 Lynn Soong1,2*
Lynn Soong1,2*- 1 Center for Tropical Diseases, Sealy Center for Vaccine Development, Department of Microbiology and Immunology, The University of Texas Medical Branch, Galveston, TX, USA
- 2 Department of Pathology, The University of Texas Medical Branch, Galveston, TX, USA
Infection with Leishmania amazonensis and other members of the Leishmania mexicana complex can lead to diverse clinical manifestations, some of which are relatively difficult to control, even with standard chemotherapy. Diffuse cutaneous leishmaniasis (CL) is a rare but severe form, and its clinical hallmark is excessive parasitic growth in infected cells accompanied by profound impairments in host immune responses to the parasites. Since these parasites also cause non-healing CL in most inbred strains of mice, these animals are valuable models for dissecting the mechanisms of persistent infection and disease pathogenesis. In comparison to other Leishmania species, L. amazonensis infections are most remarkable for their ability to repress the activation and effector functions of macrophages, dendritic cells, and CD4+ T cells, implying discrete mechanisms at work. In addition to this multilateral suppression of host innate and adaptive immunity, the activation of types I and II interferon-mediated responses and autophagic/lipid metabolic pathways actually promotes rather than restrains L. amazonensis infection. These seemingly contradictory findings reflect the remarkable adaptation of the parasites to the ancient defense machinery of the host, as well as the complex parasite–host interactions at different stages of infection, which collectively contribute to non-healing leishmaniasis in the New World. This review article highlights new evidence that reveals the strategies utilized by L. amazonensis parasites to subvert or modulate host innate defense machinery in neutrophils and macrophages, as well as the regulatory roles of host innate responses in promoting parasite survival and replication within the huge parasitophorous vacuoles. A better understanding of unique features in host responses to these parasites at early and late stages of infection is important for the rational design of control strategies for non-healing leishmaniasis.
Introduction
Leishmania amazonensis is a member of the Leishmania mexicana complex and the etiological agent for a broad-spectrum of leishmaniasis in South American countries. It can cause mild cutaneous leishmaniasis (CL), diffuse CL (DCL), and deadly visceral leishmaniasis (Silveira et al., 2004). DCL is a rare but severe form and is not limited to patients in South America. In patients with Leishmania tropica and Leishmania major infections in India and Africa, DCL is also considered a clinical indicator of HIV co-infection (Niamba et al., 2007; Khandelwal et al., 2011). Generally speaking, patients with DCL share some clinical characteristics. For example, they have defective responses for the leishmanin skin test and poor antigen-stimulated T cell proliferation in vitro, although some patients remain responsiveness to other antigens such as tuberculin and lepromin (Silveira et al., 2004). The mechanisms responsible for this antigen-specific T cell anergy remain obscure, although it is likely due to alterations in activation of dendritic cells (DC) and macrophages (MΦ) following infection by L. amazonensis. These profound impairments in cell-mediated immune responses partially explain the relatively poor responses of DCL patients to available anti-Leishmania chemotherapy.
Mouse models are important in understanding the biology of leishmaniasis. The Th1/Th2 polarization of IFN-γ/IL-4 production seen in L. major infection between resistant C57BL/6 and susceptible BALB/c mice has provided valuable insights into protective and pathogenic mechanisms; however, this dogma is not fully applicable to human diseases and to some other Leishmania infection models (McMahon-Pratt and Alexander, 2004). L. amazonensis infections in mice have several distinguished features. All inbred strains of mice are genetically susceptible to L. amazonensis, developing progressive non-healing lesions that contain characteristic parasitophorous vacuoles (PV) and heavy parasite loads (Soong et al., 1997). This generalized susceptibility is not linked to a polarized expansion of IL-4-producing Th2 cells, but rather to the activation of pathogenic CD4+ T cells that produce relatively low levels of IFN-γ, IL-10, IL-17, and other cytokines (Soong et al., 1997; Xin et al., 2007). While knockout of endogenous IFN-γ or IL-10 alone is insufficient to change host susceptibility to this infection, depletion of endogenous CD4+ T cells or adoptive transfer of primed CD4+ T cells can deprive or exacerbate lesion pathogenesis, respectively (Soong et al., 1997; Xin et al., 2007). The low and mixed Th1/Th2 responses seen in L. amazonensis-infected mice are very similar to those observed in human infections (Silveira et al., 2009), validating the biological relevance of these mouse models for the study of human diseases.
Among the Leishmania species that are pathogenic to humans, only L. amazonensis and other members in the L. mexicana complex are capable of forming huge communal PV. It has been proposed that the large PVs may serve as a “shield,” by diluting the leishmanicidal effects of nitric oxide (NO) and reactive oxygen species (ROS) in infected cells (Wilson et al., 2008); however, additional studies are needed to verify this possibility. Comparative studies with L. amazonensis promastigotes generated from DCL and CL patients have revealed that DCL-derived parasites tend to form big PVs with heavy infection in mouse MΦs, suggesting a positive correlation between parasite virulence and PV sizes (Franca-Costa et al., submitted). Under the cell-free condition, L. amazonensis parasites (especially the amastigote form) are also more resistant to neutrophil- or MΦ-derived leishmanicidal molecules than are their counterparts of other species (Mukbel et al., 2007; Wang et al., 2011), possibly indicative of biological features that are unique and intrinsic to these parasites.
The purpose of this review is not to provide a comprehensive summary for the biology of Leishmania parasites or the pathogenic roles of adaptive immune responses, as there are excellent reviews covering these areas in this Special Topics issue for leishmaniasis. Instead, this review will focus on new evidence that reveals the strategies utilized by L. amazonensis parasites to down-modulate and subvert host innate defense machinery in neutrophils and MΦ, as well as the regulatory roles of host innate responses in promoting parasite survival and infection in the host. These studies indicate complex roles of host immune responses to Leishmania infections and open areas for further investigation.
L. amazonensis Infection Can Trigger Broad-Range Immunosuppression
As successful intracellular parasites that have a unique replication niche, it is not surprising that Leishmania have developed multifaceted strategies to subvert or suppress MΦ’s defense machinery (Osorio y Fortea et al., 2007; McConville and Naderer, 2011). It is commonly thought that L. amazonensis parasites have evolved additional mechanisms, some of which account for their uncontrolled replication in humans and in various strains of mice. Although promastigotes of L. amazonensis can transiently trigger DC and MΦ activation, the magnitude of cell activation, and the spectrum of cytokine production are often much lower than those triggered by promastigotes of L. major and Leishmania braziliensis (Xin et al., 2007; Vargas-Inchaustegui et al., 2008). Since these species-related differences are evident even at 12 h of infection in vitro (Xin et al., 2007) or at 1 week of infection in mice (Ji et al., 2003), several scenarios may account for the deficient immune responses against L. amazonensis: these parasites are either intrinsically potent in immune repression, antigenically weak in immune activation, or both. These hypotheses are supported by the findings that promastigote antigens of L. amazonensis (but not L. braziliensis) markedly inhibit mitogen- and anti-CD3-induced T cell proliferation, and that such T cell anergy cannot be reverted by exogenous cytokines (Pinheiro et al., 2004). Likewise, vaccination with antigens of L. amazonensis (but not L. braziliensis) preferentially stimulate IL-10 and TGF-β production, leading to an increased susceptibility of vaccinated BALB/c mice to L. amazonensis infection (Pinheiro et al., 2005). The mechanisms underlying the promastigote species-specific differences remain ambiguous.
The in situ and in vivo measurement of a large panel of cytokines and chemokines and their corresponding receptors during L. amazonensis infection has confirmed that the early immunosuppressive status in antigen-presenting cells sets the stage for impaired inflammatory responses and T cell responses. For example, IL-1-related cytokines (IL-1α, IL-1β, and IL-1Rα) are rapidly induced and tightly regulated in L. major-infected C57BL/6 (self-healing) mice; however, these cytokines are barely induced even at 4 weeks of infection with L. amazonensis (Ji et al., 2003). Surprisingly, while IL-1βtreatment in vitro promotes the activation of L. amazonensis-infected DCs and CD4+ T cells, such treatment actually exacerbates lesion progression and parasite growth in mice (Xin et al., 2007), suggesting that the pathogenic condition may not be readily reprogrammed by exogenous cytokines. To better understand these pathogenic T cells, the Soong group compared the frequency of cytokine-producing cells and their T cell receptor (TCR) repertoires for CD4+ T cells isolated from non-healing (L. amazonensis-infected) and self-healing (L. braziliensis-infected) mice during primary and secondary infections. These different sources of CD4+ T cells differ at the level of IFN-γ, IL-10, and IL-17 production by the same TCR Vβ-barring subsets, implying that the low magnitude of CD4+ T cell activation (rather than the altered TCR diversity) is the determining factor for non-healing disease caused by L. amazonensis (Xin et al., 2011).
Leishmania amazonensis amastigotes are superior in silent invasion, forming huge PV, and preventing the activation of NK, DC, and MΦ triggered by exogenous stimuli such as LPS (Soong, 2008; Bhardwaj et al., 2010). The intracellular events triggered by L. amazonensis infection in MΦs have been described in detail (Bhardwaj et al., 2010; McConville and Naderer, 2011). The suppression of host cell activation can be initiated at the stage of amastigote–host contact. On one hand, L. amazonensis amastigotes can infect human DCs through multiple receptor–ligand interactions such as antibodies/Fc receptors, complement components/component receptors, and proteoglycans/heparin-binding proteins (Bosetto and Giorgio, 2007), leading to altered DC activation and impaired responsiveness to exogenous stimuli through ERK/MAP kinase-mediated mechanisms (Boggiatto et al., 2009). On the other hand, L. amazonensis amastigotes can expose phosphatidylserine (PS) on their surface, leading to enhanced parasite growth and host suppression through IL-10 and TGF-β production by host cells (Wanderley et al., 2006). The levels of PS exposure are relatively high on lesion- and MΦ-derived amastigotes, but nearly absent on axenic amastigotes, and are positively correlated to host immune responses (unpublished data). Although the origin of and the conditions inducing PS exposure on amastigote surface are matters of investigation, modulating PS exposure is fundamental for repressing host immune responses and granting survival for Leishmania parasites (Wanderley and Barcinski, 2010). This “apoptotic mimicry” strategy is also important for other pathogens, including Toxoplasma gondii and Vibrio cholerae (Thanawastien et al., 2009; Dos Santos et al., 2011). No doubt, L. amazonensis can elicit a broad-spectrum suppression against host innate and adaptive immune responses, but low levels of host immune responses are required for disease progression.
L. amazonensis Parasites are Proficient in Utilizing Host Innate Immune Responses for Their Own Growth
Several lines of new evidence have revealed that L. amazonensis parasites take advantage of host innate defense machinery in neutrophils and MΦs for their own infection. The first one is related to the utilization of host type I interferons (IFN). The Soong group has shown that signaling through type I IFNs and IFN-α receptor (IFNAR) actually favors L. amazonensis infection, rather than promotes its control (Xin et al., 2010). As shown in Figure 1, L. amazonensis-infected IFNAR−/− mice developed much smaller lesions that contained lower numbers of parasites than did wild-type controls. Even at 3 days post-infection, parasite growth was restrained in IFNAR−/− mice but exacerbated in IFN-α-treated mice, implying a regulatory role for type I IFNs in this infection model. Along the same line, it is known that IFNAR−/− mice are also more resistant to intracellular bacteria such as Listeria monocytogenes and Mycobacterium tuberculosis (Rayamajhi et al., 2010a,b). In the case of L. amazonensis infection, the disease-promoting effect of type I IFNs is multifarious, correlating with the functions and interactions of neutrophils and MΦs. For instance, IFN-α prolongs the life span of un-infected and infected neutrophils, whereas the deletion of IFNAR decreases the survival of neutrophils (and most likely the survival of promastigotes; Xin et al., 2010). Importantly, the lack of IFNAR signaling promotes the differential recruitment and activation of neutrophils and inflammatory monocytes, as well as restrains parasite infection in MΦs that are co-cultured with neutrophils (Xin et al., 2010). Therefore, New World leishmaniasis caused by L. amazonensis and L. guyanensis infections are exacerbated by the production of type I IFNs (Xin et al., 2010; Ives et al., 2011). During Leishmania infection, type I IFN production can be triggered by TLR9-dependent responses to Leishmania genomic DNA or TLR3-dependent responses to Leishmania RNA virus-1 or exposure to other dsRNA viruses (Liese et al., 2007; Ives et al., 2011). The pathogenic role of type I IFNs is supported by the findings that exogenous IFN-β stimulates superoxide dismutase 1 (SOD1) and impairs the superoxide-dependent killing of L. amazonensis and L. braziliensis in human MΦ, playing a deleterious role in CL (Khouri et al., 2009). Since type I IFNs are known to play a protective role in infections against L. major , L. braziliensis, and Leishmania infantum infections (Liese et al., 2008), careful evaluation is needed for the functions of type I IFNs in murine and human leishmaniasis.
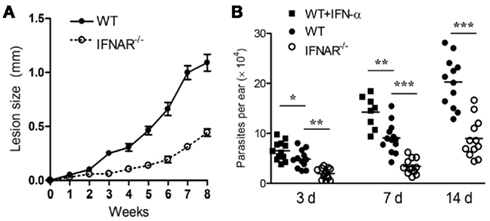
Figure 1. The IFNAR signaling promotes L. amazonensis infection in mice. (A) IFNAR−/− mice developed smaller lesions that contained lower parasite loads than did wild-type controls. (B) The infection-promoting effect of IFN-a was detectable at 3 days post-infection. The lack of IFNAR signaling restrained parasite infection at early stages of the infection. Adapted from Xin et al. (2010).
The second line of evidence is related to the utilization of TLR2-mediated responses for parasite growth. While MyD88 is required for the effective control of L. amazonensis and L. braziliensis infections, signaling throughTLR2 seems to have a regulatory role, favoring these infections (Vargas-Inchaustegui et al., 2009; Guerra et al., 2010). In the absence of TLR2, DC, and CD4+ T cells display an elevated activation and enhanced production of IL-12, IL-10, and IFN-γcytokines. Lesions of TLR2−/− mice had reduced numbers of neutrophils and MΦs, but increased numbers of eosinophils. As a consequence, TLR2−/− mice have attenuated lesions following infection with L. braziliensis (Vargas-Inchaustegui et al., 2009) and L. amazonensis (Guerra et al., 2010; unpublished data).
Mechanistically, the disease-promoting effects of IFNAR and TLR2 appear to be linked to the initial intracellular events triggered by parasite infection. The elegant studies conducted by the Lopes group have shown that infection with promastigotes of L. amazonensis leads to the activation of an interferon-dependent expression of dsRNA-dependent protein kinase (PKR), a classic antiviral protein, and that treatment of infected mouse MΦs with the synthetic dsRNA poly(I:C), a potent PKR inductor, increases parasite intracellular growth through an IL-10-mediated mechanism (Pereira et al., 2010). More recently, the same group also found that L. amazonensis and lipophosphoglycan, the major surface component of promastigotes, induce the activation of the PKR promoter by a TLR2-dependent manner, and that TLR2−/− MΦs express low IFN-β and PKR levels with reduced parasite loads, while IFN-α treatment promotes the growth of L. amazonensis promastigotes (de Carvalho Vivarini et al., 2011). Importantly, lesions of DCL patients displayed higher levels of PKR/type I IFN-expressing cells compared with those from single CL patients, indicating an important and regulatory role for type I IFNs and TLR2 in human leishmaniasis (de Carvalho Vivarini et al., 2011).
How type I IFNs and TLR2 execute their regulatory roles at the early and late stages of L. amazonensis infection remain unclear. In the cases of L. monocytogenes and M. tuberculosis infections, host susceptibility is known to be associated with the antagonistic crosstalk between type I and type II IFNs, as IFN-α/β can impair MΦ activation and their responsiveness to IFN-γ (Rayamajhi et al., 2010b). Also, type I IFN singling can influence the phenotypes of MΦ subsets, as well as their responsiveness to a range of cytokines that use STAT1 (IFN-γ, IL-10, IL-6, M-CSF, CCL5, and CXCL10; Fleetwood et al., 2009). Given that exogenous pro-inflammatory cytokines (IFN-α/β, IFN-γ, IL-1β), regulatory cytokines (TGF-β, IL-10), and insulin-like growth factor-1 all have the potential to promote L. amazonensis infection in vitro and/or in mice (Vendrame et al., 2007; Wanasen et al., 2007; Xin et al., 2007), it is reasonable to speculate that the low level and crosstalk of these host cytokines create a microenvironment, which is exploited by these parasites for efficient entry and intracellular growth.
L. amazonensis Parasites Can Exploit Host’s Lipid Metabolism and Endocytic/Autophagic Pathways
Autophagy is an ancient mechanism for recycling aged organelles or proteins, and an intrinsic antimicrobial defense for pathogen killing and antigen presentation. Signaling pathways triggered through TLR3/4/7/8 and pro-inflammatory cytokines (IFN-α/β/γ, TNF-α, IL-1β) can induce canonical macro-autophagy characterized by the double-membrane autophagosome, leading to the clearance of most viruses and bacteria and the presentation of microbial antigens (Delgado et al., 2009; Deretic, 2011). However, some pathogens can override or exploit this innate defense machinery for their favored growth (Deretic and Levine, 2009). The successful intracellular pathogens often have multipronged strategies to avoid autophagolysosomal degradation and/or dampen autophagy-dependent activation of host immune responses. In Leishmania research, autophagy is known to be critically important for parasite stage differentiation and development (Besteiro et al., 2006; Williams et al., 2009); however, it is largely unclear as to how host cell autophagy affects Leishmania infection and how replicating parasites manipulate host autophagic pathways. Several recent studies have suggested a unique ability of L. amazonensis to circumvent the host’s lipid metabolism and endocytic/autophagic pathways.
First, L. amazonensis parasites can selectively manipulate host cell lipid biosynthesis pathways. Wilson et al. (2008) have reported that L. amazonensis promastigote infection can up-regulate the lysosomal trafficking regulator gene (LYST/Beige) in MΦs, and that up- or down-regulation of the LYST/Beige gene in MΦs and fibroblasts directly modulates PV sizes and parasite growth. At present, the definitive roles for and regulation of LYST/Beige expression during in vivo infection have not been demonstrated. Recently, Osorio y Fortea et al. (2009) conducted large-scale, transcriptional signature analyses of amastigote-free MΦ and amastigote-harboring MΦ at 24 h after the uptake of nude mouse-derived L. amazonensis amastigotes. Their data indicate that amastigote infection preferentially up-regulates the function of lipid metabolism, especially the canonical metabolic pathway of biosynthesis of steroids, as well as the polyamine pathways. Therefore, while these replicating amastigotes repress host cell activation in general, they can selectively exploit the host cell’s lysosomal trafficking and sterol biosynthesis machinery for their own cell membrane sterols and for the sterol-dependent remodeling and expansion of the PV membrane. However, these transcriptomes profiles represent early host–parasite interactions in the absence of host cytokines and components that are often detected in the lesion microenvironment or on the amastigote surface.
To address the functional role of host phospholipids and ceramides in parasite infection, the Zhang group generated Leishmania parasites that are deficient in the degradation of host sphingolipid, due to the deletion of the inositol phosphosphingolipid phospholipase C-like (ISCL) protein. They found that the iscl-null mutants of L. major completely abolished their virulence in BALB/c mice, partially due to parasite’s defects in survival under an acid pH condition; however, a low number of mutant parasites could persist in the injection site for several months (Zhang et al., 2009; Xu et al., 2011). In sharp contrast, the iscl-deficient L. amazonensis are fully virulent in BALB/c mice, but are partially attenuated in C57BL/6 mice (K. Zhang, personal communication). The striking differences in disease outcomes following infection with iscl-deficient L. major and L. amazonensis in BALB/c mice suggest that ISCL is an essential virulence factor for L. major; however, L. amazonensis may have additional, ISCL-independent mechanisms for survival within the acidified PV.
Second, L. amazonensis amastigote-harboring PVs are enriched with nutrients, but are not augmented with microbicidal function of the lysosome. Osorio y Fortea et al. (2009) reported the infection-induced gene expression of eight isoforms of the V0 and V1 sub-units of the MΦ vacuolar proton ATPase, supporting the notion that amastigote-harboring PVs are highly acidified and optimal for amastigote nutrient acquisition (McConville and Naderer, 2011). Most notably, L. amazonensis amastigote-harboring PVs are quite special, as they can fuse with vacuoles that carry L. major promastigote but not amastigotes. L. major promastigotes within these chimeric PVs can temporarily survive and multiply, but fail to differentiate into amastigotes (Real et al., 2010). Therefore, the mature PV of L. amazonensis is not the compartment with a license to kill. Furthermore, L. amazonensis amastigote-harboring PVs can efficiently fuse with vacuoles that carry Coxiella burnetii, the causative agent of human Q fever. Importantly, L. amazonensis amastigotes, as well as wild-type and mutant C. burnetii deficient in their ability of lysosomal renovation, can replicate comfortably within these chimeric vacuoles (Rabinovitch and Veras, 1996; Beare et al., 2011). The findings that L. amazonensis and C. burnetii replicate in fusogenic vacuoles are intriguing. It is plausible that both pathogens can block the microbicidal function of the lysosome and acquire nutrients by continuous fusion and/or membrane transport with other compartments in the secretory and endolysosomal systems (McConville and Naderer, 2011; Newton and Roy, 2011). At this stage, it remains to be a big challenge and a big prize to cell biologists to isolate and study the protein and lipid composition of Leishmania PV. One hypothesis may be incompleteness of PV membrane for ROS producing machinery after L. amazonensis infection. An assessment of absolute NO or ROS levels in Leishmania-carrying PVs versus lysosomes and phagosomes formed after engulfment of bacteria and latex beads will shield some light in this area.
Lastly, factors that induce MΦ autophagy can promote L. amazonensis infection. In mammalian cells, autophagy induction is regulated by sequential activation of regulators, the formation of Atg complexes and autophagosomes, and the maturation of autolysosomes. Although IFN-γ treatment (10 ∼ 100 ng/ml) is known to increase PV sizes and L. amazonensis amastigote loads in mouse MΦs in an arginase-independent manner (Wanasen et al., 2007), the underlying mechanism is ambiguous. Recently, Pinheiro et al. (2009) have shown that autophagy induction via IFN-γ or starvation increased L. amazonensis but not L. major infection in BALB/c MΦs; however, this was not the case in C57BL/6 MΦs. Since L. amazonensis amastigotes do not co-localize with IFN-γ-induced, double-membrane vacuoles (Pinheiro et al., 2009), it is plausible that IFN-γ, starvation, or autophagy-inducing drugs may create a vacuolar environment that is generally permissive for engulfment and replication of the parasites. Information on whether and how Leishmania parasites alter, modulate, and utilize host autophagic pathways is needed. Additional studies with mutant parasites, in the context of genetic and/or pharmacological perturbation in the host autophagic pathway and lipid metabolism, will uncover the parasites’ intrinsic ability of utilizing host cell resources and exploiting host defense mechanisms for their favored intracellular growth.
Concluding Remarks
The emerging evidence from recent studies has uncovered the complex host–Leishmania interactions. A detrimental regulatory loop at very early stages of the infection with L. amazonensis can lead to the activation and expansion of pathogenic CD4+ T and B cells and non-healing lesions. Figure 2 is a schematic diagram, illustrating key regulatory events at the level of neutrophils and MΦs that collectively contribute to the exacerbated L. amazonensis infection. Firstly, the IFNAR-mediated signaling augments the differential recruitment and activation of neutrophils and inflammatory monocytes, as well as the prolonged survival of neutrophils, the potential Trojan horses for parasite spread to MΦ (Jochim and Teixeira, 2009). Secondly, type I IFNs contribute to parasite infection by impairing the superoxide-dependent killing of parasites in MΦ and simulating PKR-mediated production of IL-10 in infected MΦs. Thirdly, pro-inflammatory cytokines such as IFN-γcan turn on the host cell autophagic pathway and lipid metabolism, thereby creating a microenvironment that favors parasite entry or replication. Further validation of the cross-talking by using immunological, genetic, and pharmacological approaches will be important. For the study of innate immune responses in vivo, another issue of consideration is the differences in local responses triggered by needle- versus sand fly-delivered parasites (see other reviews in this Special Topic). A better understanding of how different species and stages of Leishmania parasites can selectively utilize host innate defense machinery and adoptive immunity for immune evasion, intracellular growth, and immunopathogenesis will lead to the rational design of control strategies for this and other related infections.
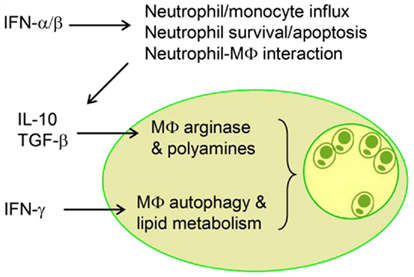
Figure 2. A schematic diagram illustrates the interplays between innate immune cells and the regulatory roles of key cytokines during infection with Leishmania amazonensis. Modulation of intracellular pathways by host and/or parasite factors can lead to enlarged vacuoles and enhanced parasite growth.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I thank Jiaren Sun for helpful discussion and Mardelle Susman for proofreading this article. This work was supported by NIH grants AI043003 and AI076849.
Abbreviations
Atg, autophagy-related genes; CL, cutaneous leishmaniasis; DCL, diffuse cutaneous leishmaniasis; IFNAR, type I interferon-α receptor; ISCL, inositol phosphosphingolipid phospholipase C-like; LYST/Beige, lysosomal trafficking regulator gene; PKR, dsRNA-dependent protein kinase; PS, phosphatidylserine; PV, parasitophorous vacuoles; SOD1, superoxide dismutase 1; TCR, T cell receptor.
References
Beare, P. A., Gilk, S. D., Larson, C. L., Hill, J., Stead, C. M., Omsland, A., Cockrell, D. C., Howe, D., Voth, D. E., and Heinzen, R. A. (2011). Dot/Icm type IVB secretion system requirements for Coxiella burnetii growth in human macrophages. MBio 2, e00175–00111.
Besteiro, S., Williams, R. A., Morrison, L. S., Coombs, G. H., and Mottram, J. C. (2006). Endosome sorting and autophagy are essential for differentiation and virulence of Leishmania major. J. Biol. Chem. 281, 11384–11396.
Bhardwaj, S., Srivastava, N., Sudan, R., and Saha, B. (2010). Leishmania interferes with host cell signaling to devise a survival strategy. J. Biomed. Biotechnol. 109189, 1–3.
Boggiatto, P. M., Jie, F., Ghosh, M., Gibson-Corley, K. N., Ramer-Tait, A. E., Jones, D. E., and Petersen, C. A. (2009). Altered dendritic cell phenotype in response to Leishmania amazonensis amastigote infection is mediated by MAP kinase, ERK. Am. J. Pathol. 174, 1818–1826.
Bosetto, M. C., and Giorgio, S. (2007). Leishmania amazonensis: multiple receptor-ligand interactions are involved in amastigote infection of human dendritic cells. Exp. Parasitol. 116, 306–310.
de Carvalho Vivarini, A., Pereira, R. D., Dias Teixeira, K. L., Calegari-Silva, T. C., Bellio, M., Laurenti, M. D., Corbett, C. E., de Castro Gomes, C. M., Soares, R. P., Mendes Silva, A., Silveira, F. T., and Lopes, U. G. (2011). Human cutaneous leishmaniasis: interferon-dependent expression of double-stranded RNA-dependent protein kinase (PKR) via TLR2. FASEB J. 25, 4162–4173.
Delgado, M., Singh, S., De Haro, S., Master, S., Ponpuak, M., Dinkins, C., Ornatowski, W., Vergne, I., and Deretic, V. (2009). Autophagy and pattern recognition receptors in innate immunity. Immunol. Rev. 227, 189–202.
Deretic, V. (2011). Autophagy in immunity and cell-autonomous defense against intracellular microbes. Immunol. Rev. 240, 92–104.
Deretic, V., and Levine, B. (2009). Autophagy, immunity, and microbial adaptations. Cell Host Microbe 5, 527–549.
Dos Santos, T. A., Portes Jde, A., Damasceno-Sa, J. C., Caldas, L. A., de Souza, W., Damatta, R. A., and Seabra, S. H. (2011). Phosphatidylserine exposure by Toxoplasma gondii is fundamental to balance the immune response granting survival of the parasite and of the host. PLoS ONE 6, e27867. doi:10.1371/journal.pone.0027867
Fleetwood, A. J., Dinh, H., Cook, A. D., Hertzog, P. J., and Hamilton, J. A. (2009). GM-CSF- and M-CSF-dependent macrophage phenotypes display differential dependence on type I interferon signaling. J. Leukoc. Biol. 86, 411–421.
Guerra, C. S., Silva, R. M., Carvalho, L. O., Calabrese, K. S., Bozza, P. T., and Corte-Real, S. (2010). Histopathological analysis of initial cellular response in TLR-2 deficient mice experimentally infected by Leishmania (L.) amazonensis. Int. J. Exp. Pathol. 91, 451–459.
Ives, A., Ronet, C., Prevel, F., Ruzzante, G., Fuertes-Marraco, S., Schutz, F., Zangger, H., Revaz-Breton, M., Lye, L. F., Hickerson, S. M., Beverley, S. M., Acha-Orbea, H., Launois, P., Fasel, N., and Masina, S. (2011). Leishmania RNA virus controls the severity of mucocutaneous leishmaniasis. Science 331, 775–778.
Ji, J., Sun, J., and Soong, L. (2003). Impaired expression of inflammatory cytokines and chemokines at early stages of infection with Leishmania amazonensis. Infect. Immun. 71, 4278–4288.
Jochim, R. C., and Teixeira, C. (2009). Leishmania commandeers the host inflammatory response through neutrophils. Trends Parasitol. 25, 145–147.
Khandelwal, K., Bumb, R. A., Mehta, R. D., Kaushal, H., Lezama-Davila, C., Salotra, P., and Satoskar, A. R. (2011). A patient presenting with diffuse cutaneous leishmaniasis (DCL) as a first indicator of HIV infection in India. Am. J. Trop. Med. Hyg. 85, 64–65.
Khouri, R., Bafica, A., Silva Mda, P., Noronha, A., Kolb, J. P., Wietzerbin, J., Barral, A., Barral-Netto, M., and Van Weyenbergh, J. (2009). IFN-beta impairs superoxide-dependent parasite killing in human macrophages: evidence for a deleterious role of SOD1 in cutaneous leishmaniasis. J. Immunol. 182, 2525–2531.
Liese, J., Schleicher, U., and Bogdan, C. (2007). TLR9 signaling is essential for the innate NK cell response in murine cutaneous leishmaniasis. Eur. J. Immunol. 37, 3424–3434.
Liese, J., Schleicher, U., and Bogdan, C. (2008). The innate immune response against Leishmania parasites. Immunobiology 213, 377–387.
McConville, M. J., and Naderer, T. (2011). Metabolic pathways required for the intracellular survival of Leishmania. Annu. Rev. Microbiol. 65, 543–561.
McMahon-Pratt, D., and Alexander, J. (2004). Does the Leishmania major paradigm of pathogenesis and protection hold for New World cutaneous leishmaniases or the visceral disease? Immunol. Rev. 201, 206–224.
Mukbel, R. M., Patten, C. Jr., Gibson, K., Ghosh, M., Petersen, C., and Jones, D. E. (2007). Macrophage killing of Leishmania amazonensis amastigotes requires both nitric oxide and superoxide. Am. J. Trop. Med. Hyg. 76, 669–675.
Newton, H. J., and Roy, C. R. (2011). The Coxiella burnetii Dot/Icm system creates a comfortable home through lysosomal renovation. MBio. e00226-11, 1–3.
Niamba, P., Goumbri-Lompo, O., Traore, A., Barro-Traore, F., and Soudre, R. T. (2007). Diffuse cutaneous leishmaniasis in an HIV-positive patient in western Africa. Australas. J. Dermatol. 48, 32–34.
Osorio y Fortea, J., de La Llave, E., Regnault, B., Coppee, J. Y., Milon, G., Lang, T., and Prina, E. (2009). Transcriptional signatures of BALB/c mouse macrophages housing multiplying Leishmania amazonensis amastigotes. BMC Genomics 10, 119. doi:10.1186/1471-2164-10-119
Osorio y Fortea, J., Prina, E., de La Llave, E., Lecoeur, H., Lang, T., and Milon, G. (2007). Unveiling pathways used by Leishmania amazonensis amastigotes to subvert macrophage function. Immunol. Rev. 219, 66–74.
Pereira, R. M., Teixeira, K. L., Barreto-de-Souza, V., Calegari-Silva, T. C., De-Melo, L. D., Soares, D. C., Bou-Habib, D. C., Silva, A. M., Saraiva, E. M., and Lopes, U. G. (2010). Novel role for the double-stranded RNA-activated protein kinase PKR: modulation of macrophage infection by the protozoan parasite Leishmania. FASEB J. 24, 617–626.
Pinheiro, R. O., Nunes, M. P., Pinheiro, C. S., D’Avila, H., Bozza, P. T., Takiya, C. M., Corte-Real, S., Freire-de-Lima, C. G., and DosReis, G. A. (2009). Induction of autophagy correlates with increased parasite load of Leishmania amazonensis in BALB/c but not C57BL/6 macrophages. Microbes Infect. 11, 181–190.
Pinheiro, R. O., Pinto, E. F., Benedito, A. B., Lopes, U. G., and Rossi-Bergmann, B. (2004). The T-cell anergy induced by Leishmania amazonensis antigens is related with defective antigen presentation and apoptosis. An. Acad. Bras. Cienc. 76, 519–527.
Pinheiro, R. O., Pinto, E. F., Lopes, J. R., Guedes, H. L., Fentanes, R. F., and Rossi-Bergmann, B. (2005). TGF-β-associated enhanced susceptibility to leishmaniasis following intramuscular vaccination of mice with Leishmania amazonensis antigens. Microbes Infect. 7, 1317–1323.
Rabinovitch, M., and Veras, P. S. (1996). Cohabitation of Leishmania amazonensis and Coxiella burnetii. Trends Microbiol. 4, 158–161.
Rayamajhi, M., Humann, J., Penheiter, K., Andreasen, K., and Lenz, L. L. (2010a). Induction of IFN-α/β enables Listeria monocytogenes to suppress macrophage activation by IFN-γ. J. Exp. Med. 207, 327–337.
Rayamajhi, M., Humann, J., Kearney, S., Hill, K. K., and Lenz, L. L. (2010b). Antagonistic crosstalk between type I and II interferons and increased host susceptibility to bacterial infections. Virulence 1, 418–422.
Real, F., Mortara, R. A., and Rabinovitch, M. (2010). Fusion between Leishmania amazonensis and Leishmania major parasitophorous vacuoles: live imaging of coinfected macrophages. PLoS Negl. Trop. Dis. 4, e905. doi:10.1371/journal.pntd.0000905
Silveira, F. T., Lainson, R., and Corbett, C. E. (2004). Clinical and immunopathological spectrum of American cutaneous leishmaniasis with special reference to the disease in Amazonian Brazil: a review. Mem. Inst. Oswaldo Cruz 99, 239–251.
Silveira, F. T., Lainson, R., De Castro Gomes, C. M., Laurenti, M. D., and Corbett, C. E. (2009). Immunopathogenic competences of Leishmania (V.) braziliensis and L. (L.) amazonensis in American cutaneous leishmaniasis. Parasite Immunol. 31, 423–431.
Soong, L. (2008). Modulation of dendritic cell function by Leishmania parasites. J. Immunol. 180, 4355–4360.
Soong, L., Chang, C. H., Sun, J., Longley, B. J. Jr., Ruddle, N. H., Flavell, R. A., and McMahon-Pratt, D. (1997). Role of CD4+ T cells in pathogenesis associated with Leishmania amazonensis infection. J. Immunol. 158, 5374–5383.
Thanawastien, A., Montor, W. R., Labaer, J., Mekalanos, J. J., and Yoon, S. S. (2009). Vibrio cholerae proteome-wide screen for immunostimulatory proteins identifies phosphatidylserine decarboxylase as a novel Toll-like receptor 4 agonist. PLoS Pathog. 5, e1000556. doi:10.1371/journal.ppat.1000556
Vargas-Inchaustegui, D. A., Tai, W., Xin, L., Hogg, A. E., Corry, D. B., and Soong, L. (2009). Distinct roles for MyD88 and Toll-like receptor 2 during Leishmania braziliensis infection in mice. Infect. Immun. 77, 2948–2956.
Vargas-Inchaustegui, D. A., Xin, L., and Soong, L. (2008). Leishmania braziliensis infection induces dendritic cell activation, ISG15 transcription, and the generation of protective immune responses. J. Immunol. 180, 7537–7545.
Vendrame, C. M., Carvalho, M. D., Rios, F. J., Manuli, E. R., Petitto-Assis, F., and Goto, H. (2007). Effect of insulin-like growth factor-I on Leishmania amazonensis promastigote arginase activation and reciprocal inhibition of NOS2 pathway in macrophage in vitro. Scand. J. Immunol. 66, 287–296.
Wanasen, N., MacLeod, C. L., Ellies, L. G., and Soong, L. (2007). L-arginine and cationic amino acid transporter 2B regulate growth and survival of Leishmania amazonensis amastigotes in macrophages. Infect. Immun. 75, 2802–2810.
Wanderley, J. L., and Barcinski, M. A. (2010). Apoptosis and apoptotic mimicry: the Leishmania connection. Cell. Mol. Life Sci. 67, 1653–1659.
Wanderley, J. L., Moreira, M. E., Benjamin, A., Bonomo, A. C., and Barcinski, M. A. (2006). Mimicry of apoptotic cells by exposing phosphatidylserine participates in the establishment of amastigotes of Leishmania (L) amazonensis in mammalian hosts. J. Immunol. 176, 1834–1839.
Wang, Y., Chen, Y., Xin, L., Beverley, S. M., Carlsen, E. D., Popov, V., Chang, K. P., Wang, M., and Soong, L. (2011). Differential microbicidal effects of human histone proteins H2A and H2B on Leishmania promastigotes and amastigotes. Infect. Immun. 79, 1124–1133.
Williams, R. A., Woods, K. L., Juliano, L., Mottram, J. C., and Coombs, G. H. (2009). Characterization of unusual families of ATG8-like proteins and ATG12 in the protozoan parasite Leishmania major. Autophagy 5, 159–172.
Wilson, J., Huynh, C., Kennedy, K. A., Ward, D. M., Kaplan, J., Aderem, A., and Andrews, N. W. (2008). Control of parasitophorous vacuole expansion by LYST/Beige restricts the intracellular growth of Leishmania amazonensis. PLoS Pathog. 4, e1000179. doi:10.1371/journal.ppat.1000179
Xin, L., Li, Y., and Soong, L. (2007). Role of interleukin-1β in activating the CD11chigh CD45RB- dendritic cell subset and priming Leishmania amazonensis-specific CD4+ T cells in vitro and in vivo. Infect. Immun. 75, 5018–5026.
Xin, L., Vargas-Inchaustegui, D. A., Raimer, S. S., Kelly, B. C., Hu, J., Zhu, L., Sun, J., and Soong, L. (2010). Type I IFN receptor regulates neutrophil functions and innate immunity to Leishmania parasites. J. Immunol. 184, 7047–7056.
Xin, L., Wanderley, J. L., Wang, Y., Vargas-Inchaustegui, D. A., and Soong, L. (2011). The magnitude of CD4+ T-cell activation rather than TCR diversity determines the outcome of Leishmania infection in mice. Parasite Immunol. 33, 170–180.
Xu, W., Xin, L., Soong, L., and Zhang, K. (2011). Sphingolipid degradation by Leishmania major is required for its resistance to acidic pH in the mammalian host. Infect. Immun. 79, 3377–3387.
Keywords: Leishmania amazonensis, innate immunity, parasite adaptation, immunopathogenesis
Citation: Soong L (2012) Subversion and utilization of host innate defense by Leishmania amazonensis. Front. Immun. 3:58. doi: 10.3389/fimmu.2012.00058
Received: 04 January 2012; Accepted: 06 March 2012;
Published online: 21 March 2012.
Edited by:
Nathan Peters, National Institute of Allergy and Infectious Diseases, USAReviewed by:
Abhay Satoskar, The Ohio State University, USASujoy Dutta, Rosalind Franklin University, USA
Copyright: © 2012 Soong. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Lynn Soong, Department of Microbiology and Immunology, The University of Texas Medical Branch, Medical Research Building 3.132, Galveston, TX 77555-1070, USA. e-mail:bHlzb29uZ0B1dG1iLmVkdQ==