
- Department of Biological Sciences, George Washington University, Washington, DC, USA
The California purple sea urchin, Strongylocentrotus purpuratus, is a long-lived echinoderm with a complex and sophisticated innate immune system. There are several large gene families that function in immunity in this species including the Sp185/333 gene family that has ∼50 (±10) members. The family shows intriguing sequence diversity and encodes a broad array of diverse yet similar proteins. The genes have two exons of which the second encodes the mature protein and has repeats and blocks of sequence called elements. Mosaics of element patterns plus single nucleotide polymorphisms-based variants of the elements result in significant sequence diversity among the genes yet maintains similar structure among the members of the family. Sequence of a bacterial artificial chromosome insert shows a cluster of six, tightly linked Sp185/333 genes that are flanked by GA microsatellites. The sequences between the GA microsatellites in which the Sp185/333 genes and flanking regions are located, are much more similar to each other than are the sequences outside the microsatellites suggesting processes such as gene conversion, recombination, or duplication. However, close linkage does not correspond with greater sequence similarity compared to randomly cloned and sequenced genes that are unlikely to be linked. There are three segmental duplications that are bounded by GAT microsatellites and include three almost identical genes plus flanking regions. RNA editing is detectible throughout the mRNAs based on comparisons to the genes, which, in combination with putative post-translational modifications to the proteins, results in broad arrays of Sp185/333 proteins that differ among individuals. The mature proteins have an N-terminal glycine-rich region, a central RGD motif, and a C-terminal histidine-rich region. The Sp185/333 proteins are localized to the cell surface and are found within vesicles in subsets of polygonal and small phagocytes. The coelomocyte proteome shows full-length and truncated proteins, including some with missense sequence. Current results suggest that both native Sp185/333 proteins and a recombinant protein bind bacteria and are likely important in sea urchin innate immunity.
Introduction
Sea Urchins and Echinoderms
The phylum Echinodermata is phylogenetically positioned within the deuterostome lineage of animals that includes the Chordata and a few minor invertebrate phyla. There are five extant classes of echinoderms, of which the echinoid class includes sea urchins and sand dollars (Figure 1). Immunologists initially assumed that all animals had adaptive immune functions like mammals and worked toward demonstrating these characteristics in many species of animals including invertebrates. Initial investigations to determine the immune capabilities in echinoderms employed allograft rejection assays (Hildemann and Dix, 1972; Karp and Hildemann, 1976; Coffaro and Hinegardner, 1977; Coffaro, 1980; Varadarajan and Karp, 1983). However, the rejection kinetics of first set, second set, and third party allografts in the sea urchin, Lytechinus pictus (Coffaro and Hinegardner, 1977; Coffaro, 1979), showed that the response did not demonstrate specific immune recognition and immune memory and was therefore solely innate (Smith and Davidson, 1992). The initial assumptions that immunologists held about universal functions of adaptive immunity were very wrong.

Figure 1. An adult California purple sea urchin, Strongylocentrotus purpuratus. Image kindly provided by Hung-Yen Chou and Yen-Lin Kuo.
The cellular mediators of immunity in echinoderms are coelomocytes that are present in the fluid that fills the coelomic cavity (Boolootian and Geise, 1958; Johnson, 1969a; Smith, 1981; reviewed in Smith et al., 2010). Coelomocyte immune function was first recognized by Metchnikoff (1893) when he inserted rose prickles, glass rods, and bacteria into the blastocoel of larval sea stars and observed either encapsulation or phagocytosis by the blastocoelar cells (larval equivalents of coelomocytes). More recent analysis of the clearance of foreign cells and particles from adult and larval echinoderms shows that it is swift, efficient, and is mediated by the phagocytic cells in the coelomic and blastocoelar cavities (Reinisch and Bank, 1971; Yui and Bayne, 1983; Plytycz and Seljelid, 1993; Silva, 2000; Furukawa et al., 2009) In the purple sea urchin, there are three classes of coelomocytes: phagocytes, spherule cells, and vibratile cells (Smith et al., 2010; Figure 2). The phagocytes are composed of three morphotypes that vary in size and shape. The large phagocytes include the polygonal and discoidal cells that have different morphologies when spread on glass (Henson et al., 1992, 1999; Edds, 1993). The cytoskeleton of the polygonal phagocytes is shaped by parallel bundles of actin that give the cell an angular, polygonal shape. Discoidal phagocytes, which are slightly smaller than the polygonal cells, have bundles of actin that are aligned radially from the nucleus to the periphery resulting in a disk-like or “fried egg” morphology (Figures 2 and 3). Small phagocytes are significantly smaller than the large phagocytes and have perpetual filopodial morphology (Gross et al., 2000; Brockton et al., 2008; Figures 2–4). Large phagocytes, on the other hand, readily modify their morphology and appear as lamellipodial or filopodial depending on the microenvironmental conditions of the coelomic cavity (Edds, 1977, 1979, 1993; Henson et al., 1999). The spherule cells include cells with colorless inclusions and cells with red spherules that contain echinochrome A, which has antibacterial and antifungal activity (Johnson and Chapman, 1970; Messer and Wardlaw, 1980; Service and Wardlaw, 1984; Calestani et al., 2003). The vibratile cells have a single flagellum, are highly motile, and may be involved with clotting reactions (Johnson, 1969b; Bertheussen and Seijelid, 1978; Sacchi and Smith, unpublished). Immune functions mediated by coelomocytes include phagocytic activities of the large phagocytes, encapsulation by many types of cells, degranulation of echinochrome A from red spherule cells in response to bacterial contact (Johnson, 1969b; Johnson and Chapman, 1970) and injury (Coffaro and Hinegardner, 1977), and secretion of the complement C3 homolog by a subset of the discoidal cells (Gross et al., 2000; reviewed in Smith et al., 2010). Unfractionated coelomocytes from the sea urchin Paracentrotus lividus, show cytotoxic activity against red blood cells and K562 tumor cells, a function that may be mediated by the colorless spherule cells that is augmented by the presence of phagocytes (Arizza et al., 2007).
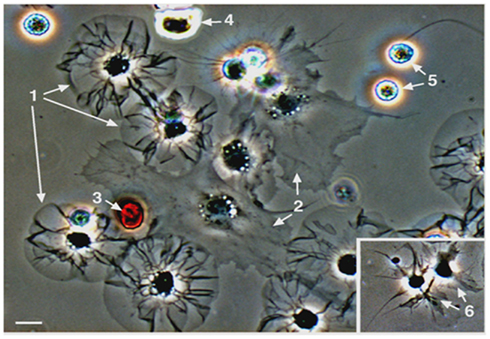
Figure 2. Sea urchin coelomocytes. Coelomocytes from the green sea urchin, Strongylocentrotus droebachiensis, were settled onto a glass coverslip and imaged live. Cell types that are typical of echinoids include discoidal phagocytes (1), polygonal phagocytes (2), red spherule cells (3), colorless spherule cells (4), vibratile cells (5; the lower cell has lost the prominent flagellum that is present in the upper cell), and small phagocytes (6, inset). Bar = 10 μm. This is Figure 2 from Smith et al. (2010), reproduced with permission from Landes Bioscience and Springer Science + Business Media.
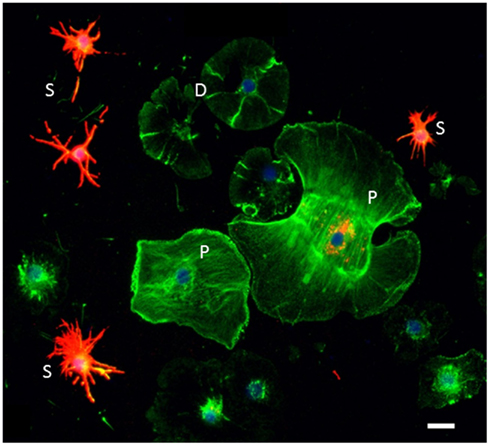
Figure 3. Sp185/333 proteins are expressed by subsets of small phagocytes and polygonal phagocytes. Phagocytes are labeled for actin (green), Sp185/333 proteins (red), and DNA (blue). Small phagocytes (S) have different filopodial morphology and actin organization than the discoidal phagocytes (D) or polygonal phagocytes (P). A subset of small phagocytes are strongly labeled for Sp185/333 proteins. The larger polygonal cell has perinuclear vesicles that are Sp185/333+. Bar = 10 μm. This is Figure 1D reproduced from Brockton et al. (2008).
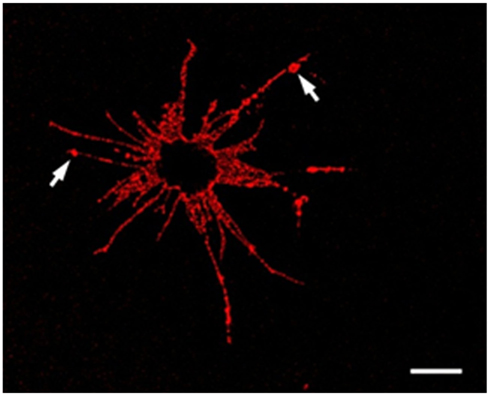
Figure 4. An Sp185/333+ Small Phagocyte. Confocal image of a Sp185/333+ small phagocyte labeled with anti-Sp185/333 sera prior to fixation shows the filopodial morphology of the cell and indicates the presence of Sp185/333 proteins on the cell surface. This includes knobs on the filopodia that are strongly positive for Sp185/333 (arrows). The dark area in the center of the cell is the location of the nucleus. Bar = 10 μm. This is Figure 5B reproduced from Brockton et al. (2008).
Early work to analyze gene expression in coelomocytes under conditions of immune challenge identified homologs of complement components including putative complement regulatory proteins, in addition to genes encoding transcription factors, lectins, ions channels, a Tie receptor homolog, lysosomal enzymes, cytoskeletal proteins, folding chaperones, mitochondrial enzymes, proteins that function in RNA splicing, signaling pathways, and secretion, plus a large number of unknowns (Smith et al., 1996, 1998; Al-Sharif et al., 1998; Rast et al., 2000; Multerer and Smith, 2004; Nair et al., 2005; Stevens et al., 2010). Many of the expressed sequence tags (ESTs) correlate with gene models annotated in the genome of the purple sea urchin, although the major finding from genome annotation was the striking level of complexity and sophistication of this invertebrate innate immune system (Hibino et al., 2006). For example, large gene families that are homologous to those in vertebrates that function in innate immune detection, such as the Toll-like receptors, NOD-like receptors and scavenger receptors are greatly expanded in the purple sea urchin (Rast et al., 2006; Rast and Messier-Solek, 2008). One gene family that is highly up-regulated in response to immune challenge is the Sp185/333 family (Rast et al., 2000; Nair et al., 2005). This family has been estimated to have about 50 ± 10 gene loci using a variety of methods including quantitative PCR of genomic DNA, a statistical estimate based on the number of unique genes cloned and sequenced from three sea urchins, and an estimate from screens of two arrayed bacterial artificial chromosome (BAC) libraries (Terwilliger et al., 2006; Buckley et al., 2008a; Ghosh et al., 2010). BLAST analysis of 1025 randomly chosen cDNA clones that were up-regulated in response to lipopolysaccharide (LPS) showed that 73% matched to two sequences on GenBank; DD185 (Rast et al., 2000) and EST333 (Smith et al., 1996). Hence the name for the set of cDNAs, Sp185/333, which was chosen because none of the sequences matched to proteins with known function and homologs have not been identified in organisms outside of echinoids.
The Sp185/333 Genes
Gene Structure
Initial speculation on the Sp185/333 gene structure was based on the extraordinary structure of the cDNAs (Nair et al., 2005). Optimal amino acid alignments of the translated cDNAs required artificial insertions of large gaps (see below), which seemed consistent with extensive alternative splicing of transcripts from one or a few large genes with many exons, similar to that for Dscam in arthropods (Schmucker et al., 2000; Watson et al., 2005; Dong et al., 2006; Brites et al., 2008; Schmucker and Chen, 2009). This prediction of large genes with many exons might have been consistent with Southern blots if all of the positive bands had been large (Figure 5). However, genomic DNA digested with PstI released small fragments that were hybridized by probes from both the 5′ and 3′ ends of the cDNAs (Terwilliger et al., 2006). Furthermore, small amplicons were produced by PCR of genomic DNA using primers that hybridized to the ends of the cDNAs (Buckley and Smith, 2007). These results suggested that the cDNAs were expressed from many small genes rather than a few large genes with many exons. Accordingly, preliminary genome sequences from the purple sea urchin confirmed that the Sp185/333 genes were small (1.2–2 kb) and had only two exons (Terwilliger et al., 2006; Figure 6). The first exon is very short (51 or 54 nucleotides), encodes the hydrophobic leader, and is followed by an intron of 380–413 nucleotides. The second exon encodes the mature protein and has the same unusual structure as the cDNAs, which is evident from alignments of the genes (Figure 6). Our initial speculation that alternative splicing generated the structures of the cDNAs is clearly improbable given that the genes only have two exons. Furthermore, splicing signals that might function within the intron or second exon to generate alternatively spliced transcripts are not present in the Sp185/333 sequences (Buckley et al., 2009).
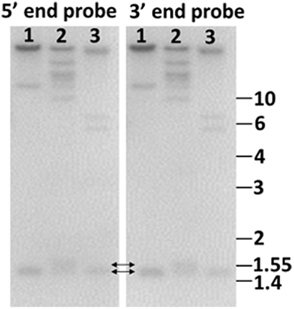
Figure 5. Genome blot of Sp185/333 genes indicates that the genes are small. Probes for the 5′ end and the 3′ end of the second exon hybridize to the same bands. This includes bands that are 1.4–1.55 kb (double headed arrows), demonstrating that the genes are small. Genomic DNA from three sea urchins (1, 2, 3) was digested to completion with PstI and separated by electrophoresis. Duplicate gels were blotted onto nylon membranes and analyzed with 32P-labeled riboprobes according to Terwilliger et al. (2006). Cloned templates used to generate the riboprobes were amplified by PCR from genomic DNA using primers that hybridized to elements 1 and 7 (5′ probe), and elements 7 and 25 (3′ probe). See Figure 6A for element positions. Size standards in kilobase are shown to the right. (Terwilliger and Smith, unpublished).
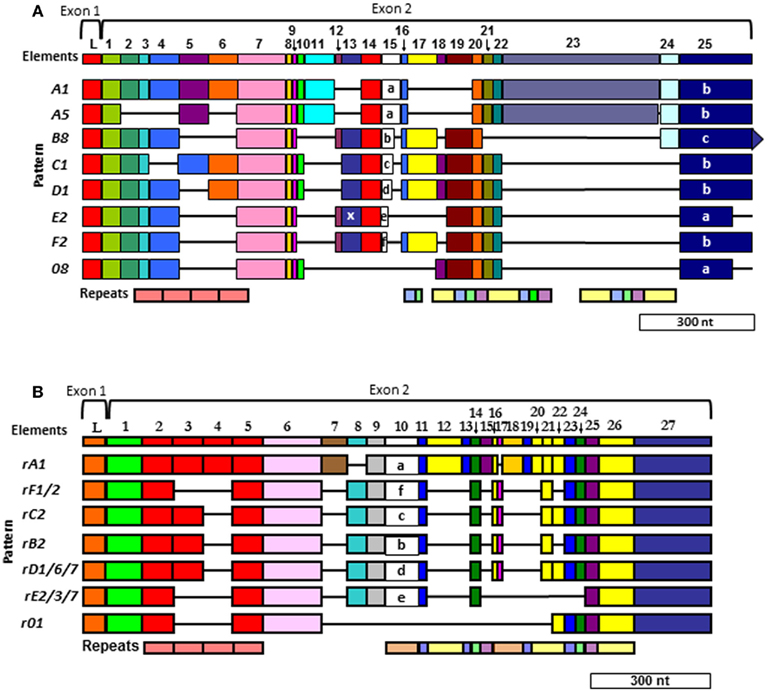
Figure 6. Two different alignments for Sp185/333 sequences are equally optimal. (A) The cDNA alignment was initially done with ESTs and full-length cDNA sequences (Terwilliger et al., 2006, 2007). (B) The Repeat-based alignment optimizes correspondence between repeats and elements whenever possible (Buckley and Smith, 2007). Optimal alignments require the insertion of artificial gaps (horizontal black lines) that delineate individual elements shown as colored blocks (the consensus of all elements are numbered across the top of each alignment; L, leader). Different element patterns are based on the variable presence or absence of elements. Designations of element patterns are listed to the left of each alignment. There are three types of element 25 in (A) (a, b, and c), which are defined by the location of three possible stop codons. A common, single nucleotide RNA edit in element pattern E2 alters a glycine codon to a stop (X) in element 13 and is denoted as an E2.1 pattern. Repeats, shown at the bottom of (A,B), are shows as different colors and occur as tandem repeats and interspersed tandem repeats. The genes have two exons (brackets at the top of each alignment), of which the first encodes the leader and the second encodes the mature protein including all of the elements. A single intron of ∼400 nt is positioned between the leader and the first element and is not shown. Modified from Ghosh et al. (2010) with permission from Elsevier.
Two Alignments
The gaps that are required to align optimally the Sp185/333 genes and cDNAs define recognizable blocks of sequence called elements (Figure 6). There are 25–27 different elements depending on the alignment that are shared among genes and cDNAs. They are variably present or absent within different genes and cDNAs resulting in recognizable mosaics of elements, called element patterns. Note that elements are not the result of alternative splicing during processing of the mRNAs, but are present within the second exon of the genes. Based on alignments of 121 unique gene sequences (of 171 sequenced genes), 31 different element patterns have been identified from three sea urchins (Buckley and Smith, 2007). When both genes and cDNA sequences from 16 individuals are evaluated together, 51 different element patterns can be identified. Although each type of element has a recognizable sequence, there are variants of each type that show sequence diversity. There can be between 1 and 28 (average = 11/element) different sequence versions for a given element based on genes cloned from individual animals (Buckley et al., 2008a). In addition to the complexity of the elements and element patterns, there are six types of repeats in the second exon of the genes (Nair et al., 2005; Terwilliger et al., 2006; Buckley et al., 2008a). Depending on the gene, there are two to four imperfect type 1 repeats near the 5′ end of the second exon that are direct, tandem repeats (Figure 6). Toward the 3′ end of the exon, there are five additional types of repeats that occur in an interspersed and partially duplicated pattern. Because of the repeats, the Sp185/333 sequences can be aligned in two different ways that are equally optimal. Initially, the alignment was done with cDNA sequences and no attempt was made to correlate the elements with the repeats (Terwilliger et al., 2006, 2007; Figure 6A). The second alignment was done with both cDNA and gene sequences and efforts were made to make the repeats and elements correspond as much as possible (Buckley and Smith, 2007; Figure 6B). The two alignments are somewhat similar for the type 1 tandem repeats, but are quite different in the region of the interspersed repeats near the 3′ end of the exon with different numbers of interspersed repeats in the two alignments. Comparisons between the two alignments shows that some of the sequences could be interdigitated, essentially collapsing some of the repeats together, which reduced their numbers in the repeat-based alignment. (Compare the interspersed repeats shown in Figures 6A,B. For a detailed illustration, see Buckley and Smith, 2007 and Figure 7 within that publication.) Although the number of elements in the two alignments differ because of the collapse of some of the repeats (25 vs. 27 elements), the overall length of the two alignments is similar and both are about equally optimal (Buckley and Smith, 2007).
Origins of Gene Structure
The extraordinary sequence diversity of the Sp185/333 gene family is due primarily to the mosaic element patterns in the second exon. Sequence variations within individual elements adds a second level of sequence diversity to the genes. The diverse structure of the genes is extraordinary and comparisons of full-length sequences among three sea urchins has not identified an identical shared gene (Buckley et al., 2008a). Yet surprisingly, 28% of individual element sequences are shared among genes from two or more animals (Figure 7). This paradox of element sequences shared among genes, but genes that are not shared among individuals, is quite remarkable and hints at an intriguing mechanism to generate the diversity within this gene family. Initial efforts to determine whether the type 1 repeats might be involved in generating diversity predicted three theoretical ancestral type 1 repeats (5′ end of the second exon; Buckley et al., 2008a). A computational estimate of the evolutionary history suggests that the extant type 1 repeats may have been derived from these ancestral sequences through modification by recombination, duplication, deletion, and point mutations. In the repeat-based alignment, the five types of interspersed repeats in the 3′ end of the exon are present in a block that is repeated twice with an extra type 4 repeat suggesting origins from duplication events (Figure 6B). Because there is no correlation between the patterns of tandem type 1 repeats and the patterns of the interspersed repeats in the 3′ end of the exon, this suggests swift recombination among genes plus recombination among the repeats within the genes. Several examples of putative gene recombination could be identified because different regions of a few genes matched optimally to corresponding regions of two other genes. Because the recombination rate among Sp185/333 genes may be quite high, identifying the ancestral genes that contributed sequence to a given recombinant gene is quite difficult. Consequently, sequences evaluation to identify recombination sites or hotspots using incongruence length difference (ILD; Michevich and Ferris, 1981) and incongruence permutation (IP; Huelsenbeck and Bull, 1996) both show that recombination is likely to occur at any position within and between elements and repeats and that there are no recombination hotspots (Buckley et al., 2008a). The level of putative gene recombination among members of the Sp185/333 gene family is swift, appearing surprisingly similar to somatic recombination of gene segments that is known to occur for T cell receptor genes (Siu et al., 1984). Given that these genes appear to encode proteins with immune effector function, ongoing diversification, based in part by gene recombination, will be advantageous for the host in the arms race with pathogens.
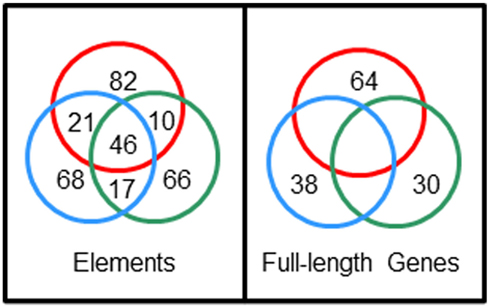
Figure 7. Sp185/333 gene sequences are not shared among sea urchins but element sequences are shared among genes. Full-length genes were cloned and sequenced from three sea urchins; 38 unique genes from animal 10 (blue circle), 64 unique genes from animal 2 (red circle), 30 unique genes from animal 4 (green circle). Nucleotide sequences were compared among full-length genes and among individual elements, and the numbers of shared unique sequences are shown in the intersections of the circles. Unpublished figure provided by Katherine Buckley.
Age Estimate for the Gene Family
Given the apparently swift recombination of the Sp185/333 genes, molecular clock analysis of the evolutionary history of the Sp185/333 family suggests that the last common ancestral Sp185/333 sequence of the extant genes may have been present in the population of purple sea urchins 2.7–10 million years ago (Ghosh et al., 2010). This time frame overlaps with the estimated time period when Strongylocentrotus purpuratus diverged from its sister species (Grula et al., 1982; Biermann, 1998; Lee, 2003; Buckley et al., 2008a). When trace sequences of 185/333 genes from the genomes of two other sea urchins, S. franciscanus and Allocentrotus fragilis are compared to the Sp185/333 sequences from S. purpuratus, molecular clock analysis shows that the divergence times for these three gene families appear older than the three species of sea urchins in which they function. This apparent paradox is another illustration of a rapid rate of evolution of the 185/333 genes in all three species, a characteristic that is consistent with immune function, participation in an arms race with pathogens, and mechanisms to promote sequence diversification. All three of these echinoid species live in the marine environment at the intersection of water and sediment. This habitat is estimated to have 105–106 microbes per milliliter of sea water with two or three orders of magnitude more in the sediment (Massana et al., 1997; Llobet-Brossa et al., 1998; Whitman et al., 1998). It is likely that some of the microbes that co-habit with sea urchins are pathogens or opportunists and function as selection pressure to drive the diversification of the Sp185/333 gene family and the complexity of the innate immune system in these invertebrates.
Size and Organization of the Sp185/333 Gene Family
The three different approaches used to estimate the size of the Sp185/333 family included qPCR of genomic DNA, a computational likelihood method to evaluate sequenced genes, and screens of two BAC libraries. Results from each evaluation was in general agreement and provided a family size of ∼50 ± 10 genes (Buckley et al., 2008a; Ghosh et al., 2010). Quantitative PCR of genomic DNA samples from three sea urchins indicated 40–60 genes (Terwilliger et al., 2006). A computational likelihood method evaluates the probability of sequencing the same gene twice given the total number of genes cloned and sequenced and resulted in an estimate of 45–71 genes (Buckley et al., 2008a). Screens of two BAC libraries for clones with Sp185/333 sequences estimated a haploid genomic region of 200–250 kb in which the genes might be located, given the fold coverage of the genome for each library, the average insert size for the BACs, and the number of positive clones obtained (Buckley et al., 2008a). For a region of 200–250 kb, if all the genes (∼2 kb) in the haploid genome are closely linked and the intergenic region is ∼3 kb, this estimates ∼40–50 genes. However, evaluation of the purple sea urchin genome assembly (including build versions 0.5, 2.1, and 3.1) shows only five or six Sp185/333 genes, depending on the version, that are positioned on a few separate scaffolds. Based on the level of sequence diversity that has been identified from 171 genes cloned and sequenced from three individuals (Buckley and Smith, 2007), six Sp185/333 genes assembled in the genome is insufficient to explain this diversity, and does not agree with the our estimated number of ∼50 genes in the family. The under-representation of Sp185/333 genes assembled in the genome is likely the result of a computational collapse of similar sequence reads into consensus genes based on the shortcomings of shotgun assembly methods that incorrectly assemble regions that appear as repeats (Sodergren et al., 2006; Ghosh et al., 2010; Miller et al., 2010).
To begin to understand the Sp185/333 gene family structure, one BAC clone (GenBank accession number AC178508.1) was re-sequenced and independently assembled (GenBank accession number 7096) with careful attention given to avoiding artifacts introduced by the assembly methods (Miller et al., 2010). The 7096 version of the BAC assembly that was experimentally validated, shows six Sp185/333 genes closely linked within 34 kb near one end of the BAC insert (Figure 8). Five of the genes are tightly clustered within 20 kb and the sixth is located 14 kb away. The outer genes are oriented in the same direction, while the four internal genes are oriented in the opposite direction with intergenic regions as short as 3.2 kb. The element patterns of the genes in the cluster are, in order, A2γ, B8β, three D1α and E2δ (Greek letters indicate intron class; see Buckley and Smith, 2007). These genes do not match the genes assembled in the sea urchin genome. If gene duplication and/or gene conversion functions in generating the diversity observed within the Sp185/333 gene family, closely linked genes would be expected to show more sequence similarity than unlinked genes. However, the diversity of the linked genes on the 7096 BAC is not different from the diversity of 121 unique genes that have been randomly isolated and sequenced from three sea urchins (Miller et al., 2010). Proximity is not associated with sequence similarity, which also holds true for the three linked D1 genes in the BAC compared to randomly cloned and sequenced D1 genes from three different animals. This suggests that if gene conversion is involved with sequence diversification, genes may be sharing sequences from the entire family rather than just those that are most tightly linked and/or that conversion tracts may be shorter than the length of a gene.
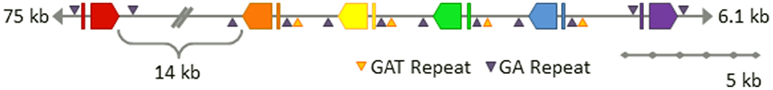
Figure 8. Six Sp185/333 genes are linked on the 7096 BAC insert. The finished-level assembly of the region containing the Sp185/333 genes was experimentally confirmed by PCR, pulsed field gel electrophoresis, AseI digests, and subclone sequences. The six Sp185/333 genes include one gene with the A2 element pattern (red), one B8 gene (orange), three D1 genes (yellow, green, blue), and one E2 gene (purple; see Figure for element patterns). All are located near the 3′ end of the BAC insert. Gene orientations are indicated and spacing is to scale unless otherwise noted. GA microsatellites flank each gene and GAT microsatellites flank segmental duplications that are positioned on 5′ side of B8 and include the three D1 genes. This is Figure 4 from Miller et al. (2010) reproduced with permission from BioMed Central.
With the availability of the sequence for the intergenic regions between the Sp185/333 genes on the BAC assembly, additional oddities, and similarities were discovered. All six genes are flanked by microsatellites on both the 5′ side (30–60 GA repeats) and the 3′ side (140–165 GA repeats; Figure 8; Miller et al., 2010). In addition, there are GAT microsatellites that surround the D1 genes and are positioned at the edges of three ∼13.5 kb tandem segmental duplications. These duplications have very low sequence diversity including the three D1 genes that are 99.7% identical. This not only suggests a recent duplication event of the regions bounded by GAT microsatellites, but it also infers the potential of an unknown number of identical Sp185/333 genes in any given genome. Because previous estimates of family size (see above) have assumed that all genes have different sequences, the demonstration of genes on the 7096 BAC that are almost identical indicates that our estimates of family size are likely are likely to be too low.
Speculation on diversification mechanisms that act on the Sp185/333 gene family is based on a number of unusual aspects of the clustered genes including sequence similarity, repeats, tight linkage, and microsatellites. Instability has been documented for regions of genomes that harbor tandem gene arrays, microsatellites, minisatellites, and other types of repeats (Gendrel et al., 2000; Wilson et al., 2000; Trifonov, 2003; Bagshaw et al., 2008; Smith, 2010; Despons et al., 2011). It is noteworthy that D1 is the most common element pattern identified from randomly cloned and sequenced genes (Buckley and Smith, 2007), which corresponds with multiple D1 genes on the BAC insert that are positioned within segmental duplications. This suggests that the D1 genes in other regions of the genome may also be surrounded by GAT microsatellites that drive segmental duplications. The sequence between the GA microsatellites, which includes the Sp185/333 genes and their flanking regions, has much lower sequence diversity than the intergenic regions positioned outside of the microsatellites (Miller et al., 2010). The sequence conservation may be driven by the presence of the microsatellites on either side of the genes plus element sequences that are shared within the second exons. The shared elements may promote gene recombination, conversion, and crossover, whereas the microsatellites may block the progression of DNA strand exchange during gene conversion, thereby protecting the region of the genome harboring clusters of Sp185/333 genes from sequence homogenization (Gendrel et al., 2000; Miller et al., 2010). This model predicts that the exchanged sequences would be limited to the regions located between microsatellites, which would show higher sequence conservation. In general, the genomic structure of the family predicts mechanisms for sequence homogenization through gene duplication and/or conversion, yet maintains sequence diversity among the members of the family. Other families of clustered genes tend to generate pseudogenes, such as primate odorant receptors, sea urchin TLRs, and fish aquaporins (Gilad et al., 2003; Rast and Messier-Solek, 2008; Dong et al., 2009; Tingaud-Sequeira et al., 2010) however, only a single pseudogene has been identified from the 171 sequenced Sp185/333 genes (Buckley and Smith, 2007). The Sp185/333 gene family appears to employ intriguing mechanisms for sharing sequences, maintaining diversification, and blocking or correcting disruption of the open reading frame.
The Diversity of the Sp185/333 mRNAs
The striking up-regulation of Sp185/333 gene expression in response to a variety of immune challenges was the initial basis for our interest in investigating this immune response system. The genes are expressed in coelomocytes within 3–6 h of challenge from LPS, β-1,3-glucan, double stranded RNA (dsRNA), peptidoglycan (PDG), and heat-killed marine bacteria (Rast et al., 2000; Nair et al., 2005; Terwilliger et al., 2007). Blastocoelar cells, larval phagocytes in S. purpuratus, express the Sp185/333 genes when cultured with marine microbes (Ghosh et al., 2010). The coding regions of the Sp185/333 mRNAs range in size from 0.16 to 1.5 kb, depending on the element pattern. In addition to the wide variety of element patterns encoded in the genes, half of the mRNAs show changes to the reading frame, compared to the genes, and appear as single nucleotide polymorphisms (SNPs) that encode non-synonymous amino acids or introduce early stop codons. Furthermore, small indels shift the reading frame to missense sequence, typically ending with an early stop (Terwilliger et al., 2007; Ghosh et al., 2010). Unexpectedly, rather than being degraded as non-functional mRNAs, at least some are translated because both truncated and missense Sp185/333 proteins are produced (Dheilly et al., 2009). The most common element pattern observed in the cDNAs is E2, which is also present in a truncated version, E2.1, that has a SNP in element 13 that changes a glycine to a stop and deletes about half of the protein (Terwilliger et al., 2006, 2007; see Figure 6A). Prior to immune challenge, 81% of the sequenced cDNAs encode a truncated protein with the E2.1 pattern, whereas after challenge this is decreased to 58% (Ghosh et al., 2011). This variation can be interpreted as directed or regulated RNA editing rather than a change in gene expression because no gene has been identified with a stop codon in element 13.
Although many mRNAs encode truncated and missense proteins (Terwilliger et al., 2007), this is strikingly different from the sequenced genes of which all but one (of 171) have perfect open reading frames (Buckley and Smith, 2007). More detailed comparisons of genes and mRNAs from three individual sea urchins show a number of additional unexpected findings (Buckley et al., 2008b). The sequences of the genes and mRNAs both show high sequence diversity, which is present throughout and is not concentrated in diversity hotspots. However, for individual animals, the level of sequence diversity for genes is different from that for mRNAs. Similarly, the level of sequence diversity for elements that are shared between genes and mRNAs is also different. This means that the most variable nucleotide positions in the genes do not correspond with the most variable positions in the mRNAs. Very few of the mRNAs isolated from an individual animal match identically to any of the genes cloned from the same animal. Furthermore, estimates to identify the most likely gene as the source for sets of messages that share the same element pattern (based on the fewest number of substitutions and indels) show that 67–98% of the messages from individual sea urchins match best to a single gene with an E2 element pattern. Comparisons between the variable positions in the gene and its most likely set of transcribed messages indicate that 73% of the changes are transitions, significantly higher than the expected 33% that would occur randomly. Furthermore, 30% of these transitions change a cytidine in the gene to uridine the mRNA. This is consistent with RNA editing by a cytidine deaminase, a family of enzymes for which several gene models have been identified in the genome (Hibino et al., 2006). In addition to possible RNA editing, transcription of the Sp185/333 genes may be performed by a low fidelity polymerase, such as polμ, which has also been annotated in the genome (Hibino et al., 2006).
The Sp185/333 Proteins
Structure
The structure of the Sp185/333 proteins, as deduced from the genes and mRNAs, have an N-terminal hydrophobic leader that is encoded by the first exon and is assumed to be cleaved during processing. The mature proteins are encoded by the second exon and have a generic structure even though there is significant sequence diversity among them (Terwilliger et al., 2006; Figure 9). The N-terminal region of the mature, full-length proteins is glycine-rich and includes two to four imperfect tandem repeats, and the central region has an integrin binding motif; arginine–glycine–aspartic acid (RGD). In addition to the interspersed, imperfect, tandem repeats, the C-terminal region is histidine-rich with patches of poly-histidines that are interspersed with proline, glycine, arginine, and glutamine, and patches of acidic amino acids composed of short stretches of glutamic acid often combined with aspartic acid (Terwilliger et al., 2006). Up to three stop codons are present in conserved positions at the 3′ end of the genes, and alter the length of the C-terminus for different proteins. Conserved, predicted N-linked glycosylation sites are present throughout the length of the proteins, and O-linked sites are predicted in those sequences that employ the third stop codon and have the longest C-terminal region (Terwilliger et al., 2006, 2007). The deduced proteins have no recognizable transmembrane region, no conserved motif for a glycosylphosphatidylinositol (GPI) linkage, and no apparent means to be associated with membranes. The native proteins have no predictable secondary structure or folding characteristics, and consequently may be present in an extended, unfolded, or intrinsically disordered conformation. The amino acid sequence predictions from the genes indicates that the proteins have no cysteines, although frame shifts introduced by RNA editing predicts missense cysteines in some truncated proteins (Terwilliger et al., 2007), which have been confirmed for native Sp185/333 proteins by proteomic analysis (Dheilly et al., 2009; Table 1).
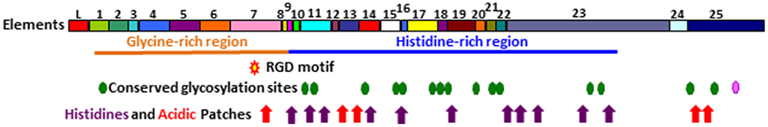
Figure 9. Representation of Sp185/333 protein structure showing all possible elements. The deduced protein contains a leader (L), a glycine-rich region (orange line), and a histidine-rich region (blue line). Symbols indicate the presence of an RGD motif within element 7 (red star with a yellow center), N-linked glycosylation sites (green heptagons), an O-linked glycosylation site (pink heptagon), patches of acidic amino acids (red arrows), and histidines (purple arrows). Modified from Terwilliger et al. (2006).

Table 1. Mass spectrometric identification of native Sp185/333 proteins from coelomic fluid*.
Diversity
The combination of the mosaic structure of the elements plus RNA editing and the introduction of early stop codons results in a deduced size range of the Sp185/333 proteins of 4–55 kDa (Terwilliger et al., 2006, 2007). This includes varying numbers of histidine and acidic patches, the presence or absence of the RGD motif, and the deletion of the entire C-terminal histidine-rich region. However, the observed size range of the native proteins is unexpectedly large relative to deduced predictions, and likely results in part, from multimerization (Brockton et al., 2008). The presence of truncated and missense proteins in coelomic fluid is demonstrated on two dimensional (2D) Western blots by fewer spots for antisera that recognize the central and C-terminal regions of the proteins (Dheilly et al., 2009; Figure 10). Multimerization and putative post-translational modifications are likely underlying mechanisms for generating up to 264 different Sp185/333-positive (Sp185/333+) spots for individual sea urchins (Figure 11). Variations in the arrays of Sp185/333 proteins evaluated from different animals are striking (Figure 12) and suggestive of variable gene expression, perhaps differences in the numbers and/or versions of the genes in different individuals, in addition to mRNA editing and post-translational modifications to the proteins. It is apparent that the array of proteins from individual sea urchins shows more variation than can likely be encoded by a gene family of ∼50 members.
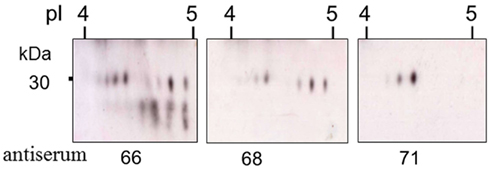
Figure 10. Truncated Sp185/333 proteins are present in the coelomic fluid. Enlarged regions of three different two dimensional Western blots of coelomic fluid proteins from the same sea urchin were analyzed with different anti-Sp185/333 sera; anti-66, anti-68, or anti-71. Anti-66 recognizes AHAQRDFNERRGKENDTER from element 1; anti-68 recognizes GGRRGDGEEETDAAQQIGDGLC from element 7; anti-71 recognizes TEEGSPRRDGQRRPYGNR from element 25 (see Figure 6A for element positions). Decreasing numbers of spots in blots analyzed with antisera that recognize peptides in more C-terminal regions of the proteins suggests that many are either truncated or have missense sequence toward the C-terminus. Reprinted from Dheilly et al. (2009) with permission from the American Association of Immunologists, Inc., copyright 2009.
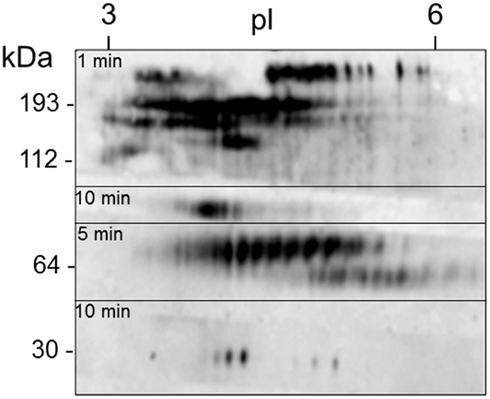
Figure 11. A single sea urchin can have as many as 264 spots that are Sp185/333+. Coelomic fluid proteins (200 μg) from sea urchin 12 (see Figure 12) were separated by 2D electrophoresis and transferred to a filter. The filter was immunostained with an equal mixture of the three anti-Sp185/333 sera (see legend to Figure 10) and exposed to autoradiographic film for 1, 5, or 10 min. The different exposures were merged to give a final composite image. Isoelectric points (pI) are shown at the top and the molecular weight standards (kDa) are shown to the left. Reprinted from Dheilly et al. (2009) with permission from the American Association of Immunologists, Inc., copyright 2009.
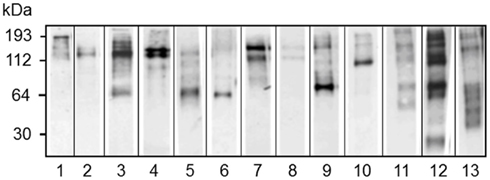
Figure 12. Different sea urchins express different arrays of Sp185/333 proteins. Western blots of coelomic fluid from 13 different sea urchins sampled 96 h after challenge with LPS show different arrays of Sp185/333 proteins. The blots were immunostained with an equal mixture of the three different anti-Sp185/333 sera (see legend for Figure 10). Reprinted from Dheilly et al. (2009) with permission from the American Association of Immunologists, Inc., copyright 2009.
Expression
Subsets of the polygonal and small phagocytes express Sp185/333 proteins, which are present in the trans cisternae of the Golgi Apparatus, in perinuclear vesicles, and are associated with the membrane of transport vesicles that appear to fuse with the plasma membrane and are present on the extracellular surface of the small phagocytes (Brockton et al., 2008; Dheilly et al., 2011; Figures 3 and 4). It is noteworthy and although the Sp185/333 proteins do not have a predicted transmembrane region, they are also associated with the membranes of internal organelles and vesicles, and with the plasma membrane. This localization may be mediated by interactions between the RGD motif and one or more of the integrin family members in the purple sea urchin (Whittaker et al., 2006) that are expressed in coelomocytes. A second sea urchin species, Heliocidaris erythrogramma, expresses He185/333 proteins, which are very similar to those in S. purpuratus, and are present in phagocytes and in colorless spherule cells (Dheilly et al., 2011).
Function
The putative immune function of the Sp185/333 proteins has been predicted based on the diversity of the genes, mRNAs, and proteins, and the striking up-regulation of gene expression in response to immunological challenge (Rast et al., 2000; Nair et al., 2005; Brockton et al., 2008; Dheilly et al., 2009). The amount of Sp185/333 protein in coelomic fluid increases in response to injection of LPS, which is reflected by an increase in the number of Sp185/333+ cells (Brockton et al., 2008; Dheilly et al., 2009). These changes appear to be due, in part, to an increase in the number of small phagocytes that includes an increase in cells that are Sp185/333+. On the other hand, while there is no increase in the number of polygonal cells in response to immune challenge, there is an increase in the percentage of Sp185/333+ polygonal cells. Organs also have Sp185/333+ cells including esophagus, gut, testis, ovary, pharynx, and axial organ (Majeske and Smith, unpublished). It is noteworthy that the number of Sp185/333+ cells in the axial organ increases significantly in response to injections of LPS, whereas the other tissues do not show a similar response. The axial organ is located at the axis of the somewhat spherical echinoid (Hyman, 1955) and has no clearly defined function. However, cells in the axial organ of the sea star, Asterias rubens, proliferate in response to LPS and concanavalin A (Holm et al., 2008). Some gut associated cells in the sea urchin H. erythrogramma, are He185/333+ and may be phagocytes (Dheilly et al., 2011). To date, it appears Sp185/333+ cells are present throughout echinoid tissues, but it is not known whether they are wandering coelomocytes or stromal cells of the organs. The increase in Sp185/333+ cells and the increase in protein content post-immune challenge is consistent with an immune function of this protein family.
Speculation that the Sp185/333 proteins have antibacterial activity has been based on the diversity of the proteins and their expression kinetics in response to challenge from bacterial molecular patterns or from heat-killed marine gram negative bacterial species, Vibrio diazotrophicus. Native Sp185/333 proteins bind tightly to V. diazotrophicus (Figure 13), are not eluted by high or low pH, and in some cases, show an increase in size upon binding the target cells (Schrankel and Smith, unpublished). Perhaps the oligomerization that is commonly observed on Western blots is an aspect of binding to targets. Because predictions of Sp185/333 folding suggests that they may be unordered and unfolded, this has led to questions of whether the proteins conform to the target for binding and whether binding is specific. Testing these functions with a recombinant Sp185/333 protein shows that it binds to V. diazotrophicus and Baker’s yeast in the absence of the coelomic fluid, but does not bind to the gram positive species, Bacillus cereus or B. subtilis. Binding to V. diazotrophicus appears to be saturable (Figure 14) and preliminary competition assays suggest that the binding site may be specific (Lun and Smith, unpublished). Future evaluation of different versions of recombinant Sp185/333 proteins with different element patterns, different numbers of repeats and histidine patches (see Figure 9), or that are truncated may have a variety of anti-pathogen functions besides binding such as opsonization and/or agglutination of bacteria to augment phagocytosis, plus bacteriostatic or bactericidal activity.
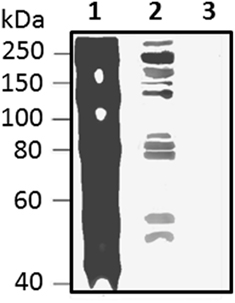
Figure 13. Native Sp185/333 proteins bind Vibrio diazotrophicus (Vd), a gram negative marine bacterial species. Whole coelomic fluid (wCF) lysate from a sea urchin was incubated with Vd. Bacteria were pelleted, washed, and analyzed by Western blot using equal amounts of all three anti-Sp185/333 sera (see legend for Figure 10). wCF and Vd alone are shown for comparison. Lane 1, wCF; lane 2, wCF proteins bound to Vd; lane 3, Vd. Protein standard is to the left. Unpublished figure provided by Catherine Schrankel.
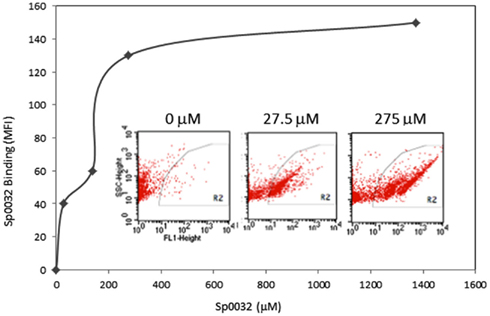
Figure 14. A recombinant Sp185/333 protein (rSp0032) binds Vibrio diazotrophicus (Vd). Biotinylated rSp0032 incubated with 109Vd cells and post-labeled with Neutravidin–FITC shows increased binding with increased protein concentration within the gate area (R2). Binding plateaus at about 400 mM of rSp0032 indicating the saturation point. MFI, mean fluorescence intensity. Results from flow cytometry (inserted images) show fluorescence (X-axis; fluorescent events measuring FITC) associated with bacterial cells (Y-axis; side scatter or cell counts) for increasing concentrations of rSp0032. Unpublished figure provided by Catherine Schrankel.
Conclusion
The rapid evolution of pathogens imposes significant burdens on metazoan immune systems to prevent or combat effectively all possible infections. Vertebrate adaptive immune systems employ somatic recombination of the immunoglobulin gene family (Hozumi and Tonegawa, 1976; Brack et al., 1978; Weigert et al., 1978) or assembly of the variable lymphocyte receptor genes (Nagawa et al., 2007; Rogozin et al., 2007; Kishishita et al., 2010) as mechanisms that enable specific recognition and neutralization of pathogens. Until recently, immune diversification mechanisms were thought to be restricted to the vertebrates and that invertebrates relied on innate immunity based entirely on receptors and antimicrobial peptides with broad specificities for pathogen detection and antibacterial capabilities. However, this may not be adequate for host protection, and several highly variable immune response gene families have been identified in a few invertebrate groups that may significantly expand capabilities for non-self detection and protection against pathogen attack (reviewed in Ghosh et al., 2011). The central question that emerges from evidence of invertebrate immune gene diversity is – what are the mechanisms that function to enhance diversification in these groups? DSCAM variability in arthropods is based on extensive alternative splicing (Watson et al., 2005; Brites et al., 2008; Schmucker and Chen, 2009; Lee et al., 2010), which may be function in hemocytes to generate proteins that bind specifically to the invading pathogen (Graveley, 2005; Dong and Dimopoulos, 2009). Some immune genes in invertebrates are members of multi-gene families and include those encoding the variable chitin binding proteins in protochordates (Cannon et al., 2004; Dishaw et al., 2008, 2010) and fibrinogen related proteins (FRePs) in mollusks (Stout et al., 2009; Hanington and Zhang, 2010; Loker, 2010). Other invertebrate immune genes are single copy, such as fuhc in compound tunicates, in which the diversity is encoded in the many hundreds of alleles in the population and is enhanced by alternative splicing (De Tomaso et al., 2005; McKitrick and De Tomaso, 2010). Except for alternative splicing of Dscam mRNAs, diversification mechanisms beyond gene duplication are not understood for invertebrate immunity, and mechanisms to generate and/or maintain diversity may be different for each group of organisms or even for individual species. The beginnings for understanding invertebrate immune diversification has come from investigations of the FReP3 gene subfamily (Loker et al., 2004; Zhang et al., 2004). The number of FReP3 loci in the genome of an individual snail is insufficient to account for the level of sequence diversity observed in the FReP3 mRNAs. Consequently, FReP3 genes may undergo point mutations and/or somatic recombinatorial diversity, which is predicted to result from the concatenation of pairs of gene segments in the hemocytes.
Whether sea urchins have specific mechanisms to manipulate and diversify the members of the Sp185/333 gene family, or whether diversification is the result of genomic instability and DNA repair, or both, is not known. However, multiple levels sequence diversification in the Sp185/333 system appears to act on the genes, the mRNAs, and the proteins (Figure 15). Gene diversification may occur because region(s) of the genome that harbor clusters of Sp185/333 genes (assuming that they have structure similar to that observed for the 7096 BAC sequence; Figure 8) are highly unusual due of the preponderance of various sized repeats. These include (i) large repeats in the form of segmental duplications and the genes themselves based on their sequence similarity and tight linkage, (ii) medium repeats that appear as shared elements and both tandem and interspersed repeats within the genes, and (iii) small repeats or microsatellites that tightly flank the genes in addition to simple repeats that also appear within the genes (Buckley and Smith, 2007; Miller et al., 2010). This range of clustered repeats may act together, perhaps synergistically, to drive genomic instability that appears as gene duplication, deletion, conversion, and recombination. This may change the sequences of the genes including changes in the mosaic patterns of the elements plus SNPs within the elements. In addition, the Sp185/333 family may show size variation among individuals because the genomic structure that harbors the family may promote both meiotic mispairing of genes on sister chromatids and drive unequal crossovers. On the other hand, instability and diversification may be controlled so that gene or regional sequence homogenization from gene conversion is blocked from progressing through the microsatellites. Furthermore, only a single pseudogenes and no gene fragments have been detected from the 171 Sp185/333 genes that have been cloned and sequenced, which suggests mechanisms to either block pseudogene formation or to correct or delete them once formed.
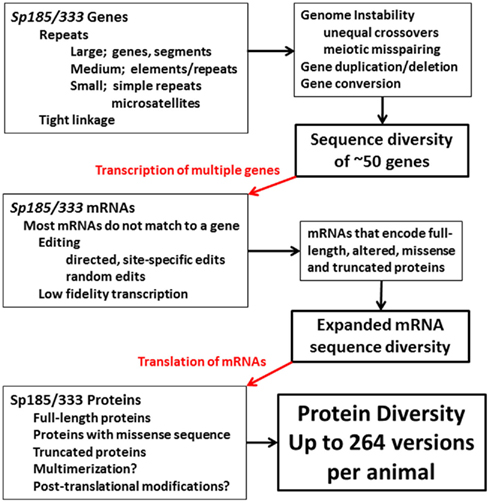
Figure 15. Diversification in the Sp185/333 system in the purple sea urchin. Putative diversification mechanisms based on genomic instability may function to increase gene sequence diversity and may also vary the size of the gene family among individuals. mRNA sequence diversity imparted by the genes is expanded by editing some of the mRNAs in addition to possible low fidelity transcription. The mRNAs are translated to generate a broad array of proteins that may multimerize and be post-translationally modified. The end result is protein diversity that is much broader than what is encoded by the genes.
The diversity that is encoded within the members of the gene family is expanded by apparent RNA editing that changes the nucleotide sequence at single positions to alter codons and insert stops, in addition to introducing small indels that change the reading frame to encode missense sequence (Figure 15). Although RNA editing is typically observed for a single or a few nucleotide positions in other systems (Chan et al., 1997; Maas and Rich, 2000), editing of the Sp185/333 mRNAs is a combination of random edits throughout the entire length plus site specific edits to certain nucleotides. The overall result broadens the array of expressed full-length proteins to include some that are altered at single amino acids, some with missense sequence, and many that are truncated. This is a noteworthy deviation from the central dogma in biology in which the proteins are only partially encoded by the genes.
The Sp185/333 proteins are larger than predicted from the cDNAs and genes suggesting post-translational diversification (Figure 15). This is evident for both native proteins and for individual recombinant proteins expressed in bacteria or insect cells (Brockton et al., 2008). The proteins may multimerize with themselves and/or with other Sp185/333 variants, both full-length and truncated, and possibly with other proteins in the coelomic fluid. Post-translational modifications may also act to diversify the proteins, perhaps as an underlying mechanism to produce a preponderance of acidic proteins (Dheilly et al., 2009) rather than the predicted full spectrum of isoelectric points. Given this dizzying array of putative diversification mechanisms, one must keep in mind that the proteins functioning in the immune response of the sea urchin must withstand selection pressures from pathogens and not be altered to autoreactive forms. The arms race between pathogen virulence and host self protection takes different forms in this coevolutionary process and its direction depends on the capabilities of the pathogens to evade or counteract the immune system and the diversification mechanisms employed by the host to identify, deter, or destroy the invaders.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author is appreciative of the preliminary and unpublished results provided by Katherine Buckley, Catherine Schrankel, and Cheng Man Lun. Julie Ghosh, Cheng Man Lun, and three reviewers kindly provided improvements to the manuscript. Lauren Sherman, Preethi Golconda, and Hung-Yen Chou helped with the early organization of the information for this review. Work on sea urchin immunology is supported by the National Science Foundation.
References
Al-Sharif, W. Z., Sunyer, J. O., Lambris, J. D., and Smith, L. C. (1998). Sea urchin coelomocytes specifically express a homologue of the complement component C3. J. Immunol. 160, 2983–2997.
Arizza, V., Giaramita, F. T., Parrinello, D., Cammarata, M., and Parrinello, N. (2007). Cell cooperation in coelomocyte cytotoxic activity of Paracentrotus lividus coelomocytes. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 147, 389–394.
Bagshaw, A. T., Pitt, J. P., and Gemmell, N. J. (2008). High frequency of microsatellites in S. cerevisiae meiotic recombination hotspots. BMC Genomics 9, 49. doi:10.1186/1471-2164-9-49
Bertheussen, K., and Seijelid, R. (1978). Echinoid phagocytes in vitro. Exp. Cell Res. 111, 401–412.
Biermann, C. H. (1998). The molecular evolution of sperm binding in six species of sea urchins (Echinoidea: Strongylocentrotidae). Mol. Biol. Evol. 15, 1761–1771.
Boolootian, R. A., and Geise, C. A. (1958). Coelomic corpuscles of echinoderms. Biol. Bull. 15, 53–56.
Brack, C., Hirama, M., Lenhard-Schuuler, R., and Tonegawa, S. (1978). A complete immunoglobulin gene is created by somatic recombination. Cell 15, 1–14.
Brites, D., Mctaggart, S., Morris, K., Anderson, J., Thomas, K., Colson, I., Fabbro, T., Little, T. J., Ebert, D., and Du Pasquier, L. (2008). The Dscam homologue of the crustacean Daphnia is diversified by alternative splicing like in insects. Mol. Biol. Evol. 25, 1429–1439.
Brockton, V., Henson, J. H., Raftos, D. A., Majeske, A. J., Kim, Y. O., and Smith, L. C. (2008). Localization and diversity of 185/333 proteins from the purple sea urchin – unexpected protein-size range and protein expression in a new coelomocyte type. J. Cell. Sci. 121, 339–348.
Buckley, K. M., Florea, L. D., and Smith, L. C. (2009). A method of identifying alternative or cryptic splice sites within gene and mRNA sequences. Comparisons among sequences from vertebrates, echinoderms and other groups. BMC Genomics 10, 318. doi:10.1186/1471-2164-10-318
Buckley, K. M., Munshaw, S., Kepler, T. B., and Smith, L. C. (2008a). The 185/333 gene family is a rapidly diversifying host-defense gene cluster in the purple sea urchin, Strongylocentrotus purpuratus. J. Mol. Biol. 379, 912–928.
Buckley, K. M., Terwilliger, D., and Smith, L. C. (2008b). Sequence variations in 185/333 messages from the purple sea urchin suggest posttranscriptional modifications to increase immune diversity. J. Immunol. 181, 8585–8594.
Buckley, K. M., and Smith, L. C. (2007). Extraordinary diversity among members of the large gene family, 185/333, from the purple sea urchin, Strongylocentrotus purpuratus. BMC Mol. Biol. 8, 68. doi:10.1186/1471-2199-8-68
Calestani, C., Rast, J. P., and Davidson, E. H. (2003). Isolation of pigment cell specific genes in the sea urchin embryo by differential macroarray screening. Development 130, 4587–4596.
Cannon, J. P., Haire, R. N., Schnitker, N., Mueller, M. G., and Litman, G. W. (2004). Individual protochordates have unique immune-type receptor repertoires. Curr. Biol. 14, R465–R466.
Chan, L., Chang, B. H.-J., Makamuta, M., Li, W.-H., and Smith, L. C. (1997). Apobec-1 and apolipoprotein B mRNA editing. Biochim. Biophys. Acta 1345, 11–26.
Coffaro, K. A. (1979). Memory and Specificity in the Sea Urchin Lytechinus pictus. Doctoral Dissertation, University of California, Santa Cruz.
Coffaro, K. A. (1980). Memory and Specificity in the Sea Urchin Lytechinus pictus. New York, NY: Elsevier/North Holland Biomedical Press.
Coffaro, K. A., and Hinegardner, R. T. (1977). Immune response in the sea urchin Lytechinus pictus. Science 197, 1389–1390.
De Tomaso, A. W., Nyholm, S. V., Palmeri, K. J., Ishizuka, K. J., Ludington, W. B., Mitchel, K., and Weissman, I. L. (2005). Isolation and characterization of a protochordate histocompatibility locus. Nature 438, 454–945.
Despons, L., Uzunov, Z., and Louis, V. L. (2011). Tandem gene arrays, plastic chromosomal organizations. C. R. Biol. 334, 639–646.
Dheilly, N., Nair, S. V., Birch, D., and Raftos, D. A. (2011). Ultrastructural localization of the highly variable immune response protein (He185/333) within coelomocytes and the gut tissues of the sea urchin, Heliocidaris erythrogramma. Immunol. Cell Biol. 89, 861–869.
Dheilly, N. M., Nair, S. V., Smith, L. C., and Raftos, D. A. (2009). Highly variable immune-response proteins (185/333) from the sea urchin Strongylocentrotus purpuratus: proteomic analysis identifies diversity within and between individuals. J. Immunol. 182, 2203–2212.
Dishaw, L., Ota, T., Mueller, M., Cannon, J., Haire, R., Gwatney, N., Litman, R., and Litman, G. (2010). The basis for haplotype complexity in VCBPs, an immune-type receptor in amphioxus. Immunogenetics 62, 623–631.
Dishaw, L. J., Mueller, M. G., Gwatney, N., Cannon, J. P., Haire, R. N., Litman, R. T., Amemiya, C. T., Ota, T., Rowen, L., Glusman, G., and Litman, G. W. (2008). Genomic complexity of the variable region-containing chitin-binding proteins in amphioxus. BMC Genet. 9, 78. doi:10.1186/1471-2156-9-78
Dong, D., He, G., Zhang, S., and Zhang, Z. (2009). Evolution of olfactory receptor genes in primates dominated by birth-and-death process. Genome Biol. Evol. 1, 258–264.
Dong, Y., and Dimopoulos, G. (2009). Anopheles fibrinogen-related proteins provide expanded pattern recognition capacity against bacteria and malaria parasites. J. Biol. Chem. 284, 9835–9844.
Dong, Y., Taylor, H. E., and Dimopoulos, G. (2006). AgDscam, a hypervariable immunoglobulin domain-containing receptor of the Anopheles gambiae innate immune system. PLoS Biol. 4, e229. doi:10.1371/journal.pbio.0040229
Edds, K. T. (1977). Dynamic aspects of filopodial formation by reorganization of microfilaments. J. Cell Biol. 73, 479–491.
Edds, K. T. (1979). Isolation and characterization of two forms of a cytoskeleton. J. Cell Biol. 83, 109–115.
Furukawa, R., Takahashi, Y., Kanajima, Y., Dan-Sohkawa, M., and Kaneko, H. (2009). Defense system by mesenchyme cells in bipinnaria larvae of the starfish, Asterina pectinifera. Dev. Comp. Immunol. 33, 205–215.
Gendrel, C.-G., Boulet, A., and Dutreix, M. (2000). (CA/GT)n microsatellites affect homologous recombination during yeast meiosis. Genes Dev. 14, 1261–1268.
Ghosh, J., Lun, C. M., Majeske, A. J., Sacchi, S., Schrankel, C. S., and Smith, L. C. (2011). Invertebrate immune diversity. Dev. Comp. Immunol. 35, 959–974.
Ghosh, J. G., Buckley, K. M., Nair, S. V., Raftos, D. A., Miller, C. A., Majeske, A. J., Hibino, T., Rast, J. P., Roth, M., and Smith, L. C. (2010). Sp185/333: a novel family of genes and proteins involved in the purple sea urchin immune response. Dev. Comp. Immunol. 34, 235–245.
Gilad, Y., Man, O., Paabo, S., and Lancet, D. (2003). Human specific loss of olfactory receptor genes. Proc. Natl. Acad. Sci. U.S.A. 100, 3324–3327.
Graveley, B. R. (2005). Mutually exclusive splicing of the insect Dscam pre-mRNA directed by competing intronic RNA secondary structures. Cell 123, 65–73.
Gross, P. S., Clow, L. A., and Smith, L. C. (2000). SpC3, the complement homologue from the purple sea urchin, Strongylocentrotus purpuratus, is expressed in two subpopulations of the phagocytic coelomocytes. Immunogenetics 51, 1034–1044.
Grula, J. W., Hall, T. J., Hunt, J. A., Guiugni, D. F., Graham, G. J., Davidson, E. H., and Britten, R. J. (1982). Sea urchin DNA sequence variation and reduced interspecies differences of the less variable DNA sequences. Evolution 36, 655–676.
Hanington, P. C., and Zhang, S.-M. (2010). The primary role of fibrinogen-related proteins in invertebrates is defense, not coagulation. J. Innate Immun. 3, 17–27.
Henson, J. H., Nesbitt, D., Wright, B. D., and Scholey, J. M. (1992). Immunolocalization of kinesin in sea urchin coelomocytes. Association of kinesin with intracellular organelles. J. Cell. Sci. 103(Pt 2), 309–320.
Henson, J. H., Svitkina, T. M., Burns, A. R., Hughes, H. E., Macpartland, K. J., Nazarian, R., and Borisy, G. G. (1999). Two components of actin-based retrograde flow in sea urchin coelomocytes. Mol. Biol. Cell 10, 4075–4090.
Hibino, T., Loza-Coll, M., Messier, C., Majeske, A. J., Cohen, A., Terwilliger, D. P., Buckley, K. M., Brockton, V., Nair, S., Berney, K., Fugmann, S. D., Anderson, M. K., Pancer, Z., Cameron, R. A., Smith, L. C., and Rast, J. P. (2006). The immune gene repertoire encoded in the purple sea urchin genome. Dev. Biol. 300, 349–365.
Hildemann, W. H., and Dix, T. G. (1972). Transplantation reactions of tropical Australian echinoderms. Transplantation 14, 624–633.
Holm, K., Dupont, S., Skold, H., Stenius, A., Thorndyke, M., and Hernroth, B. (2008). Induced cell proliferation in putative haematopoietic tissues of the sea star, Asterias rubens (L.). J. Exp. Biol. 211, 2551–2558.
Hozumi, N., and Tonegawa, S. (1976). Evidence for somatic rearrangement of immunoglobulin genes coding for variable and constant regions. Proc. Natl. Acad. Sci. U.S.A. 73, 3628–3632.
Huelsenbeck, J. P., and Bull, J. J. (1996). A likelihood ratio test to detect conflicting phylogenetic signal. Syst. Biol. 45, 92–98.
Hyman, L. H. (1955). The Invertebrates: Echinodermata, the Coelomate Bilateria, Vol. 4. New York, NY: McGraw-Hill Book Co., Inc.
Johnson, P. T. (1969a). The coelomic elements of sea urchins (Strongylocentrotus). I. The normal coelomocytes; their morphology and dynamics in hanging drops. J. Invertebr. Pathol. 13, 25–41.
Johnson, P. T. (1969b). The coelomic elements of the sea urchins (Strongylocentrotus) III. In vitro reaction to bacteria. J. Invertebr. Pathol. 13, 42–62.
Johnson, P. T., and Chapman, F. A. (1970). Abnormal epithelial growth in sea urchin spines (Strongylocentrotus franciscanus). J. Invertebr. Pathol. 16, 116–122.
Karp, R. D., and Hildemann, W. H. (1976). Specific allograft reactivity in the sea star Dermasterias imbricata. Transplantation 22, 434–439.
Kishishita, N., Matsuno, T., Takahashi, Y., Takaba, H., Nishizumi, H., and Nagawa, F. (2010). Regulation of antigen-receptor gene assembly in hagfish. EMBO Rep. 11, 126–132.
Lee, C., Kim, N., Roy, M., and Graveley, B. R. (2010). Massive expansions of Dscam splicing diversity via staggered homologous recombination during arthropod evolution. RNA 16, 91–105.
Lee, Y. H. (2003). Molecular phylogenies and divergence times of sea urchin species of Strongylocentrotidae, Echinoidea. Mol. Biol. Evol. 20, 1211–1221.
Llobet-Brossa, E., Rossello-Mora, R., and Amann, R. (1998). Microbial community composition of Wadden sea sediments as revealed by fluorescence in situ hybridization. Appl. Environ. Microbiol. 64, 2691–2696.
Loker, E. S. (2010). “Gastropod immunobiology,” in Invertebrate Immunity, ed. K. Soderhall (Austin: Landes Bioscience and Springer Science+Business Media), 17–43.
Loker, E. S., Adema, C. M., Zhang, S.-M., and Kepler, T. B. (2004). Invertebrate immune systems – not homogeneous, not simple, not well understood. Immunol. Rev. 198, 10–24.
Maas, S., and Rich, A. (2000). Changing genetic information through RNA editing. Bioessays 22, 790–802.
Massana, R., Murray, A. E., Preston, C. M., and Delong, E. F. (1997). Vertical distribution and phylogenetic characterization of marine palnktonic Archaea in the Santa Barbara chanel. Appl. Environ. Microbiol. 63, 50–56.
McKitrick, T. R., and De Tomaso, A. W. (2010). Molecular mechanisms of allorecognition in a basal chordate. Semin. Immunol. 22, 34–38.
Messer, L. I., and Wardlaw, A. C. (1980). “Separation of coelomocytes of Echinus esculentus by density gradient centrifugation,” in Proceedings of the European Colloquium on Echinoderms, ed. M. Jangoux (Rotterdam: A. A. Balkema), 319–323.
Metchnikoff, E. (1893). Lectures on the Comparative Pathology of Inflammation: Delivered at the Pasteur Institute in 1891. Kegan Paul, Trench, Trubner & Co. Ltd, London.
Michevich, M. F., and Ferris, J. S. (1981). The implications of congruence in Mendida. Syst. Zool. 30, 351–370.
Miller, C. A., Buckley, K. M., Easley, R. L., and Smith, L. C. (2010). An Sp185/333 gene cluster from the purple sea urchin and putative microsatellite-mediated gene diversification. BMC Genomics 11, 575. doi:10.1186/1471-2164-11-575
Multerer, K. A., and Smith, L. C. (2004). Two cDNAs from the purple sea urchin, Strongylocentrotus purpuratus, encoding mosaic proteins with domains found in factor H, factor I, and complement components C6 and C7. Immunogenetics 56, 89–106.
Nagawa, F., Kishishita, N., Shimizu, K., Hirose, S., Miyoshi, M., Nezu, J., Nishimura, T., Nishizumi, H., Takahashi, Y., Hashimoto, S., Takeuchi, M., Miyajima, A., Takemori, T., Otsuka, A. J., and Sakano, H. (2007). Antigen-receptor genes of the agnathan lamprey are assembled by a process involving copy choice. Nat. Immunol. 8, 206–213.
Nair, S. V., Del Valle, H., Gross, P. S., Terwilliger, D. P., and Smith, L. C. (2005). Macroarray analysis of coelomocyte gene expression in response to LPS in the sea urchin. Identification of unexpected immune diversity in an invertebrate. Physiol. Genomics 22, 33–47.
Plytycz, B., and Seljelid, R. (1993). Bacterial clearance by the sea urchin, Strongylocentrotus droebachiensis. Dev. Comp. Immunol. 17, 283–289.
Rast, J. P., and Messier-Solek, C. (2008). Marine invertebrate genome sequences and our evolving understanding of animal immunity. Biol. Bull. 214, 274–283.
Rast, J. P., Pancer, Z., and Davidson, E. H. (2000). New approaches towards an understanding of deuterostome immunity. Curr. Top. Microbiol. Immunol. 248, 3–16. doi:10.1186/1471-2199-8-16
Rast, J. P., Smith, L. C., Loza-Coll, M., Hibino, T., and Litman, G. W. (2006). Genomic insights into the immune system of the sea urchin. Science 314, 952–956.
Reinisch, C. L., and Bank, F. B. (1971). Cell recognition: reactions of the sea star (Asterias vulgaris) to the injection of amebocytes of sea urchin (Arbacia punctulata). Cell. Immunol. 2, 496–503.
Rogozin, I. B., Iyer, L. M., Liang, L., Glazko, G. V., Liston, V. G., Pavlov, Y. I., Aravind, L., and Pancer, Z. (2007). Evolution and diversification of lamprey antigen receptors: evidence for involvement of an AID-APOBEC family cytosine deaminase. Nat. Immunol. 8, 647–656.
Schmucker, D., and Chen, B. (2009). Dscam and DSCAM: complex genes in simple animals, complex animals yet simple genes. Genes Dev. 23, 147–156.
Schmucker, D., Clemens, J. C., Shu, H., Worby, C. A., Xiao, J., Muda, M., Dixon, J. E., and Zipursky, S. L. (2000). Drosophila Dscam is an axon guidance receptor exhibiting extraordinary molecular diversity. Cell 101, 671–684.
Service, M., and Wardlaw, A. C. (1984). Echinochrome-A as a bactericidal substance in the coelomic fluid of Echinus esculentus (L). Comp. Biochem. Physiol. B Biochem. Mol. Biol. 79, 161–165.
Silva, J. R. (2000). The onset of phagocytosis and identity in the embryo of Lytechinus variegatus. Dev. Comp. Immunol. 24, 733–739.
Siu, G., Clark, S. P., Yoshikai, Y., Malissen, M., Yanagi, Y., Strauss, E., Mak, T. W., and Hood, L. (1984). The human T cell antigen receptor is encoded by variable, diversity, and joining gene segments that rearrange to generate a complete V gene. Cell 37, 393–401.
Smith, L. C. (2010). Diversification of innate immune genes: lessons from the purple sea urchin. Dis. Model Mech. 3, 274–279.
Smith, L. C., Chang, L., Britten, R. J., and Davidson, E. H. (1996). Sea urchin genes expressed in activated coelomocytes are identified by expressed sequence tags. Complement homologues and other putative immune response genes suggest immune system homology within the deuterostomes. J. Immunol. 156, 593–602.
Smith, L. C., and Davidson, E. H. (1992). The echinoid immune system and the phylogenetic occurrence of immune mechanisms in deuterostomes. Immunol. Today 13, 356–362.
Smith, L. C., Ghosh, J., Buckley, K. M., Clow, L. A., Dheilly, N. M., Haug, T., Henson, J. H., Li, C., Lun, C. M., Majeske, A. J., Matranga, V., Nair, S. V., Rast, J. P., Raftos, D. A., Roth, M., Sacchi, S., Schrankel, C. S., and Stensvag, K. (2010). “Echinoderm immunity,” in Invertebrate Immunity, ed. K. Soderhall (Austin: Landes Bioscience and Spring Science+Business Media), 260–301.
Smith, L. C., Shih, C. S., and Dachenhausen, S. G. (1998). Coelomocytes express SpBf, a homologue of factor B, the second component in the sea urchin complement system. J. Immunol. 161, 6784–6793.
Smith, V. J. (1981). “The echinoderms,” in Invertebrate Blood Cells, eds N. A. Ratcliffe and A. F. Rowley (New York, NY: Academic Press), 513–562.
Sodergren, E., Weinstock, G. M., Davidson, E. H., Cameron, R. A., Gibbs, R. A., Angerer, R. C., Angerer, L. M., Arnone, M. I., Burgess, D. R., Burke, R. D., Coffman, J. A., Dean, M., Elphick, M. R., Ettensohn, C. A., Foltz, K. R., Hamdoun, A., Hynes, R. O., Klein, W. H., Marzluff, W., Mcclay, D. R., Morris, R. L., Mushegian, A., Rast, J. P., Smith, L. C., Thorndyke, M. C., Vacquier, V. D., Wessel, G. M., Wray, G., Zhang, L., Elsik, C. G., Ermolaeva, O., Hlavina, W., Hofmann, G., Kitts, P., Landrum, M. J., Mackey, A. J., Maglott, D., Panopoulou, G., Poustka, A. J., Pruitt, K., Sapojnikov, V., Song, X., Souvorov, A., Solovyev, V., Wei, Z., Whittaker, C. A., Worley, K., Durbin, K. J., Shen, Y., Fedrigo, O., Garfield, D., Haygood, R., Primus, A., Satija, R., Severson, T., Gonzalez-Garay, M. L., Jackson, A. R., Milosavljevic, A., Tong, M., Killian, C. E., Livingston, B. T., Wilt, F. H., Adams, N., Belle, R., Carbonneau, S., Cheung, R., Cormier, P., Cosson, B., Croce, J., Fernandez-Guerra, A., Geneviere, A. M., Goel, M., Kelkar, H., Morales, J., Mulner-Lorillon, O., Robertson, A. J., Goldstone, J. V., Cole, B., Epel, D., Gold, B., Hahn, M. E., Howard-Ashby, M., Scally, M., Stegeman, J. J., Allgood, E. L., Cool, J., Judkins, K. M., Mccafferty, S. S., Musante, A. M., Obar, R. A., Rawson, A. P., Rossetti, B. J., Gibbons, I. R., Hoffman, M. P., Leone, A., Istrail, S., Materna, S. C., Samanta, M. P., Stolc, V., Tongprasit, W., Tu, Q., Bergeron, K. F., Brandhorst, B. P., Whittle, J., Berney, K., Bottjer, D. J., Calestani, C., Peterson, K., Chow, E., Yuan, Q. A., Elhaik, E., Graur, D., Reese, J. T., Bosdet, I., Heesun, S., Marra, M. A., Schein, J., Anderson, M. K., Brockton, V., Buckley, K. M., Cohen, A. H., Fugmann, S. D., Hibino, T., Loza-Coll, M., Majeske, A. J., Messier, C., Nair, S. V., Pancer, Z., Terwilliger, D. P., Agca, C., Arboleda, E., Chen, N., Churcher, A. M., Hallböök, F., Humphrey, G. W., Idris, M. M., Kiyama, T., Liang, S., Mellott, D., Mu, X., Murray, G., Olinski, R. P., Raible, F., Rowe, M., Taylor, J. S., Tessmar-Raible, K., Wang, D., Wilson, K. H., Yaguchi, S., Gaasterland, T., Galindo, B. E., Gunaratne, H. J., Juliano, C., Kinukawa, M., Moy, G. W., Neill, A. T., Nomura, M., Raisch, M., Reade, A., Roux, M. M., Song, J. L., Su, Y. H., Townley, I. K., Voronina, E., Wong, J. L., Amore, G., Branno, M., Brown, E. R., Cavalieri, V., Duboc, V., Duloquin, L., Flytzanis, C., Gache, C., Lapraz, F., Lepage, T., Locascio, A., Martinez, P., Matassi, G., Matranga, V., Range, R., Rizzo, F., Röttinger, E., Beane, W., Bradham, C., Byrum, C., Glenn, T., Hussain, S., Manning, G., Miranda, E., Thomason, R., Walton, K., Wikramanayke, A., Wu, S. Y., Xu, R., Brown, C. T., Chen, L., Gray, R. F., Lee, P. Y., Nam, J., Oliveri, P., Smith, J., Muzny, D., Bell, S., Chacko, J., Cree, A., Curry, S., Davis, C., Dinh, H., Dugan-Rocha, S., Fowler, J., Gill, R., Hamilton, C., Hernandez, J., Hines, S., Hume, J., Jackson, L., Jolivet, A., Kovar, C., Lee, S., Lewis, L., Miner, G., Morgan, M., Nazareth, L. V., Okwuonu, G., Parker, D., Pu, L. L., Thorn, R., and Wright, R. (2006). The genome of the sea urchin, Strongylocentrotus purpuratus. Science 314, 941–952.
Stevens, M. E., Dhillon, J., Miller, C. A., Messier-Solek, C., Rast, J. P., Majeske, A. J., Zuelke, D., and Smith, L. C. (2010). SpTie1/2 from the sea urchin Strongylocentrotus purpuratus, is an orthologue of vertebrate Tie1 and Tie2 and is expressed in coelomocytes, axial organ and embryos. Dev. Comp. Immunol. 34, 884–895.
Stout, B. A., Adema, C. M., Zhang, S. M., and Loker, E. S. (2009). “The biology of FREPs: diversified lectins with fibrinogen-related domains from the freshwater snail Biomphalaria glabrata,” in Animal Lectins: A Functional View, ed. V. A. Ahmen (New York: CRC Press, Taylor & Francis), 475–491.
Terwilliger, D. P., Buckley, K. M., Brockton, V., Ritter, N. J., and Smith, L. C. (2007). Distinctive expression patterns of 185/333 genes in the purple sea urchin, Strongylocentrotus purpuratus: an unexpectedly diverse family of transcripts in response to LPS, beta-1,3-glucan, and dsRNA. BMC Mol. Biol. 8, 16.
Terwilliger, D. P., Buckley, K. M., Mehta, D., Moorjani, P. G., and Smith, L. C. (2006). Unexpected diversity displayed in cDNAs expressed by the immune cells of the purple sea urchin, Strongylocentrotus purpuratus. Physiol. Genomics 26, 134–144.
Tingaud-Sequeira, A., Calusinska, M., Finn, R. M., Chauvigne, F., Lozano, J., and Cerda, J. (2010). The zebrafish genome encodes the largest vertebrate repertoire of functional aquaporins with dual paralogy and substrate specificities similar to mammals. BMC Evol. Biol. 10, 38. doi:10.1186/1471-2148-10-38
Trifonov, E. N. (2003). “Tuning function of tandemly repeating sequences: a molecular device for fast adaptation,” in Evolutionary Theory and Processes: Modern Horizons, Papers in Honor of Eviatar Nevo, ed. S. P. Wassser (Amsterdam: Kluwer Academic Publishers), 1–24.
Varadarajan, J., and Karp, R. D. (1983). Histological versus morphological assessment of graft rejection in invertebrates. Transplantation 36, 629–631.
Watson, F. L., Puttmann-Holgado, R., Thomas, F., Lamar, D. L., Hughes, M., Kondo, M., Rebel, V. I., and Schmucker, D. (2005). Extensive diversity of Ig-superfamily proteins in the immune system of insects. Science 309, 1874–1878.
Weigert, M., Gatmaitan, L., Loh, E., Schilling, J., and Hood, L. (1978). Rearrangement of genetic information may produce immunoglobulin diversity. Nature 276, 785–790.
Whitman, W. B., Coleman, D. C., and Wiebe, W. J. (1998). Prokaryotes: the unseen majority. Proc. Natl. Acad. Sci. U.S.A. 95, 6578–6583.
Whittaker, C. A., Bergeron, K. F., Whittle, J., Brandhorst, B. P., Burke, R. D., and Hynes, R. O. (2006). The echinoderm adhesome. Dev. Biol. 300, 252–266.
Wilson, M. J., Torkar, M., Haude, A., Milne, S., Jones, T., Sheer, D., Beck, S., and Trowsdale, J. (2000). Plasticity in the organization and sequences of human KIR/ILT gene families. Proc. Natl. Acad. Sci. U.S.A. 97, 4778–4783.
Yui, M., and Bayne, C. (1983). Echinoderm immunity:bacterial clearance by the sea urchin Strongylocentrotus purpuratus. Biol. Bull. 165, 473–485.
Keywords: echinoid, invertebrate, evolution, innate immunity, coelomocyte, microsatellites, RNA editing, gene family
Citation: Smith LC (2012) Innate immune complexity in the purple sea urchin: diversity of the Sp185/333 system. Front. Immun. 3:70. doi: 10.3389/fimmu.2012.00070
Received: 01 December 2011; Accepted: 21 March 2012;
Published online: 12 April 2012.
Edited by:
Larry J. Dishaw, University of South Florida, USAReviewed by:
Yuko Ota, University of Maryland, USAPhilip Rosenstiel, University Kiel, Germany
Tatsuya Ota, The Graduate University for Advanced Studies, Japan
Copyright: © 2012 Smith. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: L. Courtney Smith, Department of Biological Sciences, George Washington University, 340 Lisner Hall, 2023 G Street NorthWest, Washington, DC 20052, USA. e-mail:Y3NtaXRoQGd3dS5lZHU=