
 F. Bossi
F. Bossi
- Department of Life Sciences, University of Trieste, Trieste, Italy
An inflammatory-like process and vascular remodeling represent the main changes that occur in decidua in the early phase of pregnancy. These changes are partly induced by trophoblast cells that colonize the decidua and are also contributed by the complement system, which can easily be activated as a result of tissue remodeling. Local control by several complement regulators including surface-bound and soluble molecules is critical to prevent complement-mediated tissue damage in normal pregnancy. C7 expressed on the endothelial cells (ECs) surface has been recognized as a novel complement regulator involved in the control of the proinflammatory effect of the terminal complement complex. The protective role of placental complement regulators in pregnancy is underscored by the recent finding of an association of preeclampsia with mutations in the genes encoding for some of these proteins. Complement components produced at feto-maternal interface serve an important function in placental development. C1q synthesized by decidual ECs and expressed on the cell surface is particularly important in this regard because it acts as a molecular bridge between endovascular trophoblast and ECs. C1q is also produced by extravillous trophoblast and is used to favor trophoblast migration through the decidua. Defective expression of C1q by trophoblast is associated with impaired trophoblast invasion of decidua and may have important implications in pregnancy disorders such as preeclampsia characterized by reduced vascular remodeling.
Introduction
During the early phase of pregnancy the endometrium of the uterus undergoes profound changes and transforms into decidua, a newly formed tissue that plays a critical role for successful embryo implantation and regular fetal growth. The decidua provides a physical anchorage for the implanted embryo tightly attached to the maternal tissue by means of anchoring villi surrounded by an outer layer of trophoblasts that form cell columns. Some of these cells leave the villi and invade the decidua as extravillous trophoblasts (EVTs) reaching the inner third of the myometrium where they form multinucleated giant cells (Plaisier, 2011). A distinct group of EVTs penetrates the uterine spiral arteries and migrates upward against the blood flow partially replacing the endothelial cells (ECs) to form mosaic vessels (Bulla et al., 2005; Figure 1).
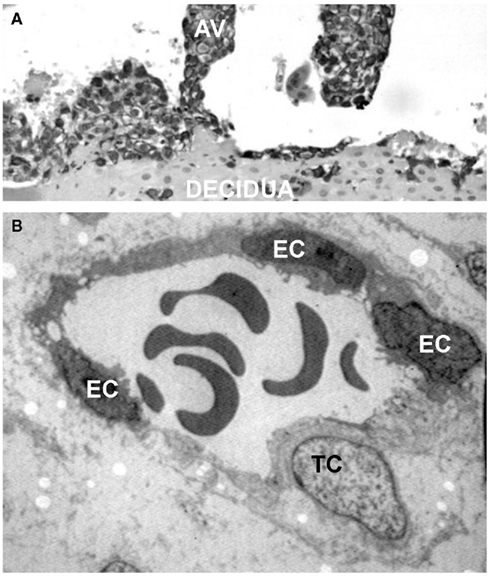
Figure 1. Interstitial and endovascular invasion of trophoblast cells. (A) Histological section of first trimester placenta stained for cytokeratin 7, showing an anchoring villi (AV). (B) Transmission electron microscope image of a remodeled vessel in which a trophoblast cell (TC) lines the vascular wall together with endothelial cells (ECs).
Being of fetal origin, villous trophoblasts exposed to maternal blood circulating in the intervillous space and EVTs present in human decidua represent a real challenge for the maternal immune system. As a result, a vigorous response may be mounted against paternal antigens expressed on trophoblasts leading to potential deleterious effects on fetal survival. To avoid these undesired consequences, different strategies are developed to prevent an immunologic attack of the mother against the fetus while keeping intact the protective function against harmful pathogens. Failure to express classical polymorphic MHC class I and class II molecules is used by villous trophoblasts to evade recognition by T cells. Unlike villous trophoblasts, EVTs express MHC molecules, but these are restricted to class I classical HLA-C and non-classical HLA-E, HLA-F, and HLA-G proteins that are not usually used to stimulate T cells.
The mother adopts a different strategy to protect the fetus organizing a protective environment at site of embryo implantation with the aim to neutralize harmful attack of the fetus by the maternal immune response. Maternal tolerance of the fetus is maintained primarily by CD4+ CD25+ regulatory T cells (Aluvihare et al., 2004) with the contribution of CD8+ T cells (Tilburgs et al., 2010) and NK cells (Hanna et al., 2006), which have recently been shown to induce formation of T regulatory cells as a result of physical interaction with decidual myelomonocytic CD14+ (Vacca et al., 2010).
Embryo implantation is associated with an inflammatory process that develops in decidua in response to trophoblast cells that make their way through the stroma by means of the proteases of the matrix metalloproteinase and plasminogen activator/plasmin systems (Salamonsen, 1999; Pepper, 2001) to invade the maternal tissue. Trophoblast invasion of decidua results in recruitment and activation of leukocytes and precursors of ECs and in the release of various cytokines, chemokines, growth factors that promote tissue remodeling (Plaisier, 2011). Hormonal changes contribute to the development of inflammation in maternal decidua in the early pregnancy as suggested by the observation that a similar process, though less intense, occurs in non-pregnant uterus during the mid-luteal phase of the menstrual cycle. It should be pointed out, however, that this process rather than having deleterious effects helps to promote fetal survival and somehow resembles the inflammation developing within the tumor microenvironment that stimulates tumor progression, tissue remodeling and angiogenesis, and down-modulates adaptive immune response (Sica and Bronte, 2007; Le Bitoux and Stamenkovic, 2008).
The cell infiltrate present in decidua during normal pregnancy includes leukocytes that constitute 30% of stromal cells and comprise approximately 70% NK cells, 20–25% macrophages, and 1.7% dendritic cells, while T lymphocytes represent about 3–10% of the decidual immune cells (Vacca et al., 2010). These cells communicate with each other and talk to EVTs contributing in this way to the important changes that occur in decidua. Thus, decidual NK cells have been found to regulate trophoblast invasion (Hanna et al., 2006) while EVTs recruit decidual NK through the release of CXCL12 (Hanna et al., 2003; Wu et al., 2005).
A cross-talk is also established between EVTs and other cells of both innate and adaptive immunity leading to the differentiation of immune cells into a trophoblast-supporting phenotype through the action of HLA-G expressed by invading trophoblasts (Shakhawat et al., 2010; Mor et al., 2011). A special interaction occurs between trophoblasts and macrophages that represent approximately 20–25% of the leukocyte population in decidua. These cells are stimulated by trophoblasts to remove apoptotic cells in order to prevent the release of harmful proinflammatory and pro-immunogenic intracellular contents (Abrahams et al., 2004) and are also educated to secrete cytokines and chemokines that support trophoblast growth and survival (Fest et al., 2007).
Vascular changes represent an important feature of the decidual tissue in the early phase of pregnancy and are characterized by active angiogenesis and vascular remodeling involving the spiral arteries. These changes ensure a regular supply of maternal blood to the developing placenta at an optimal flow rate and are made possible by the transformation of the spiral arteries into low-resistance dilated thin-walled vessels as a result of loss of vascular smooth muscle cells and partial replacement of ECs by endovascular trophoblasts (Lyall et al., 2001; Pijnenborg et al., 2006). Data collected both in mice (Guimond et al., 1998; Greenwood et al., 2000), and humans (Smith et al., 2009) suggest that decidual NK cells clustered around spiral arteries in early pregnancy play a role in vessel remodeling. Perivascular trophoblasts have also been shown to contribute to this process inducing degradation of the internal elastic lamina and medial elastin fibers of the vessel wall through the action of metalloproteinases MMP-12 (Harris et al., 2010).
The original idea that pregnancy represents an anti-inflammatory state is no longer tenable in the light of evidence supporting the concept that a proinflammatory environment is required for embryo implantation and trophoblast invasion (Mor et al., 2011). The data so far available indicate that the inflammatory state is sustained by cytokines and chemokines released by the cells that accumulate in the decidua. Less attention has been paid to the contribution of the complement (C) system to the success of embryo implantation and also to the regular development of the fetus. In this review we shall examine the double role that C plays in the pathophysiology of pregnancy focusing in particular on the local effect of C components and C activation products in placenta.
Production of C Components and Regulators at the Feto-Maternal Interface
Although the liver represents the major source of circulating C components (Morris et al., 1982), evidence collected over the last three decades indicates that they can also be synthesized at extrahepatic sites in various tissues and organs (Morgan and Gasque, 1997). Several cell types have been found to produce C components including macrophages (Johnson and Hetland, 1988), fibroblasts (Garred et al., 1990), ECs (Langeggen et al., 2000) as well as tissue specific cells (Brooimans et al., 1991; Dovezenski et al., 1992; Khirwadkar et al., 1993; Gulati et al., 1994; Andoh et al., 1998; Griffiths et al., 2009; Pattrick et al., 2009). As a result of the contribution of different cell types to local synthesis of C components, an entire C system can be organized at tissue sites that can be readily available for local needs. The C proteins are secreted locally in limited amount under physiologic conditions, but their level is expected to meet the special requirements elicited by pathologic situations that regulate their biosynthesis through the action of cytokines and other stimuli (Colten and Garnier, 1998).
Although locally synthesized C components may contribute to tissue damage, as is the case of C3 produced by renal tubular epithelium involved in the development of inflammation leading to transplant rejection (Pratt et al., 2002), they play a critical role in host defense at tissue and organ level (Morgan and Gasque, 1997; Laufer et al., 2001). The protective function of the C system is particularly important in the female reproductive tract during pregnancy since the implanted embryo is vulnerable to attack by pathogens that colonize the cervico-vaginal cavity. It is therefore not surprising that a local protection is needed to prevent the access of infectious agents to the feto-placental unit. The recognition molecules of the C system MBL, C1q, and C3 are present in the cervico-vaginal cavity where they provide a first line of defense against infectious agents (Pellis et al., 2005). Interestingly, the vaginal epithelium is the main local source of MBL which is secreted under hormonal control reaching the highest value in the post-ovulatory phase of the menstrual cycle in close correlation with the increased level of progesterone.
Several C components of both classical and alternative pathways and C regulators are secreted by chorionic tissue and chorion-derived cells and their synthesis is differently regulated by various cytokines (Goldberg et al., 2007). The ability of some of these cytokines to up-regulate local production of C components and regulators in normal pregnancy is critical to neutralize invading pathogens and also to protect the fetus from attack by the maternal immune system.
Trophoblasts constitute a major component of decidual cells and contribute to local synthesis of C components in maternal decidua representing the primary source of C3 and C4 at feto-maternal interface (Bulla et al., 2009). The expression of these C components by trophoblast cells is not modulated by IL-1α and TNF-α but is up-regulated by IFNγ. The latter cytokine produced at placental level mainly by decidual NK (Lash et al., 2006) controls trophoblast activation (Gagioti et al., 2000) and invasion into the decidua (Hu et al., 2006; Lash et al., 2006), and plays a critical role in remodeling spiral arteries in the mouse (Ashkar et al., 2000). The finding that trophoblast cells secrete C3 and C4 involved in opsonophagocytosis further supports previous observations suggesting that these cells help to promote the innate immune response at the placental level during pregnancy and potentially contribute to placental immune defense against pathogens (Amarante-Paffaro et al., 2004). In addition to C3 and C4, trophoblast cells have been found to secrete C1q (Agostinis et al., 2010), which is relatively abundant in maternal decidua being synthesized by other cells including macrophages, fibroblast, and ECs. It is important to note that the decidua is the only tissue where ECs are able to produce C1q in physiologic conditions (Bulla et al., 2008). Interestingly, the molecule synthesized by decidual ECs (DECs) is expressed to a large extent on the cell surface and only marginally secreted. The high avidity binding of C1q to the cell membrane is not due to its interaction with the receptors for the collagen tail or the globular head of the molecule identified on ECs (Peerschke et al., 1996). Removal of a substantial portion of bound C1q from the cell membrane by treatment with heparinases rather suggests that surface expressed heparan sulfates offer the binding site for the molecule (Bulla et al., 2008). C7 is another C component synthesized by ECs (Langeggen et al., 2000) and is also detected on the membrane of DECs (Figure 2; Bossi et al., 2009). However, the expression of C7 on the cell surface, unlike that of C1q, is not restricted to DECs but it is observed also on the vessel endothelium of several tissues including endometrium of non-pregnant uterus, skin, kidney, and brain. The expression level of membrane bound C7 is up-regulated by the proinflammatory cytokines I-1α, TNF-α, and IFN-γ. In contrast with bound C1q, surface expressed C7 cannot be removed by treatment with heparinases and is associated with vimentin.
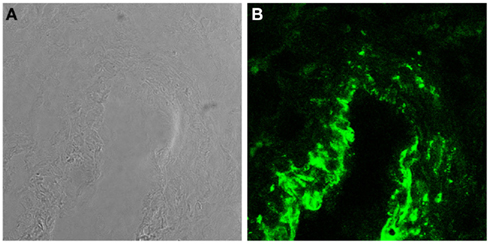
Figure 2. C7 is synthesized and expressed by DECs. (A) Light microscopy image of a decidual vessel from human first trimester placenta. (B) Confocal analysis of the same decidual vessel stained for C7.
Non-Canonical Role of the C System in Human Placenta
For a long time C has been viewed as an effector system that, once recognized the target to neutralize, acts by promoting the inflammatory process or by inducing cell cytotoxicity. Data accumulated over the last few years indicate that C components and C activation products may exhibit alternative functions. The increased expression of C1q and C4 mRNA in some areas of the rat brain during the embryonic life is suggestive of a novel role of C in brain development (Johnson et al., 1994). C3 and C5 have been implicated in lens regeneration (Kimura et al., 2003), while C3a and C5a were shown to promote liver regeneration through the intermediate action of IL-6 and TNFα (Strey et al., 2003).
The decidua is another tissue where C has been found to contribute to tissue remodeling and vascular changes (Bulla et al., 2008). The recognition that the C system serves non-canonical functions in human placenta stems from studies showing that C1q is involved in placental development (Bulla et al., 2008). More specifically, C1q was found to play an important role in the replacement of DECs by endovascular trophoblasts migrating along the decidual spiral arteries and in trophoblast invasion of maternal decidua. The original observation that prompted these studies was the detection of C1q on the surface of DECs in normal pregnancy in the absence of immunoglobulins and C4. This finding was surprising because deposits of C1q are usually observed in pathologic conditions associated with C activation. Interestingly, C1q was found to be localized at the contact site between endovascular trophoblasts and DECs suggesting its implication in the physical interaction between the two cell types. In vitro experiments of adhesion assay confirmed this hypothesis showing that the adhesion of purified trophoblasts to a confluent monolayer of DECs is prevented by antibodies to C1q. This cell bound C component acts as a bridge between endovascular trophoblasts and decidual endothelium by virtue of its interaction with a receptor for the globular head of C1q (gC1qR) expressed on trophoblasts. Binding of trophoblasts to DECs is a rather complex process promoted by several molecules including C1q and VE-cadherin (Bulla et al., 2005) and is followed by transendothelial migration of trophoblasts that eventually reach the basal membrane and partially replace ECs.
The finding that C1q is actively synthesized by EVTs and is widely distributed in decidual stroma led to the recognition of an additional function of the early C component in promoting trophoblast migration through the decidua. Trophoblast cells were found to adhere to C1q and to migrate through an insert of a transwell coated with C1q that recognizes cell-expressed gC1qR, as well as a4 and β1 integrins and triggers activation of the MAP kinases pathway. The in vivo relevance of these findings is supported by the observation of impaired labyrinth development and decreased remodeling of decidual vessel associated with increased fetal resorption rate, reduced fetal weight, and smaller litter size observed in C1q KO mice as compared with WT animals (Agostinis et al., 2010).
Regulatory Functions of the C System at the Feto-Maternal Interface
Extensive remodeling of decidua favors C activation that may lead to tissue damage and adverse pregnancy outcome. C-dependent damage of trophoblasts may promote cell destruction or more likely increased permeability of the barrier, opening the way to bacteria, virus, and other toxic molecules that compromise fetal survival. No wonder if decidual cells have developed strategies to prevent C-mediated damage consisting in the expression of C regulatory proteins (CRPs) DAF or CD55, MCP or CD46, and CD59.
Syncytiotrophoblasts that form the outer layer of villous trophoblasts require a particular protection against C activation products because they are directly exposed to maternal blood and are therefore at special risk of being attacked by activated C. These cells express all three regulatory proteins (Holmes et al., 1990, 1992) and are further protected by the binding of vitronectin or S protein, a soluble inhibitor of the assembling membrane attack complex (MAC) of the C system acquired from the maternal blood (Tedesco et al., 1990). Villous cytotrophoblasts form a continuous layer of cells adhering to the basal membrane of the villi below the syncytiotrophoblasts and express MCP and CD59, while controversy exists on the expression of DAF (Holmes et al., 1990, 1992; Hsi et al., 1991). EVTs also express CRPs on their surface, in particular CD59, while DAF and MCP are mainly detected on decidual giant cells (Hsi et al., 1991; Bulla et al., 2003). Endovascular trophoblast cells that colonize decidual spiral arteries are similarly exposed to maternal blood and are a potential C target. Damage may also involve ECs and other cells of maternal origin in decidua, since locally released C effector molecules do not distinguish between foreign agents and self components and cause tissue alterations through the destructive effect of MAC or via promotion of the inflammatory process.
Evidence for the protective function of the regulatory molecules expressed on the surface of syncytiotrophoblasts was obtained in studies analyzing the susceptibility of these cells to C-dependent killing induced by antibodies directed against steroid producing cell that recognize syncytiotrophoblasts (Tedesco et al., 1993). Antibody-mediated neutralization of CD46 and CD59 expressed on syncytiotrophoblasts led to a substantial increase in cell killing emphasizing the important role of the two C regulators in trophoblast protection while CD55 seems to be less critical being expressed in only 30% of these cells (Tedesco et al., 1993).
Endothelial cells are another potential target of C attack in decidua and are protected by several CRPs expressed on the cell surface or acquired from the fluid phase (Fischetti and Tedesco, 2006). Evidence has been collected supporting an additional protective mechanism used by ECs to control the proinflammatory effects of the terminal complex SC5b-9. This complex is assembled in the fluid phase as a result of C activation and can be detected both in plasma and in the extravascular fluids as a cytolytically inactive complex (Bossi et al., 2009). We have previously shown that this inactive complex, despite its failure to cause cell damage, is still able to bind to ECs and stimulate the cells to express adhesion molecules and tissue factor (TF; Tedesco et al., 1997), to release chemokines (Dobrina et al., 2002), to induce transendothelial migration of PMN (Dobrina et al., 2002), and to promote vascular leakage (Bossi et al., 2004). Protection against the damaging effect of the assembling terminal C complex is provided by EC-bound C7, that acts as a trapping molecule for excess C5b6 complex released in the fluid phase under conditions of unrestricted C activation during acute phase response. Subsequent binding of C8 and C9 leads to the assembly of a cytolytically inactive membrane bound terminal complex (mTCC), that contains also S protein and clusterin and is cytolytically inactive. However, unlike soluble SC5b-9, mTCC fails to activate ECs, and in addition down-regulates cell activation induced by SC5b-9 modulating the expression of adhesion molecules, the release of IL-8, and EC permeability (Bossi et al., 2009). ECs normally utilize GPI-anchored molecule CD59 to neutralize the C-mediated cell lysis caused by the assembling MAC, but this C regulator has no effect on the damaging effect of the soluble complex SC5b-9 which can only be controlled by mTCC. Tight control of C activation by several regulators is particularly important at feto-placental site where direct C-mediated damage and indirect effect associated with excessive inflammation may impair fetal growth and possibly lead to miscarriage (Christiansen et al., 2006).
The importance of C regulation at the embryo implantation site is emphasized by the data obtained in murine models of pregnancy failure. Xu et al. (2000) made the original observation that Crry deficiency in utero leads to progressive embryonic lethality and Crry−/− embryos are surrounded by activated C3 fragments and PMN. Crry is expressed on fetal trophoblast cells and maternal decidua, where it controls deposition of C3 and C4 on the cell surface, preventing formation of the cytolytic MAC (Weigle et al., 1983; Molina et al., 1992; Kim et al., 1995). The critical role played by C activation products in causing fetal loss in Crry−/− mice is supported by the finding that the embryos are completely rescued from lethality and live pups are born at a normal Mendelian frequency when Crry+/− parents are intercrossed with C3−/− mice to generate C3−/−, Crry−/− embryos (Xu et al., 2000). This outcome is a genetic proof that Crry−/− embryos die in utero due to their inability to suppress C activation and tissue damage mediated by C3. The C regulator Crry has also been shown to control fetal resorption in a murine model of antibody-mediated anti-phospholipid syndrome (APS; Holers et al., 2002), as suggested by the ability of human IgG containing anti-phospholipid antibodies (aPL) injected into pregnant mice to cause fetal resorption in C sufficient animal, but not in C3 deficient mice. Protection against fetal loss was also observed in mice that received recombinant Crry-Ig to prevent C activation through the inhibition of C3 convertase.
There is some indication that C regulators contribute to protect the feto-placental unit also in human APS. Francis et al. (2006) reported an impaired endometrial expression of DAF/CD55 in patients with recurrent pregnancy loss associated with APS suggesting that the abnormal expression of this C regulator before conception may compromise implantation and predisposes to C-mediated pregnancy failure.
A protective role for C regulators has also been established in animal models of PE developed in CBA/J females mated to DBA/J males. These mice manifest many features of human PE, such as primigravidity, albuminuria, glomerular endotheliosis, increased sensitivity to angiotensin II and increased plasma leptin levels, correlated with bad pregnancy outcome (Ahmed et al., 2010). Administration of Crry to pregnant DBA/2 CBA/J mice helped to reduce the level of proteinuria, and blood urea nitrogen, fibrin deposition, and glomerular endotheliosis (Qing et al., 2011), supporting the concept that local C activation is deleterious to the developing fetus.
The importance of the C regulators in patients with PE has recently been underscored by Salmon et al. (2011), who performed a prospective study of 250 pregnant patients with SLE and/or APL antibodies to identify predictors of pregnancy outcome. They sequenced the genes encoding three C control proteins, MCP, factor H (CFH), and Factor I (CFI) and found heterozygous mutations in 7 out of 40 patients who developed PE. Based on these findings, they concluded that mutations in MCP and CFI predispose to the development of PE, as already observed in patients with atypical hemolytic uremic syndrome, and that dysregulation of C activation represents a risk factor for PE.
Role of C in Pathologic Pregnancy
It is commonly believed that C contributes to tissue damage in pathologic conditions through the destructive activity of MAC or the proinflammatory effect of C5a and other activation products including the non-cytolytic complex SC5b-9. Placenta is particularly susceptible to C-dependent damage because trophoblast exposure to maternal blood and extensive tissue remodeling in maternal decidua promote C activation during physiologic pregnancy. Unrestricted local activation of the system may overcome the control of CRPs resulting in pregnancy failure or poor pregnancy outcome.
Anti-Phospholipid Syndrome
Anti-phospholipid syndrome has received special attention in the last few years with the establishment of animal models that have revealed the involvement of C in the onset of the two main clinical manifestations of the syndrome represented by fetal loss and vascular thrombosis (Holers et al., 2002; Fischetti et al., 2005; Girardi et al., 2006). Pregnancy failure in the mouse model of APS has been attributed to the release of C5a and recruitment of PMN followed by TF-dependent promotion of coagulation as a result of C activation induced by APL (Girardi et al., 2003; Ritis et al., 2006; Redecha et al., 2007). The serum protein β2-glycoprotein I (β2GPI) represents the main target of these antibodies once bound to the cell surface of trophoblasts and ECs (Pierangeli et al., 2008; Meroni et al., 2011). Binding of β2GPI to trophoblasts and ECs in placenta occurs spontaneously, but its interaction with the endothelium of other tissues such as brain and gut requires priming with LPS (Agostinis et al., 2011a).
While the contribution of the C system to fetal loss and thrombosis in animal models is well established, the role played by C in mediating tissue damage in human APS is less clear, though this is suggested by the finding of C deposition in placental tissue from patients with aPL (Girardi et al., 2006). Analysis of placentae obtained from a large number of APS patients by Shamonki et al. (2007) revealed increased deposition of C4d in the cytoplasm of villous trophoblasts correlated with the histologic alterations of APS placentae. An essentially similar conclusion was reached by Cohen et al. (2011), who found diffuse C4d deposits on villous syncytiotrophoblasts of placentae from patients with SLE and/or APS suggestive of adverse pregnancy outcome. The relevance of C5b-9 deposits in APS patients is somewhat controversial since the local level of this complex was found to be lower in APS patients compared with that of the control group (Shamonki et al., 2007). In a recent prospective study on full-term placentae from patients with primary APS and normal control we were able to reveal marked C5b-9 deposits that were only weak or undetectable in the control group, although the presence of the complex was not always related to pregnancy outcome or therapy (Meroni et al., 2011).
Preeclampsia
This is a multifactorial pathological condition of pregnancy that affects 3%–5% of all pregnancies and is characterized by hypertension and proteinuria. The C system has been implicated in the development of PE placental alterations together with other pathogenic factors. Activation products of both early and late C components have been detected in the circulation of PE patients and the split product Bb has been proposed as a marker for high risk of PE (Lynch et al., 2008), though there is no general agreement on this issue (Derzsy et al., 2010). Deposits of C components including late components and their activation products responsible for promotion of inflammation and tissue damage have been documented in PE placenta, in particular in chorionic villi, in inter- and peri-villus fibrin surrounding syncytiotrophoblasts, and in maternal decidua (Sinha et al., 1984; Tedesco et al., 1990; Girardi et al., 2006). Despite the failure to detect increased activity of the MBL–MASP2 complex in the PE patients (Csuka et al., 2010), two activators of the lectin pathway (LP) H-ficolin and L-ficolin have been detected on syncytiotrophoblasts of PE placentae (Wang et al., 2007) suggesting an association of the placental abnormalities with the activation of this pathway. The involvement of LP of C activation is further supported by the finding that MBL-A, one of the two murine MBL proteins, is deposited on the implantation sites of CBA/J females mice mated with DBA/2 males, a abortion-prone mating combination recognized as a murine model of PE (Ahmed et al., 2010).
Besides inducing tissue damage, the C system has been shown to exert a physiologic role contributing to promote tissue remodeling in maternal decidua. This process is defective in PE characterized by the persistence of unremodeled spiral arteries and poor placentation (Redman and Sargent, 2010). Following the observation that C1q promotes trophoblast invasion of maternal decidua and that C1q deficiency is associated with impaired labyrinth development and decidual vessel remodeling in pregnant mice, Singh et al. (2011) have recently shown that pregnant C1q−/− deficient mice manifest the essential features of human PE including hypertension, albuminuria, endotheliosis, decreased placental vascular endothelial growth factor (VEGF), and elevated levels of soluble VEGF receptor 1 (sFlt-1). Our preliminary data showing failure of trophoblasts surrounding unremodelled spiral arteries in human PE decidua to synthesize and secrete C1q suggest that the observations in C1q−/− deficient mice may be relevant to human PE (Agostinis et al., 2011b).
Conclusion
The contribution of the C system to host defense and tissue damage mediated through the inflammatory process and cell cytotoxicity is well established. More recently, alternative functions on cells and tissues have been identified involving both native components and C activation products. The C system has been shown to contribute to tissue regeneration and brain development through the action of C3, C5, and their activation fragments. C1q is emerging as one of the C components able to exert functions independent on C activation. Several cell targets have been found to respond to C1q including cell of innate and acquired immunity as well as other cells not directly involved in the immune response. Most of the information available on this issue have been obtained in in vitro studies using purified C1q. The pitfall of these studies is that C1q detected in plasma and in extravascular fluid is bound to C1r and C1s to form a complex. Cells synthesizing C1q at tissue level represent a source of free C component that may exert alternative functions. This is the case of trophoblasts and DECs that secrete C1q at the embryo implantation site and contribute to its widespread distribution in decidual stroma. C1q has been shown to play a critical role in placental development favoring interaction between trophoblasts and DECs and promoting EVT migration. The relevance of these in vitro effects is underscored by the findings of impaired labyrinth development and decidual vessel remodeling in C1q−/− pregnant mice that have important implications in pregnancy disorders such as PE associated with defective trophoblast invasion.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the European NoE “EMBIC” within FP6 (contract number LSHN-CT-2004-512040) and by a research grant of the Ministry of Health (Ricerca Finalizzata RC41/08 e RC 01/09), Italian Association of Cancer Research (AIRC), Fondazione Casali – Trieste, and Italian Ministry of University and Research (PRIN2009 MFXE7L_004).
References
Abrahams, V. M., Kim, Y. M., Straszewski, S. L., Romero, R., and Mor, G. (2004). Macrophages and apoptotic cell clearance during pregnancy. Am. J. Reprod. Immunol. 51, 275–282.
Agostinis, C., Biffi, S., Garrovo, C., Durigutto, P., Lorenzon, A., Bek, A., Bulla, R., Grossi, C., Borghi, M. O., Meroni, P., and Tedesco, F. (2011a). In vivo distribution of beta2 glycoprotein I under various pathophysiologic conditions. Blood 118, 4231–4238.
Agostinis, C., Bulla, R., Tripodo, C. F. D. S., Tonon, M., Spessotto, P., Botto, M., and Tedesco, F. (2011b). “Preeclampsia is associated with defective production of C1q by invasive trophoblast,” in XIII European Meeting on Complement in Human Disease, Vol. 48, eds J. A. Villadangos and M. R. Daha (Leiden: Elsevier), 1678–1679.
Agostinis, C., Bulla, R., Tripodo, C., Gismondi, A., Stabile, H., Bossi, F., Guarnotta, C., Garlanda, C., De Seta, F., Spessotto, P., Santoni, A., Ghebrehiwet, B., Girardi, G., and Tedesco, F. (2010). An alternative role of C1q in cell migration, and tissue remodeling: contribution to trophoblast invasion, and placental development. J. Immunol. 185, 4420–4429.
Ahmed, A., Singh, J., Khan, Y., Seshan, S. V., and Girardi, G. (2010). A new mouse model to explore therapies for preeclampsia. PLoS ONE 5, e13663. doi:10.1371/journal.pone.0013663
Aluvihare, V. R., Kallikourdis, M., and Betz, A. G. (2004). Regulatory T cells mediate maternal tolerance to the fetus. Nat. Immunol. 5, 266–271.
Amarante-Paffaro, A., Queiroz, G. S., Correa, S. T., Spira, B., and Bevilacqua, E. (2004). Phagocytosis as a potential mechanism for microbial defense of mouse placental trophoblast cells. Reproduction 128, 207–218.
Andoh, A., Fujiyama, Y., Sakumoto, H., Uchihara, H., Kimura, T., Koyama, S., and Bamba, T. (1998). Detection of complement C3 and factor B gene expression in normal colorectal mucosa, adenomas and carcinomas. Clin. Exp. Immunol. 111, 477–483.
Ashkar, A. A., Di Santo, J. P., and Croy, B. A. (2000). Interferon gamma contributes to initiation of uterine vascular modification, decidual integrity, and uterine natural killer cell maturation during normal murine pregnancy. J. Exp. Med. 192, 259–270.
Bossi, F., Fischetti, F., Pellis, V., Bulla, R., Ferrero, E., Mollnes, T. E., Regoli, D., and Tedesco, F. (2004). Platelet-activating factor and kinin-dependent vascular leakage as a novel functional activity of the soluble terminal complement complex. J. Immunol. 173, 6921–6927.
Bossi, F., Rizzi, L., Bulla, R., Debeus, A., Tripodo, C., Picotti, P., Betto, E., Macor, P., Pucillo, C., Wurzner, R., and Tedesco, F. (2009). C7 is expressed on endothelial cells as a trap for the assembling terminal complement complex and may exert anti-inflammatory function. Blood 113, 3640–3648.
Brooimans, R. A., Stegmann, A. P., van Dorp, W. T., van der Ark, A. A., van der Woude, F. J., van Es, L. A., and Daha, M. R. (1991). Interleukin 2 mediates stimulation of complement C3 biosynthesis in human proximal tubular epithelial cells. J. Clin. Invest. 88, 379–384.
Bulla, R., Agostinis, C., Bossi, F., Rizzi, L., Debeus, A., Tripodo, C., Radillo, O., De Seta, F., Ghebrehiwet, B., and Tedesco, F. (2008). Decidual endothelial cells express surface-bound C1q as a molecular bridge between endovascular trophoblast and decidual endothelium. Mol. Immunol. 45, 2629–2640.
Bulla, R., Bossi, F., Agostinis, C., Radillo, O., Colombo, F., De Seta, F., and Tedesco, F. (2009). Complement production by trophoblast cells at the feto-maternal interface. J. Reprod. Immunol. 82, 119–125.
Bulla, R., Bossi, F., Radillo, O., de Seta, F., and Tedesco, F. (2003). Placental trophoblast and endothelial cells as target of maternal immune response. Autoimmunity 36, 11–18.
Bulla, R., Villa, A., Bossi, F., Cassetti, A., Radillo, O., Spessotto, P., De Seta, F., Guaschino, S., and Tedesco, F. (2005). VE-cadherin is a critical molecule for trophoblast-endothelial cell interaction in decidual spiral arteries. Exp. Cell Res. 303, 101–113.
Christiansen, O. B., Nielsen, H. S., and Kolte, A. M. (2006). Inflammation and miscarriage. Semin. Fetal Neonatal Med. 11, 302–308.
Cohen, D., Buurma, A., Goemaere, N. N., Girardi, G., le Cessie, S., Scherjon, S., Bloemenkamp, K. W., de Heer, E., Bruijn, J. A., and Bajema, I. M. (2011). Classical complement activation as a footprint for murine and human antiphospholipid antibody-induced fetal loss. J. Pathol. 225, 502–511.
Colten, H. R., and Garnier, G. (1998). “Regulation of the complement system,” in The Human Complement System in Health and Disease, eds J. E. Volanakis and M. M. Frank (New York: Marcel Dekker, INC.), 217–240.
Csuka, D., Molvarec, A., Derzsy, Z., Varga, L., Fust, G., Rigo, J. Jr., and Prohaszka, Z. (2010). Functional analysis of the mannose-binding lectin complement pathway in normal pregnancy, and preeclampsia. J. Reprod. Immunol. 87, 90–96.
Derzsy, Z., Prohaszka, Z., Rigo, J. Jr., Fust, G., and Molvarec, A. (2010). Activation of the complement system in normal pregnancy and preeclampsia. Mol. Immunol. 47, 1500–1506.
Dobrina, A., Pausa, M., Fischetti, F., Bulla, R., Vecile, E., Ferrero, E., Mantovani, A., and Tedesco, F. (2002). Cytolytically inactive terminal complement complex causes transendothelial migration of polymorphonuclear leukocytes in vitro and in vivo. Blood 99, 185–192.
Dovezenski, N., Billetta, R., and Gigli, I. (1992). Expression and localization of proteins of the complement system in human skin. J. Clin. Invest. 90, 2000–2012.
Fest, S., Aldo, P. B., Abrahams, V. M., Visintin, I., Alvero, A., Chen, R., Chavez, S. L., Romero, R., and Mor, G. (2007). Trophoblast-macrophage interactions: a regulatory network for the protection of pregnancy. Am. J. Reprod. Immunol. 57, 55–66.
Fischetti, F., Durigutto, P., Pellis, V., Debeus, A., Macor, P., Bulla, R., Bossi, F., Ziller, F., Sblattero, D., Meroni, P., and Tedesco, F. (2005). Thrombus formation induced by antibodies to beta2-glycoprotein I is complement dependent and requires a priming factor. Blood 106, 2340–2346.
Fischetti, F., and Tedesco, F. (2006). Cross-talk between the complement system and endothelial cells in physiologic conditions and in vascular diseases. Autoimmunity 39, 417–428.
Francis, J., Rai, R., Sebire, N. J., El-Gaddal, S., Fernandes, M. S., Jindal, P., Lokugamage, A., Regan, L., and Brosens, J. J. (2006). Impaired expression of endometrial differentiation markers and complement regulatory proteins in patients with recurrent pregnancy loss associated with antiphospholipid syndrome. Mol. Hum. Reprod. 12, 435–442.
Gagioti, S., Scavone, C., and Bevilacqua, E. (2000). Participation of the mouse implanting trophoblast in nitric oxide production during pregnancy. Biol. Reprod. 62, 260–268.
Garred, P., Hetland, G., Mollnes, T. E., and Stoervold, G. (1990). Synthesis of C3, C5, C6, C7, C8, and C9 by human fibroblasts. Scand. J. Immunol. 32, 555–560.
Girardi, G., Berman, J., Redecha, P., Spruce, L., Thurman, J. M., Kraus, D., Hollmann, T. J., Casali, P., Caroll, M. C., Wetsel, R. A., Lambris, J. D., Holers, V. M., and Salmon, J. E. (2003). Complement C5a receptors and neutrophils mediate fetal injury in the antiphospholipid syndrome. J. Clin. Invest. 112, 1644–1654.
Girardi, G., Bulla, R., Salmon, J. E., and Tedesco, F. (2006). The complement system in the pathophysiology of pregnancy. Mol. Immunol. 43, 68–77.
Goldberg, M., Luknar-Gabor, N., Keidar, R., and Katz, Y. (2007). Synthesis of complement proteins in the human chorion is differentially regulated by cytokines. Mol. Immunol. 44, 1737–1742.
Greenwood, J. D., Minhas, K., di Santo, J. P., Makita, M., Kiso, Y., and Croy, B. A. (2000). Ultrastructural studies of implantation sites from mice deficient in uterine natural killer cells. Placenta 21, 693–702.
Griffiths, M. R., Gasque, P., and Neal, J. W. (2009). The multiple roles of the innate immune system in the regulation of apoptosis and inflammation in the brain. J. Neuropathol. Exp. Neurol. 68, 217–226.
Guimond, M. J., Wang, B., and Croy, B. A. (1998). Engraftment of bone marrow from severe combined immunodeficient (SCID) mice reverses the reproductive deficits in natural killer cell-deficient tg epsilon 26 mice. J. Exp. Med. 187, 217–223.
Gulati, P., Guc, D., Lemercier, C., Lappin, D., and Whaley, K. (1994). Expression of the components and regulatory proteins of the classical pathway of complement in normal and diseased synovium. Rheumatol. Int. 14, 13–19.
Hanna, J., Goldman-Wohl, D., Hamani, Y., Avraham, I., Greenfield, C., Natanson-Yaron, S., Prus, D., Cohen-Daniel, L., Arnon, T. I., Manaster, I., Gazit, R., Yutkin, V., Benharroch, D., Porgador, A., Keshet, E., Yagel, S., and Mandelboim, O. (2006). Decidual NK cells regulate key developmental processes at the human fetal-maternal interface. Nat. Med. 12, 1065–1074.
Hanna, J., Wald, O., Goldman-Wohl, D., Prus, D., Markel, G., Gazit, R., Katz, G., Haimov-Kochman, R., Fujii, N., Yagel, S., Peled, A., and Mandelboim, O. (2003). CXCL12 expression by invasive trophoblasts induces the specific migration of CD16- human natural killer cells. Blood 102, 1569–1577.
Harris, L. K., Smith, S. D., Keogh, R. J., Jones, R. L., Baker, P. N., Knofler, M., Cartwright, J. E., Whitley, G. S., and Aplin, J. D. (2010). Trophoblast- and vascular smooth muscle cell-derived MMP-12 mediates elastolysis during uterine spiral artery remodeling. Am. J. Pathol. 177, 2103–2115.
Holers, V. M., Girardi, G., Mo, L., Guthridge, J. M., Molina, H., Pierangeli, S. S., Espinola, R., Xiaowei, L. E., Mao, D., Vialpando, C. G., and Salmon, J. E. (2002). Complement C3 activation is required for antiphospholipid antibody-induced fetal loss. J. Exp. Med. 195, 211–220.
Holmes, C. H., Simpson, K. L., Okada, H., Okada, N., Wainwright, S. D., Purcell, D. F., and Houlihan, J. M. (1992). Complement regulatory proteins at the feto-maternal interface during human placental development: distribution of CD59 by comparison with membrane cofactor protein (CD46) and decay accelerating factor (CD55). Eur. J. Immunol. 22, 1579–1585.
Holmes, C. H., Simpson, K. L., Wainwright, S. D., Tate, C. G., Houlihan, J. M., Sawyer, I. H., Rogers, I. P., Spring, F. A., Anstee, D. J., and Tanner, M. J. (1990). Preferential expression of the complement regulatory protein decay accelerating factor at the fetomaternal interface during human pregnancy. J. Immunol. 144, 3099–3105.
Hsi, B. L., Hunt, J. S., and Atkinson, J. P. (1991). Differential expression of complement regulatory proteins on subpopulations of human trophoblast cells. J. Reprod. Immunol. 19, 209–223.
Hu, Y., Dutz, J. P., MacCalman, C. D., Yong, P., Tan, R., and von Dadelszen, P. (2006). Decidual NK cells alter in vitro first trimester extravillous cytotrophoblast migration: a role for IFN-gamma. J. Immunol. 177, 8522–8530.
Johnson, E., and Hetland, G. (1988). Mononuclear phagocytes have the potential to synthesize the complete functional complement system. Scand. J. Immunol. 27, 489–493.
Johnson, S. A., Pasinetti, G. M., and Finch, C. E. (1994). Expression of complement C1qB and C4 mRNAs during rat brain development. Brain Res. Dev. Brain Res. 80, 163–174.
Khirwadkar, K., Zilow, G., Oppermann, M., Kabelitz, D., and Rother, K. (1993). Interleukin-4 augments production of the third complement component by the alveolar epithelial cell line A549. Int. Arch. Allergy Immunol. 100, 35–41.
Kim, Y. U., Kinoshita, T., Molina, H., Hourcade, D., Seya, T., Wagner, L. M., and Holers, V. M. (1995). Mouse complement regulatory protein Crry/p65 uses the specific mechanisms of both human decay-accelerating factor and membrane cofactor protein. J. Exp. Med. 181, 151–159.
Kimura, Y., Madhavan, M., Call, M. K., Santiago, W., Tsonis, P. A., Lambris, J. D., and Del Rio-Tsonis, K. (2003). Expression of complement 3 and complement 5 in newt limb and lens regeneration. J. Immunol. 170, 2331–2339.
Langeggen, H., Pausa, M., Johnson, E., Casarsa, C., and Tedesco, F. (2000). The endothelium is an extrahepatic site of synthesis of the seventh component of the complement system. Clin. Exp. Immunol. 121, 69–76.
Lash, G. E., Otun, H. A., Innes, B. A., Kirkley, M., De Oliveira, L., Searle, R. F., Robson, S. C., and Bulmer, J. N. (2006). Interferon-gamma inhibits extravillous trophoblast cell invasion by a mechanism that involves both changes in apoptosis and protease levels. FASEB J. 20, 2512–2518.
Laufer, J., Katz, Y., and Passwell, J. H. (2001). Extrahepatic synthesis of complement proteins in inflammation. Mol. Immunol. 38, 221–229.
Le Bitoux, M. A., and Stamenkovic, I. (2008). Tumor-host interactions: the role of inflammation. Histochem. Cell Biol. 130, 1079–1090.
Lyall, F., Bulmer, J. N., Duffie, E., Cousins, F., Theriault, A., and Robson, S. C. (2001). Human trophoblast invasion and spiral artery transformation: the role of PECAM-1 in normal pregnancy, preeclampsia, and fetal growth restriction. Am. J. Pathol. 158, 1713–1721.
Lynch, A. M., Murphy, J. R., Byers, T., Gibbs, R. S., Neville, M. C., Giclas, P. C., Salmon, J. E., and Holers, V. M. (2008). Alternative complement pathway activation fragment Bb in early pregnancy as a predictor of preeclampsia. Am. J. Obstet. Gynecol. 198, e381–e389.
Meroni, P. L., Borghi, M. O., Raschi, E., and Tedesco, F. (2011). Pathogenesis of antiphospholipid syndrome: understanding the antibodies. Nat. Rev. Rheumatol. 7, 330–339.
Molina, H., Wong, W., Kinoshita, T., Brenner, C., Foley, S., and Holers, V. M. (1992). Distinct receptor and regulatory properties of recombinant mouse complement receptor 1 (CR1) and Crry, the two genetic homologues of human CR1. J. Exp. Med. 175, 121–129.
Mor, G., Cardenas, I., Abrahams, V., and Guller, S. (2011). Inflammation and pregnancy: the role of the immune system at the implantation site. Ann. N. Y. Acad. Sci. 1221, 80–87.
Morgan, B. P., and Gasque, P. (1997). Extrahepatic complement biosynthesis: where, when and why? Clin. Exp. Immunol. 107, 1–7.
Morris, K. M., Aden, D. P., Knowles, B. B., and Colten, H. R. (1982). Complement biosynthesis by the human hepatoma-derived cell line HepG2. J. Clin. Invest. 70, 906–913.
Pattrick, M., Luckett, J., Yue, L., and Stover, C. (2009). Dual role of complement in adipose tissue. Mol. Immunol. 46, 755–760.
Peerschke, E. I., Smyth, S. S., Teng, E. I., Dalzell, M., and Ghebrehiwet, B. (1996). Human umbilical vein endothelial cells possess binding sites for the globular domain of C1q. J. Immunol. 157, 4154–4158.
Pellis, V., De Seta, F., Crovella, S., Bossi, F., Bulla, R., Guaschino, S., Radillo, O., Garred, P., and Tedesco, F. (2005). Mannose binding lectin and C3 act as recognition molecules for infectious agents in the vagina. Clin. Exp. Immunol. 139, 120–126.
Pepper, M. S. (2001). Role of the matrix metalloproteinase and plasminogen activator-plasmin systems in angiogenesis. Arterioscler. Thromb. Vasc. Biol. 21, 1104–1117.
Pierangeli, S. S., Chen, P. P., Raschi, E., Scurati, S., Grossi, C., Borghi, M. O., Palomo, I., Harris, E. N., and Meroni, P. L. (2008). Antiphospholipid antibodies and the antiphospholipid syndrome: pathogenic mechanisms. Semin. Thromb. Hemost. 34, 236–250.
Pijnenborg, R., Vercruysse, L., and Hanssens, M. (2006). The uterine spiral arteries in human pregnancy: facts and controversies. Placenta 27, 939–958.
Plaisier, M. (2011). Decidualisation and angiogenesis. Best Pract. Res. Clin. Obstet. Gynaecol. 25, 259–271.
Pratt, J. R., Basheer, S. A., and Sacks, S. H. (2002). Local synthesis of complement component C3 regulates acute renal transplant rejection. Nat. Med. 8, 582–587.
Qing, X., Redecha, P. B., Burmeister, M. A., Tomlinson, S., D’Agati, V. D., Davisson, R. L., and Salmon, J. E. (2011). Targeted inhibition of complement activation prevents features of preeclampsia in mice. Kidney Int. 79, 331–339.
Redecha, P., Tilley, R., Tencati, M., Salmon, J. E., Kirchhofer, D., Mackman, N., and Girardi, G. (2007). Tissue factor: a link between C5a and neutrophil activation in antiphospholipid antibody induced fetal injury. Blood 110, 2423–2431.
Redman, C. W., and Sargent, I. L. (2010). Immunology of pre-eclampsia. Am. J. Reprod. Immunol. 63, 534–543.
Ritis, K., Doumas, M., Mastellos, D., Micheli, A., Giaglis, S., Magotti, P., Rafail, S., Kartalis, G., Sideras, P., and Lambris, J. D. (2006). A novel C5a receptor-tissue factor cross-talk in neutrophils links innate immunity to coagulation pathways. J. Immunol. 177, 4794–4802.
Salmon, J. E., Heuser, C., Triebwasser, M., Liszewski, M. K., Kavanagh, D., Roumenina, L., Branch, D. W., Goodship, T., Fremeaux-Bacchi, V., and Atkinson, J. P. (2011). Mutations in complement regulatory proteins predispose to preeclampsia: a genetic analysis of the PROMISSE cohort. PLoS Med. 8, e1001013. doi:10.1371/journal.pmed.1001013
Shakhawat, A., Shaikly, V., Elzatma, E., Mavrakos, E., Jabeen, A., and Fernandez, N. (2010). Interaction between HLA-G and monocyte/macrophages in human pregnancy. J. Reprod. Immunol. 85, 40–46.
Shamonki, J. M., Salmon, J. E., Hyjek, E., and Baergen, R. N. (2007). Excessive complement activation is associated with placental injury in patients with antiphospholipid antibodies. Am. J. Obstet. Gynecol. 196, e161–e165.
Sica, A., and Bronte, V. (2007). Altered macrophage differentiation and immune dysfunction in tumor development. J. Clin. Invest. 117, 1155–1166.
Singh, J., Ahmed, A., and Girardi, G. (2011). Role of complement component C1q in the onset of preeclampsia in mice. Hypertension 58, 716–724.
Sinha, D., Wells, M., and Faulk, W. P. (1984). Immunological studies of human placentae: complement components in pre-eclamptic chorionic villi. Clin. Exp. Immunol. 56, 175–184.
Smith, S. D., Dunk, C. E., Aplin, J. D., Harris, L. K., and Jones, R. L. (2009). Evidence for immune cell involvement in decidual spiral arteriole remodeling in early human pregnancy. Am. J. Pathol. 174, 1959–1971.
Strey, C. W., Markiewski, M., Mastellos, D., Tudoran, R., Spruce, L. A., Greenbaum, L. E., and Lambris, J. D. (2003). The proinflammatory mediators C3a and C5a are essential for liver regeneration. J. Exp. Med. 198, 913–923.
Tedesco, F., Narchi, G., Radillo, O., Meri, S., Ferrone, S., and Betterle, C. (1993). Susceptibility of human trophoblast to killing by human complement and the role of the complement regulatory proteins. J. Immunol. 151, 1562–1570.
Tedesco, F., Pausa, M., Nardon, E., Introna, M., Mantovani, A., and Dobrina, A. (1997). The cytolytically inactive terminal complement complex activates endothelial cells to express adhesion molecules and tissue factor procoagulant activity. J. Exp. Med. 185, 1619–1627.
Tedesco, F., Radillo, O., Candussi, G., Nazzaro, A., Mollnes, T. E., and Pecorari, D. (1990). Immunohistochemical detection of terminal complement complex and S protein in normal and pre-eclamptic placentae. Clin. Exp. Immunol. 80, 236–240.
Tilburgs, T., Schonkeren, D., Eikmans, M., Nagtzaam, N. M., Datema, G., Swings, G. M., Prins, F., van Lith, J. M., van der Mast, B. J., Roelen, D. L., Scherjon, S. A., and Claas, F. H. (2010). Human decidual tissue contains differentiated CD8+ effector-memory T cells with unique properties. J. Immunol. 185, 4470–4477.
Vacca, P., Cantoni, C., Vitale, M., Prato, C., Canegallo, F., Fenoglio, D., Ragni, N., Moretta, L., and Mingari, M. C. (2010). Crosstalk between decidual NK and CD14+ myelomonocytic cells results in induction of Tregs and immunosuppression. Proc. Natl. Acad. Sci. U.S.A. 107, 11918–11923.
Wang, C. C., Yim, K. W., Poon, T. C., Choy, K. W., Chu, C. Y., Lui, W. T., Lau, T. K., Rogers, M. S., and Leung, T. N. (2007). Innate immune response by ficolin binding in apoptotic placenta is associated with the clinical syndrome of preeclampsia. Clin. Chem. 53, 42–52.
Weigle, W. O., Goodman, M. G., Morgan, E. L., and Hugli, T. E. (1983). Regulation of immune response by components of the complement cascade and their activated fragments. Springer Semin. Immunopathol. 6, 173–194.
Wu, X., Jin, L. P., Yuan, M. M., Zhu, Y., Wang, M. Y., and Li, D. J. (2005). Human first-trimester trophoblast cells recruit CD56brightCD16- NK cells into decidua by way of expressing and secreting of CXCL12/stromal cell-derived factor 1. J. Immunol. 175, 61–68.
Keywords: complement, C1q, C7, inflammation, placenta, pregnancy disorders
Citation: Bulla R Bossi F and Tedesco F (2012) The complement system at the embryo implantation site: friend or foe? Front. Immun. 3:55. doi: 10.3389/fimmu.2012.00055
Received: 21 December 2011; Paper pending published: 12 January 2012;
Accepted: 02 March 2012; Published online: 19 March 2012.
Edited by:
Berhane Ghebrehiwet, Stonybrook University, USAReviewed by:
Berhane Ghebrehiwet, Stonybrook University, USAMin Wu, University of North Dakota, USA
Copyright: © 2012 Bulla, Bossi and Tedesco. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: R. Bulla, Department of Life Sciences, University of Trieste, Via Valerio 28, 34127 Trieste, Italy. e-mail:cmJ1bGxhQHVuaXRzLml0