


- 1 Department of Neurology and Neurological Sciences, Stanford University, Stanford, CA, USA
- 2 Department of Biology, Stanford University, Stanford, CA, USA
Historically, small heat shock proteins (sHSPs) have been extensively studied in the context of being intracellular molecular chaperones. However, recent studies looking at the role of sHSPs in neurological diseases have demonstrated a near universal upregulation of certain sHSPs in damaged and diseased brains. Initially, it was thought that sHSPs are pathological in these disease states because they are found in the areas of damage. However, transgenic overexpression and exogenous administration of sHSPs in various experimental disease paradigms have shown just the contrary – that sHSPs are protective, not pathological. This review examines sHSPs in neurological diseases and highlights the potential for using these neuroprotective sHSPs as novel therapeutics. It first addresses the endogenous expression of sHSPs in a variety of neurological disorders. Although many studies have examined the expression of sHSPs in neurological diseases, there are no review articles summarizing these data. Furthermore, it focuses on recent studies that have investigated the therapeutic potential of sHSPs for neurological diseases. Finally, it will explain what we think is the function of endogenous sHSPs in neurological diseases.
Overview of Small Heat Shock Proteins
Small heat shock proteins (sHSPs) have molecular weights between 12 and 43 kDa, distinguishing them in size from large heat shock proteins (Ganea, 2001; Arrigo et al., 2007). There are 10 human sHSPs: HSPB1–HSPB10 (Kappé et al., 2003). They share common structural characteristics, including a highly conserved 90 amino acid long HSP20 domain, often referred to as the alpha-crystallin domain, and the capacity to form large dynamic oligomers (Poulain et al., 2010). sHSPs are intracellular molecular chaperones (Horwitz, 1992; Van Montfort et al., 2001). As chaperone proteins, sHSPs bind misfolded proteins and prevent them from aggregating. However, they are unable to actively re-fold the protein themselves due to their lack of ATPase activity. Instead, sHSPs sequester the misfolded proteins within the cell to prevent aggregation until a large heat shock protein can assist in refolding (Jakob et al., 1993).
Although sHSPs share both common structural and functional characteristics, they differ in tissue distribution and expression patterns (Table 1). HSPB1, HSPB5, HSPB6, HSPB7, and HSPB8 are ubiquitously expressed, and are constitutively present in the brain at low levels (Quraishe et al., 2008). HSPB4 is expressed in the lens of the eye, composing nearly 50% of the protein mass in the human lens (Andley, 2007). HSPB2 and HSPB3 are expressed in muscle and heart (Quraishe et al., 2008), although a recent study indicates that they also have some expression in the brain (Kirbach and Golenhofen, 2011). HSPB9 and HSPB10 are expressed in the testes (Quraishe et al., 2008). Notably, only three members of the sHSP family (HSPB1, HSPB5, and HSPB8) are induced in response to challenges such as heat (Morimoto and Santoro, 1998; Zhang et al., 2002), glucocorticoids (Nédellec et al., 2002), prostaglandins (Ito et al., 1997), and interferon-gamma (Oba et al., 2008), rendering them true sHSPs that are upregulated in response to cellular stress.
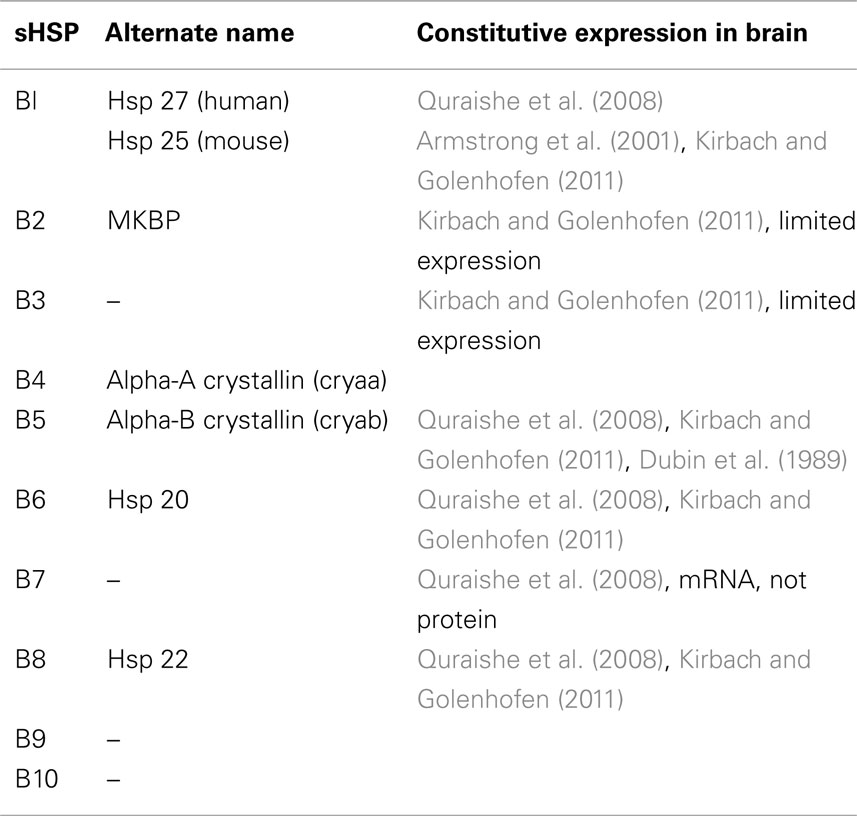
Table 1. Endogenous expression of sHSPs in the brain.
Biochemical, biophysical, and crystallography studies have elucidated the structure of HSPB5 and key residues important for quaternary structure and chaperone function (Bagnéris et al., 2009; Jehle et al., 2009, 2010, 2011). Naturally occurring mutations in conserved regions in several human sHSPs have functional consequences including myopathies (Vicart et al., 1998; Simon et al., 2007), cataracts (Litt et al., 1998), and Charcot Marie Tooth disease (Evgrafov et al., 2004; Ackerley et al., 2006). The crystal structures of sHSPs give us insights into understanding how some of these mutations have pathological consequences. However, mounting evidence over the past two decades suggests that sHSPs may not only play a role in maintaining a healthy body, but that they also have protective functions in disease or injury to the central nervous system (CNS; Sun and MacRae, 2005; Arrigo et al., 2007; Steinman, 2008). This insight has illuminated the possibility of using sHSPs as a novel class of neuroprotective agents.
Role of Endogenous sHSPs in Neurological Diseases
Altered regulation of sHSPs has been seen in many neurodegenerative and neuroinflammatory diseases in both human and rodent brain tissue. A summary of published studies is shown in Table 2.

Table 2. Small heat shock protein expression in human and rodent models of neurological diseases.
Tauopathies: Alzheimer’s Disease and Pick’s Complex
Tauopathies are neurological diseases that involve abnormal aggregation of the tau protein in the brain (Ballatore et al., 2011). The healthy tau protein stabilizes microtubules, which are necessary for the proper transportation of proteins and neurotransmitters along neuronal axons. However, when tau becomes defective and hyperphosphorylated, it can aggregate and forms neurofibrillary tangles (NFTs) that interfere with normal neuronal function and ultimately lead to cell death.
The most common tauopathy is Alzheimer’s disease, a neurodegenerative disease characterized by NFTs and amyloid-rich plaques in the brain that ultimately results in cognitive decline and dementia (Ballard et al., 2011). The endogenous regulation of sHSPs in Alzheimer’s disease has been well examined. HSPB1 levels are elevated in the cortex of Alzheimer’s patients, with higher levels corresponding to increased severity and duration of dementia (Renkawek et al., 1994a). In fact, HSPB1 levels correlate significantly with levels of phosphorylated tau (Shimura et al., 2004; Björkdahl et al., 2008), suggesting that it may play a role in protecting the cell from the pathological effects of hyperphosphorylated tau.
HSPB5 is also upregulated in Alzheimer’s disease (Iwaki et al., 1992; Shinohara et al., 1993; Renkawek et al., 1994b) and highly expressed in ballooned neurons (Lowe et al., 1992). Mao et al. (2001) found that HSPB5 is highly expressed in neurons in the vicinity of extracellular NFTs, but less so in classical senile plaques, diffuse plaques, and intracellular NFTs.
Three other sHSPs have been shown to be elevated in Alzheimer’s disease: HSPB2, HSPB6, and HSPB8. Using immunohistochemistry, Wilhelmus et al. (2006b) found small, but not significant, elevations in HSPB2 and HSPB6 in Alzheimer brains. HSPB6 localizes to both diffuse and classic senile plaques, whereas HSPB2 was only present in the classic senile plaques. An additional study by Wilhelmus et al. (2006a) showed that HSPB8 is found in classic senile plaques from AD brains. These data collectively indicate that certain members of sHSPs are elevated in Alzheimer’s disease, but it is unknown whether they are playing a protective or pathological role in the disease process and there are no studies to date that investigate the therapeutic potential of sHSPs in Alzheimer’s disease.
Less common tauopathies include Pick’s complex, which is now more commonly referred to as frontotemporal dementia (FTD; Weder et al., 2007). This collection of tauopathies includes frontotemporal lobar degeneration, corticobasal degeneration (CBD), progressive supranuclear palsy (PSP), and familial tauopathy FTD with parkinsonism linked to chromosome 17 (FTDP-17; Kertesz, 2003; Tolnay and Probst, 2003).
In many of these conditions, tau pathology is not limited to neurons and instead extends to glial cells such as astrocytes and oligodendrocytes (Komori, 1999). Both HSPB1 and HSPB5 were found to be elevated in brains of patients with olivary hypertrophy, a condition characterized by the enlargement of neurons and neuronal loss in response to a lesion in the dentatoolivary pathway. This can occur as a result of trauma, tumors, cerebrovascular disease, and PSP, a component of Pick’s Complex (Hanihara et al., 1998). HSPB5 has been shown to be upregulated early in disease progression in neurons and later in astrocytes, but HSPB1 was only elevated in astrocytes later in the disease course (Fukushima et al., 2006). However, another study did not find upregulation of HSPB1 or HSPB5 in neurons, but rather that localization of these sHSPs is specific to glial cells in sporadic CBD and PSP and familial FTDP-17 (Dabir et al., 2004). The specificity to astrocytes was confirmed by a study that found elevated levels of HSPB1 in astrocytes in PSP and CBD (Schwarz et al., 2010). It is currently unknown why there are elevated levels of sHSPs in these diseases; there have been no published studies determining the effects that a loss of sHSP (i.e., using genetically deficient mice) or a gain of sHSP (i.e., exogenous administration or overexpression) would have on these diseases.
Other Protein Aggregation Diseases: Parkinson’s and ALS
Parkinson’s disease is a neurodegenerative disease that is caused by the loss of dopamine-producing neurons in the substantia nigra. The decrease in dopamine is accompanied by an accumulation of alpha-synuclein protein, forming inclusions called Lewy bodies (Burn, 2006). Clinical features of Parkinson’s disease are described by the acronym TRAP: tremor at rest, rigidity, akinesia or bradykinesia (loss of movement and slowness of movement, respectively), and postural instability (Jankovic, 2008).
HSPB1 and HSPB5 are upregulated in the cortex of Parkinson’s disease patients (Renkawek et al., 1994a, 1999). These sHSPs are associated with an increased number of tangles in the hippocampus of these patients. Mouse data corroborated the human data, and showed similar elevations in both HSPB1 and HSPB5 using the transgenic Parkinson’s model, alpha-SynA53T (Wang et al., 2008).
Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig’s disease, is a neurodegenerative disease characterized by the progressive loss of motor neurons. ALS is associated with the formation of intraneuronal proteinaceous inclusions that are non-amyloid, many of which include hyperphosphorylated and ubiquitinated TAR DNA-binding protein 43 (TDP-43; Perry et al., 2010). Approximately 95% of ALS cases appear to be sporadic and the remaining 5% are familial (Traub et al., 2011). Twenty percent of the familial cases can be attributed to a mutation in the superoxide dismutase 1 (SOD1) enzyme (Rosen et al., 1993).
Iwaki et al. (1992) found that eight human ALS brains had higher immunoreactivity against HSPB5 compared with healthy brains. This result is corroborated in a study using the mutant SOD mouse model of familial ALS, in which SOD1G93A mice exhibited higher levels of HSPB5 in the cytoplasm of reactive glial cells (Vleminckx et al., 2002). This study found that levels of HSPB1 were upregulated in mouse neurons and glial cells as well. However, the story is complicated by the fact that Maatkamp et al. (2004) found that protein levels of HSPB1 were downregulated just before the degeneration of motoneurons in the mutant SOD mouse model. They also found that mRNA levels of HSPB1 remained unchanged despite lower protein levels, highlighting the need for further studies to clarify what the role of sHSPs might be in ALS.
Poly-Glutamine Diseases: Huntington’s and Spinocerebellar Ataxias
Poly-Q diseases are caused by genetic mutations that lead to a trinucleotide repeat of CAG, the triplet that encodes the amino acid glutamine. This increases the number of glutamines in the protein from as few as 20 to as many as 306 residues depending on the disease. These diseases are known as poly-Q diseases based on the one letter amino acid abbreviation of glutamine, and they include Huntington’s disease (HD), dentatorubral-pallidoluysian atrophy (DRPLA), spinobulbar muscular atrophy (SBMA), and the spinocerebellar ataxias (SCAs; Paulson et al., 2000).
Huntington’s disease is a neurodegenerative disease caused by a poly-glutamine expansion of the huntingtin (Htt) gene (Eidelberg and Surmeier, 2011). It typically appears during middle age and is characterized by chorea, or involuntary, explosive, fidgeting movements.
The SCA diseases are a group of neurodegenerative disorders that are also caused by an expanded poly-glutamine repeat. They manifest as a loss of gait and coordination difficulties. There are distinct subtypes, each caused by a specific mutation in a gene encoding an ataxin protein; around 30 different genes have been identified to date (Di Donato et al., 2001).
Contrary to findings in other protein aggregation diseases of the CNS, elevation of sHSPs have not been observed in the poly-Q diseases. One study using the R6/2 mouse model of HD found reduced levels of HSPB5 at the end of the disease course (Zabel et al., 2002). However, studies on HSPB1 in SCA found that HSPB1 is downregulated early in the disease (Wen et al., 2003; Chang et al., 2005; Tsai et al., 2005), but upregulated during later stages of the disease (Chang et al., 2005). These controversial data might uncover a dynamic property in the expression of sHSPs, indicating the need to examine sHSP expression at multiple time points in all diseases discussed above.
Infectious Protein Aggregation Disease: Prions
Misfolded prions are infectious proteins that are responsible for the transmissible spongiform encephalopathies (Prusiner, 1998; DeArmond and Prusiner, 2003). These include bovine spongiform encephalopathy (BSE, commonly referred to as “mad cow disease”) in cattle, scrapie in sheep, and Creutzfeldt–Jakob disease (CJD) in humans.
Small heat shock proteins are elevated in prion disease. HSPB1 is increased in scrapie in sheep (Vidal et al., 2009) and in a mouse model of BSE (Tortosa et al., 2008). Research on HSPB5 has been limited to humans; HSPB5 has been shown to be dramatically elevated in both glia and neurons from CJD brains (Kato et al., 1992; Renkawek et al., 1992). Given the similarities in protein aggregation between tauopathies and prions, it is likely that sHSPs are playing similar roles in both disease states.
Leukodystrophies: Alexander’s Disease
Alexander’s disease is a rare genetic disease, a leukodystrophy with abnormal development of the myelin sheath, resulting from a mutation in the glial fibrillary acidic protein (GFAP; Messing et al., 2010). Alexander’s disease is typified by the appearance of Rosenthal fibers – fibrous, eosinophilic deposits in the brain that are involved in the pathogenesis of the disease. It is a progressive, neurodegenerative disease that is usually fatal.
Both HSPB1 and HSPB5 are upregulated in Alexander’s disease. Head et al. (1993) showed that both mRNA and protein levels of HSPB1 and HSPB5 were elevated in Rosenthal fibers in astrocytes taken from human patients, which was corroborated by Iwaki et al. (1989, 1993). Additionally, mouse models of Alexander’s disease that lacked HSPB5 demonstrated greater mortality, indicating that the presence of alpha-B crystallin in the astrocytes is protective (Hagemann et al., 2009).
Autoimmune Diseases: Multiple Sclerosis
Multiple sclerosis (MS) is an autoimmune demyelinating disease of the CNS that manifests as lesions predominantly in the white matter (Rejdak et al., 2010). It is characterized by a T cell mediated attack on the myelin sheath surrounding the axons of neurons. Although protein aggregation has not been observed in MS, sHSPs are elevated and appear to play a protective role during the course of the disease.
van Noort et al. (1995) first identified that HSPB5 is involved in MS pathogenesis when they pinpointed this molecule as the most immunodominant myelin T cell antigen in this disease. These findings suggested that HSPB5 might be an autoantigen in MS and that immune cells attacked endogenous HSPB5 as part of the pathology of the disease. This theory was supported by data that showed high levels of HSPB5 in astrocytes and oligodendrocytes in MS lesions (Iwaki et al., 1992; Bajramovic et al., 1997). Although subsequent studies found that HSPB5 was the most abundant transcript in MS lesions (Chabas et al., 2001), attempts to induce experimental autoimmune encephalomyelitis (EAE), the predominant mouse model of MS, with HSPB5 as an antigen, rather than using a myelin antigen, were never successful (Verbeek et al., 2007). Further research indicated that although HSPB5 is upregulated during the course of the disease, its purpose is protective rather than pathological. A 2007 study conducted by Ousman et al. (2007) found that mice deficient in HSPB5 suffered from more severe EAE than wild type mice and that treatment with exogenous HSPB5 ameliorated EAE. This study demonstrated that the absence of HSPB5 results in a more pro-inflammatory state of immune cells and a higher level of immune cell infiltration into the brain. Furthermore, treatment of HSPB5 deficient EAE mice or WT EAE mice with exogenous HSPB5 decreases immune infiltration into the brain and shifts the phenotype of these immune cells to an anti-inflammatory state. Additional studies have validated the initial reports that HSPB5 is elevated in the MS brain (Sinclair et al., 2005) and that HSPB5 is elevated in the blood of MS patients (Rothbard et al., 2012).
HSPB1 has also been shown to be upregulated in MS. Using immunohistochemistry, HSPB1 was found to be elevated in astrocytes and oligodendrocytes in the plaques (Aquino et al., 1997). A recent study found elevated levels of HSPB1 circulating in the blood of MS patients, peaking during relapses (Ce et al., 2011).
Acute Neurological Insult and Inflammation: Stroke and Epilepsy
Small heat shock proteins are not just upregulated in long-term, chronic diseases of the CNS; recent studies have shown an upregulation in acute inflammatory conditions such as stroke and epilepsy.
Stroke is the result of the lack of blood supply to the brain, leading to brain injury. The most common type is ischemic stroke, which is defined as a blocked blood vessel to the brain. Inflammatory mediators exacerbate acute stroke. These mediators infiltrate the injured area upon reperfusion. Several studies have been conducted that examine the expression of HSPB5 in human and mouse models of stroke. HSPB5 is found in ballooned neurons at the edge of cerebral infarcts (Lowe et al., 1992) and elevated in 68% of human stroke brains, specifically in the neurons (Minami et al., 2003). Studies using rodent models of cerebral ischemia indicate that HSPB5 is transiently upregulated in neurons a few hours after reperfusion and followed by gradual sustained increase in astrocytes (Piao et al., 2005). HSPB5 has also been shown to be elevated in human and mouse plasma post-stroke (Arac et al., 2011). Notably, HSPB1 is also upregulated in both rat and mouse models of ischemia (Imura et al., 1999) and overexpression of HSPB1 is neuroprotective in cerebral ischemia models (van der Weerd et al., 2010).
Epilepsy is a neurological disorder characterized by seizures. These are due to abnormal, excessive, or synchronous neuronal activity in the brain (Vezzani et al., 2011). Only one study has found that HSPB1 is elevated in epileptic human neocortex (Bidmon et al., 2004). Using immunohistochemistry, they found that HSPB1 was located in both astrocytes and the walls of blood vessels.
Proof of Concept Therapeutic Experiments
Many studies have shown that endogenous sHSPs are elevated in neurological diseases, with some studies showing the lack of sHSPs leads to worse disease, implying a protective role for these molecules. This has led to the examination of these molecules as potential novel therapeutics with proof of concept experiments in mice using three routes of administration: transgenic overexpression, viral administration, and exogenous treatment (Table 3).
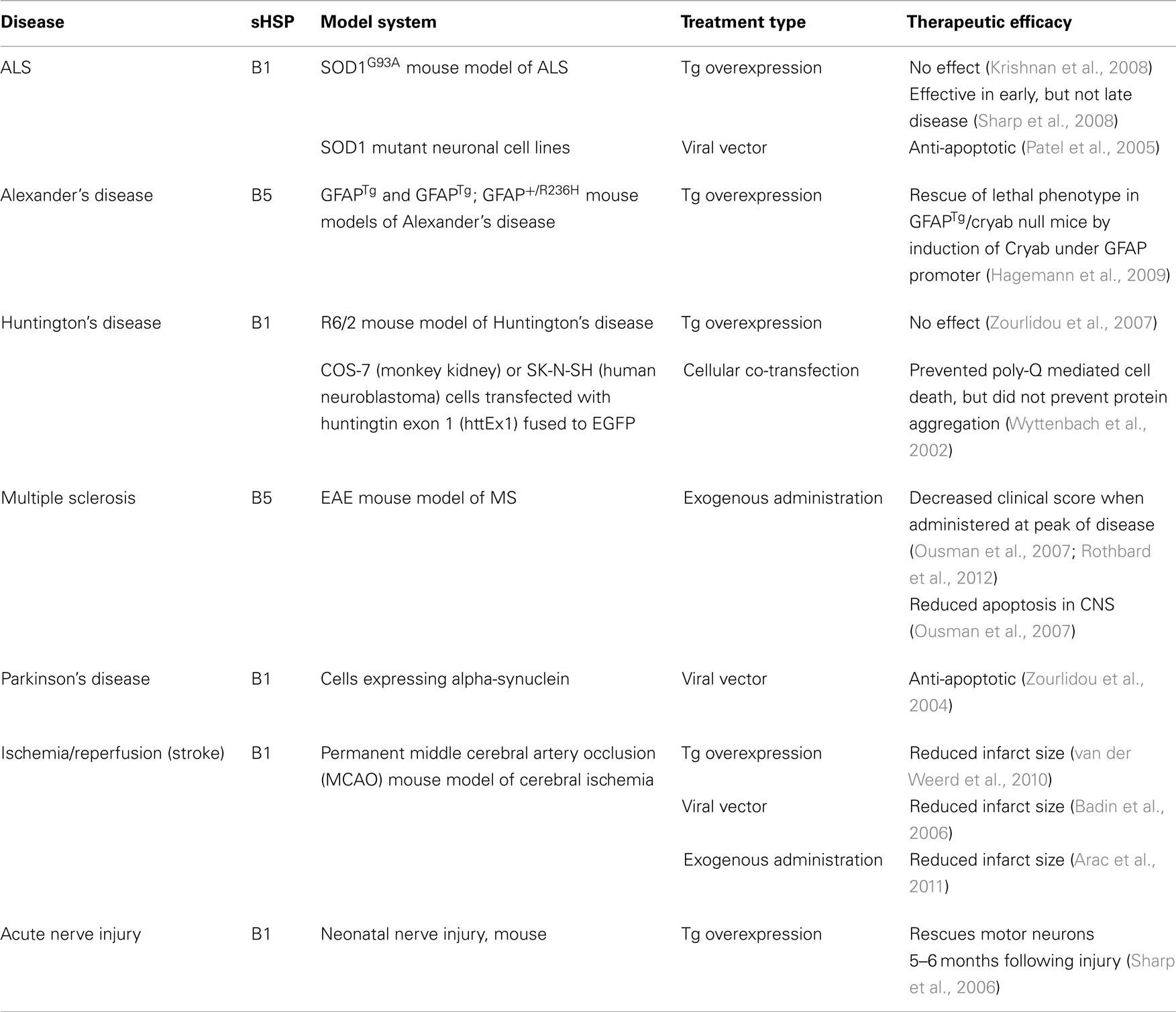
Table 3. Overexpression and exogenous administration of sHSPs.
Parkinson’s Disease and ALS
Using a viral vector, HSPB1 has been shown to have a positive effect in an in vitro model of Parkinson’s disease by protecting against alpha-synuclein induced cell death (Zourlidou et al., 2004). However, no studies have been conducted on the therapeutic effects of HSPB1 on in vivo models of PD.
Overexpression studies using transgenic mouse models of ALS have shown mixed results. One study demonstrates that overexpression of transgenic HSPB1 in the SOD1G93A mouse model of ALS by crossing SOD1G93A mice with HSPB1 overexpressing mice did not delay disease onset or decrease disease severity (Krishnan et al., 2008), despite the fact that HSPB1 overexpression was protective in acute motor neuron injury (Sharp et al., 2006). However, another study by Sharp et al. (2008) indicates that HSPB1 does have a positive effect early in the disease course in the same mouse model of ALS. They show that SOD1G93A/HSP27 double transgenic mice had delayed decline in motor strength and increased survival of spinal motor neurons compared to SOD1G93A single transgenics during the early phase of disease. In vitro experiments using SOD1 mutant cells demonstrate that HSPB1 has an anti-apoptotic function (Patel et al., 2005). This suggests that HSPB1 does have an effect, but the levels in the overexpressing mouse may not be high enough to combat the chronic disease state. It may also suggest that the cellular upregulation of sHSPs represents only one part of the protective response and that the presence of sHSPs in the plasma may be a crucial factor in ameliorating disease.
Alexander’s Disease
A study conducted by Hagemann et al. (2009) using a mouse model of Alexander’s disease demonstrated that mice lacking HSPB5 exhibited increased mortality, but restoring HSPB5 specifically in astrocytes using a GFAP promoter reversed this effect. Additionally, transgenic overexpression of HSPB5 protects mice from death in a second model of Alexander’s disease that typically causes the mice to die at one month of age (Hagemann et al., 2009).
Multiple Sclerosis
Mice lacking HSPB5 were found to suffer from more severe disease in the experimental autoimmune encephalomyelitis (EAE) model of MS, and intravenous administration of exogenous HSPB5 ameliorated disease (Ousman et al., 2007). EAE mice treated with HSPB5 showed a dampened immune response, including less proliferation of immune cells and a lower production of pro-inflammatory cytokines (Ousman et al., 2007). Furthermore, we believe that this anti-inflammatory effect is based on HSPB5’s ability to act as a chaperone extracellularly, binding pro-inflammatory molecules, including members of the acute phase, coagulation, and complement pathways (Rothbard et al., 2012). Additional experiments using other members of the sHSP family have indicated that all members are therapeutic in EAE and they act through anti-inflammatory effects (unpublished data). This further expands the research supporting the therapeutic possibilities of sHSPs in MS (Steinman, 2008).
Stroke
Transgenic mice that overexpress HSPB1 exhibited 30% smaller lesion sizes after undergoing a permanent MCAO model of stroke (van der Weerd et al., 2010). This corroborated previous work conducted in a cardiac ischemia model that showed that transgenic overexpression of HSPB1 was protective (Hollander et al., 2004). Virally administered HSPB1 was also shown to be protective in cerebral ischemia, although the mechanism was not clear (Badin et al., 2006). The researchers of that study speculated that the protective effect of HSPB1 might be due to its chaperone function. Finally, recent work has shown that HSPB5 is therapeutic when treating mouse models of stroke 12 h post-insult (Arac et al., 2011).
Mechanisms of sHSP Neuroprotection
A substantial number of studies have indicated that endogenous sHSPs, particularly HSPB1 and HSPB5, are upregulated in CNS injury and disease. Studies investigating both sHSP deficiency and overexpression support the conclusion that these molecules are serving neuroprotective roles rather than pathological ones.
A caveat to the studies done using the HSPB5 deficient mice: these mice also lack the sHSP HSPB2. HSPB2 is not thought to be inducible, so the levels of HSPB2 are typically not elevated in neurological diseases (for an exception see Wilhelmus et al., 2006b). Although we believe that the phenotype we see in HSPB5 deficient mice is due to the lack of HSPB5 alone, it is possible that the lack of HSPB2 is contributing to the effects observed in these mice. However, HSPB5/HSPB2 deficient mice have worse disease, which supports the idea that sHSPs are serving protective roles whether that is due to HSPB2 or HSPB5. An additional point to consider is whether the phenotype observed in HSPB5/HSPB2 deficient mice is due to the lack of the sHSPs or the altered levels of another molecule that is dependent on normal sHSP function. If isogenic mouse strains are not used as controls, then it is possible that polymorphisms in other genes could be the cause of differences seen between the WT and HSPB5/HSPB2 deficient mice.
How sHSPs are neuroprotective is still a subject of debate. It could be due to its molecular chaperone properties that prevent protein aggregation, in particular for protein aggregation diseases such as Alzheimer’s or Parkinson’s. However, sHSPs are also protective in non-aggregation diseases such as acute ischemia. Several studies have indicated an anti-apoptotic role for sHSPs, which might be the reason they are protective. HSPB5 and HSPB1 have been shown to be anti-apoptotic when overexpressed by either a transgene or virus (Akbar et al., 2003; Kalwy et al., 2003; Li et al., 2005). HSPB5 confers protection against apoptosis through the regulation of caspase-3, a proapoptotic factor (Shin et al., 2009) and has also been shown to sequester the p53 tumor suppressor, thus preventing apoptosis (Liu et al., 2007).
However, in light of recent studies that have indicated that sHSPs have an anti-inflammatory role (Ousman et al., 2007; Arac et al., 2011), the anti-apoptotic effects of sHSPs are unlikely to be the whole story. Although historically the brain was thought to be immune privileged, research over the past few decades has shown that the immune system is an important factor in many neurological diseases previously thought to be independent of the immune system, including Alzheimer’s and stroke. Endogenously upregulated sHSPs may be playing dual roles as both anti-apoptotic and anti-inflammatory molecules in these diseases. In fact, Arac et al. (2011) investigated the relative effects of the deficiency of HSPB5 in the immune system and the brain post-stroke by doing a bone marrow chimera experiment. By irradiating WT and HSPB5−/− mice, depleting their immune systems, and reconstituting their immune systems with immune cells from either WT or HSPB5 deficient mice, they created four different types of mice: WT mice with WT immune cells, WT mice with HSPB5−/− immune cells, HSPB5−/− mice with WT immune cells, and HSPB5−/− mice with HSPB5−/− immune cells. After inducing stroke and comparing infarct size, they found that deficiencies of HSPB5 in either the brain or immune system alone increases infarct size and together result in a synergistic effect (Arac et al., 2011). They did not demonstrate any specificity to particular brain cells and this is an area of future investigation to fully understand the contribution of HSPB5 in the brain compared to the immune system.
The mechanism of action by which sHSPs are anti-inflammatory is currently under investigation. A study conducted by Rothbard et al. (2012) suggests that HSPB5 acts extracellularly as a molecular chaperone, binding acute inflammatory molecules. Notably, they showed that HSPB5 binding was temperature-dependent and binding increases with an increase in temperature, making HSPB5 more effective at sites of inflammation. Exactly how endogenous HSPB5 is released from cells is currently a matter of speculation. HSPB5 does not possess a signal sequence, so it is not secreted by the normal secretory pathway. However, a recent paper showed that HSPB5 could be released via exosomes (Gangalum et al., 2011). Additionally, if damage is occurring in the brain, it is very likely that cells are undergoing apoptosis or necrosis and releasing HSPB5 upon death. However, whether HSPB5 is released in a regulated manner through a specific process or leaked out of damaged or dying cells, the specific mechanism by which it becomes extracellular does not affect our interpretation that it can act as an extracellular chaperone.
Although we do see higher endogenous levels of sHSPs in neurological diseases, in particular at the site of damage, studies investigating the therapeutic effect of sHSPs using exogenous administration of sHSPs have been focused only on mouse models of MS and stroke. Using varying concentrations of human recombinant HSPB5 administered both intravenously and intraperitoneally, a therapeutic effect of HSPB5 has been shown in mouse models of MS (Ousman et al., 2007; unpublished data). This therapeutic effect has been demonstrated in EAE mice with different genetic backgrounds and we have seen a similar therapeutic effect from the administration of other members of the sHSP family (unpublished data). HSPB5 has also been shown to be therapeutic when administered intraperitoneally in a mouse model of stroke (Arac et al., 2011). We believe that it is likely that exogenous administration of HSPB5 will be therapeutic in other neurological diseases, but the experiments have yet to be done.
The vast literature indicating that endogenous sHSPs, particularly HSPB1 and HSPB5, are protective in neurological diseases opens the door for the possibility that these molecules could be developed as novel therapeutics. To date, therapeutic strategies utilizing sHSPs have been conducted solely in mice; however, the data generated from these experiments have provided the foundation to pursue this exciting avenue of therapy.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ackerley, S., James, P. A., Kalli, A., French, S., Davies, K. E., and Talbot, K. (2006). A mutation in the small heat-shock protein HSPB1 leading to distal hereditary motor neuronopathy disrupts neurofilament assembly and the axonal transport of specific cellular cargoes. Hum. Mol. Genet. 15, 347–354.
Akbar, M. T., Lundberg, A. M., Liu, K., Vidyadaran, S., Wells, K. E., Dolatshad, H., Wynn, S., Wells, D. J., Latchman, D. S., and de Belleroche, J. (2003). The neuroprotective effects of heat shock protein 27 overexpression in transgenic animals against kainate-induced seizures and hippocampal cell death. J. Biol. Chem. 278, 19956–19965.
Andley, U. P. (2007). Crystallins in the eye: function and pathology. Prog. Retin. Eye Res. 26, 78–98.
Aquino, D. A., Capello, E., Weisstein, J., Sanders, V., Lopez, C., Tourtellotte, W. W., Brosnan, C. F., Raine, C. S., and Norton, W. T. (1997). Multiple sclerosis: altered expression of 70- and 27-kDa heat shock proteins in lesions and myelin. J. Neuropathol. Exp. Neurol. 56, 664–672.
Arac, A., Brownell, S. E., Rothbard, J. B., Chen, C., Ko, R. M., Pereira, M. P., Albers, G. W., Steinman, L., and Steinberg, G. K. (2011). Systematic augmentation of alpha B crystallin, a novel immunomodulatory target, provides therapeutic benefit twelve hours post-stroke onset. Proc. Natl. Acad. Sci. U.S.A. 108, 13287–13292.
Armstrong, C. L., Krueger-Naug, A. M., Currie, R. W., and Hawkes, R. (2001). Constitutive expression of heat shock protein HSP25 in the central nervous system of the developing and adult mouse. J. Comp. Neurol. 434, 262–274.
Arrigo, A. P., Simon, S., Gibert, B., Kretz-Remy, C., Nivon, M., Czekalla, A., Guillet, D., Moulin, M., Diaz-Latoud, C., and Vicart, P. (2007). Hsp27 (HspB1) and alphaB-crystallin (HspB5) as therapeutic targets. FEBS Lett. 581, 3665–3674.
Badin, R. A., Lythgoe, M. F., van der Weerd, L., Thomas, D. L., Gadian, D. G., and Latchman, D. S. (2006). Neuroprotective effects of virally delivered HSPs in experimental stroke. J. Cereb. Blood Flow Metab. 26, 371–381.
Bagnéris, C., Bateman, O. A., Naylor, C. E., Cronin, N., Boelens, W. C., Keep, N. H., and Slingsby, C. (2009). Crystal structures of alpha-crystallin domain dimers of alphaB-crystallin and Hsp20. J. Mol. Biol. 392, 1242–1252.
Bajramovic, J. J., Lassmann, H., and van Noort, J. M. (1997). Expression of alphaB-crystallin in glia cells during lesional development in multiple sclerosis. J. Neuroimmunol. 78, 143–151.
Ballard, C., Gauthier, S., Corbett, A., Brayne, C., Aarsland, D., and Jones, E. (2011). Alzheimer’s disease. Lancet 377, 1019–1031.
Ballatore, C., Brunden, K. R., Trojanowski, J. Q., Lee, V. M., Smith AB, I. I., and Huryn, D. M. (2011). Modulation of protein-protein interactions as a therapeutic strategy for the treatment of neurodegenerative tauopathies. Curr. Top. Med. Chem. 11, 317–330.
Bidmon, H. J., Görg, B., Palomero-Gallagher, N., Behne, F., Lahl, R., Pannek, H. W., Speckmann, E. J., and Zilles, K. (2004). Heat shock protein-27 is upregulated in the temporal cortex of patients with epilepsy. Epilepsia 45, 1549–1559.
Björkdahl, C., Sjögren, M. J., Zhou, X., Concha, H., Avila, J., Winblad, B., and Pei, J. J. (2008). Small heat shock proteins Hsp27 or alphaB-crystallin and the protein components of neurofibrillary tangles: tau and neurofilaments. J. Neurosci. Res. 86, 1343–1352.
Burn, D. J. (2006). Cortical Lewy body disease and Parkinson’s disease dementia. Curr. Opin. Neurol. 19, 572–579.
Ce, P., Erkizan, O., and Gedizlioglu, M. (2011). Elevated HSP27 levels during attacks in patients with multiple sclerosis. Acta Neurol. Scand. 124, 317–320.
Chabas, D., Baranzini, S. E., Mitchell, D., Bernard, C. C., Rittling, S. R., Denhardt, D. T., Sobel, R. A., Lock, C., Karpuj, M., Pedotti, R., Heller, R., Oksenberg, J. R., and Steinman, L. (2001). The influence of the proinflammatory cytokine, osteopontin, on autoimmune demyelinating disease. Science 294, 1731–1735.
Chang, W. H., Cemal, C. K., Hsu, Y. H., Kuo, C. L., Nukina, N., Chang, M. H., Hu, H. T., Li, C., and Hsieh, M. (2005). Dynamic expression of Hsp27 in the presence of mutant ataxin-3. Biochem. Biophys. Res. Commun. 336, 258–267.
Dabir, D. V., Trojanowski, J. Q., Richter-Landsberg, C., Lee, V. M., and Forman, M. S. (2004). Expression of the small heat-shock protein alphaB-crystallin in tauopathies with glial pathology. Am. J. Pathol. 164, 155–166.
DeArmond, S. J., and Prusiner, S. B. (2003). Perspectives on prion biology, prion disease pathogenesis, and pharmacologic approaches to treatment. Clin. Lab. Med. 23, 1–41.
Di Donato, S., Gellera, C., and Mariotti, C. (2001). The complex clinical and genetic classification of inherited ataxias. II. Autosomal recessive ataxias. Neurol. Sci. 22, 219–228.
Dubin, R. A., Wawrousek, E. F., and Piatigorsky, J. (1989). Expression of the murine alpha B-crystallin gene is not restricted to the lens. Mol. Cell. Biol. 9, 1083–1091.
Eidelberg, D., and Surmeier, D. J. (2011). Brain networks in Huntington disease. J. Clin. Invest. 121, 484–492.
Evgrafov, O. V., Mersiyanova, I., Irobi, J., Van Den Bosch, L., Dierick, I., Leung, C. L., Schagina, O., Verpoorten, N., Van Impe, K., Fedotov, V., Dadali, E., Auer-Grumbach, M., Windpassinger, C., Wagner, K., Mitrovic, Z., Hilton-Jones, D., Talbot, K., Martin, J. J., Vasserman, N., Tverskaya, S., Polyakov, A., Liem, R. K., Gettemans, J., Robberecht, W., De Jonghe, P., and Timmerman, V. (2004). Mutant small heat-shock protein 27 causes axonal Charcot-Marie-tooth disease and distal hereditary motor neuropathy. Nat. Genet. 36, 602–606.
Fukushima, K., Mizuno, Y., Takatama, M., and Okamoto, K. (2006). Increased neuronal expression of alpha B-crystallin in human olivary hypertrophy. Neuropathology 26, 196–200.
Ganea, E. (2001). Chaperone-like activity of alpha-crystallin and other small heat shock proteins. Curr. Protein Pept. Sci. 2, 205–225.
Gangalum, R. K., Atanasov, I. C., Zhou, Z. H., and Bhat, S. P. (2011). AlphaB-crystallin is found in detergent-resistant membrane microdomains and is secreted via exosomes from human retinal pigment epithelial cells. J. Biol. Chem. 286, 3261–3269.
Hagemann, T. L., Boelens, W. C., Wawrousek, E. F., and Messing, A. (2009). Suppression of GFAP toxicity by alphaB-crystallin in mouse models of Alexander disease. Hum. Mol. Genet. 18, 1190–1199.
Han, M. H., Hwang, S. I., Roy, D. B., Lundgren, D. H., Price, J. V., Ousman, S. S., Fernald, G. H., Gerlitz, B., Robinson, W. H., Baranzini, S. E., Grinnell, B. W., Raine, C. S., Sobel, R. A., Han, D. K., and Steinman, L. (2009). Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature 451, 1076–1081.
Hanihara, T., Amano, N., Takahashi, T., Itoh, Y., and Yagishita, S. (1998). Hypertrophy of the inferior olivary nucleus in patients with progressive supranuclear palsy. Eur. Neurol. 39, 97–102.
Head, M. W., Corbin, E., and Goldman, J. E. (1993). Overexpression and abnormal modification of the stress proteins alpha B-crystallin and HSP27 in Alexander disease. Am. J. Pathol. 143, 1743–1753.
Hollander, J. M., Martin, J. L., Belke, D. D., Scott, B. T., Swanson, E., Krishnamoorthy, V., and Dillmann, W. H. (2004). Overexpression of wild-type heat shock protein 27 and a nonphosphorylatable heat shock protein 27 mutant protects against ischemia/reperfusion injury in a transgenic mouse model. Circulation 110, 3544–3552.
Horwitz, J. (1992). Alpha-crystallin can function as a molecular chaperone. Proc. Natl. Acad. Sci. U.S.A. 89, 10449–10453.
Imura, T., Shimohama, S., Sato, M., Nishikawa, H., Madono, K., Akaike, A., and Kimura, J. (1999). Differential expression of small heat shock proteins in reactive astrocytes after focal ischemia: possible role of beta-adrenergic receptor. J. Neurosci. 19, 9768–9779.
Ito, H., Okamoto, K., and Kato, K. (1997). Prostaglandins stimulate the stress-induced synthesis of hsp27 and alpha B crystallin. J. Cell. Physiol. 170, 255–262.
Iwaki, T., Iwaki, A., Tateishi, J., Sakaki, Y., and Goldman, J. E. (1993). Alpha B-crystallin and 27-kd heat shock protein are regulated by stress conditions in the central nervous system and accumulate in Rosenthal fibers. Am. J. Pathol. 143, 487–495.
Iwaki, T., Kume-Iwaki, A., Liem, R. K., and Goldman, J. E. (1989). Alpha B-crystallin is expressed in non-lenticular tissues and accumulates in Alexander’s disease brain. Cell 57, 71–78.
Iwaki, T., Wisniewski, T., Iwaki, A., Corbin, E., Tomokane, N., Tateishi, J., and Goldman, J. E. (1992). Accumulation of alpha B-crystallin in central nervous system glia and neurons in pathologic conditions. Am. J. Pathol. 140, 345–356.
Jakob, U., Gaestel, M., Engel, K., and Buchner, J. (1993). Small heat shock proteins are molecular chaperones. J. Biol. Chem. 268, 1517–1520.
Jankovic, J. (2008). Parkinson’s disease: clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatr. 79, 368–376.
Jehle, S., Rajagopal, P., Bardiaux, B., Markovic, S., Kühne, R., Stout, J. R., Higman, V. A., Klevit, R. E., van Rossum, B. J., and Oschkinat, H. (2010). Solid-state NMR and SAXS studies provide a structural basis for the activation of alphaB-crystallin oligomers. Nat. Struct. Mol. Biol. 17, 1037–1042.
Jehle, S., van Rossum, B., Stout, J. R., Noguchi, S. M., Falber, K., Rehbein, K., Oschkinat, H., Klevit, R. E., and Rajagopal, P. (2009). alphaB-crystallin: a hybrid solid-state/solution-state NMR investigation reveals structural aspects of the heterogeneous oligomer. J. Mol. Biol. 385, 1481–1497.
Jehle, S., Vollmar, B. S., Bardiaux, B., Dove, K. K., Rajagopal, P., Gonen, T., Oschkinat, H., and Klevit, R. E. (2011). N-terminal domain of alphaB-crystallin provides a conformational switch for multimerization and structural heterogeneity. Proc. Natl. Acad. Sci. U.S.A. 108, 6409–6414.
Kalwy, S. A., Akbar, M. T., Coffin, R. S., de Belleroche, J., and Latchman, D. S. (2003). Heat shock protein 27 delivered via a herpes simplex virus vector can protect neurons of the hippocampus against kainic-acid-induced cell loss. Brain Res. Mol. Brain Res. 111, 91–103.
Kappé, G., Franck, E., Verschuure, P., Boelens, W. C., Leunissen, J. A., and de Jong, W. W. (2003). The human genome encodes 10 alpha-crystallin-related small heat shock proteins: HspB1-10. Cell Stress Chaperones 8, 53–61.
Kato, S., Hirano, A., Umahara, T., Kato, M., Herz, F., and Ohama, E. (1992). Comparative immunohistochemical study on the expression of alpha B crystallin, ubiquitin and stress-response protein 27 in ballooned neurons in various disorders. Neuropathol. Appl. Neurobiol. 18, 335–340.
Kertesz, A. (2003). Pick complex: an integrative approach to frontotemporal dementia: primary progressive aphasia, corticobasal degeneration, and progressive supranuclear palsy. Neurologist 9, 311–317.
Kirbach, B. B., and Golenhofen, N. (2011). Differential expression and induction of small heat shock proteins in rat brain and cultured hippocampal neurons. J. Neurosci. Res. 89, 162–175.
Komori, T. (1999). Tau-positive glial inclusions in progressive supranuclear palsy, corticobasal degeneration and Pick’s disease. Brain Pathol. 9, 663–679.
Krishnan, J., Vannuvel, K., Andries, M., Waelkens, E., Robberecht, W., and Van Den Bosch, L. (2008). Over-expression of Hsp27 does not influence disease in the mutant SOD1 (G93A) mouse model of amyotrophic lateral sclerosis. J. Neurochem. 106, 2170–2183.
Li, D. W., Liu, J. P., Mao, Y. W., Xiang, H., Wang, J., Ma, W. Y., Dong, Z., Pike, H. M., Brown, R. E., and Reed, J. C. (2005). Calcium-activated RAF/MEK/ERK signaling pathway mediates p53-dependent apoptosis and is abrogated by alpha B-crystallin through inhibition of RAS activation. Mol. Biol. Cell 16, 4437–4453.
Litt, M., Kramer, P., LaMorticella, D. M., Murphey, W., Lovrien, E. W., and Weleber, R. G. (1998). Autosomal dominant congenital cataract associated with a missense mutation in the human alpha crystallin gene CRYAA. Hum. Mol. Genet. 7, 471–474.
Liu, S., Li, J., Tao, Y., and Xiao, X. (2007). Small heat shock protein alphaB-crystallin binds to p53 to sequester its translocation to mitochondria during hydrogen peroxide-induced apoptosis. Biochem. Biophys. Res. Commun. 354, 109–114.
Lowe, J., Errington, D. R., Lennox, G., Pike, I., Spendlove, I., Landon, M., and Mayer, R. J. (1992). Ballooned neurons in several neurodegenerative diseases and stroke contain alpha B crystallin. Neuropathol. Appl. Neurobiol. 18, 341–350.
Maatkamp, A., Vlug, A., Haasdijk, E., Troost, D., French, P. J., and Jaarsma, D. (2004). Decrease of Hsp25 protein expression precedes degeneration of motoneurons in ALS-SOD1 mice. Eur. J. Neurosci. 20, 14–28.
Mao, J. J., Katayama, S., Watanabe, C., Harada, Y., Noda, K., Yamamura, Y., and Nakamura, S. (2001). The relationship between alphaB-crystallin and neurofibrillary tangles in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 27, 180–188.
Messing, A., Daniels, C. M., and Hagemann, T. L. (2010). Strategies for treatment in Alexander disease. Neurotherapeutics 7, 507–515.
Minami, M., Mizutani, T., Kawanishi, R., Suzuki, Y., and Mori, H. (2003). Neuronal expression of alphaB crystallin in cerebral infarction. Acta Neuropathol. 105, 549–554.
Morimoto, R. I., and Santoro, M. G. (1998). Stress-inducible responses and heat shock proteins: new pharmacologic targets for cytoprotection. Nat. Biotechnol. 16, 833–838.
Nédellec, P., Edling, Y., Perret, E., Fardeau, M., and Vicart, P. (2002). Glucocorticoid treatment induces expression of small heat shock proteins in human satellite cell populations: consequences for a desmin-related myopathy involving the R120G alpha B-crystallin mutation. Neuromuscul. Disord. 12, 457–465.
Oba, M., Yano, S., Shuto, T., Suico, M. A., Eguma, A., and Kai, H. (2008). IFN-gamma down-regulates Hsp27 and enhances hyperthermia-induced tumor cell death in vitro and tumor suppression in vivo. Int. J. Oncol. 32, 1317–1324.
Ousman, S. S., Tomooka, B. H., van Noort, J. M., Wawrousek, E. F., O’Connor, K. C., Hafler, D. A., Sobel, R. A., Robinson, W. H., and Steinman, L. (2007). Protective and therapeutic role for alphaB-crystallin in autoimmune demyelination. Nature 448, 474–479.
Patel, Y. J., Payne Smith, M. D., de Belleroche, J., and Latchman, D. S. (2005). Hsp27 and Hsp70 administered in combination have a potent protective effect against FALS-associated SOD1-mutant-induced cell death in mammalian neuronal cells. Brain Res. Mol. Brain Res. 134, 256–274.
Paulson, H. L., Bonini, N. M., and Roth, K. A. (2000). Polyglutamine disease and neuronal cell death. Proc. Natl. Acad. Sci. U.S.A. 97, 12957–12958.
Perry, J., Shin, D. S., Pratt, A. J., and Getzoff, E. D. (2010). Amyotrophic lateral sclerosis. Adv. Exp. Med. Biol. 685, 9–20.
Piao, C. S., Kim, S. W., Kim, J. B., and Lee, J. K. (2005). Co-induction of alphaB-crystallin and MAPKAPK-2 in astrocytes in the penumbra after transient focal cerebral ischemia. Exp. Brain Res. 163, 421–429.
Poulain, P., Gelly, J. C., and Flatters, D. (2010). Detection and architecture of small heat shock protein monomers. PLoS ONE 5, e9990. doi:10.1371/journal.pone.0009990
Quraishe, S., Asuni, A., Boelens, W. C., O’Connor, V., and Wyttenbach, A. (2008). Expression of the small heat shock protein family in the mouse CNS: differential anatomical and biochemical compartmentalization. Neuroscience 153, 483–491.
Rejdak, K., Jackson, S., and Giovannoni, G. (2010). Multiple sclerosis: a practical overview for clinicians. Br. Med. Bull. 95, 79–104.
Renkawek, K., Bosman, G. J., and de Jong, W. W. (1994a). Expression of small heat-shock protein hsp 27 in reactive gliosis in Alzheimer disease and other types of dementia. Acta Neuropathol. 87, 511–519.
Renkawek, K., Voorter, C. E., Bosman, G. J., van Workum, F. P., and de Jong, W. W. (1994b). Expression of alpha B-crystallin in Alzheimer’s disease. Acta Neuropathol. 87, 155–160.
Renkawek, K., de Jong, W. W., Merck, K. B., Frenken, C. W., van Workum, F. P., and Bosman, G. J. (1992). alpha B-crystallin is present in reactive glia in Creutzfeldt-Jakob disease. Acta Neuropathol. 83, 324–327.
Renkawek, K., Stege, G. J., and Bosman, G. J. (1999). Dementia, gliosis and expression of the small heat shock proteins hsp27 and alpha B-crystallin in Parkinson’s disease. Neuroreport 10, 2273–2276.
Rosen, D. R., Siddique, T., Patterson, D., Figlewicz, D. A., Sapp, P., Hentati, A., Donaldson, D., Goto, J., O’Regan, J. P., Deng, H. X., Rahmani, Z., Krizus, A., McKenna-Yasek, D., Cayabyab, A., Gaston, S. M., Berger, R., Tanzi, R. E., Halperin, J. J., Herzfeldt, B., den Bergh R, V., Hung, W.-Y., Bird, T., Deng, G., Mulder, D. W., Smyth, C., Laing, N. G., Soriano, E., Pericak–Vance, M. A., Haines, J., Rouleau, G. A., Gusella, J. S., Horvitz, H. R., and Brown, R. H. Jr. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62.
Rothbard, J. B., Kurnellas, M. P., Brownell, S., Adams, C. M., Su, L., Axtell, R. C., Chen, R., Fathman, C. G., Robinson, W. H., and Steinman, L. (2012). Therapeutic effects of systemic administration of chaperone αB-crystallin associated with binding proinflammatory plasma proteins. J. Biol. Chem. 287, 9708–9721.
Schwarz, L., Vollmer, G., and Richter-Landsberg, C. (2010). The small heat shock protein HSP25/27 (HspB1) is abundant in cultured astrocytes and associated with astrocytic pathology in progressive supranuclear palsy and corticobasal degeneration. Int. J. Cell Biol. 2010, 717520.
Sharp, P., Krishnan, M., Pullar, O., Navarrete, R., Wells, D., and de Belleroche, J. (2006). Heat shock protein 27 rescues motor neurons following nerve injury and preserves muscle function. Exp. Neurol. 198, 511–518.
Sharp, P. S., Akbar, M. T., Bouri, S., Senda, A., Joshi, K., Chen, H. J., Latchman, D. S., Wells, D. J., and de Belleroche, J. (2008). Protective effects of heat shock protein 27 in a model of ALS occur in the early stages of disease progression. Neurobiol. Dis. 30, 42–55.
Shimura, H., Miura-Shimura, Y., and Kosik, K. S. (2004). Binding of tau to heat shock protein 27 leads to decreased concentration of hyperphosphorylated tau and enhanced cell survival. J. Biol. Chem. 279, 17957–17962.
Shin, J. H., Kim, S. W., Lim, C. M., Jeong, J. Y., Piao, C. S., and Lee, J. K. (2009). alphaB-crystallin suppresses oxidative stress-induced astrocyte apoptosis by inhibiting caspase-3 activation. Neurosci. Res. 64, 355–361.
Shinohara, H., Inaguma, Y., Goto, S., Inagaki, T., and Kato, K. (1993). Alpha B crystallin and HSP28 are enhanced in the cerebral cortex of patients with Alzheimer’s disease. J. Neurol. Sci. 119, 203–208.
Simon, S., Fontaine, J. M., Martin, J. L., Sun, X., Hoppe, A. D., Welsh, M. J., Benndorf, R., and Vicart, P. (2007). Myopathy-associated alphaB-crystallin mutants: abnormal phosphorylation, intracellular location, and interactions with other small heat shock proteins. J. Biol. Chem. 282, 34276–34287.
Sinclair, C., Mirakhur, M., Kirk, J., Farrell, M., and McQuaid, S. (2005). Up-regulation of osteopontin and alpha beta-crystallin in the normal-appearing white matter of multiple sclerosis: an immunohistochemical study utilizing tissue microarrays. Neuropathol. Appl. Neurobiol. 31, 292–303.
Sun, Y., and MacRae, T. H. (2005). The small heat shock proteins and their role in human disease. FEBS J. 272, 2613–2627.
Tolnay, M., and Probst, A. (2003). The neuropathological spectrum of neurodegenerative tauopathies. IUBMB Life 55, 299–305.
Tortosa, R., Vidal, E., Costa, C., Alamillo, E., Torres, J. M., Ferrer, I., and Pumarola, M. (2008). Stress response in the central nervous system of a transgenic mouse model of bovine spongiform encephalopathy. Vet. J. 178, 126–129.
Traub, R., Mitsumoto, H., and Rowland, L. P. (2011). Research advances in amyotrophic lateral sclerosis, 2009 to 2010. Curr. Neurol. Neurosci. Rep. 11, 67–77.
Tsai, H. F., Lin, S. J., Li, C., and Hsieh, M. (2005). Decreased expression of Hsp27 and Hsp70 in transformed lymphoblastoid cells from patients with spinocerebellar ataxia type 7. Biochem. Biophys. Res. Commun. 334, 1279–1286.
van der Weerd, L., Tariq Akbar, M., Aron Badin, R., Valentim, L. M., Thomas, D. L., Wells, D. J., Latchman, D. S., Gadian, D. G., Lythgoe, M. F., and de Belleroche, J. S. (2010). Overexpression of heat shock protein 27 reduces cortical damage after cerebral ischemia. J. Cereb. Blood Flow Metab. 30, 849–856.
Van Montfort, R., Slingsby, C., and Vierling, E. (2001). Structure and function of the small heat shock protein/alpha-crystallin family of molecular chaperones. Adv. Protein Chem. 59, 105–156.
van Noort, J. M., van Sechel, A. C., Bajramovic, J. J., el Ouagmiri, M., Polman, C. H., Lassmann, H., and Ravid, R. (1995). The small heat-shock protein alpha B-crystallin as candidate autoantigen in multiple sclerosis. Nature 375, 798–801.
Verbeek, R., van Dongen, H., Wawrousek, E. F., Amor, S., and van Noort, J. M. (2007). Induction of EAE by T cells specific for alpha B-crystallin depends on prior viral infection in the CNS. Int. Immunol. 19, 277–285.
Vezzani, A., French, J., Bartfai, T., and Baram, T. Z. (2011). The role of inflammation in epilepsy. Nat. Rev. Neurol. 7, 31–40.
Vicart, P., Caron, A., Guicheney, P., Li, Z., Prévost, M. C., Faure, A., Chateau, D., Chapon, F., Tomé, F., Dupret, J. M., Paulin, D., and Fardeau, M. (1998). A missense mutation in the alphaB-crystallin chaperone gene causes a desmin-related myopathy. Nat. Genet. 20, 92–95.
Vidal, E., Acín, C., Foradada, L., Monzón, M., Márquez, M., Monleón, E., Pumarola, M., Badiola, J. J., and Bolea, R. (2009). Immunohistochemical characterisation of classical scrapie neuropathology in sheep. J. Comp. Pathol. 141, 135–146.
Vleminckx, V., Van Damme, P., Goffin, K., Delye, H., Van Den Bosch, L., and Robberecht, W. (2002). Upregulation of HSP27 in a transgenic model of ALS. J. Neuropathol. Exp. Neurol. 61, 968–974.
Wang, J., Martin, E., Gonzales, V., Borchelt, D. R., and Lee, M. K. (2008). Differential regulation of small heat shock proteins in transgenic mouse models of neurodegenerative diseases. Neurobiol. Aging 29, 586–597.
Weder, N. D., Aziz, R., Wilkins, K., and Tampi, R. R. (2007). Frontotemporal dementias: a review. Ann. Gen. Psychiatry 6, 15.
Wen, F. C., Li, Y. H., Tsai, H. F., Lin, C. H., Li, C., Liu, C. S., Lii, C. K., Nukina, N., and Hsieh, M. (2003). Down-regulation of heat shock protein 27 in neuronal cells and non-neuronal cells expressing mutant ataxin-3. FEBS Lett. 546, 307–314.
Wilhelmus, M. M., Boelens, W. C., Otte-Höller, I., Kamps, B., Kusters, B., Maat-Schieman, M. L., de Waal, R. M., and Verbeek, M. M. (2006a). Small heat shock protein HspB8: its distribution in Alzheimer’s disease brains and its inhibition of amyloid-beta protein aggregation and cerebrovascular amyloid-beta toxicity. Acta Neuropathol. 111, 139–149.
Wilhelmus, M. M., Otte-Höller, I., Wesseling, P., de Waal, R. M., Boelens, W. C., and Verbeek, M. M. (2006b). Specific association of small heat shock proteins with the pathological hallmarks of Alzheimer’s disease brains. Neuropathol. Appl. Neurobiol. 32, 119–130.
Wyttenbach, A., Sauvageot, O., Carmichael, J., Diaz-Latoud, C., Arrigo, A. P., and Rubinsztein, D. C. (2002). Heat shock protein 27 prevents cellular polyglutamine toxicity and suppresses the increase of reactive oxygen species caused by huntingtin. Hum. Mol. Genet. 11, 1137–1151.
Zabel, C., Chamrad, D. C., Priller, J., Woodman, B., Meyer, H. E., Bates, G. P., and Klose, J. (2002). Alterations in the mouse and human proteosome caused by Huntington’s disease. Mol. Cell Proteomics 1, 366–375.
Zhang, Y., Huang, L., Zhang, J., Moskophidis, D., and Mivechi, N. F. (2002). Targeted disruption of hsf1 leads to lack of thermotolerance and defines tissue-specific regulation for stress-inducible Hsp molecular chaperones. J. Cell. Biochem. 86, 376–393.
Zourlidou, A., Gidalevitz, T., Kristiansen, M., Landles, C., Woodman, B., Wells, D. J., Latchman, D. S., de Belleroche, J., Tabrizi, S. J., Morimoto, R. I., and Bates, G. P. (2007). Hsp27 overexpression in the R6/2 mouse model of Huntington’s disease: chronic neurodegeneration does not induce Hsp27 activation. Hum. Mol. Genet. 16, 1078–1090.
Keywords: small heat shock proteins, neuroinflammation, neurological diseases, HSPB1, HSPB5, sHSPs
Citation: Brownell SE, Becker RA and Steinman L (2012) The protective and therapeutic function of small heat shock proteins in neurological diseases. Front. Immun. 3:74. doi: 10.3389/fimmu.2012.00074
Received: 20 December 2011; Paper pending published: 10 January 2012;
Accepted: 23 March 2012; Published online: 01 May 2012.
Edited by:
Willem Van Eden, Utrecht University, NetherlandsReviewed by:
Ruurd Van Der Zee, Universiteit Utrecht, NetherlandsHarm Kampinga, University Medical Center Groningen, Netherlands
N. H. Lubsen, Radboud University Nijmegen, Netherlands
Copyright: © 2012 Brownell, Becker and Steinman. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Lawrence Steinman, Department of Neurology and Neurological Sciences, Beckman Center for Molecular Medicine, Campus Drive, Room B002, Stanford, CA 94305-5316, USA. e-mail:c3RlaW5tYW5Ac3RhbmZvcmQuZWR1