 Dennis J. Horvath Jr1
Dennis J. Horvath Jr1
- 1 Department of Medicine, Vanderbilt University School of Medicine, Nashville, TN, USA
- 2 Department of Pathology, Vanderbilt University School of Medicine, Nashville, TN, USA
- 3 Department of Veteran’s Affairs, Veterans Affairs Tennessee Valley Healthcare System, Nashville, TN, USA
The immune response to Helicobacter pylori involves a mixed T helper-1, T helper-2, and T helper-17 response. It has been suggested that T helper cells contribute to the gastric inflammatory response during infection, and that T helper 1 (Th1) and T helper 17 (Th17) subsets may be required for control of H. pylori colonization in the stomach. The relative contributions of these subsets to gastritis and control of infection are still under investigation. IL-23 plays a role in stabilizing and expanding Th17 cell cytokine expression. Expression of IL-23, which is induced in dendritic cells and macrophages following co-culture with H. pylori, has also been reported to increase during H. pylori infection in humans and animal models. To investigate the role of IL-23 in H. pylori, we infected IL-23p19 deficient mice (IL-23−/−) and wild-type littermates with H. pylori strain SS1. At various time points post-infection, we assessed colonization, gastric inflammation, and cytokine profiles in the gastric tissue. Specifically, H. pylori-infected IL-23−/− mice have higher levels of H. pylori in their stomachs, significantly less chronic gastritis, and reduced expression of IL-17 and IFNγ compared to H. pylori-infected wild-type mice. While many of these differences were significant, the H. pylori infected IL-23−/− had mild increases in our measurements of disease severity. Our results indicate that IL-23 plays a role in the activation of the immune response and induction of gastritis in response to H. pylori by contributing to the control of infection and severity of gastritis.
Introduction
The hallmark of Helicobacter pylori infection is the development of gastric inflammation, referred to as gastritis. It is believed that all H. pylori infected individuals develop gastritis. In animal models of disease, the development of gastritis is dependent on the presence of T lymphocytes (T cells). Severe combined immunodeficiency mice, which lack B and T cells, require adoptive transfer of T cells to develop gastritis (Eaton et al., 1999, 2001).
T cells play a decisive role in initiating and shaping pathological responses in many tissues. Classical examples of T-cell mediated diseases are inflammatory bowel disease (IBD), diabetes, psoriasis, rheumatoid arthritis, and multiple sclerosis. Interplay, between T cells and other immune and non-immune cells, is critical in driving pathologic processes, and T cell-derived cytokines are essential mediators of this cross-talk. CD4+ T cells were traditionally categorized as Th1 or Th2, but over the last decade, research has characterized a number of new distinct CD4+ T cell subsets including Th17 cells, T regulatory cells, T follicular helper cells (Tfh), and T helper-9 cells. A fundamental function of T helper cells is to provide “help” to B cells and regulate their proliferation and immunoglobulin class switching. But CD4+ cells do more than just help B cells; their cytokine production also regulates antimicrobial/antiviral responses. For example, IFNγ activates macrophages and stimulates CD8+ cytotoxic responses, while IL-17 induces neutrophil migration for clearance of extracellular pathogens, such as H. pylori. TGF-β, which can be produced by natural Tregs, also controls inflammation by inducing Foxp3 expression in T helper cells stimulating the production of inducible Tregs.
The development of the Th17 lineage may be important for the control of H. pylori infection and the development of gastritis during H. pylori infection. Induction of Th17 cells is attributed to several H. pylori factors including Urease B (Zhang et al., 2011). Several studies have indicated that H. pylori stimulated macrophages produce IL-6, TGF-β, IL-23, and CCL20 (Meyer et al., 2000; Mandell et al., 2004; Odenbreit et al., 2006; Zhuang et al., 2011).
The gastritis associated with H. pylori infection involves both acute and chronic inflammation suggesting that T cell derived cytokines such as those produced by Th17 cells may be responsible for the ongoing acute inflammatory response associated with the recruitment of neutrophils to the stomach. Th17 lineage develops in a pathway independent from Th1 and Th2 differentiation (Bettelli et al., 2008). A hallmark of Th17 cells is the production of IL-17, IL-21, and IL-22, which are pro-inflammatory cytokines. Th17 cells are known to have an important role in a growing list of immune-mediated diseases, including IBD, experimental autoimmune encephalopathy (EAE), and collagen-induced arthritis (CIA; reviewed in McKenzie et al., 2006; Furuzawa-Carballeda et al., 2007; Sarkar et al., 2007). In addition, in several animal models of infectious diseases, the IL-23/IL-17 axis promotes cell migration to the site of infection to kill microorganisms and activates the bactericidal activity of macrophages (reviewed in Curtis and Way, 2009). The host’s ability to control the proliferation of Klebsiella pneumoniae, Citrobacter rodentium, Mycoplasma pneumoniae, Bordetella pertussis, Pseudomonas aeruginosa, Porphyromonas gingivalis, Escherichia coli, Listeria monocytogenes, and Salmonella enterica is at least partially dependent on IL-23 and IL-17 (Ye et al., 2001; Mangan et al., 2006; Chen et al., 2007a; Dubin and Kolls, 2007; Shibata et al., 2007; Wu et al., 2007; Yu et al., 2007; Hamada et al., 2008; Schulz et al., 2008a). Several studies from infection models in IL-23p19 deficient mice demonstrate IL-23 contributes to deficient neutrophil recruitment (Cruz et al., 2006; Meeks et al., 2009), reduction in IL-17 (Happel et al., 2005; Khader et al., 2005; Chackerian et al., 2006; Kleinschek et al., 2006; Schulz et al., 2008b), increase in both disease susceptibility and organism burden in some infectious models (Happel et al., 2005; Khader et al., 2005; Meeks et al., 2009), whereas other animal models exhibit no differences in neither disease susceptibility nor organism burden (Khader et al., 2005; Chackerian et al., 2006; Schulz et al., 2008b). IL-17 activates gastric epithelial cells to produce neutrophil attracting chemokines (Algood et al., 2009). In some studies, IL-17 is required for control of bacterial load, while in other studies it is only required for recruitment of neutrophils (Shiomi et al., 2008; Algood et al., 2009; DeLyria et al., 2009; Otani et al., 2009).
The development of a Th17 response is promoted by TGF-β, IL-1β, and IL-6 and further expanded and activated by IL-23. IL-23 is a heterodimer comprised of an IL-23p19 subunit and an IL-12p40 subunit. It is produced by a number of immune cells including macrophages, dendritic cells, and neutrophils. Broadly speaking, the function of IL-23 resembles the function of IL-12 in linking innate responses and adaptive immunity. IL-23 regulates the secretion of IL-17 through at STAT3 dependent pathway (Caruso et al., 2008). IL-23 induced activation of STAT3 leads to direct binding of phosphorylated STAT3 to IL-17A and IL-17F promoters (Chen et al., 2006). Moreover, STAT3 up-regulates the expression of Retinoic Acid Receptor-Related Orphan Receptor Gamma-T (ROR-γ), a Th17 specific transcriptional regulator that is critical for the expression of two members of IL-17A and IL-17F (Chen et al., 2007b; Laurence and O’Shea, 2007; Yang et al., 2007).
Although IL-23 is a marker of H. pylori infection (Caruso et al., 2008), levels of IL-23 alone do not correlate with chronic gastritis, duodenal, or gastric ulcers (Koussoulas et al., 2009). However, positive correlations were found between levels of IL-23 and the degree of infiltration of neutrophils and monocytes in patients with H. pylori infection, and the kinetics of IL-23 expression correlates with those of IL-1β (Koussoulas et al., 2009).
In the current study, we investigated the role of IL-23p19 on the development of the Th17 and Th1 adaptive immune responses during H. pylori infection in a mouse model of infection. Moreover, this study investigates the requirement of IL-23p19 in the development of an effective immune response (control of H. pylori colonization) and the development of gastritis. Our results indicate that IL-23 contributes to the control of H. pylori infection and severity of gastritis, playing a role in the activation of the immune response and induction of gastritis in response to H. pylori.
Materials and Methods
Animals
Male and female IL-23+/− mice, on a mixed background (Strain name B6; 129S5-Il-23atm1Lex/Mmcd) were obtained from the NIH Consortium (Stock number 011725-UCD MMRC, UC Davis) for the establishment of a breeding colony. The targeted mouse gene is IL-23a (interleukin 23, alpha subunit p19), an ortholog of human IL-23A. Other aliases of IL-23 include P19, SGRF, IL-23, IL-23A, IL-23P19, interleukin 23p19 subunit, and JKA3 induced upon T-cell activation. Coding exon 1 was targeted by homologous recombination. The IL23+/− breeding pairs tested negative for intestinal Helicobacter. Helicobacter-free male mice, 8–10 weeks old, were used in all experiments. The Vanderbilt University Institutional Animal Care and Use Committee approved all animal protocols used in this study. Feces from sentinel mice housed in the same room were routinely tested by PCR for intestinal Helicobacter, pinworms, mouse parvovirus, and several other murine pathogens, and consistently tested negative for each of these infections.
Culture of H. pylori
A mouse-passaged derivative of H. pylori strain SS1 was used in all animal experiments. For cell culture experiments several strains were used, including PM-SS1, SS1, 60190, X47, 26695, and 7.13. Bacteria were grown on trypticase soy agar (TSA) plates containing 5% sheep blood. Alternatively, bacteria were grown in Brucella broth containing 10% heat-inactivated fetal bovine serum (FBS) and 10 μg/ml vancomycin. Cultures were grown at 37°C in either room air supplemented with 5% CO2, or under microaerobic conditions generated by a CampyPak Plus* Hydrogen + CO2 with Integral Palladium Catalyst (BD).
Bone Marrow Preparation
After CO2 euthanasia and cervical dislocation, femurs, and tibiae from male mice strains were separated from muscle tissue. Cleaned bones were collected in 50 ml conical tubes on ice with ice-cold PBS without calcium and magnesium (Cellgro by Mediatech, Manassas, VA, USA). Bones were washed by inverting the tube several times with 1X PBS and then followed by a wash with ice-cold RPMI 1640 media (Mediatech). Bone marrow was flushed from the bones with a 21G needle with ice-cold RPMI 1640. Aggregates within the bone marrow suspension were disrupted by pipetting until single cell suspensions were obtained. Single cell bone marrow suspension was passed through a sterile 70 μm nylon cell strainer (Fisherbrand, Pittsburgh, PA, USA) to remove remaining cellular debris. Bone marrow cells were pelleted by centrifugation, red blood cells were removed using water lysis, and bone marrow cells were then diluted 5× with ice-cold RPMI 1640, passed through a nylon strainer and then centrifuged. Viable bone marrow cells were enumerated and were adjusted to a concentration of 1 × 106 cells/ml in complete RPMI 1640 medium supplemented with 5% heat-inactivated FBS (Atlanta Biologicals, Miami, FL, USA), 55 μM 2-mercaptoethanol, 1 mM sodium pyruvate (GIBCO), and 20 ng/ml mouse granulocyte-macrophage colony stimulating factor (GM-CSF) premium grade (Miltenyi Biotec, Boston, MA, USA). Cells were cultured in T75 tissue culture flasks at 37°C with 5% atmospheric CO2. After 3 days of culture, 10 ml of fresh complete medium containing GM-CSF were added to the flasks. On day 6 of culture, cells were resuspended by repeated pipetting and after washing, viable cells were enumerated as previously described.
Positive Selection of CD11c Bone Marrow-Derived Dendritic Cells by AutoMACS
Bone marrow cells were magnetically labeled with CD11c Microbeads (Miltenyi Biotec). CD11c+ bone marrow derived dendritic cells (BMDCs) were positively selected using the positive selection sensitive program on the autoMACS machine (Miltenyi Biotec) according to manufacturer’s protocol.
Bone Marrow-Derived Dendritic Cell Co-Culture with H. pylori Strains
Bone marrow derived dendritic cells were centrifuged at 200 × g for 10 min at 4°C, enumerated, and adjusted to a concentration of 2 × 106 cells/ml in RPMI 1640 media supplemented with 5% heat-inactivated FBS, 55 μM 2-mercaptoethanol, and 1 mM sodium pyruvate. One milliliter, 2 × 106 cells, were transferred into each well of a 24 well plate and incubated 1 h at 37°C with 5% atmospheric CO2 prior to infection with H. pylori strains. BMDCs were infected at a MOI of ∼50 with given H. pylori strains in duplicate or triplicate wells as indicated. To facilitate contact between BMDCs and H. pylori, the 24 well plate was centrifuged at 200 × g for 10 min at 4°C. Infected BMDCs were incubated at 37°C with 5% atmospheric CO2 for 6 h. After 6 h of culture cells were collected, resuspended in TRIZOL and RNA was isolated as described below for real-time rtPCR analysis. Alternatively, supernatants were collected from these cultures at 24 h. The supernatant medium was centrifuged and filtered. Supernatants were analyzed for expression of IL-23p19, IL-1β, IL-6, by using the Searchlight System (Aushon, Billerica, MA, USA).
Infection of Mice with H. pylori
One day prior to infection of mice, H. pylori from plate cultures were inoculated into liquid medium and were cultured for 18 h under microaerobic conditions, as described above. Mice were orogastrically inoculated with a suspension of 5 × 108 CFU of H. pylori in 0.5 ml of Brucella broth. Mice were inoculated twice over 5 days.
Processing of Mouse Stomachs
The stomach was removed from each mouse by excising between the esophagus and the duodenum. The forestomach (non-glandular portion) was removed from the glandular stomach and discarded. The glandular stomach was opened, rinsed gently in cold PBS, and cut into three longitudinal strips that were used for bacterial culture, RNA analysis, and histology. For culturing of H. pylori from the stomach, gastric tissue was placed into Brucella broth-10% FBS for immediate processing. Gastric tissue was stored in RNALater (Ambion) solution for subsequent RNA isolation. A longitudinal strip from the greater curvature of the stomach was excised and placed in 10% normal buffered formalin for 24 h, embedded in paraffin and processed routinely for hematoxylin and eosin (H&E) staining. Indices of inflammation and injury were scored by a single pathologist (KW) who was blinded to the identity of the mice. Acute and chronic inflammation in the gastric antrum and corpus were graded on a 0-3 scale. Acute inflammation was graded based on density of neutrophils and chronic inflammation was graded based on the density of lamina propria mononuclear cell infiltration independent of lymphoid follicles. The total inflammation score is the sum of the chronic and acute inflammation scores in the antrum and the corpus.
Culture of H. pylori from Mouse Stomach
Gastric tissue was homogenized using the Tissue Tearor (BioSpec Products, Bartlesville, OK, USA). Serial dilutions of the homogenate were plated on trypticase soy agar, containing 5% sheep blood, 10 μg/ml nalidixic acid, 100 μg/ml vancomycin, 2 μg/ml amphotericin, and 200 μg/ml bacitracin (all antibiotics were purchased from Sigma). After 5–7 days of culture under microaerobic conditions, H. pylori colonies were counted and the number of colony forming units per gram of tissue calculated (CFU/g).
RNA Extraction and Real-Time rtPCR
RNA was isolated from the stomach using the TRIZOL isolation protocol (Invitrogen, Carlsbad, CA, USA) with slight modifications. Stomach tissue was homogenized in 1 ml of TRIZOL reagent and then two chloroform extractions were performed. Following an isopropanol precipitation, the RNA was washed with 70% ethanol and treated with RNase Inhibitor (Applied Biosystems, Foster City, CA, USA) for 30 min. The RNA was resuspended at 65°C for 15 min and RNA preparations were further purified using the Qiagen RNA isolation kit as directed by the manufacturer (Qiagen Inc., Valencia, CA, USA). RNA was reverse transcribed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). For real time rtPCR, we used the relative gene expression method (Giulietti et al., 2001). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) served as the normalizer, and tissue from uninfected mouse stomachs served as the calibrator. All cDNA samples were analyzed in triplicate, along with “no reverse transcriptase” controls, using an Applied Biosystems StepOne Plus real time PCR instrument. Levels of cytokine expression are indicated as “normalized expression,” based on comparison of tissue from H. pylori-infected mice with tissue from uninfected mice (calibrator tissue; Giulietti et al., 2001). Relative units (or normalized expression) were calculated as 2^−ΔΔCt (Ct, cycle threshold) where ΔΔCt is equal to the difference between the ΔCt of the gene of interest of the experimental sample and the ΔCt of the gene of interest of the calibrator tissue. The ΔCt of the gene of interest is calculated as the difference between the cycle threshold of the gene of interest and the cycle threshold of GAPDH (our normalizer). Primer and probe sets were purchased as Taqman Gene Expression Assays from Applied Biosystems [IL-1β (accession number Mm00434227_g1), IL-6 (Mm99999064_m1), IL-10 (Mm01288386_m1), IL-12a (Mm00434165_m1), IL-17a (Mm00439619_m1), IL-23a (Mm01160011_g1), Foxp3 (Mm00475162_m1), GAPDH (Mm99999915_g1), IFNγ (Mm999 99071_m1)].
Statistical Analysis
Four to six mice per group per time point were used for all of the studies. To compare results obtained with different groups of mice, statistical analysis was performed using an unpaired student’s T test utilizing GraphPad Prism software. For analyses of bacterial numbers and cell numbers, the data were normalized by log transformation prior to statistical analysis. For histology scores, the Mann-Whitney U-test was applied to compare results between IL23+/+ and IL-23−/− mice.
Results
IL-23p19 and Th17 Inducing Cytokines are Stimulated in Dendritic Cells by H. pylori
The ability of DCs to sense infection and to differentially secrete both IL-12 and IL-23 are critical to the initiation of both Th1 and Th17 responses to pathogens. To determine whether Th17 inducing cytokines were upregulated in dendritic cells upon exposure to H. pylori, BMDC were generated from wild-type mice (both IL-23+/+ mice which are on a mixed background, and C57Bl/6 mice) and co-cultured with H. pylori. The results indicate that H. pylori co-culture with BMDCs induces mRNA expression of Th17 inducing cytokines, IL-1β and IL-6, and IL-23p19 (Figure 1A), which likely enhances IL-17 expression through a STAT3 dependent mechanism. The increase in IL-1β and IL-6, and IL-23p19 was confirmed using a protein based multiplex assay on supernatants from 24 h co-cultures (Figure 1C). Moreover, Th1 inducing cytokine, IL-12p40, and regulatory cytokine, IL-10, also increase in expression relative to untreated dendritic cells (Figure 1B). Several strains were used to investigate whether different virulence factors would influence the dendritic cell’s cytokine response. For example PM-SS1 and SS1 were compared to investigate the influence of a functional type IV secretion system (the Cag pathogenicity island CagPAI). PM-SS1 (pre-mouse SS1) contains a functional CagPAI, whereas in SS1 the CagPAI is no longer intact. Our data indicate that the cytokine expression by the dendritic cells is independent of a functional CagPAI (Figures 1A,B). Several additional strains of H. pylori (including 26695, 60190, 7.13, and X47) were also used in this co-culture system and induce these cytokines in dendritic cells with no significant differences between the dendritic cell cytokine responses with these H. pylori strains (data not shown). Performing similar experiment to compare cytokine expression at 6 h of co-culture in IL-23+/+ verse IL-23−/− BMDC, we observed no consistent statistical differences in the expression of these cytokines (data not shown) with the exception of IL-23 which as expected was not detected in the IL-23−/− BMDC.
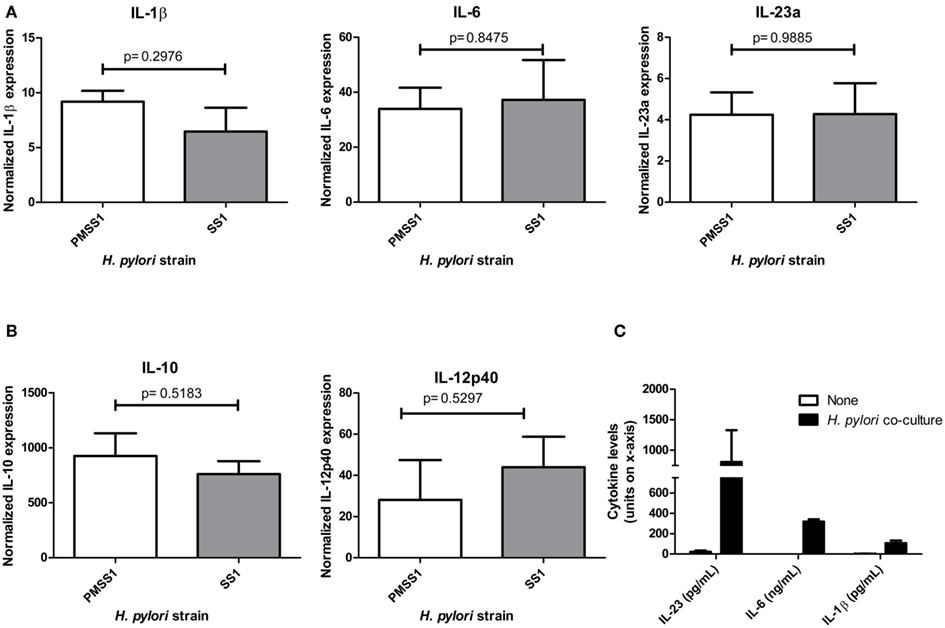
Figure 1. Dendritic cell co-cultures with H. pylori. When cultured with H. pylori strains SS1 or PM-SS1, bone marrow derived dendritic cells from wild-type mice produce (A). Th17 influencing cytokines (IL-1β, IL-6, and IL-23a), (B). Regulatory cytokine IL-10 and Th1 inducing cytokine IL-12. Real time rtPCR was performed to measure cytokine expression at 6 h of co-culture with and without H. pylori. Graphs are representative of four independent experiments. (C) Th17 influencing cytokines (IL-1β, IL-6, and IL-23a) protein levels in the supernatants from dendritic cell- H. pylori co-cultures at 24 h (units for each cytokine are noted on the x-axis).
IL-23p19 Deficient Mice Carry Higher H. pylori Bacterial Loads during Chronic Infection
To investigate whether a deficiency in IL-23p19 leads to a change in the ability of the mice to control H. pylori colonization, we infected IL-23−/− mice and IL-23+/+ mice with H. pylori strain SS1 and followed the course of infection for up to 4 months. At 1 month post-infection, there was no significant difference in the bacterial burden in the IL-23−/− mice compared to the IL-23+/+ mice (data not shown). By 3 and 4 months post infection, the IL-23−/− mice did not control the bacterial burden as well as the IL-23+/+ mice (Figure 2).
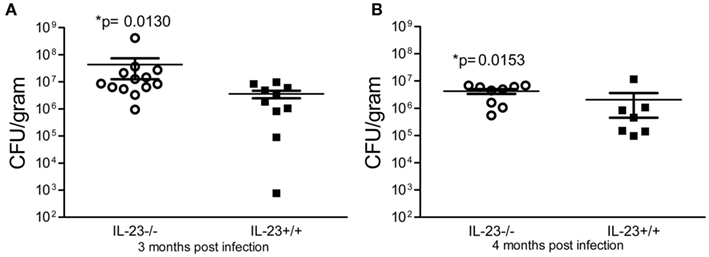
Figure 2. Colonization of IL-23p19 deficient mice with H. pylori. IL-23−/− mice and IL-23+/+ littermates were infected with H. pylori strain SS1 for 3 and 4 months. Levels of colonization were measured by plating serial dilutions of stomach homogenates. The number of colony forming units (CFU) per gram of stomach tissue is presented on the graphs for 3 months post-infection (A) and 4 months post-infection (B).
IL-23p19 is Required for High Levels of T Helper Cytokine Expression during H. pylori Infection
To determine whether IL-23p19 deficient mice have reduced expression of T cell derived cytokines, IL-17, and IFNγ, we performed real time rtPCR on stomach tissue of IL-23−/− and IL-23+/+ mice during chronic H. pylori infection. Our data indicate that H. pylori infected IL-23−/− mice have significantly reduced expression of IL-17 and IFNγ by 3 months post infection compared to H. pylori infected IL23+/+ mice (Figure 3).
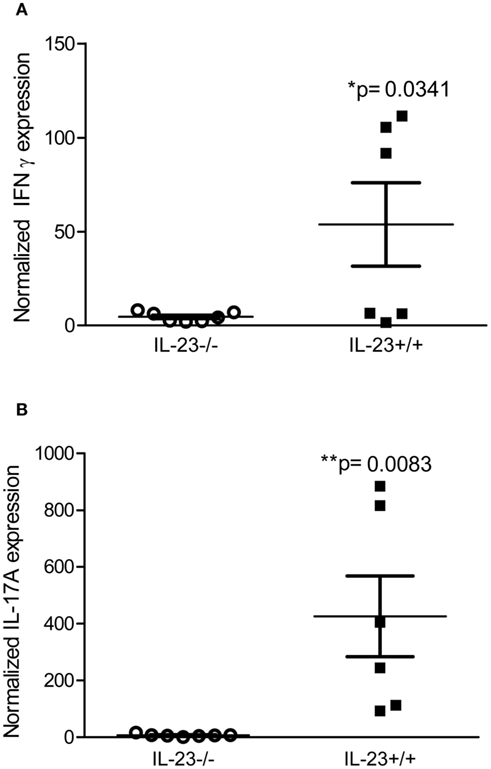
Figure 3. Th17 and Th1 responses are reduced in H. pylori infected IL-23−/− mice compared to H. pylori infected IL-23+/+ mice. Real time rtPCR was performed on stomach tissue of H. pylori infected mice. Relative units of IFNγ (A) and IL-17A (B) were measured at 3 months post-infection. Graphs are representative of three independent experiments.
To investigate whether the decrease in pro-inflammatory T cell cytokines was a result of an increase in Treg cells, we assayed for expression of Foxp3 in the tissue. We were unable to detect any significant differences in expression of Foxp3 comparing H. pylori infected IL-23−/− mice and H. pylori infected IL-23+/+ mice at 3 or 4 months post infection (data not shown).
IL-23p19-Deficient Mice Have Minimal Chronic Gastritis
To investigate the impact of the IL-23 deficiency, increased bacterial burden and decreased T helper responses in the stomachs of the IL-23−/− mice on the inflammatory response, we scored the stomach tissue for both chronic and acute inflammation (see methods for how these types of inflammation are defined). Our data indicate that there is no significant difference in total inflammation (sum of acute and chronic inflammation in the corpus and antrum) between IL-23−/− mice and IL-23+/+ mice (p = 0.0521; Figure 4A) at 3 months post infection. However, there is a significant decrease in the chronic inflammatory scores of H. pylori-infected IL-23−/− mice compared to H. pylori infected IL-23+/+ littermates at 3 months post infection (p = 0.0074; Figure 4B). At 4 months post infection, while there was a trend toward decreased total inflammation in the H. pylori-infected IL-23−/− mice compared to H. pylori-infected wild-type mice, the differences were not significant (p = 0.20, data not shown).
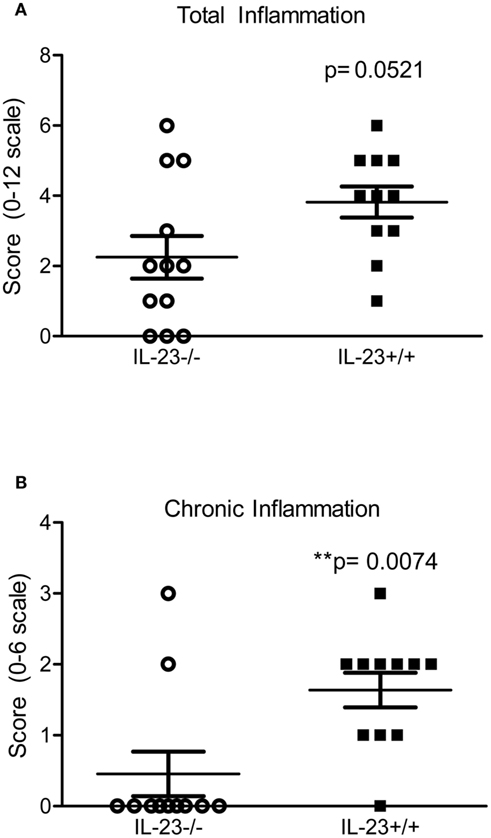
Figure 4. Inflammation is reduced in H. pylori infected IL-23−/− mice compared to H. pylori infected IL-23+/+ mice. Levels of acute and chronic inflammation were scored on stomach tissue (in the corpus and antrum) at 3 months post infection. Total inflammation (A) was scored on a scale of 0–12. Chronic inflammation (B) in the corpus and antrum was scored on a scale of 0–6.
Peyer’s Patches from IL-23−/− Mice Have Differential Expression of IL-12p40
In a mouse model of infection it has been demonstrated that Peyer’s patches are required for the development of gastritis in the stomach (Nagai et al., 2007). To investigate whether defective priming accounted for the reduced level of chronic inflammation observed in the IL-23 deficient mice, we measured the expression of cytokines known to influence T cell priming (IL-23p19, and IL-12p40) in the Peyer’s Patches of both H. pylori infected IL-23−/− and IL-23+/+ mice. Moreover we measured expression of IFNγ, IL-17, and Foxp3 as a measure of Th responses in the Peyer’s patches.
While expression of IL-17, IFNγ, and Foxp3 was similar in H. pylori-infected IL-23−/− and IL23+/+ mice during chronic infection (Figure 5A), the levels of IL-12p40 were significantly higher (relative to uninfected wild type mice) in IL-23−/− mice when compared to IL-23+/+ mice (Figure 5B). Analysis of IL-12p40 gene expression in the stomach and in the spleen was similar in H. pylori infected IL-23−/− and H. pylori infected IL-23+/+ mice (Figure 5B). To investigate whether differences in baseline expression of IL-12p40 mRNA accounted for this difference in the Peyer’s patches, we measured IL-12p40 mRNA expression in uninfected IL-23−/− v. uninfected IL-23+/+. The data indicate that IL-23−/− mice exhibit about twofold higher levels of IL-12 than uninfected IL-23+/+ mice (data not shown).
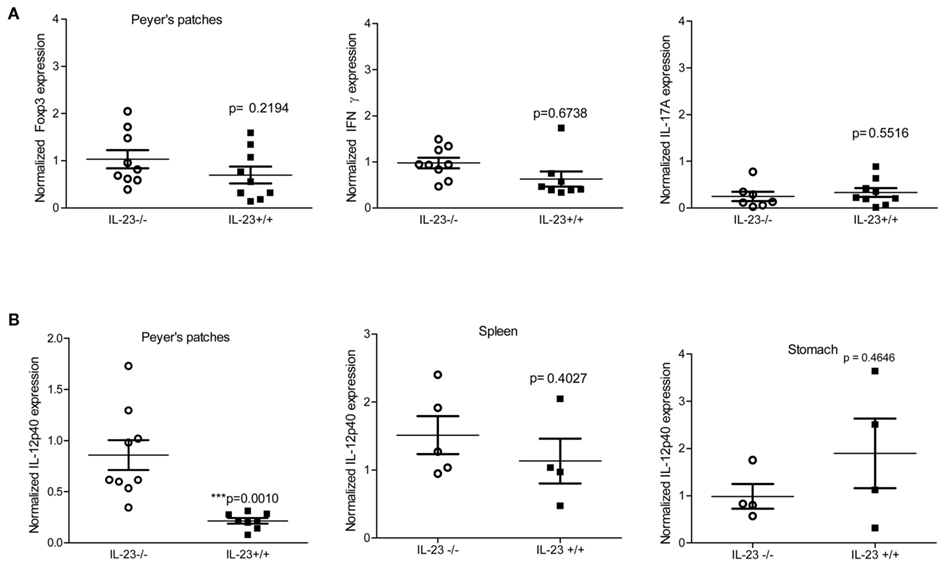
Figure 5. Peyer’s patches express T cell skewing cytokines during chronic infection. Levels of Foxp3, IFNγ, and IL-17A (A), IL-12p40 (B) were measured by real time rtPCR at 3 and 4 months post infection. Normalized expression also known as relative units are the relative expression calibrated to uninfected wild-type with GAPDH as the endogenous control. Due to differences in IL-12p40 in the Peyer’s patches, IL-12p40 expression was also measured in the spleen and gastric mucosa (B).
Discussion
The T helper response, which develops toward H. pylori infection, is a mixed response. The level of gastritis and bacterial colonization may be dependent on the balance between Th1, Th17, and Treg responses. Because Th1 and Th17 cells likely contribute to protective immunity to H. pylori, understanding the contributions of cytokines produced by innate immune responses which influence the T helper responses is of great interest to understanding this balance. Our study specifically investigated the contribution of IL-23 on the development of T cell responses and gastritis.
IL-23 drives intestinal pathology in experimental models of T cell-dependent and innate colitis (Hue et al., 2006; Kullberg et al., 2006; Uhlig et al., 2006; Yen et al., 2006; Elson et al., 2007); therefore we hypothesized that it would also drive H. pylori-induced pathology in the stomach. Our studies on BMDCs indicate that culture of dendritic cells with H. pylori (at an MOI of 50) leads to increased expression of IL-23, IL-1β, IL-6, IL-12, and IL-10 by 6 h post infection. We have observed the increased expression of these cytokines with several strains of H. pylori suggesting that the major determinants of dendritic cell activation are not dependent on the CagPAI or VacA since these strains vary in their expression of the Type IV secretion system and express various isoforms of the vacuolating toxin, VacA (if any). These data are consistent with data published by Kao et al. (2010) which indicated that CagA deletion did not affect the Treg skewing of H. pylori treated-DCs. Contrary to our results, Tanaka et al. (2010) present data which suggests the presence of CagA contributes to regulation of cytokine production in DCs co-cultured with H. pylori.
In the mouse model of infection, our data suggests that IL-23 makes a minor contribution to the development of chronic gastritis in this model of H. pylori infection. For instance, differences in gastritis were only observed at chronic time points (3 and 4 months post infection) and in chronic inflammation (when data is stratified, no differences were observed in acute inflammation).
It is clear that IL-23 plays a role in expanding and maintaining the Th17 subset of CD4+ T helper cells. In addition, there is evidence in mouse models of M. tuberculosis and H. hepaticus that IL-23 is involved in inducing IFNγ production by CD4+ T-cell (Khader et al., 2005; Kullberg et al., 2006). Our studies indicate that IL-23 makes a contribution to both Th1 and Th17 responses during H. pylori infection especially during the chronic stage of infection. By 3 months post infection, IL-23−/− mice have significantly reduced IL-17 and IFNγ expression compared to H. pylori infected wild-type mice. It is important to recognize that Th17 cells are likely still developing in these IL-23−/− mice, but overall expression of IL-17a is lower. We were unable to detect differences in neither IL-21 nor IL-17f in these studies (data not shown). Due to the increased bacterial burden in the stomachs of IL-23−/− mice compared to wild-type mice, we speculate that chronic bacterial exposure in conjunction with ongoing stimulation of antigen presenting cells and continual expression of IL-1β, IL-6, and TGFβ would drive Th17 differentiation even in the absence of IL-23 expanding and maintaining the population of cells.
IL-23 has been reported to inhibit the accumulation of Foxp3+ Tregs in the intestines. IL-23 receptor deficient (IL-23R−/−) T cells adoptively transferred into Rag−/− recipients demonstrate an increased ability to develop into Tregs compared to wild-type T cells adoptively transferred into Rag−/− recipients. Moreover, there is evidence that IL-23 may inhibit IL-10 production. To investigate the effects of IL-23 on the development of Tregs in our mouse model of H. pylori infection, we measured Foxp3 expression in both the stomach and the Peyer’s patches. Our data indicate there is no significant difference in expression of Foxp3 comparing H. pylori infected IL-23−/− and wild-type mice.
Our findings that the levels of chronic inflammation were significantly reduced in the H. pylori infected IL-23−/− mice compared to H. pylori infected wild-type mice has led us to question how efficient priming is in the IL-23−/− mice. Unfortunately, this is a very difficult question to answer with few defined antigens for H. pylori. Based on the evidence that in the absence of Peyer’s patches there is no gastritis (Nagai et al., 2007), we focused on this site to investigate the activation of innate T helper driving cytokines, IL-12 and IL-23, and measured expression of Foxp3 as a marker for Treg development. Our findings suggest that in the absence of IL-23, there may be an increase in the baseline expression (in uninfected mice) of IL-12, which suggests that there may be inherited differences in the ability of these mice to develop Th1 responses. We would have expected IL-23−/− mice to have lower IL-12 in the Peyer’s patches since their Th1 responses are depressed in the gastric mucosa. Previously, Becker et al. (2006) demonstrated that IL-23 can downregulate TLR-induced IL-12 expression suggesting that IL-23 can indirectly inhibit Th1 responses, but our data suggests that in the absence of IL-23, both Th1 and Th17 responses are depressed in the gastric mucosa. We speculate that while the Peyer’s patches may be a site for T cell priming a potential surrogate marker for what is happening in the gastric mucosa, primed T cells must still receive the correct signals to migrate into the gastric mucosa from the Peyer’s patches. Therefore, IL-23 and the Th17 response may be necessary for inflammation to initiate a cascade of chemokines which would then also recruit Th1 cells to the site of infection.
The study was performed on mice which were obtained from the NIH Consortium and they are described as B6:129 mice, therefore they are on a mixed background. We used wild-type littermates as controls for the IL-23−/− mice. The B6:129 mice exhibit a milder gastritis response compared to C57Bl/6 mice (total inflammatory scores are often higher in WT C57Bl/6 mice than these B6:129 WT mice, Algood et al., 2009). We also observed more variation within groups which may be a result of the mixed background of the mice. While it may be somewhat cumbersome, there is a need for a study which would experimentally measure T cell priming and activation in various secondary lymphatic tissues over time, with multiple strains of H. pylori, in several strains of inbred mice. It is likely that the differences in inflammation observed in mice of different backgrounds could be correlated with differences in the magnitude of T cell priming in the lymphatics, the ratio of Th1:Th17:Treg development, and the timing of the response.
In this study in particular, we have demonstrated that IL-23 plays a minor role in control of H. pylori induced gastritis. Reduced chronic inflammation correlates with reduced Th17 and Th1 cytokine expression in the stomachs of the H. pylori-infected IL-23−/− mice compared to wild-type mice. In future studies, it will be of interest to investigate T cell priming early during infection and the role IL-23 plays at the sites of priming.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the NIH Consortium for providing IL-23+/− mice and Dr. Kokoye at Vanderbilt Division of Animal Care for help breeding Helicobacter free mice. We thank Tim Cover, Jana Radin, and Mark Boothby for helpful discussions. We thank Jennifer Gaddy and Jana Radin for carefully reading this manuscript.
Footnote
- ^This work was supported by the Medical Research Service of the Department of Veterans Affairs. The VMC Flow Cytometry Shared Resource is supported by the Vanderbilt Ingram Cancer Center (P30 CA68485) and the Vanderbilt Digestive Disease Research Center (P30 DK058404). Research Histology Core Services were supported by the Vanderbilt Digestive Disease Research Center.
References
Algood, H. M., Allen, S. S., Washington, M. K., Peek, R. M. Jr., Miller, G. G., and Cover, T. L. (2009). Regulation of gastric B cell recruitment is dependent on IL-17 receptor A signaling in a model of chronic bacterial infection. J. Immunol. 183, 5837–5846.
Becker, C., Dornhoff, H., Neufert, C., Fantini, M. C., Wirtz, S., Huebner, S., Nikolaev, A., Lehr, H. A., Murphy, A. J., Valenzuela, D. M., Yancopoulos, G. D., Galle, P. R., Karow, M., and Neurath, M. F. (2006). Cutting edge: IL-23 cross-regulates IL-12 production in T cell-dependent experimental colitis. J. Immunol. 177, 2760–2764.
Bettelli, E., Korn, T., Oukka, M., and Kuchroo, V. K. (2008). Induction and effector functions of T(H)17 cells. Nature 453, 1051–1057.
Caruso, R., Fina, D., Paoluzi, O. A., Del Vecchio Blanco, G., Stolfi, C., Rizzo, A., Caprioli, F., Sarra, M., Andrei, F., Fantini, M. C., MacDonald, T. T., Pallone, F., and Monteleone, G. (2008). IL-23-mediated regulation of IL-17 production in Helicobacter pylori-infected gastric mucosa. Eur. J. Immunol. 38, 470–478.
Chackerian, A. A., Chen, S. J., Brodie, S. J., Mattson, J. D., McClanahan, T. K., Kastelein, R. A., and Bowman, E. P. (2006). Neutralization or absence of the interleukin-23 pathway does not compromise immunity to mycobacterial infection. Infect. Immun. 74, 6092–6099.
Chen, X., Howard, O. M., and Oppenheim, J. J. (2007a). Pertussis toxin by inducing IL-6 promotes the generation of IL-17-producing CD4 cells. J. Immunol. 178, 6123–6129.
Chen, Z., Laurence, A., and O’Shea, J. J. (2007b). Signal transduction pathways and transcriptional regulation in the control of Th17 differentiation. Semin. Immunol. 19, 400–408.
Chen, Z., Laurence, A., Kanno, Y., Pacher-Zavisin, M., Zhu, B. M., Tato, C., Yoshimur, A., Hennighausen, L., and O’Shea, J. J. (2006). Selective regulatory function of Socs3 in the formation of IL-17-secreting T cells. Proc. Natl. Acad. Sci. U.S.A. 103, 8137–8142.
Cruz, A., Khader, S. A., Torrado, E., Fraga, A., Pearl, J. E., Pedrosa, J., Cooper, A. M., and Castro, A. G. (2006). Cutting edge: IFN-gamma regulates the induction and expansion of IL-17-producing CD4 T cells during mycobacterial infection. J. Immunol. 177, 1416–1420.
Curtis, M. M., and Way, S. S. (2009). Interleukin-17 in host defence against bacterial, mycobacterial and fungal pathogens. Immunology 126, 177–185.
DeLyria, E. S., Redline, R. W., and Blanchard, T. G. (2009). Vaccination of mice against H. pylori induces a strong Th-17 response and immunity that is neutrophil dependent. Gastroenterology 136, 247–256.
Dubin, P. J., and Kolls, J. K. (2007). IL-23 mediates inflammatory responses to mucoid Pseudomonas aeruginosa lung infection in mice. Am. J. Physiol. Lung Cell Mol. Physiol. 292, L519–L528.
Eaton, K. A., Mefford, M., and Thevenot, T. (2001). The role of T cell subsets and cytokines in the pathogenesis of Helicobacter pylori gastritis in mice. J. Immunol. 166, 7456–7461.
Eaton, K. A., Ringler, S. R., and Danon, S. J. (1999). Murine splenocytes induce severe gastritis and delayed-type hypersensitivity and suppress bacterial colonization in Helicobacter pylori-infected SCID mice. Infect. Immun. 67, 4594–4602.
Elson, C. O., Cong, Y., Weaver, C. T., Schoeb, T. R., McClanahan, T. K., Fick, R. B., and Kastelein, R. A. (2007). Monoclonal anti-interleukin 23 reverses active colitis in a T cell-mediated model in mice. Gastroenterology 132, 2359–2370.
Furuzawa-Carballeda, J., Vargas-Rojas, M. I., and Cabral, A. R. (2007). Autoimmune inflammation from the Th17 perspective. Autoimmun. Rev. 6, 169–175.
Giulietti, A., Overbergh, L., Valckx, D., Decallonne, B., Bouillon, R., and Mathieu, C. (2001). An overview of real-time quantitative PCR: applications to quantify cytokine gene expression. Methods 25, 386–401.
Hamada, S., Umemura, M., Shiono, T., Tanaka, K., Yahagi, A., Begum, M. D., Oshiro, K., Okamoto, Y., Watanabe, H., Kawakami, K., Roark, C., Born, W. K., O’Brien, R., Ikuta, K., Ishikawa, H., Nakae, S., Iwakura, Y., Ohta, T., and Matsuzaki, G. (2008). IL-17A produced by gammadelta T cells plays a critical role in innate immunity against Listeria monocytogenes infection in the liver. J. Immunol. 181, 3456–3463.
Happel, K. I., Dubin, P. J., Zheng, M., Ghilardi, N., Lockhart, C., Quinton, L. J., Odden, A. R., Shellito, J. E., Bagby, G. J., Nelson, S., and Kolls, J. K. (2005). Divergent roles of IL-23 and IL-12 in host defense against Klebsiella pneumoniae. J. Exp. Med. 202, 761–769.
Hue, S., Ahern, P., Buonocore, S., Kullberg, M. C., Cua, D. J., McKenzie, B. S., Powrie, F., and Maloy, K. J. (2006). Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J. Exp. Med. 203, 2473–2483.
Kao, J. Y., Zhang, M., Miller, M. J., Mills, J. C., Wang, B., Liu, M., Eaton, K. A., Zou, W., Berndt, B. E., Cole, T. S., Takeuchi, T., Owyang, S. Y., and Luther, J. (2010). Helicobacter pylori immune escape is mediated by dendritic cell-induced Treg skewing and Th17 suppression in mice. Gastroenterology 138, 1046–1054.
Khader, S. A., Pearl, J. E., Sakamoto, K., Gilmartin, L., Bell, G. K., Jelley-Gibbs, D. M., Ghilardi, N., deSauvage, F., and Cooper, A. M. (2005). IL-23 compensates for the absence of IL-12p70 and is essential for the IL-17 response during tuberculosis but is dispensable for protection and antigen-specific IFN-gamma responses if IL-12p70 is available. J. Immunol. 175, 788–795.
Kleinschek, M. A., Muller, U., Brodie, S. J., Stenzel, W., Kohler, G., Blumenschein, W. M., Straubinger, R. K., McClanahan, T., Kastelein, R. A., and Alber, G. (2006). IL-23 enhances the inflammatory cell response in Cryptococcus neoformans infection and induces a cytokine pattern distinct from IL-12. J. Immunol. 176, 1098–1106.
Koussoulas, V., Vassiliou, S., Giamarellos-Bourboulis, E. J., Tassias, G., Kotsaki, A., Barbatzas, C., and Tzivras, M. (2009). Implications for a role of interleukin-23 in the pathogenesis of chronic gastritis and of peptic ulcer disease. Clin. Exp. Immunol. 156, 97–101.
Kullberg, M. C., Jankovic, D., Feng, C. G., Hue, S., Gorelick, P. L., McKenzie, B. S., Cua, D. J., Powrie, F., Cheever, A. W., Maloy, K. J., and Sher, A. (2006). IL-23 plays a key role in Helicobacter hepaticus-induced T cell-dependent colitis. J. Exp. Med. 203, 2485–2494.
Laurence, A., and O’Shea, J. J. (2007). T(H)-17 differentiation: of mice and men. Nat. Immunol. 8, 903–905.
Mandell, L., Moran, A. P., Cocchiarella, A., Houghton, J., Taylor, N., Fox, J. G., Wang, T. C., and Kurt-Jones, E. A. (2004). Intact gram-negative Helicobacter pylori, Helicobacter felis, and Helicobacter hepaticus bacteria activate innate immunity via toll-like receptor 2 but not toll-like receptor 4. Infect. Immun. 72, 6446–6454.
Mangan, P. R., Harrington, L. E., O’Quinn, D. B., Helms, W. S., Bullard, D. C., Elson, C. O., Hatton, R. D., Wahl, S. M., Schoeb, T. R., and Weaver, C. T. (2006). Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 441, 231–234.
McKenzie, B. S., Kastelein, R. A., and Cua, D. J. (2006). Understanding the IL-23-IL-17 immune pathway. Trends Immunol. 27, 17–23.
Meeks, K. D., Sieve, A. N., Kolls, J. K., Ghilardi, N., and Berg, R. E. (2009). IL-23 is required for protection against systemic infection with Listeria monocytogenes. J. Immunol. 183, 8026–8034.
Meyer, F., Wilson, K. T., and James, S. P. (2000). Modulation of innate cytokine responses by products of Helicobacter pylori. Infect. Immun. 68, 6265–6272.
Nagai, S., Mimuro, H., Yamada, T., Baba, Y., Moro, K., Nochi, T., Kiyono, H., Suzuki, T., Sasakawa, C., and Koyasu, S. (2007). Role of Peyer’s patches in the induction of Helicobacter pylori-induced gastritis. Proc. Natl. Acad. Sci. U.S.A. 104, 8971–8976.
Odenbreit, S., Linder, S., Gebert-Vogl, B., Rieder, G., Moran, A. P., and Haas, R. (2006). Interleukin-6 induction by Helicobacter pylori in human macrophages is dependent on phagocytosis. Helicobacter 11, 196–207.
Otani, K., Watanabe, T., Tanigawa, T., Okazaki, H., Yamagami, H., Watanabe, K., Tominaga, K., Fujiwara, Y., Oshitani, N., and Arakawa, T. (2009). Anti-inflammatory effects of IL-17A on Helicobacter pylori-induced gastritis. Biochem. Biophys. Res. Commun. 382, 252–258.
Sarkar, S., Tesmer, L. A., Hindnavis, V., Endres, J. L., and Fox, D. A. (2007). Interleukin-17 as a molecular target in immune-mediated arthritis: immunoregulatory properties of genetically modified murine dendritic cells that secrete interleukin-4. Arthritis Rheum. 56, 89–100.
Schulz, S. M., Kohler, G., Holscher, C., Iwakura, Y., and Alber, G. (2008a). IL-17A is produced by Th17, gammadelta T cells and other CD4- lymphocytes during infection with Salmonella enterica serovar enteritidis and has a mild effect in bacterial clearance. Int. Immunol. 20, 1129–1138.
Schulz, S. M., Kohler, G., Schutze, N., Knauer, J., Straubinger, R. K., Chackerian, A. A., Witte, E., Wolk, K., Sabat, R., Iwakura, Y., Holscher, C., Muller, U., Kastelein, R. A., and Alber, G. (2008b). Protective immunity to systemic infection with attenuated Salmonella enterica serovar enteritidis in the absence of IL-12 is associated with IL-23-dependent IL-22, but not IL-17. J. Immunol. 181, 7891–7901.
Shibata, K., Yamada, H., Hara, H., Kishihara, K., and Yoshikai, Y. (2007). Resident Vdelta1+ gammadelta T cells control early infiltration of neutrophils after Escherichia coli infection via IL-17 production. J. Immunol. 178, 4466–4472.
Shiomi, S., Toriie, A., Imamura, S., Konishi, H., Mitsufuji, S., Iwakura, Y., Yamaoka, Y., Ota, H., Yamamoto, T., Imanishi, J., and Kita, M. (2008). IL-17 is involved in Helicobacter pylori-Induced gastric inflammatory responses in a mouse model. Helicobacter 13, 518–524.
Tanaka, H., Yoshida, M., Nishiumi, S., Ohnishi, N., Kobayashi, K., Yamamoto, K., Fujita, T., Hatakeyama, M., and Azuma, T. (2010). The CagA protein of Helicobacter pylori suppresses the functions of dendritic cell in mice. Arch. Biochem. Biophys. 498, 35–42.
Uhlig, H. H., McKenzie, B. S., Hue, S., Thompson, C., Joyce-Shaikh, B., Stepankova, R., Robinson, N., Buonocore, S., Tlaskalova-Hogenova, H., Cua, D. J., and Powrie, F. (2006). Differential activity of IL-12 and IL-23 in mucosal and systemic innate immune pathology. Immunity 25, 309–318.
Wu, Q., Martin, R. J., Rino, J. G., Breed, R., Torres, R. M., and Chu, H. W. (2007). IL-23-dependent IL-17 production is essential in neutrophil recruitment and activity in mouse lung defense against respiratory Mycoplasma pneumoniae infection. Microbes Infect. 9, 78–86.
Yang, X. O., Panopoulos, A. D., Nurieva, R., Chang, S. H., Wang, D., Watowich, S. S., and Dong, C. (2007). STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J. Biol. Chem. 282, 9358–9363.
Ye, P., Garvey, P. B., Zhang, P., Nelson, S., Bagby, G., Summer, W. R., Schwarzenberger, P., Shellito, J. E., and Kolls, J. K. (2001). Interleukin-17 and lung host defense against Klebsiella pneumoniae infection. Am. J. Respir. Cell Mol. Biol. 25, 335–340.
Yen, D., Cheung, J., Scheerens, H., Poulet, F., McClanahan, T., McKenzie, B., Kleinschek, M. A., Owyang, A., Mattson, J., Blumenschein, W., Murphy, E., Sathe, M., Cua, D. J., Kastelein, R. A., and Rennick, D. (2006). IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J. Clin. Invest. 116, 1310–1316.
Yu, J. J., Ruddy, M. J., Wong, G. C., Sfintescu, C., Baker, P. J., Smith, J. B., Evans, R. T., and Gaffen, S. L. (2007). An essential role for IL-17 in preventing pathogen-initiated bone destruction: recruitment of neutrophils to inflamed bone requires IL-17 receptor-dependent signals. Blood 109, 3794–3802.
Zhang, J. Y., Liu, T., Guo, H., Liu, X. F., Zhuang, Y., Yu, S., Chen, L., Wu, C., Zhao, Z., Tang, B., Luo, P., Mao, X. H., Guo, G., Shi, Y., and Zou, Q. M. (2011). Induction of a Th17 cell response by Helicobacter pylori urease subunit B. Immunobiology 216, 803–810.
Keywords: Helicobacter pylori, T lymphocytes, IL-23, IL-17, cytokines, gastritis
Citation: Horvath DJ, Jr, Washington MK, Cope VA and Algood HMS (2012) IL-23 contributes to control of chronic Helicobacter pylori infection and the development of T helper responses in a mouse model. Front. Immun. 3:56. doi: 10.3389/fimmu.2012.00056
Received: 13 December 2011; Paper pending published: 06 January 2012;
Accepted: 02 March 2012; Published online: 26 March 2012.
Edited by:
Thomas Blanchard, University of Maryland School of Medicine, USAReviewed by:
Diane Bimczok, University of Alabama at Birmingham, USAAnne Hosmalin, Cochin Institute, France
Copyright: © 2012 Horvath Jr, Washington, Cope and Algood. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Holly M. Scott Algood, Department of Medicine, Vanderbilt University School of Medicine, Nashville, TN 37212, USA. e-mail:aG9sbHkubS5hbGdvb2RAdmFuZGVyYmlsdC5lZHU=