support the possibility that angiogenesis inhibitors have potential as therapeutics in the future.
Endothelial Cell to Mesenchymal Transition: Possible Final End Result of Leukocyte-Induced or Pathological Angiogenesis
The Kalluri group demonstrated that EC within cardiac allografts appear to undergo a process of mesenchymal transition to fibroblasts in association with inflammation and chronic rejection (Zeisberg et al., 2007a). This process, called EndMT by several laboratories results from the dedifferentiation of EC, such that they lose their endothelial phenotype and gain the expression of mesenchymal markers (illustrated in Figure 2). In this manner, the process of EndMT is characterized by the loss of well-established EC molecules including CD31 and CD34, and the gain in expression of fibroblast-specific protein 1 (FSP1), alpha-smooth muscle actin (Zeisberg et al., 2007a,b), as well as other non-EC molecules (Kokudo et al., 2008). In vitro, it is reported, that EndMT occurs in response to both TGFβ1 (Arciniegas et al., 1992) and TGFβ2 (Medici et al., 2010), but the stimuli for mesenchymal transition in vivo are poorly understood. The process of EndMT occurs in association with pericyte loss, when EC are denuded from their basement membrane. Denuded EC and pericytes migrate into the surrounding tissue, where they are exposed to TGFβ that may be produced locally by multiple cell types and/or delivered into the graft by monocyte/macrophages (Jain et al., 2002; Booth and Bishop, 2010), thus leading to their differentiation into fibroblasts (illustrated in Figure 2).
Endothelial-to-mesenchymal transition is well-established to occur during embryonic development of the heart, in normal wound healing and in several different cancers (Markwald et al., 1975; Zeisberg et al., 2007b). It has also been implicated in chronic fibrotic disease states including atherosclerosis, pulmonary hypertension, cardiac fibrosis, and diabetic nephropathy (Zeisberg et al., 2007a; Li et al., 2009; Hashimoto et al., 2010). In cancer, EndMT has been reported to account for up to 40% of cancer-associated fibroblasts, and the process has been found to alter the microenvironment in several ways (Zeisberg et al., 2007b). EC that have undergone EndMT produce collagen, deposit extracellular matrix molecules, and secrete pro-fibrotic factors including TGFβ, leading to a self-perpetuating cycle of events. In contrast, its role in allograft rejection has not been well characterized, and it remains controversial whether EndMT is primarily related to pericyte loss and their migration into the local tissue, or whether it is truly related to EC dedifferentiation (Humphreys et al., 2010). Regardless of whether the pericyte and/or the EC dedifferentiates into the collagen-producing cell, the process, and long-term consequence of microvascular destruction is fibrosis.
Since the intragraft microenvironment has similarities to the tumor microenvironment (i.e., abnormal microvessels, local hypoxia, TGFβ, high VEGF, mononuclear cell infiltrates), we have speculated that pathological and/or leukocyte-induced angiogenesis will precede EndMT within allografts (illustrated in Figure 2). Consistent with this possibility, proliferating EC have been shown to be associated with alloimmune targeting and chronic rejection (Sis et al., 2009; Osasan et al., 2011). In addition, we have found that proliferating EC are most susceptible to EndMT in vitro in the presence of inflammatory cytokines, TGFβ, and hypoxia. In contrast, we find that EC that have not undergone multiple rounds of proliferation are not as susceptible to EndMT (Woda and Briscoe, unpublished observations). It will be important to understand if intragraft cytokines or growth factors that activate EC can initiate this pathological process and how VEGF functions in the EndMT process. An alternate hypothesis is that the intragraft cytokine milieu and/or hypoxia within allografts at times of leukocyte-induced angiogenesis alters pericyte biology which becomes a central determinant of both the loss of the microvasculature and differentiation into fibroblasts. Thus, there is a need for research in this area as it is a potential target of future therapeutics.
Vascular Endothelial Growth Factor: A Central Player in the Chronic Rejection Intragraft Microenvironment
By definition, the development of acute allograft rejection involves a marked inflammatory reaction, characterized by the recruitment of leukocytes and an intense cellular and humoral attack on the graft (Ingulli et al., 2009). By contrast, chronic rejection is a more insidious process involving delayed type hypersensitivity mechanisms and is characterized by mononuclear cell infiltration, an obliterative vasculopathy, and progressive fibrosis (Libby and Pober, 2001; Ingulli et al., 2009). Over the past 10–15 years, VEGF has emerged as an important player in the rejection process, and its expression has been reported by several groups to be associated with both acute and chronic allograft rejection (Torry et al., 1995; Pilmore et al., 1999; Lemstrom et al., 2002; Reinders et al., 2003b, 2006; Malmstrom et al., 2008). In one study (Pilmore et al., 1999), VEGF expression was found to be most striking in the interstitium of human renal allografts in association with CD68+ monocyte/macrophage infiltrates and evidence of chronic rejection. In another study (Torry et al., 1995), the expression of VEGF was found to be associated with fibrin deposition, and was confined to areas with monocyte/macrophage infiltrates. In our own analyses, we observed intense VEGF expression within human cardiac allografts localized to both inflammatory cell infiltrates as well as to vascular EC in association with acute and chronic rejection (Reinders et al., 2003b). Moreover, we found that persistent intragraft VEGF overexpression identified patients at high risk for the development of chronic allograft vasculopathy/chronic rejection (Reinders et al., 2003b). Furthermore, human transplant recipients with genotypes encoding high VEGF production are at increased risk for the development of rejection (Shahbazi et al., 2002; Girnita et al., 2008). Levels of VEGF increase significantly in the serum and urine of patients in association with cardiac and renal allograft rejection respectively, and in most cases return to baseline after effective treatment of the rejection episode (Abramson et al., 2002; Peng et al., 2008). It is thus possible that serum levels of VEGF may serve as a reliable biomarker of the development of allograft vasculopathy following human cardiac transplantation (Daly et al., 2011).
VEGF may be delivered into allografts in the course of rejection by infiltrating monocytes and by activated T cells (Leibovich et al., 1987; Leibovich and Wiseman, 1988; Freeman et al., 1995; Melter et al., 2000). Alternatively, VEGF may be induced locally within the allograft as a result of cellular interactions among activated platelets and EC (Chiodoni et al., 2006; Dormond et al., 2008). Nevertheless, the local overexpression of VEGF within allografts results in the development of chronic rejection and allograft vasculopathy (Lemstrom et al., 2002). Taken together, there is extensive data to support the hypothesis that VEGF is mechanistic in the process of chronic allograft rejection.
The major stimulus for VEGF expression is hypoxia (Shweiki et al., 1992; Mukhopadhyay et al., 1995; Goel et al., 2011), but other factors that can upregulate local tissue VEGF production include paracrine effects of hormones, glucose and prostaglandins, as well as cytokine/growth factor modulators of protein kinase C, nitric oxide, and stimulators of adenylate cyclase (Brown et al., 1997). Several cytokines, including IL-1, TNF, and IL-6, have been found to induce the expression of VEGF and/or VEGF receptors (Leibovich et al., 1987; Giraudo et al., 1998; Amano et al., 2004; Huang et al., 2004). In addition, we have demonstrated that the ligation of CD40 by CD154 (CD40 ligand, expressed by activated platelets and T cells) is potent for local tissue induction of VEGF expression (Melter et al., 2000; Dormond et al., 2008). In this manner, it is not surprising that cell-mediated immune inflammation is associated with VEGF–VEGFR biological responses. Furthermore, since multiple VEGF-inducing factors are present within allografts at different times post transplantation, one might conclude that VEGF expression, and VEGF-dependent biological responses should be characteristic features of both the initiation and the maintenance of chronic rejection.
Biology of VEGF–VEGFR Interactions
As its name suggests, VEGF classically functions as a potent angiogenesis factor, and as such it was originally proposed to facilitate microvascular repair following ischemic injury (Kang et al., 2001a,b; Mayer, 2011) as well as injury following inflammatory insults (Choi et al., 2000; Reinders et al., 2006). VEGF is a 45-kDa protein produced by most cell types including cells of the immune system such as monocyte/macrophages and activated T cell subsets (Freeman et al., 1995; Brown et al., 1997; Polverini, 1997; Melter et al., 2000; Basu et al., 2010). It is a heparin-binding homodimeric glycoprotein with several protein variants of 206, 189, 164, 145, and 121 amino acids that arise from the alternative splicing of a single gene (Leung et al., 1989; Tischer et al., 1991). Intense research beyond the scope of this review, has clarified the function of VEGF in EC, where it mediates migration, growth, survival as well as activation responses including the expression of adhesion molecules and chemokines (Alon et al., 1995; Klagsbrun and D’Amore, 1996; Melder et al., 1996; Brown et al., 1997; Gerber et al., 1999; Kim et al., 2001; Ferrara, 2005; Boulday et al., 2006; Lee et al., 2007).
The biological activities of VEGF are mediated via interactions with its receptors, Flt-1 (VEGF receptor 1), KDR (VEGF receptor 2), and neuropilin-1 (Shalaby et al., 1995; Klagsbrun and D’Amore, 1996; Brown et al., 1997; Miao and Klagsbrun, 2000; Ferrara, 2005; Takahashi and Shibuya, 2005; Matsumoto and Mugishima, 2006). Several studies have shown that signaling mediated via KDR is critical for the VEGF-induced response (Zeng et al., 2001). For instance, many of the biological properties of VEGF can be inhibited by neutralization of KDR; and inhibition of KDR has similar effects as neutralization of VEGF in vivo in inflammatory diseases (Brown et al., 1997; Watanabe et al., 2004, 2005; Ferrara, 2005), including allograft rejection (Reinders et al., 2003a; Sho et al., 2005). Also, knockout of either VEGF or KDR results in embryonic lethality due to inhibition of angioblast differentiation and vasculogenesis (Shalaby et al., 1995; Ferrara et al., 1996). Classically, it is thought that neuropilin-1 serves as an accessory co-receptor for KDR to bind VEGF and mediate crosslinking to KDR (Klagsbrun and D’Amore, 1996; Matsumoto and Mugishima, 2006). However, some studies have suggested that neuropilin-1 might also mediate signaling directly in response to the semaphorin 3 family of proteins (Wang et al., 2003; Catalano et al., 2006; Mizui et al., 2009; Suzuki et al., 2008). VEGF also signals through Flt-1 to initiate a direct signaling response, but its interaction with Flt-1 may inhibit KDR-induced responses in EC (Zeng et al., 2002). Thus, VEGF inducible responses in different cells can be determined according to the profile of expression of its receptors and thus, the select VEGFR-dependent signal.
All VEGF receptors are expressed by EC, but individual receptors are also expressed by different leukocyte subsets indicating that VEGF may have direct effects on the immune response (Barleon et al., 1996; Bruder et al., 2004; Sarris et al., 2008; Shin et al., 2009; Basu et al., 2010; Suzuki et al., 2010). Flt-1 and neuropilin-1 are expressed by human monocytes and APCs (Barleon et al., 1996; Romeo et al., 2002; Bourbie-Vaudaine et al., 2006; Chapoval et al., 2009), and VEGF is known to induce activation responses and chemotactic activity in monocytes in part via interactions with Flt-1 (Barleon et al., 1996; Clauss et al., 1996; Laxmanan et al., 2005; Chapoval et al., 2009). VEGF–VEGFR interactions also promote the differentiation of monocytes into pro-inflammatory (Chapoval et al., 2009) or immunoregulatory (Gabrilovich et al., 1998; Laxmanan et al., 2005) APCs, but its effect is likely dependent on individual VEGFR(s) expressed by the APC (Laxmanan et al., 2005; Chapoval et al., 2009).
Moreover, several recent studies have indicated that T cell subsets can express VEGFRs, including Flt-1, KDR, and neuropilin-1 (Dias et al., 2000; Tordjman et al., 2002; Mor et al., 2004; Sarris et al., 2008; Basu et al., 2010; Edelbauer et al., 2010; Ziogas et al., 2011). In our studies (Basu et al., 2010; Edelbauer et al., 2010), we find low negligible levels of expression of all VEGFRs on freshly isolated populations of unactivated CD4+ and CD8+ T cells. However, we have observed that the expression of Flt-1 and KDR increase following mitogen-dependent activation. In addition, we have found high levels of KDR on memory CD45RO+ populations of CD4+ T cells (Basu et al., 2010). In general, KDR and Flt-1 are reported to be the dominant VEGFRs expressed on T effector cells (Basu et al., 2010; Edelbauer et al., 2010; Zhang et al., 2010; Ziogas et al., 2011). In contrast, it is reported that neuropilin-1 is selectively expressed at high levels on populations of CD4+CD25+FoxP3+ T regulatory cells (Bruder et al., 2004; Sarris et al., 2008). One study indicated that T regulatory cells may also express KDR (Suzuki et al., 2010). Neuropilin-1 has also been reported to be expressed on populations of human naïve T cells, where it functions to support the initiation of T cell activation in primary immune responses (Tordjman et al., 2002).
Collectively, these findings support the possibility that intragraft VEGF may interact with different T cell subsets in the course of the rejection process. VEGF may function as a potent chemoattractant for effector and memory CD4+ and CD8+ T cells, and may thus promote inflammation in association with acute and chronic allograft rejection. Consistent with this possibility, the blockade of VEGF or VEGFR interactions with T cells inhibits intragraft lymphocyte trafficking (Reinders et al., 2003a; Sho et al., 2005; Edelbauer et al., 2010; Zhang et al., 2010) as well as reactivation responses (Basu et al., 2010) in human model systems. In experimental animal models, the local overexpression of VEGF, and VEGF–VEGFR interactions also facilitate lymphocyte trafficking (Reinders et al., 2003a; Lee et al., 2004; Kim et al., 2009; Edelbauer et al., 2010; Zhang et al., 2010) and activation including the augmentation of Th1 (Chapoval et al., 2009), Th2 (Lee et al., 2004), and Th17 (Kim et al., 2009) effector responses. The mechanism(s) by which local tissue VEGF elicits signals for migration and activation/reactivation responses is the subject of ongoing investigations.
Pro-Inflammatory Effects of VEGF–VEGF Receptor Interactions
As discussed above, VEGF may act as a pro-inflammatory cytokine in vivo in several chronic diseases (Fava et al., 1994; Koch et al., 1994; Duh and Aiello, 1999; Ferrara and Alitalo, 1999; Griga et al., 1999; Hoshino et al., 2001; Kanazawa et al., 2001; McDonald, 2001; Walsh and Pearson, 2001; Kim et al., 2009; Detoraki et al., 2010), and its ability to function as a pro-inflammatory cytokine in part relates to its interactions with its receptors expressed on leukocytes (Barleon et al., 1996; Shin et al., 2009; Basu et al., 2010; Edelbauer et al., 2010; Suzuki et al., 2010; Zhang et al., 2010). Its ability to interact directly with monocytes (via Flt-1), NK T cells, and CD3+ T cells (via Flt-1 and KDR) to facilitate chemotactic activity is likely of great pathophysiological importance in rejection (Edelbauer et al., 2010; Zhang et al., 2010). In addition, VEGF induces the expression of adhesion molecules and chemokines in EC (Melder et al., 1996; Marumo et al., 1999; Kim et al., 2001; Reinders et al., 2003a; Boulday et al., 2006), and classically enhances vascular permeability (Brown et al., 1997; Basu et al., 2001). All of these events are characteristic of acute and chronic inflammation (Cotran, 1994).
In models of chronic rejection, it was demonstrated that overexpression of VEGF mobilizes bone marrow derived monocyte/macrophages and accelerates the development of allograft vasculopathy (Lemstrom et al., 2002; Zhao et al., 2002). Further, it was found that blockade of VEGF receptor 2 (KDR) signaling decreases monocyte recruitment into vascular lesions and attenuates the development of graft arteriosclerosis (Lemstrom et al., 2002; Zhao et al., 2002). We found that KDR is expressed on CD3+ infiltrates within rejecting human allografts in vivo, and that both anti-VEGF and anti-KDR antibodies inhibit the transmigration of CD4+ and CD8+ T cells across activated EC using an in vitro live-time transmigration model (Edelbauer et al., 2010). Using a huSCID mouse model of lymphocyte trafficking, we also demonstrated that KDR-expressing lymphocytes migrate into human skin in vivo, and that migration is reduced in mice treated with a blocking anti-VEGF antibody (Edelbauer et al., 2010). Collectively, these observations demonstrate that induced expression of KDR on subsets of T cells, and locally expressed VEGF facilitate lymphocyte chemotaxis. They support a model whereby intragraft VEGF mediates the localization of T cells in association with chronic rejection.
In another recent report, Zhang et al. (2010) used an anti-human VEGF antibody to study the effect of VEGF blockade on the development of allograft vasculopathy in a humanized model in SCID mice. They found that anti-VEGF inhibited intragraft accumulation of T cells without affecting T cell activation. In addition, they observed that anti-VEGF treatment inhibited neointimal formation within human coronary artery grafts in the humanized mouse. The authors suggested that the T cell chemoattractive effect of VEGF was mediated in part via interactions with a subpopulation of Flt-1-expressing CD3+ T cells. Thus, intragraft VEGF may also contribute to vascular remodeling and allograft vasculopathy by enhancing T cell recruitment into the intima of large vessels.
Collectively, these observations indicate that VEGF has potent pro-inflammatory properties under pathological conditions in association with the development of chronic rejection and allograft vasculopathy.
Overall Summary and Perspective
In this review, we have discussed a paradigm where leukocyte-induced angiogenesis, local tissue hypoxia, and the overexpression of VEGF sustain an intragraft microenvironment that fosters the development of chronic allograft rejection. We have also defined EC-based events within the allograft microenvironment that are associated with chronic rejection. The first are changes in the microcirculation resulting from destruction of the microvasculature. The second are changes in the microcirculation resulting from EC proliferation and leukocyte-induced angiogenesis. Both events are likely to disrupt normal blood flow patterns within the graft and result in local areas of tissue hypoxia. In addition, EC undergoing proliferation express adhesion molecules and chemokines that support pro-inflammation, providing another mechanism whereby the angiogenesis reaction may sustain ongoing tissue injury. A third intragraft determinant of chronic disease is the development of EndMT. While the mechanism underlying EndMT is controversial, there is sufficient evidence to suggest that inflammation and EC proliferation is associated with pericyte loss, which likely precedes microvascular capillary loss and the development of fibrosis. Whether the pericyte or the EC, or both, ultimately dedifferentiate into collagen-producing cells is being debated. Nevertheless, it has been reported that cytokines and growth factors, including TGFβ (Booth and Bishop, 2010), can accelerate this fibrotic reaction. Although beyond the scope of this review, TGFβ may also be a key aspect of the chronic rejection intragraft microenvironment (Jain et al., 2000). Finally, we propose that a fourth and key determinant of chronic rejection is intragraft overexpression of VEGF, which functions to facilitate both inflammation and pathological EC proliferation/angiogenesis. While VEGF may be delivered into allografts by inflammatory infiltrates, such as monocytes and activated T cells, the immune response can also elicit the local overproduction of VEGF within the graft. Overall, in this review we provide insight into novel aspects of the intragraft microenvironment that contribute to the development of chronic rejection and long-term attrition of allografts following transplantation. The importance of this paradigm is that it identifies key areas for future therapeutic targeting to prevent the progression of chronic rejection following solid organ transplantation.
Conflict of Interest Statement
David M. Briscoe has received research funding from the Roche Organ Transplantation Research Foundation, Astellas, Wyeth, and Pfizer Pharmaceuticals, and is an inventor on US Patent #6218361. He has given seminars at educational CME-approved conferences and grand rounds sponsored by pharmaceutical companies. All other authors do not have any disclosures.
Acknowledgments
The work cited in this review was supported by NIH Grants R01AI046756, RO1HL074436, R01AI092305-01, R21HL104602-01, T32DK007726-28, T32HL07572, and UL1RR025758 (Harvard Catalyst). Sarah Bruneau was supported by a Fellowship grant from the American Society of Transplantation. Alan Contreras was supported by a Fellowship grant from the American Society of Transplant Surgeons. The authors wish to acknowledge past members of the laboratory who worked on these projects, and we are grateful for the support of technicians for their ongoing efforts. We are also indebted to the support of our collaborators, most especially Dr. Rakesh Jain for stimulating our interest in this area of research, and to Drs. Debabrata Mukhopadhyay and Michael Klagsbrun for ongoing scientific interactions.
References
Abramson, L. P., Pahl, E., Huang, L., Stellmach, V., Rodgers, S., Mavroudis, C., Backer, C. L., Arensman, R. M., and Crawford, S. E. (2002). Serum vascular endothelial growth factor as a surveillance marker for cellular rejection in pediatric cardiac transplantation. Transplantation 73, 153–156.
Alon, T., Hemo, I., Itin, A., Pe’Er, J., Stone, J., and Keshet, E. (1995). Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurity. Nat. Med. 1, 1024–1028.
Amano, K., Okigaki, M., Adachi, Y., Fujiyama, S., Mori, Y., Kosaki, A., Iwasaka, T., and Matsubara, H. (2004). Mechanism for IL-1 beta-mediated neovascularization unmasked by IL-1 beta knock-out mice. J. Mol. Cell. Cardiol. 36, 469–480.
Arciniegas, E., Sutton, A. B., Allen, T. D., and Schor, A. M. (1992). Transforming growth factor beta 1 promotes the differentiation of endothelial cells into smooth muscle-like cells in vitro. J. Cell. Sci. 103(Pt 2), 521–529.
Arora, S., Ueland, T., Wennerblom, B., Sigurdadottir, V., Eiskjaer, H., Botker, H. E., Ekmehag, B., Jansson, K., Mortensen, S. A., Saunamaki, K., Simonsen, S., Gude, E., Bendz, B., Solbu, D., Aukrust, P., and Gullestad, L. (2011). Effect of everolimus introduction on cardiac allograft vasculopathy – results of a randomized, multicenter trial. Transplantation 92, 235–243.
Atkinson, C., Southwood, M., Pitman, R., Phillpotts, C., Wallwork, J., and Goddard, M. (2005). Angiogenesis occurs within the intimal proliferation that characterizes transplant coronary artery vasculopathy. J. Heart Lung Transplant. 24, 551–558.
Auerbach, R., and Sidky, Y. A. (1979). Nature of the stimulus leading to lymphocyte-induced angiogenesis. J. Immunol. 123, 751–754.
Aydin, Z., van Zonneveld, A. J., de Fijter, J. W., and Rabelink, T. J. (2007). New horizons in prevention and treatment of ischaemic injury to kidney transplants. Nephrol. Dial. Transplant. 22, 342–346.
Babu, A. N., Murakawa, T., Thurman, J. M., Miller, E. J., Henson, P. M., Zamora, M. R., Voelkel, N. F., and Nicolls, M. R. (2007). Microvascular destruction identifies murine allografts that cannot be rescued from airway fibrosis. J. Clin. Invest. 117, 3774–3785.
Barleon, B., Sozzani, S., Zhou, D., Weich, H. A., Mantovani, A., and Marme, D. (1996). Migration of human monocytes in response to vascular endothelial growth factor (VEGF) is mediated via the VEGF receptor flt-1. Blood 87, 3336–3343.
Basu, A., Hoerning, A., Datta, D., Edelbauer, M., Stack, M. P., Calzadilla, K., Pal, S., and Briscoe, D. M. (2010). Cutting edge: vascular endothelial growth factor-mediated signaling in human CD45RO+ CD4+ T cells promotes Akt and ERK activation and costimulates IFN-gamma production. J. Immunol. 184, 545–549.
Basu, S., Nagy, J. A., Pal, S., Vasile, E., Eckelhoefer, I. A., Bliss, V. S., Manseau, E. J., Dasgupta, P. S., Dvorak, H. F., and Mukhopadhyay, D. (2001). The neurotransmitter dopamine inhibits angiogenesis induced by vascular permeability factor/vascular endothelial growth factor. Nat. Med. 7, 569–574.
Bishop, G. A., Waugh, J. A., Landers, D. V., Krensky, A. M., and Hall, B. M. (1989). Microvascular destruction in renal transplant rejection. Transplantation 48, 408–414.
Booth, A. J., and Bishop, D. K. (2010). TGF-beta, IL-6, IL-17 and CTGF direct multiple pathologies of chronic cardiac allograft rejection. Immunotherapy 2, 511–520.
Boulday, G., Haskova, Z., Reinders, M. E., Pal, S., and Briscoe, D. M. (2006). Vascular endothelial growth factor-induced signaling pathways in endothelial cells that mediate overexpression of the chemokine IFN-gamma-inducible protein of 10 kDa in vitro and in vivo. J. Immunol. 176, 3098–3107.
Bourbie-Vaudaine, S., Blanchard, N., Hivroz, C., and Romeo, P. H. (2006). Dendritic cells can turn CD4+ T lymphocytes into vascular endothelial growth factor-carrying cells by intercellular neuropilin-1 transfer. J. Immunol. 177, 1460–1469.
Briscoe, D. M., Alexander, S. I., and Lichtman, A. H. (1998). Interactions between T lymphocytes and endothelial cells in allograft rejection. Curr. Opin. Immunol. 10, 525–531.
Briscoe, D. M., Dharnidharka, V. R., Isaacs, C., Downing, G., Prosky, S., Shaw, P., Parenteau, N. L., and Hardin-Young, J. (1999). The allogeneic response to cultured human skin equivalent in the hu-PBL-SCID mouse model of skin rejection. Transplantation 67, 1590–1599.
Briscoe, D. M., and Pal, S. (2008). Assessing the vascular effects of early erythropoietin use in pediatric renal transplant recipients. Nat. Clin. Pract. Nephrol. 4, 136–137.
Briscoe, D. M., and Sayegh, M. H. (2002). A rendezvous before rejection: where do T cells meet transplant antigens? Nat. Med. 8, 220–222.
Brown, L. F., Detmar, M., Claffey, K., Nagy, J. A., Feng, D., Dvorak, A. M., and Dvorak, H. F. (1997). Vascular permeability factor/vascular endothelial growth factor: a multifunctional angiogenic cytokine. EXS 79, 233–269.
Brown, L. F., Olbricht, S. M., Berse, B., Jackman, R. W., Matsueda, G., Tognazzi, K. A., Manseau, E. J., Dvorak, H. F., and Van de Water, L. (1995). Overexpression of vascular permeability factor (VPF/VEGF) and its endothelial cell receptors in delayed hypersensitivity skin reactions. J. Immunol. 154, 2801–2807.
Bruder, D., Probst-Kepper, M., Westendorf, A. M., Geffers, R., Beissert, S., Loser, K., von Boehmer, H., Buer, J., and Hansen, W. (2004). Neuropilin-1: a surface marker of regulatory T cells. Eur. J. Immunol. 34, 623–630.
Carmeliet, P., and Jain, R. K. (2000). Angiogenesis in cancer and other diseases. Nature 407, 249–257.
Catalano, A., Caprari, P., Moretti, S., Faronato, M., Tamagnone, L., and Procopio, A. (2006). Semaphorin-3A is expressed by tumor cells and alters T-cell signal transduction and function. Blood 107, 3321–3329.
Chapoval, S. P., Lee, C. G., Tang, C., Keegan, A. D., Cohn, L., Bottomly, K., and Elias, J. A. (2009). Lung vascular endothelial growth factor expression induces local myeloid dendritic cell activation. Clin. Immunol. 132, 371–384.
Chiodoni, C., Iezzi, M., Guiducci, C., Sangaletti, S., Alessandrini, I., Ratti, C., Tiboni, F., Musiani, P., Granger, D. N., and Colombo, M. P. (2006). Triggering CD40 on endothelial cells contributes to tumor growth. J. Exp. Med. 203, 2441–2450.
Choi, Y. J., Chakraborty, S., Nguyen, V., Nguyen, C., Kim, B. K., Shim, S. I., Suki, W. N., and Truong, L. D. (2000). Peritubular capillary loss is associated with chronic tubulointerstitial injury in human kidney: altered expression of vascular endothelial growth factor. Hum. Pathol. 31, 1491–1497.
Clauss, M., Weich, H., Breier, G., Knies, U., Rockl, W., Waltenberger, J., and Risau, W. (1996). The vascular endothelial growth factor receptor Flt-1 mediates biological activities. Implications for a functional role of placenta growth factor in monocyte activation and chemotaxis. J. Biol. Chem. 271, 17629–17634.
Contreras, A. G., and Briscoe, D. M. (2007). Every allograft needs a silver lining. J. Clin. Invest. 117, 3645–3648.
Contreras, A. G., Dormond, O., Edelbauer, M., Calzadilla, K., Hoerning, A., Pal, S., and Briscoe, D. M. (2008). mTOR-understanding the clinical effects. Transplant. Proc. 40, S9–S12.
Cotran, R. S. (1994). “Inflammation and repair,” in Pathologic Basis of Disease, eds R. S. Cotran, V. Kumar, and S. L. Robbins (Philadelphia: W. B. Saunders), 51–92.
Daly, K., Seifert, M., Stack, M., Chandraker, A., Zurakowski, D., Nohria, A., Givertz, M., Karumanchi, A., and Briscoe, D. M. (2011). Identification of highly sensitive and specific biomarker panel for the diagnosis of chronic rejection/allograft vasculopathy following human cardiac transplantation. Am. J. Transplant. 11, 68–69.
Denton, M. D., Davis, S. F., Baum, M. A., Melter, M., Reinders, M. E., Exeni, A., Samsonov, D. V., Fang, J., Ganz, P., and Briscoe, D. M. (2000). The role of the graft endothelium in transplant rejection: evidence that endothelial activation may serve as a clinical marker for the development of chronic rejection. Pediatr. Transplant. 4, 252–260.
Denton, M. D., Magee, C., Melter, M., Dharnidharka, V. R., Sayegh, M. H., and Briscoe, D. M. (2004). TNP-470, an angiogenesis inhibitor, attenuates the development of allograft vasculopathy. Transplantation 78, 1218–1221.
Detmar, M., Brown, L. F., Schon, M. P., Elicker, B. M., Velasco, P., Richard, L., Fukumura, D., Monsky, W., Claffey, K. P., and Jain, R. K. (1998). Increased microvascular density and enhanced leukocyte rolling and adhesion in the skin of VEGF transgenic mice. J. Invest. Dermatol. 111, 1–6.
Detoraki, A., Granata, F., Staibano, S., Rossi, F. W., Marone, G., and Genovese, A. (2010). Angiogenesis and lymphangiogenesis in bronchial asthma. Allergy 65, 946–958.
Dias, S., Hattori, K., Zhu, Z., Heissig, B., Choy, M., Lane, W., Wu, Y., Chadburn, A., Hyjek, E., Gill, M., Hicklin, D. J., Witte, L., Moore, M. A., and Rafii, S. (2000). Autocrine stimulation of VEGFR-2 activates human leukemic cell growth and migration. J. Clin. Invest. 106, 511–521.
Dormond, O., Contreras, A. G., Meijer, E., Datta, D., Flynn, E., Pal, S., and Briscoe, D. M. (2008). CD40-induced signaling in human endothelial cells results in mTORC2- and Akt-dependent expression of vascular endothelial growth factor in vitro and in vivo. J. Immunol. 181, 8088–8095.
Dormond, O., Madsen, J. C., and Briscoe, D. M. (2007). The effects of mTOR-Akt interactions on anti-apoptotic signaling in vascular endothelial cells. J. Biol. Chem. 282, 23679–23686.
Duh, E., and Aiello, L. P. (1999). Vascular endothelial growth factor and diabetes: the agonist versus antagonist paradox. Diabetes 48, 1899–1906.
Edelbauer, M., Datta, D., Vos, I. H., Basu, A., Stack, M. P., Reinders, M. E., Sho, M., Calzadilla, K., Ganz, P., and Briscoe, D. M. (2010). Effect of vascular endothelial growth factor and its receptor KDR on the transendothelial migration and local trafficking of human T cells in vitro and in vivo. Blood 116, 1980–1989.
Ezaki, T., Baluk, P., Thurston, G., La Barbara, A., Woo, C., and McDonald, D. M. (2001). Time course of endothelial cell proliferation and microvascular remodeling in chronic inflammation. Am. J. Pathol. 158, 2043–2055.
Fava, R. A., Olsen, N. J., Spencer-Green, G., Yeo, K. T., Yeo, T. K., Berse, B., Jackman, R. W., Senger, D. R., Dvorak, H. F., and Brown, L. F. (1994). Vascular permeability factor/endothelial growth factor (VPF/VEGF): accumulation and expression in human synovial fluids and rheumatoid synovial tissue. J. Exp. Med. 180, 341–346.
Ferrara, N. (2005). The role of VEGF in the regulation of physiological and pathological angiogenesis. EXS 209–231.
Ferrara, N., and Alitalo, K. (1999). Clinical applications of angiogenic growth factors and their inhibitors. Nat. Med. 5, 1359–1364.
Ferrara, N., Carver-Moore, K., Chen, H., Dowd, M., Lu, L., O’Shea, K. S., Powell-Braxton, L., Hillan, K. J., and Moore, M. W. (1996). Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature 380, 439–442.
Fine, L. G., and Norman, J. T. (2008). Chronic hypoxia as a mechanism of progression of chronic kidney diseases: from hypothesis to novel therapeutics. Kidney Int. 74, 867–872.
Folkman, J. (1995a). Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1, 27–31.
Folkman, J. (1995b). Seminars in Medicine of the Beth Israel Hospital, Boston. Clinical applications of research on angiogenesis. N. Engl. J. Med. 333, 1757–1763.
Folkman, J., and Brem, H. (1992). “Angiogenesis and inflammation,” in Inflammation: Basic Principles and Clinical Correlates, eds J. I. Gallin, I. M. Goldstein, and R. Synderman (New York: Raven Press), 821–839.
Freeman, M. R., Schneck, F. X., Gagnon, M. L., Corless, C., Soker, S., Niknejad, K., Peoples, G. E., and Klagsbrun, M. (1995). Peripheral blood T lymphocytes and lymphocytes infiltrating human cancers express vascular endothelial growth factor: a potential role for T cells in angiogenesis. Cancer Res. 55, 4140–4145.
Fukumura, D., and Jain, R. K. (2007). Tumor microvasculature and microenvironment: targets for anti-angiogenesis and normalization. Microvasc. Res. 74, 72–84.
Gabrilovich, D., Ishida, T., Oyama, T., Ran, S., Kravtsov, V., Nadaf, S., and Carbone, D. P. (1998). Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood 92, 4150–4166.
Gerber, H. P., Hillan, K. J., Ryan, A. M., Kowalski, J., Keller, G. A., Rangell, L., Wright, B. D., Radtke, F., Aguet, M., and Ferrara, N. (1999). VEGF is required for growth and survival in neonatal mice. Development 126, 1149–1159.
Giraudo, E., Primo, L., Audero, E., Gerber, H. P., Koolwijk, P., Soker, S., Klagsbrun, M., Ferrara, N., and Bussolino, F. (1998). Tumor necrosis factor-alpha regulates expression of vascular endothelial growth factor receptor-2 and of its co-receptor neuropilin-1 in human vascular endothelial cells. J. Biol. Chem. 273, 22128–22135.
Girnita, D. M., Brooks, M. M., Webber, S. A., Burckart, G. J., Ferrell, R., Zdanowicz, G., DeCroo, S., Smith, L., Chinnock, R., Canter, C., Addonizio, L., Bernstein, D., Kirklin, J. K., Ranganathan, S., Naftel, D., Girnita, A. L., and Zeevi, A. (2008). Genetic polymorphisms impact the risk of acute rejection in pediatric heart transplantation: a multi-institutional study. Transplantation 85, 1632–1639.
Goel, S., Duda, D. G., Xu, L., Munn, L. L., Boucher, Y., Fukumura, D., and Jain, R. K. (2011). Normalization of the vasculature for treatment of cancer and other diseases. Physiol. Rev. 91, 1071–1121.
Griga, T., Voigt, E., Gretzer, B., Brasch, F., and May, B. (1999). Increased production of vascular endothelial growth factor by intestinal mucosa of patients with inflammatory bowel disease. Hepatogastroenterology 46, 920–923.
Halloran, P. F., de Freitas, D. G., Einecke, G., Famulski, K. S., Hidalgo, L. G., Mengel, M., Reeve, J., Sellares, J., and Sis, B. (2010). The molecular phenotype of kidney transplants. Am. J. Transplant. 10, 2215–2222.
Hanto, D. W., Maki, T., Yoon, M. H., Csizmadia, E., Chin, B. Y., Gallo, D., Konduru, B., Kuramitsu, K., Smith, N. R., Berssenbrugge, A., Attanasio, C., Thomas, M., Wegiel, B., and Otterbein, L. E. (2010). Intraoperative administration of inhaled carbon monoxide reduces delayed graft function in kidney allografts in Swine. Am. J. Transplant. 10, 2421–2430.
Hashimoto, N., Phan, S. H., Imaizumi, K., Matsuo, M., Nakashima, H., Kawabe, T., Shimokata, K., and Hasegawa, Y. (2010). Endothelial-mesenchymal transition in bleomycin-induced pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 43, 161–172.
Hoshino, M., Takahashi, M., and Aoike, N. (2001). Expression of vascular endothelial growth factor, basic fibroblast growth factor, and angiogenin immunoreactivity in asthmatic airways and its relationship to angiogenesis. J. Allergy Clin. Immunol. 107, 295–301.
Huang, S. P., Wu, M. S., Shun, C. T., Wang, H. P., Lin, M. T., Kuo, M. L., and Lin, J. T. (2004). Interleukin-6 increases vascular endothelial growth factor and angiogenesis in gastric carcinoma. J. Biomed. Sci. 11, 517–527.
Humphreys, B. D., Lin, S. L., Kobayashi, A., Hudson, T. E., Nowlin, B. T., Bonventre, J. V., Valerius, M. T., McMahon, A. P., and Duffield, J. S. (2010). Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am. J. Pathol. 176, 85–97.
Ingulli, E., Alexander, S. I., and Briscoe, D. M. (2009). “Transplantation immunology,” in Pediatric Nephrology, eds E. D. Avner, W. E. Harmon, P. Niaudet, and N. Yoshikawa (Berlin: Springer-Verlag), 1835–1866.
Inoue, M., Itoh, H., Ueda, M., Naruko, T., Kojima, A., Komatsu, R., Doi, K., Ogawa, Y., Tamura, N., Takaya, K., Igaki, T., Yamashita, J., Chun, T. H., Masatsugu, K., Becker, A. E., and Nakao, K. (1998). Vascular endothelial growth factor (VEGF) expression in human coronary atherosclerotic lesions: possible pathophysiological significance of VEGF in progression of atherosclerosis. Circulation 98, 2108–2116.
Jain, R. K. (2005). Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 307, 58–62.
Jain, R. K., Koenig, G. C., Dellian, M., Fukumura, D., Munn, L. L., and Melder, R. J. (1996). Leukocyte endothelial adhesion and angiogenesis in tumors. Cancer Metastasis Rev. 15, 195–204.
Jain, S., Furness, P. N., and Nicholson, M. L. (2000). The role of transforming growth factor beta in chronic renal allograft nephropathy. Transplantation 69, 1759–1766.
Jain, S., Mohamed, M. A., Sandford, R., Furness, P. N., Nicholson, M. L., and Talbot, D. (2002). Sequential protocol biopsies from renal transplant recipients show an increasing expression of active TGF beta. Transpl. Int. 15, 630–634.
Johnson, D. W., Forman, C., and Vesey, D. A. (2006). Novel renoprotective actions of erythropoietin: new uses for an old hormone. Nephrology (Carlton) 11, 306–312.
Kaminski, M., and Auerbach, R. (1988). Angiogenesis induction by CD-4 positive lymphocytes. Proc. Soc. Exp. Biol. Med. 188, 440–443.
Kanazawa, S., Tsunoda, T., Onuma, E., Majima, T., Kagiyama, M., and Kikuchi, K. (2001). VEGF, basic-FGF, and TGF-beta in Crohn’s disease and ulcerative colitis: a novel mechanism of chronic intestinal inflammation. Am. J. Gastroenterol. 96, 822–828.
Kang, D. H., Joly, A. H., Oh, S. W., Hugo, C., Kerjaschki, D., Gordon, K. L., Mazzali, M., Jefferson, J. A., Hughes, J., Madsen, K. M., Schreiner, G. F., and Johnson, R. J. (2001a). Impaired angiogenesis in the remnant kidney model: I. Potential role of vascular endothelial growth factor and thrombospondin-1. J. Am. Soc. Nephrol. 12, 1434–1447.
Kang, D. H., Hughes, J., Mazzali, M., Schreiner, G. F., and Johnson, R. J. (2001b). Impaired angiogenesis in the remnant kidney model: II. Vascular endothelial growth factor administration reduces renal fibrosis and stabilizes renal function. J. Am. Soc. Nephrol. 12, 1448–1457.
Kang, D. H., Kanellis, J., Hugo, C., Truong, L., Anderson, S., Kerjaschki, D., Schreiner, G. F., and Johnson, R. J. (2002). Role of the microvascular endothelium in progressive renal disease. J. Am. Soc. Nephrol. 13, 806–816.
Kim, I., Moon, S. O., Kim, S. H., Kim, H. J., Koh, Y. S., and Koh, G. Y. (2001). Vascular endothelial growth factor expression of intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and E-selectin through nuclear factor-kappa B activation in endothelial cells. J. Biol. Chem. 276, 7614–7620.
Kim, Y. S., Hong, S. W., Choi, J. P., Shin, T. S., Moon, H. G., Choi, E. J., Jeon, S. G., Oh, S. Y., Gho, Y. S., Zhu, Z., and Kim, Y. K. (2009). Vascular endothelial growth factor is a key mediator in the development of T cell priming and its polarization to type 1 and type 17 T helper cells in the airways. J. Immunol. 183, 5113–5120.
Klagsbrun, M., and D’Amore, P. A. (1996). Vascular endothelial growth factor and its receptors. Cytokine Growth Factor Rev. 7, 259–270.
Koch, A. E., Harlow, L. A., Haines, G. K., Amento, E. P., Unemori, E. N., Wong, W. L., Pope, R. M., and Ferrara, N. (1994). Vascular endothelial growth factor. A cytokine modulating endothelial function in rheumatoid arthritis. J. Immunol. 152, 4149–4156.
Koch, A. E., Polverini, P. J., and Leibovich, S. J. (1986). Induction of neovascularization by activated human monocytes. J. Leukoc. Biol. 39, 233–238.
Kokudo, T., Suzuki, Y., Yoshimatsu, Y., Yamazaki, T., Watabe, T., and Miyazono, K. (2008). Snail is required for TGFbeta-induced endothelial-mesenchymal transition of embryonic stem cell-derived endothelial cells. J. Cell. Sci. 121, 3317–3324.
Kreisel, D., Krupnick, A. S., Gelman, A. E., Engels, F. H., Popma, S. H., Krasinskas, A. M., Balsara, K. R., Szeto, W. Y., Turka, L. A., and Rosengard, B. R. (2002). Non-hematopoietic allograft cells directly activate CD8+ T cells and trigger acute rejection: an alternative mechanism of allorecognition. Nat. Med. 8, 233–239.
Laxmanan, S., Robertson, S. W., Wang, E., Lau, J. S., Briscoe, D. M., and Mukhopadhyay, D. (2005). Vascular endothelial growth factor impairs the functional ability of dendritic cells through Id pathways. Biochem. Biophys. Res. Commun. 334, 193–198.
Lee, C. G., Link, H., Baluk, P., Homer, R. J., Chapoval, S., Bhandari, V., Kang, M. J., Cohn, L., Kim, Y. K., McDonald, D. M., and Elias, J. A. (2004). Vascular endothelial growth factor (VEGF) induces remodeling and enhances TH2-mediated sensitization and inflammation in the lung. Nat. Med. 10, 1095–1103.
Lee, S., Chen, T. T., Barber, C. L., Jordan, M. C., Murdock, J., Desai, S., Ferrara, N., Nagy, A., Roos, K. P., and Iruela-Arispe, M. L. (2007). Autocrine VEGF signaling is required for vascular homeostasis. Cell 130, 691–703.
Leibovich, S. J., Polverini, P. J., Fong, T. W., Harlow, L. A., and Koch, A. E. (1994). Production of angiogenic activity by human monocytes requires an L-arginine/nitric oxide-synthase-dependent effector mechanism. Proc. Natl. Acad. Sci. U.S.A. 91, 4190–4194.
Leibovich, S. J., Polverini, P. J., Shepard, H. M., Wiseman, D. M., Shively, V., and Nuseir, N. (1987). Macrophage-induced angiogenesis is mediated by tumour necrosis factor-alpha. Nature 329, 630–632.
Leibovich, S. J., and Wiseman, D. M. (1988). Macrophages, wound repair and angiogenesis. Prog. Clin. Biol. Res. 266, 131–145.
Lemstrom, K. B., Krebs, R., Nykanen, A. I., Tikkanen, J. M., Sihvola, R. K., Aaltola, E. M., Hayry, P. J., Wood, J., Alitalo, K., Yla-Herttuala, S., and Koskinen, P. K. (2002). Vascular endothelial growth factor enhances cardiac allograft arteriosclerosis. Circulation 105, 2524–2530.
Leonard, E. C., Friedrich, J. L., and Basile, D. P. (2008). VEGF-121 preserves renal microvessel structure and ameliorates secondary renal disease following acute kidney injury. Am. J. Physiol. Renal Physiol. 295, F1648–F1657.
Leung, D. W., Cachianes, G., Kuang, W. J., Goeddel, D. V., and Ferrara, N. (1989). Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 246, 1306–1309.
Li, J., Qu, X., and Bertram, J. F. (2009). Endothelial-myofibroblast transition contributes to the early development of diabetic renal interstitial fibrosis in streptozotocin-induced diabetic mice. Am. J. Pathol. 175, 1380–1388.
Majno, G. (1998). Chronic inflammation: links with angiogenesis and wound healing. Am. J. Pathol. 153, 1035–1039.
Malmstrom, N. K., Kallio, E. A., Rintala, J. M., Nykanen, A. I., Raisanen-Sokolowski, A. K., Paavonen, T., Lemstrom, K. B., and Koskinen, P. K. (2008). Vascular endothelial growth factor in chronic rat allograft nephropathy. Transpl. Immunol. 19, 136–144.
Mannon, R. B. (2006). Therapeutic targets in the treatment of allograft fibrosis. Am. J. Transplant. 6, 867–875.
Markwald, R. R., Fitzharris, T. P., and Smith, W. N. (1975). Structural analysis of endocardial cytodifferentiation. Dev. Biol. 42, 160–180.
Marumo, T., Schini-Kerth, V. B., and Busse, R. (1999). Vascular endothelial growth factor activates nuclear factor-kappaB and induces monocyte chemoattractant protein-1 in bovine retinal endothelial cells. Diabetes 48, 1131–1137.
Matsumoto, T., and Mugishima, H. (2006). Signal transduction via vascular endothelial growth factor (VEGF) receptors and their roles in atherogenesis. J. Atheroscler. Thromb. 13, 130–135.
Mayer, G. (2011). Capillary rarefaction, hypoxia, VEGF and angiogenesis in chronic renal disease. Nephrol. Dial. Transplant. 26, 1132–1137.
McDonald, D. M. (2001). Angiogenesis and remodeling of airway vasculature in chronic inflammation. Am. J. Respir. Crit. Care Med. 164, S39–S45.
Medici, D., Shore, E. M., Lounev, V. Y., Kaplan, F. S., Kalluri, R., and Olsen, B. R. (2010). Conversion of vascular endothelial cells into multipotent stem-like cells. Nat. Med. 16, 1400–1406.
Melder, R. J., Koenig, G. C., Witwer, B. P., Safabakhsh, N., Munn, L. L., and Jain, R. K. (1996). During angiogenesis, vascular endothelial growth factor and basic fibroblast growth factor regulate natural killer cell adhesion to tumor endothelium. Nat. Med. 2, 992–997.
Melter, M., Reinders, M. E., Sho, M., Pal, S., Geehan, C., Denton, M. D., Mukhopadhyay, D., and Briscoe, D. M. (2000). Ligation of CD40 induces the expression of vascular endothelial growth factor by endothelial cells and monocytes and promotes angiogenesis in vivo. Blood 96, 3801–3808.
Miao, H. Q., and Klagsbrun, M. (2000). Neuropilin is a mediator of angiogenesis. Cancer Metastasis Rev. 19, 29–37.
Mizui, M., Kumanogoh, A., and Kikutani, H. (2009). Immune semaphorins: novel features of neural guidance molecules. J. Clin. Immunol. 1, 1–11.
Mor, F., Quintana, F. J., and Cohen, I. R. (2004). Angiogenesis-inflammation cross-talk: vascular endothelial growth factor is secreted by activated T cells and induces Th1 polarization. J. Immunol. 172, 4618–4623.
Moulton, K. S., Melder, R. J., Dharnidharka, V. R., Hardin-Young, J., Jain, R. K., and Briscoe, D. M. (1999). Angiogenesis in the huPBL-SCID model of human transplant rejection. Transplantation 67, 1626–1631.
Mukhopadhyay, D., Tsiokas, L., Zhou, X. M., Foster, D., Brugge, J. S., and Sukhatme, V. P. (1995). Hypoxic induction of human vascular endothelial growth factor expression through c-Src activation. Nature 375, 577–581.
Nakao, A., Choi, A. M., and Murase, N. (2006). Protective effect of carbon monoxide in transplantation. J. Cell. Mol. Med. 10, 650–671.
Nykanen, A. I., Krebs, R., Saaristo, A., Turunen, P., Alitalo, K., Yla-Herttuala, S., Koskinen, P. K., and Lemstrom, K. B. (2003). Angiopoietin-1 protects against the development of cardiac allograft arteriosclerosis. Circulation 107, 1308–1314.
Oberbauer, R., Segoloni, G., Campistol, J. M., Kreis, H., Mota, A., Lawen, J., Russ, G., Grinyo, J. M., Stallone, G., Hartmann, A., Pinto, J. R., Chapman, J., Burke, J. T., Brault, Y., and Neylan, J. F. (2005). Early cyclosporine withdrawal from a sirolimus-based regimen results in better renal allograft survival and renal function at 48 months after transplantation. Transpl. Int. 18, 22–28.
Osasan, S. A., de Freitas, D. G., Chang, J., Mengel, M., Halloran, P. F., and Sis, B. (2011). Microcirculation endothelial cell cycling is selectively increased in antibody-mediated rejection of kidney transplants, but not in other diseases. J. Am. Soc. Nephrol. 22, 103A.
Peng, W., Chen, J., Jiang, Y., Shou, Z., Chen, Y., and Wang, H. (2008). Acute renal allograft rejection is associated with increased levels of vascular endothelial growth factor in the urine. Nephrology (Carlton) 13, 73–79.
Pilmore, H. L., Eris, J. M., Painter, D. M., Bishop, G. A., and McCaughan, G. W. (1999). Vascular endothelial growth factor expression in human chronic renal allograft rejection. Transplantation 67, 929–933.
Pober, J. S., Orosz, C. G., Rose, M. L., and Savage, C. O. S. (1996). Can graft endothelial cells initiate a host anti-graft immune response? Transplantation 61, 343–349.
Pober, J. S., and Sessa, W. C. (2007). Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 7, 803–815.
Rabelink, T. J., Wijewickrama, D. C., and de Koning, E. J. (2007). Peritubular endothelium: the Achilles heel of the kidney? Kidney Int. 72, 926–930.
Reinders, M. E., Rabelink, T. J., and Briscoe, D. M. (2006). Angiogenesis and endothelial cell repair in renal disease and allograft rejection. J. Am. Soc. Nephrol. 17, 932–942.
Reinders, M. E., Sho, M., Izawa, A., Wang, P., Mukhopadhyay, D., Koss, K. E., Geehan, C. S., Luster, A. D., Sayegh, M. H., and Briscoe, D. M. (2003a). Proinflammatory functions of vascular endothelial growth factor in alloimmunity. J. Clin. Invest. 112, 1655–1665.
Reinders, M. E., Fang, J. C., Wong, W., Ganz, P., and Briscoe, D. M. (2003b). Expression patterns of vascular endothelial growth factor in human cardiac allografts: association with rejection. Transplantation 76, 224–230.
Romeo, P. H., Lemarchandel, V., and Tordjman, R. (2002). Neuropilin-1 in the immune system. Adv. Exp. Med. Biol. 515, 49–54.
Sarris, M., Andersen, K. G., Randow, F., Mayr, L., and Betz, A. G. (2008). Neuropilin-1 expression on regulatory T cells enhances their interactions with dendritic cells during antigen recognition. Immunity 28, 402–413.
Schena, F. P., Pascoe, M. D., Alberu, J., del Carmen Rial, M., Oberbauer, R., Brennan, D. C., Campistol, J. M., Racusen, L., Polinsky, M. S., Goldberg-Alberts, R., Li, H., Scarola, J., and Neylan, J. F. (2009). Conversion from calcineurin inhibitors to sirolimus maintenance therapy in renal allograft recipients: 24-month efficacy and safety results from the CONVERT trial. Transplantation 87, 233–242.
Schor, A. M., Canfield, A. E., Sutton, A. B., Arciniegas, E., and Allen, T. D. (1995). Pericyte differentiation. Clin. Orthop. Relat. Res. 81–91.
Shahbazi, M., Fryer, A. A., Pravica, V., Brogan, I. J., Ramsay, H. M., Hutchinson, I. V., and Harden, P. N. (2002). Vascular endothelial growth factor gene polymorphisms are associated with acute renal allograft rejection. J. Am. Soc. Nephrol. 13, 260–264.
Shalaby, F., Rossant, J., Yamaguchi, T. P., Gertsenstein, M., Wu, X. F., Breitman, M. L., and Schuh, A. C. (1995). Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature 376, 62–66.
Sharma, V. K., Bologa, R. M., Xu, G. P., Li, B., Mouradian, J., Wang, J., Serur, D., Rao, V., and Suthanthiran, M. (1996). Intragraft TGF-beta 1 mRNA: a correlate of interstitial fibrosis and chronic allograft nephropathy. Kidney Int. 49, 1297–1303.
Shin, J. Y., Yoon, I. H., Kim, J. S., Kim, B., and Park, C. G. (2009). Vascular endothelial growth factor-induced chemotaxis and IL-10 from T cells. Cell. Immunol. 256, 72–78.
Sho, M., Akashi, S., Kanehiro, H., Hamada, K., Kashizuka, H., Ikeda, N., Nomi, T., Kuzumoto, Y., Tsurui, Y., Yoshiji, H., Wu, Y., Hicklin, D. J., Briscoe, D. M., and Nakajima, Y. (2005). Function of the vascular endothelial growth factor receptors Flt-1 and Flk-1/KDR in the alloimmune response in vivo. Transplantation 80, 717–722.
Shweiki, D., Itin, A., Soffer, D., and Keshet, E. (1992). Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature 359, 843–845.
Sidky, Y. A., and Auerbach, R. (1975). Lymphocyte-induced angiogenesis: a quantitative and sensitive assay of the graft-vs.-host reaction. J. Exp. Med. 141, 1084–1100.
Sis, B., and Halloran, P. F. (2010). Endothelial transcripts uncover a previously unknown phenotype: C4d-negative antibody-mediated rejection. Curr. Opin. Organ Transplant. 15, 42–48.
Sis, B., Jhangri, G. S., Bunnag, S., Allanach, K., Kaplan, B., and Halloran, P. F. (2009). Endothelial gene expression in kidney transplants with alloantibody indicates antibody-mediated damage despite lack of C4d staining. Am. J. Transplant. 9, 2312–2323.
Steegh, F. M., Gelens, M. A., Nieman, F. H., van Hooff, J. P., Cleutjens, J. P., van Suylen, R. J., Daemen, M. J., van Heurn, E. L., Christiaans, M. H., and Peutz-Kootstra, C. J. (2011). Early loss of peritubular capillaries after kidney transplantation. J. Am. Soc. Nephrol. 22, 1024–1029.
Strieter, R. M., Kunkel, S. L., Arenberg, D. A., Burdick, M. D., and Polverini, P. J. (1995a). Interferon gamma-inducible protein 10 (IP-10), a member of the C-X-C chemokine family, is an inhibitor of angiogenesis. Biochem. Biophys. Res. Commun. 210, 51–57.
Strieter, R. M., Polverini, P. J., Arenberg, D. A., and Kunkel, S. L. (1995b). The role of CXC chemokines as regulators of angiogenesis. Shock 4, 155–160.
Suzuki, H., Onishi, H., Wada, J., Yamasaki, A., Tanaka, H., Nakano, K., Morisaki, T., and Katano, M. (2010). VEGFR2 is selectively expressed by FOXP3high CD4+ Treg. Eur. J. Immunol. 40, 197–203.
Suzuki, K., Kumanogoh, A., and Kikutani, H. (2008). Semaphorins and their receptors in immune cell interactions. Nat. Immunol. 9, 17–23.
Takahashi, H., and Shibuya, M. (2005). The vascular endothelial growth factor (VEGF)/VEGF receptor system and its role under physiological and pathological conditions. Clin. Sci. 109, 227–241.
Tanaka, H., Sukhova, G. K., and Libby, P. (1994). Interaction of the allogeneic state and hypercholesterolemia in arterial lesion formation in experimental cardiac allografts. Arterioscler. Thromb. 14, 734–745.
Tischer, E., Mitchell, R., Hartman, T., Silva, M., Gospodarowicz, D., Fiddes, J. C., and Abraham, J. A. (1991). The human gene for vascular endothelial growth factor. Multiple protein forms are encoded through alternative exon splicing. J. Biol. Chem. 266, 11947–11954.
Tordjman, R., Lepelletier, Y., Lemarchandel, V., Cambot, M., Gaulard, P., Hermine, O., and Romeo, P. H. (2002). A neuronal receptor, neuropilin-1, is essential for the initiation of the primary immune response. Nat. Immunol. 3, 477–482.
Torry, R. J., Labarrere, C. A., Torry, D. S., Holt, V. J., and Faulk, W. P. (1995). Vascular endothelial growth factor expression in transplanted human hearts. Transplantation 60, 1451–1457.
Valujskikh, A., and Heeger, P. S. (2003). Emerging roles of endothelial cells in transplant rejection. Curr. Opin. Immunol. 15, 493–498.
Vos, I. H., and Briscoe, D. M. (2002). Endothelial injury: cause and effect of alloimmune inflammation. Transpl. Infect. Dis. 4, 152–159.
Walsh, D. A., and Pearson, C. I. (2001). Angiogenesis in the pathogenesis of inflammatory joint and lung diseases. Arthritis Res. 3, 147–153.
Wang, L., Zeng, H., Wang, P., Soker, S., and Mukhopadhyay, D. (2003). Neuropilin-1-mediated vascular permeability factor/vascular endothelial growth factor-dependent endothelial cell migration. J. Biol. Chem. 278, 48848–48860.
Watanabe, H., Mamelak, A. J., Wang, B., Howell, B. G., Freed, I., Esche, C., Nakayama, M., Nagasaki, G., Hicklin, D. J., Kerbel, R. S., and Sauder, D. N. (2004). Anti-vascular endothelial growth factor receptor-2 (Flk-1/KDR) antibody suppresses contact hypersensitivity. Exp. Dermatol. 13, 671–681.
Watanabe, H., Mamelak, A. J., Weiss, E., Wang, B., Freed, I., Brice, A. K., Wachtman, L., Gabrielson, K. L., Yokota, N., Hicklin, D. J., Kerbel, R. S., Haas, M., and Sauder, D. N. (2005). Anti-vascular endothelial growth factor receptor-2 antibody accelerates renal disease in the NZB/W F1 murine systemic lupus erythematosus model. Clin. Cancer Res. 11, 407–409.
Wiseman, D. M., Polverini, P. J., Kamp, D. W., and Leibovich, S. J. (1988). Transforming growth factor-beta (TGF beta) is chemotactic for human monocytes and induces their expression of angiogenic activity. Biochem. Biophys. Res. Commun. 157, 793–800.
Woywodt, A., Schroeder, M., Gwinner, W., Mengel, M., Jaeger, M., Schwarz, A., Haller, H., and Haubitz, M. (2003). Elevated numbers of circulating endothelial cells in renal transplant recipients. Transplantation 76, 1–4.
Zeisberg, E. M., Tarnavski, O., Zeisberg, M., Dorfman, A. L., McMullen, J. R., Gustafsson, E., Chandraker, A., Yuan, X., Pu, W. T., Roberts, A. B., Neilson, E. G., Sayegh, M. H., Izumo, S., and Kalluri, R. (2007a). Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 13, 952–961.
Zeisberg, E. M., Potenta, S., Xie, L., Zeisberg, M., and Kalluri, R. (2007b). Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 67, 10123–10128.
Zeng, H., Dvorak, H. F., and Mukhopadhyay, D. (2001). Vascular permeability factor (VPF)/vascular endothelial growth factor (VEGF) receptor-1 down-modulates VPF/VEGF receptor-2-mediated endo-thelial cell proliferation, but not migration, through phosphatidylinositol 3-kinase-dependent path-ways. J. Biol. Chem. 276, 26969–26979.
Zeng, H., Zhao, D., and Mukhopadhyay, D. (2002). Flt-1-mediated down-regulation of endothelial cell proliferation through pertussis toxin-sensitive G proteins, beta gamma subunits, small GTPase CDC42, and partly by Rac-1. J. Biol. Chem. 277, 4003–4009.
Zhang, J., Silva, T., Yarovinsky, T., Manes, T. D., Tavakoli, S., Nie, L., Tellides, G., Pober, J. S., Bender, J. R., and Sadeghi, M. M. (2010). VEGF blockade inhibits lymphocyte recruitment and ameliorates immune-mediated vascular remodeling. Circ. Res. 107, 408–417.
Zhang, X., and Reed, E. F. (2009). Effect of antibodies on endothelium. Am. J. Transplant. 9, 2459–2465.
Zhao, Q., Egashira, K., Inoue, S., Usui, M., Kitamoto, S., Ni, W., Ishibashi, M., Hiasa Ki, K., Ichiki, T., Shibuya, M., and Takeshita, A. (2002). Vascular endothelial growth factor is necessary in the development of arteriosclerosis by recruiting/activating monocytes in a rat model of long-term inhibition of nitric oxide synthesis. Circulation 105, 1110–1115.
Ziogas, A. C., Gavalas, N. G., Tsiatas, M., Tsitsilonis, O., Politi, E., Terpos, E., Rodolakis, A., Vlahos, G., Thomakos, N., Haidopoulos, D., Antsaklis, A., Dimopoulos, M. A., and Bamias, A. (2011). VEGF directly suppresses activation of T cells from ovarian cancer patients and healthy individuals via VEGF receptor type 2. Int. J. Cancer 130, 857–864.
Keywords: endothelial cell, microvascular injury, angiogenesis, vascular endothelial growth factor, hypoxia, allograft rejection, chronic allograft rejection, allograft vasculopathy
Citation: Bruneau S, Woda CB, Daly KP, Boneschansker L, Jain NG, Kochupurakkal N, Contreras AG, Seto T and Briscoe DM (2012) Key features of the intragraft microenvironment that determine long-term survival following transplantation. Front. Immun. 3:54. doi: 10.3389/fimmu.2012.00054
Received: 17 January 2012; Accepted: 02 March 2012;
Published online: 02 April 2012.
Edited by:
Xian Chang Li, Brigham and Women’s Hospital, USAReviewed by:
Awen Gallimore, Cardiff University, UKXian Chang Li, Brigham and Women’s Hospital, USA
Zhenhua Dai, University of Texas Health Science Center, USA
Copyright: © 2012 Bruneau, Woda, Daly, Boneschansker, Jain, Kochupurakkal, Contreras, Seto and Briscoe. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: David Michael Briscoe, Division of Nephrology, Department of Medicine, Children’s Hospital Boston, 300 Longwood Avenue, Boston, MA 02115, USA. e-mail:ZGF2aWQuYnJpc2NvZUBjaGlsZHJlbnMuaGFydmFyZC5lZHU=