 Stuart K. Calderwood*
Stuart K. Calderwood* Ayesha Murshid and Jianlin Gong
Ayesha Murshid and Jianlin Gong
- Department of Radiation Oncology, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA, USA
Heat shock protein (HSP)-based anticancer vaccines have undergone successful preclinical testing and are now entering clinical trial. Questions still remain, however regarding the immunological properties of HSPs. It is now accepted that many of the HSPs participate in tumor immunity, at least in part by chaperoning tumor antigenic peptides, introducing them into antigen presenting cells such as dendritic cells (DC) that display the antigens on MHC class I molecules on the cell surface and stimulate cytotoxic lymphocytes (CTL). However, in order for activated CD8+ T cells to function as effective CTL and kill tumor cells, additional signals must be induced to obtain a sturdy CTL response. These include the expression of co-stimulatory molecules on the DC surface and inflammatory events that can induce immunogenic cytokine cascades. That such events occur is indicated by the ability of Hsp70 vaccines to induce antitumor immunity and overcome tolerance to tumor antigens such as mucin1. Secondary activation of CTL can be induced by inflammatory signaling through Toll-like receptors and/or by interaction of antigen-activated T helper cells with the APC. We will discuss the role of the inflammatory properties of HSPs in tumor immunity and the potential role of HSPs in activating T helper cells and DC licensing.
Introduction
Heat shock proteins (HSP) were first discovered as a group of polypeptides whose level of expression increases to dominate the cellular proteome after stress (Lindquist and Craig, 1988). These increases in HSPs synthesis correlate with a marked resistance to potentially toxic stresses such as heat shock (Li and Werb, 1982). The finding that such proteins have extracellular immune functions suggested that, as highly abundant intracellular proteins they could be prime candidates as danger signals to the immune response (Srivastava and Amato, 2001). There are several human HSP gene families with known immune significance and their classification is reviewed in Kampinga et al. (2009). These include the HSPA (Hsp70) family, which includes the HPA1A and HSPA1B genes encoding the two major stress-inducible Hsp70s, that together are often referred to as Hsp72. When referring to Hsp70 in this chapter, we generally refer to the products of these two genes. The Hsp70 family also includes two other members with immune function – HSPA8 and HSPA5 genes, whose protein products are known as Hsc70 the major constitutive Hsp70 family member and Grp78, a key ER-resident protein. In addition two more Hsp70 related genes have immune significance and these include HSPH2 (Hsp110) and HSPH4 the ER-resident class H protein Grp170. The Hsp90 family also has major functions in tumor immunity and these include HSPC2 and HSPC3, which encode the major cytoplasmic proteins Hsp90a and Hsp90b, and HSPC4 that encodes ER chaperone Grp94. In addition, the product of the HSPD1 gene, the mitochondrial chaperone Hsp60 has some immunological functions. Mice have been shown to encode orthologs of each of these genes (Kampinga et al., 2009).
It has been suggested that many of the HSPs have the property of damage associated molecular patterns (DAMPs), inducers of sterile inflammation and innate immunity (Kono and Rock, 2008). The additional discovery that intracellular HSPs function as molecular chaperones and can bind to a wide spectrum of intracellular polypeptides further indicated that they could play a broad role in the immune response and might mediate both innate immunity due to their status as DAMPs and adaptive immunity by chaperoning antigens.
Heat shock proteins are currently employed as vaccines in cancer immunotherapy (Tamura et al., 1997; Murshid et al., 2011a). The rationale behind the approach is that if HSPs can be extracted from tumor tissue bound to the polypeptides which they chaperone during normal metabolism, they may retain antigenic peptides specific to the tumor (Noessner et al., 2002; Srivastava, 2002; Wang et al., 2003; Enomoto et al., 2006; Gong et al., 2010). Indeed, vaccines based on Hsp70, Hsp90, Grp94, Hsp110, and Grp170 polypeptide complexes have been used successfully to immunize mice to a range of tumor types and Hsp70 and Grp94 vaccines have undergone recent clinical trials (rev: Murshid et al., 2011a). These effects of the HSP vaccines on tumor immunity appear to be mediated largely to the associated, co-isolated tumor polypeptides, although in the case of Grp94 this question is still controversial and tumor regression was observed in mice treated with the chaperone devoid of its peptide binding domain (Udono and Srivastava, 1993; Srivastava, 2002; Nicchitta, 2003; Chandawarkar et al., 2004; Nicchitta et al., 2004). Use of such HSP vaccines is potentially a powerful approach to tumor immunotherapy as the majority of the antigenic repertoire of most individual tumor cells is unknown (Srivastava and Old, 1988; Srivastava, 1996). Individual cancer cells are likely to take a lone path in accumulating a spectrum of random mutations. Although some mutations are functional, permitting cells to become transformed and to progress into a highly malignant state, many such changes are likely to be passenger mutations not required to drive tumor growth (Srivastava and Old, 1988; Srivastava, 1996). Some of these individual mutant sequences will be novel antigenic epitopes and together with the few known shared tumor antigens comprise an “antigenic fingerprint” for each individual tumor (Srivastava, 1996). Accumulation of mutations in cancer appears to be related to, and may drive the increases in HSPs observed in many tumors (Kamal et al., 2003; Whitesell and Lindquist, 2005; Trepel et al., 2010). As the mutant conformations of tumor proteins are “locked in” due to the covalent nature of the alterations, cancer cells appear to be under permanent proteotoxic stress and rich in HSP expression (Ciocca and Calderwood, 2005). For tumor immunology these conditions may offer a therapeutic opportunity as individual HSPs, whose expression is expanded in cancer will chaperone a cross-section of the “antigenic fingerprint” of the individual tumors (Murshid et al., 2011a). This approach was first utilized by Srivastava (2000, 2006) and led to the development of immunotherapy using HSP–peptide complexes.
In addition to using HSP–peptide complexes extracted from tumors, in cases where tumor antigens are known, these can be directly loaded onto purified or recombinant HSPs and the complex used as a vaccine. This procedure has been carried out successfully in the case of the “large HSPs,” Hsp110 and Grp170 (Manjili et al., 2002, 2003). A variant of this approach employs the molecular engineering of tumor antigens in order to produce molecular chaperone-fusion genes which encode products in which the HSP is fused covalently to the antigen. The fusion proteins are then employed as vaccines. This approach was pioneered by Young et al. who showed that a fusion between mycobacterial Hsp70 and ovalbumin could induced cytotoxic lymphocytes (CTL) in mice with the capacity to kill Ova-expressing cancer cells (Suzue et al., 1997). The vaccines could be used effectively without adjuvant and adjuvant properties were ascribed to the molecular chaperone component of the fusion protein. Subsequent studies have confirmed the utility of the approach in targeting common tumor antigens such as the melanoma antigen Mage3 (Wang et al., 2009).
HSPs and Immunosurveillance in Cancer
The question next arises as to the role of endogenous HSPs, with or without bound antigens in immunosurveillance of cancer cells. Although the immune system can recognize tumor antigens and generate a CTL response, most cancers evade immune cell killing by a range of strategies (van der Bruggen et al., 1991; Pardoll, 2003). These include the down-regulation of surface MHC class I molecules by individual tumor cells and release of immunosuppressive IL-10 by tumors (Moller and Hammerling, 1992; Chouaib et al., 2002). Tumors in vivo also appear to attract a range of hematopoietic cells with immunosuppressive action including regulatory CD4+CD25+FoxP3+ T cells (Treg), M2 macrophages, myeloid-derived suppressor cells (MDSC) and some classes of natural killer cells (Pekarek et al., 1995; Terabe et al., 2005; Mantovani et al., 2008; Marigo et al., 2008). The tumor milieu also contain a small fraction of cells of mesenchymal origin identified by surface fibroblast activation protein-a (FAP cells) that suppress antitumor immune responses (Kraman et al., 2010). Endogenous tumor HSPs may also participate in immune suppression. Although the majority of the HSPs function as intracellular molecular chaperones, a fraction of these proteins can be released from cells even under unstressed conditions and may participate in immune functions (rev: Murshid and Calderwood, 2012). Intracellular Hsp70 can be actively secreted from tumor cells in either free form or packaged into lipid-bounded structures called exosomes (Mambula and Calderwood, 2006b; Chalmin et al., 2010). In addition Hsp70 and Hsp90 can also be found associated with the surfaces of tumor cells where they can function as molecular chaperones or as recognition structures for immune cells (Sidera et al., 2008; Qin et al., 2010; Multhoff and Hightower, 2011). As Hsp70 was shown in a number of earlier studies to be pro-inflammatory due to its interaction with pattern recognition receptors such as Toll-like receptors 2 and 4 (TLR2 and TLR4), these findings might suggest, as mentioned above, that Hsp70 released by tumors could be pro-inflammatory and possess the properties of DAMPs (Asea et al., 2000, 2002; Vabulas et al., 2002). However, subsequent studies indicated that a portion of the TLR4 activation detected in the earlier reports, involving exposure of monocytes, macrophages, or dendritic cells (DC) to HSPs in vitro may be due to trace contamination with bacterial pathogen associated molecular patterns (PAMPs), potent TLR activators (Tsan and Gao, 2004). In spite of these drawbacks, an overwhelming amount of evidence now seems to indicate the interaction of Hsp70 and other HSPs with TLRs (particularly TLR4) in vivo – in a wide range of physiological and pathological conditions, leading to acute inflammation in many conditions (Chase et al., 2007; Wheeler et al., 2009; see Appendix for a full list of references). Thus both TLR2 and TLR4 appear to be important components of inflammatory responses to Hsp70 under many pathophysiological conditions. In cancer therapy it has been shown that autoimmunity can be triggered in mice through necrotic killing of melanocytes engineered to overexpress Hsp70; such treatment led to the concomitant immune destruction of B16 melanoma tumors that share patterns of antigen expression with the killed melanocytes (Sanchez-Perez et al., 2006). Hsp70 appears to play an adjuvant role in this form of therapy through its interaction with TLR4 and induction of the cytokine TNF-a (Sanchez-Perez et al., 2006). However, despite these findings it has also been shown that depletion of Hsp70 in cancer cells can, in the absence of other treatments lead to tumor regression by inducing antitumor immunity (Rerole et al., 2011). This effect appears to be due to the secretion by cancer cells of immunosuppressive exosomes containing Hsp70 that activate MDSC and lead to local immunosuppression (Chalmin et al., 2010). Under normal circumstances therefore, release of endogenous Hsp70 into the extracellular microenvironment may be a component of the tumor defenses against immunosurveillance. Extracellular Hsp60 has also been shown be immunomodulatory and can increase levels of FoxP3 Treg in vitro and suppress T cell-mediated immunity (de Kleer et al., 2010; Aalberse et al., 2011).
The pro-inflammatory properties of extracellular HSPs may be more evident under in vivo situations particularly in the context of tissue damage (Sanchez-Perez et al., 2006). For instance when elevated temperatures were used to boost Hsp70 release from Lewis Lung carcinoma cells in vivo, antitumor immunity was activated along with release of chemokines CCL2, CCL5, and CCL10, in a TLR4-dependent manner, leading to attraction of DC and T cells into the tumor (Chen et al., 2009). Thus under resting conditions, the tumor milieu appears to be a specialized immunosuppressive environment, rich in inhibitory cells such as Treg, MDSC, and M2 macrophages and inaccessible to “exhausted” CD8+ T cells that often fail to penetrate the tumor microcirculation. However, under inflammatory conditions involving necrotic cell killing of tumor cells, extracellular HSPs may be able to amplify the anticancer immune response, intracellular HSPs may be released to further increase such a response and CTL may triggered to penetrate the tumor milieu, inducing antigen-specific cancer cell killing (Evans et al., 2001; Mambula and Calderwood, 2006a; Sanchez-Perez et al., 2006; Chen et al., 2009).
HSP-Based Anticancer Vaccines
It is apparent that a number of HSP types, conjugated to peptide complexes (HSP.PC) from cancer cells form effective bases for immunotherapy approaches with unique properties, as mentioned above (Calderwood et al., 2008; Murshid et al., 2011a). The immunogenicity of most HSP.PC appears to involve the ability of the HSPs to sample the tumor “antigenic fingerprint,” deliver the antigens to antigen presenting cells (APC) such as DC and stimulate activation of CTL (Tamura et al., 1997; Singh-Jasuja et al., 2000b; Wang et al., 2003; Murshid et al., 2010). A number of studies show that HSPs can chaperone tumor antigens and deliver them to the appropriate destination – MHC class I molecules on the DC surface (Singh-Jasuja et al., 2000a,b; Srivastava and Amato, 2001; Delneste et al., 2002; Enomoto et al., 2006; Gong et al., 2009). In addition, Hsp70 has been shown to chaperone viral antigenic peptides and increase cross priming of human CTL under ex vivo conditions (Tischer et al., 2011). However, it is still far from clear how the process of HSP-mediated cross priming unfolds. For instance, the CD8+ expressing DC subpopulation in lymph nodes is regarded as the primary cross-presenting APC (Heath and Carbone, 2009). It is not however currently known whether the CD8+ DC subset or other peripheral or lymph-node resident, DC interact with HSP vaccines to induce cross presentation. HSPs appear to be able to enter APC, such as mouse bone marrow derived DC (BMDC) and human DC in a receptor-mediated manner (Basu et al., 2001; Delneste et al., 2002; Gong et al., 2009; Murshid et al., 2010). However, no unique endocytosing HSP receptor has emerged and HSP–antigen complexes appear instead to be taken up by proteins with “scavenger” function such as LOX-1, SRECI, and CD91 that can each take up a wide range of extracellular ligands (Basu et al., 2001; Delneste et al., 2002; Theriault et al., 2006; Murshid et al., 2010). A pathway for Hsp90–peptide (Hsp90.PC) uptake has been characterized in mouse BMDC by scavenger receptor SRECI (Murshid et al., 2010). SRECI is able to mediate the whole process of Hsp90.PC endocytosis, trafficking through the cytoplasm to the sites of antigen processing and presentation of antigens to CD8+ T lymphocytes on MHC class I molecules (Murshid et al., 2010). This process is known as antigen cross presentation (Kurts et al., 2010). It is not currently clear what the relative contribution to antigen cross presentation of the various HSP receptors might be under in vivo conditions. It may be that each receptor class contributes to an individual aspect of CTL activation by HSP peptide complexes although a definitive understanding may await studies in mice deficient in each receptor class.
HSPs and CTL Programming
It is evident that that HSPs can mediate antigen cross presentation and activate CD8+ T lymphocytes. However, presentation of tumor antigens by DC is not sufficient for CTL programming and, in the absence of co-stimulatory molecules and innate immunity, the “helpless” CD8+ cells will cease to proliferate abundantly and will most likely undergo apoptosis (Schurich et al., 2009; Kurts et al., 2010). One mechanism for enhancing CTL programming involves activation of the TLR pathways that lead to synthesis of co-stimulatory molecules (Rudd et al., 2009; Yamamoto and Takeda, 2010). The co-stimulatory molecules, including CD80 and CD86 then become expressed on the DC cell surface and amplify the signals induced by binding of the T cell receptor on CD8+ T cells to MHC class I peptide complexes on the presenting DC (Parra et al., 1995; Rudd et al., 2009). This process is important in pathogen infection in which microbially derived antigens are encountered in the presence of inflammatory PAMPs that can activate innate immune transcriptional networks. Originally it had been thought that HSPs could provide analogous stimulation through their suspected activity as DAMPs and their inbuilt ability to trigger innate immunity through TLR2 and TLR4 on DC (Asea et al., 2000, 2002; Vabulas et al., 2002). (The potential role of HSPs as DAMPs has been the subject of a recent review: van Eden et al., 2012). Subsequent studies on the capacity of HSPs to bind TLRs do not indicate avid binding of Hsp70 to either TLR2 or TLR4 when expressed in cells deficient in HSP receptors in vitro (Theriault et al., 2006). In vivo however, TLR signaling is essential for Hsp70 vaccine-induced tumor cell killing. Studies of tumor-bearing mice treated with an Hsp70 vaccine in vivo indicated that vaccine function is depleted by knockout of the TLR signaling intermediate Myd88 and completely abrogated by double knockout of TLR2 and TLR4 (Gong et al., 2009). These findings were somewhat complicated by the fact that TLR4 is involved in upstream regulation of the expression of Hsp70 receptor SRECI, but do strongly implicate a role for these receptors in amplifying immune signaling by Hsp70 vaccines and Hsp70-based immunotherapy (Sanchez-Perez et al., 2006; Gong et al., 2009). It is still not clear to what degree HSPs are capable of providing a sturdy DC maturing signal through TLR2/TLR4. The potency of HSP anticancer vaccines could potentially be improved by addition of PAMPs such as CpG DNA shown to activate TLR9, or double stranded RNA that can activate TLR3 (Murshid et al., 2011a). As mentioned, one contradictory factor in the earlier studies was that, although TLR2 and TLR4 are required for a sturdy Hsp70 vaccine-mediated immune response, direct binding of Hsp70 to these receptors was not observed (Theriault et al., 2006; Gong et al., 2009; Murshid et al., 2012). A rationale for these findings might be that HSPs can activate TLR signaling indirectly through primary binding to established HSP receptors such as LOX-1 and SRECI which secondarily recruit and activate the TLRs (Murshid et al., 2011b). Both of these scavenger receptors bind to TLR2 upon stimulation and activate TLR2-based signaling (Jeannin et al., 2005; A. Murshid and SK Calderwood, in preparation). In addition, we have found that Hsp90–SRECI complexes move to the lipid raft compartment of the cell, an environment highly enriched in TLR2 and TLR4 (Triantafilou et al., 2002; Murshid et al., 2010).
HSPs, the Class II Pathway, and DC Licensing
In addition to activating antigen cross presentation, HSPs have been shown to chaperone antigens through the MHC class II pathway and induce CD4+ T lymphocytes (Murshid et al., 2011a). Presentation of soluble or particulate antigens though the cross presentation and Class II pathways usually involves different mechanisms and primary receptors that partition the antigens toward MHC class I or MHC class II proteins in a process that is not completely understood (Burgdorf and Kurts, 2008; Amigorena and Savina, 2010). It has been shown that SRECI mediates antigen presentation in DC through both the MHC class I and MHC class II pathways in DC from mice stimulated by Hsp70 vaccines (Gong et al., 2009). However, the sorting mechanisms that distribute antigens bound to Hsp90 and SRECI between these two antigen presentation pathways are, at this moment completely unknown. CTL programming can be powerfully reinforced by a process known as DC licensing in which CD4+ T lymphocytes recognize epitopes presented on MHC class II molecules by the same DC that activates the CTL (Bennett et al., 1997, 1998; Kurts et al., 2010). Kurts et al. (2010) have likened this process to the immune response demanding a “second opinion” as to the suitability of the antigen recognized by the CTL. CD4+ T cells recognize antigens presented by MHC class II molecules on the DC and activate the “licensing” reaction which involves the CD40 ligand on the CD4+ T cell interacting with CD40 on the DC (Kurts et al., 2010; Figure 1). The molecular consequences of CD40 activation in DC are similar to the results of TLR signaling, involving expression of co-stimulatory molecules and cytokines such as TNF-a and IL-12 (Ma and Clark, 2009). This complex licensing process leads to a sturdy activation of CTL by the DC with vigorous proliferation, robust survival of CTL clones, and production of memory T cells (Castellino and Germain, 2006; Kurts et al., 2010). The coincidence and binding of three rare cell types: antigen-bearing DC and antigen-responsive CD4+ and CD8+ T cells might seem an unlikely event (Castellino and Germain, 2006). However it seems that the interaction does not need to be concurrent and, for instance licensing Th1 CD4+ T cells may find and interact with the DC prior to the CD8+ T cells, and that the DC can store the “licensing signal” prior to secondary encounter with CD8+ T cells entering the lymph nodes. In addition it has been shown that CD8+ T cells express CCR5 chemokine receptors on their surfaces, permitting them to home toward “licensed DCs” resident in the lymph nodes (Oppermann, 2004). These interactions are strongly stimulated by inflammatory CC chemokines such as CCL3, CCL4 (Castellino and Germain, 2006). It is currently unclear whether HSP vaccines trigger such a chemokine response. However, in mouse Lewis Lung carcinoma cells in vivo, antitumor immunity was activated along with release of chemokines CCL2, CCL5, and CCL10, in an Hsp70- and TLR4-dependent manner (Chen et al., 2009). In addition, it has been shown that microbial Hsp70 can bind directly to CCR5 and stimulate IL-12 and TNF-a synthesis (Whittall et al., 2006). In addition to stimulating Th help, Hsp70 has also been shown to induce cytotoxic CD4+ cells (Figueiredo et al., 2009). Treatment of CD4+ helper T cells with Hsp70 led to increased proliferation, elevation in Granzyme B levels and target-independent enhancement in cytotoxicity (Figueiredo et al., 2009).
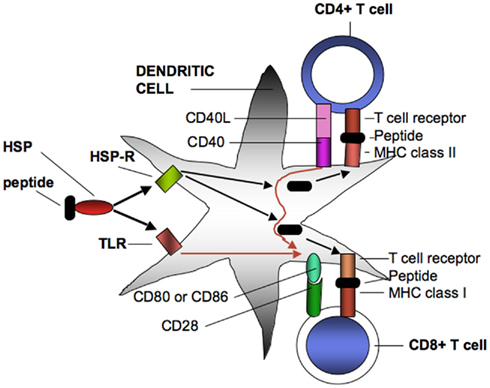
Figure 1. Heat shock protein–peptide complexes extracted from tumor cells interact with endocytosing receptors (HSP-R) such as SRECI or signaling receptors (TLR) such as TLR4 on DC. SREC1 mediates uptake and intracellular processing of antigens and the presentation of resulting peptides on surface MHC class I and MHC class II proteins. MHC class II receptor–peptide complexes then bind to T cell receptors on CD4+ cells. One consequence of binding is interaction of CD40 ligand on the MHC class II cell with CD40 on the DC leading to the licensing interaction that results in enhanced expression of co-stimulatory proteins on the DC cell surface. The licensed DC may then interact with CD8+ T cells through T cell interaction with MHC class I peptide complexes. This effect will be enhanced by simultaneous interaction of CD80 or Cd86 co-stimulatory complexes on the DC with CD28 on the CD8+ cells, leading to effective CD8+ CTL that can lyse tumor cells. T cell programming can also be amplified by signals emanating from activated TLR that can boost levels of CD80 and CD86 as well as inflammatory cytokines (not shown).
Hsp70, Cell Damage, and Inflammation
The question of whether Hsp70 acts as DAMP and could by itself induce an inflammatory response in cancer patients in vivo is still open. However, some recent studies by Vile et al. using a gene therapy approach may shed some light on the inflammatory role of Hsp70 in tumor therapy. In this approach, as mentioned above, normal murine tissues were engineered to express high Hsp70 levels then subjected to treatments that lead to necrotic killing. The aim was to stimulate an autoimmune response that could lead to bystander immune killing of tumor cells that share the antigenic repertoire as the killed normal cells (Sanchez-Perez et al., 2006). In the initial studies, normal melanocytes were preloaded with Hsp70 plasmids and then necrotic cell death was triggered (Daniels et al., 2004). This treatment led to T cell-mediated immune killing of syngeneic B16 melanoma cells transplanted at a distant site in the mouse, presumably in response to antigens shared by the killed normal melanocytes and melanoma cell (Daniels et al., 2004). This effect only occurred when melanocytes were induced to undergo necrosis and Hsp70 levels were elevated, indicating a role for high levels of Hsp70 in the tumor specific immune response. Interestingly, these conditions did not lead to a prolonged autoimmune response, an effect mediated by the induction of a delayed Treg response (Srivastava, 2003; Daniels et al., 2004). It is notable that some early studies of chaperone-based tumor vaccines in animal models demonstrated a primary CTL response to tumors in response to treatment followed by delayed activation of a Treg reaction, and that chaperone levels must be carefully titrated for effective induction of tumor immunity (Udono and Srivastava, 1993; Liu et al., 2009). The role of Hsp70 in autoimmune rejection of tumors was also investigated in prostate cancer (Kottke et al., 2007). Ablation of normal prostate cells by necrotic killing with fusogenic viruses in the absence of Hsp70 elevation led to the induction of the cytokines IL-10 and TGF-b in the mouse prostate and a Treg response. However, when Hsp70 levels were elevated in these cells, IL-10, TGF-b, and IL-6 were induced simultaneously, the IL-6 component leading to further induction of IL-17, a profound Th17 response and tumor rejection (Kottke et al., 2007). Thus elevated levels of Hsp70, presumably released from cells undergoing necrosis can influence the local cytokine patterns and lead to an inflammatory state in vivo. Interestingly, these results seem to be tissue specific as inflammatory killing of pancreatic cells even in the presence of elevated Hsp70 did not provoke IL-6 release, a Th17 response or tumor rejection and the Treg response dominated under these conditions (Kottke et al., 2009). Thus the role of Hsp70 in tissue inflammation and tumor rejection seems to require elevated concentrations of extracellular chaperones, significant levels of necrotic cell killing, and tissue specific cytokine release.
Conclusion
(1) Earlier studies investigating HSP vaccines considered such structures to be the “Swiss penknives” of immunology able to deliver antigens directly to APC and confer a maturing signal that could render DC able to effectively program CTL (Srivastava and Amato, 2001; Noessner et al., 2002). It is well established now that Hsp70, Hsp90, Hsp110, and GRP170 can chaperone tumor antigens and activate antigen cross presentation (Murshid et al., 2011a). In addition, HSPs were thought to be DAMPs with ability to strongly activate TLR signaling and innate immunity (Asea et al., 2000). However, although there is compelling evidence to indicate that Hsp70, for instance can interact with TLR4 under a number of pathological situations (see Appendix, Sanchez-Perez et al., 2006), it remains unclear whether free Hsp70 binds directly to the Toll-like receptor and induces innate immunity in the absence of other treatments in vitro (Tsan and Gao, 2004).
(2) Elevated levels of extracellular HSPs appear to have the capacity to amplify the effects of inflammatory signals emanating from necrotic cells in vivo in a TLR4-dependent manner (Daniels et al., 2004; Sanchez-Perez et al., 2006; Kottke et al., 2007). In the presence of cell injury and death, elevated levels of Hsp70 appear to increase the production of inflammatory signals that involve cytokines such as IL-6 and IL-17 and lead to a specific T cell-mediated immune response to tumor cells sharing antigens with the dying cells (Kottke et al., 2007). The mechanisms involved in these processes are not clear although one possibility is that HSPs can induce the engulfment of necrotic cells. Hsp70 has been shown to increase bystander engulfment of a variety of structures (Wang et al., 2006a,b). In addition, tumor cells treated with elevated temperatures release inflammatory chemokines in an Hsp70 and TLR4-dependent mechanisms and this effect may be significant in CTL programming and tumor cell killing (Chen et al., 2009). Our studies indicate that CTL induction by Hsp70 vaccines in vivo has an absolute requirement for TLR2 and TLR4 suggesting that at least in vivo HSPs can trigger innate immunity through TLR signaling (Gong et al., 2009).
(3) HSPs appear also to be able to direct antigen presentation through the class II pathway in DC and may stimulate T helper cells (Gong et al., 2009). It may thus be possible that HSPs participate in DC licensing and reinforce CTL programming during exposure to HSP vaccines. Future studies will address these questions.
(4) A further interesting consideration is whether HSPs released from untreated tumor cells enhance or depress tumor immunity. One initial study shows that Hsp70 released from tumor cells in exosomes can strongly decrease tumor immunity through effects on MDSC (Chalmin et al., 2010). Further studies will be required to make a definitive statement on these questions.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by NIH research grants RO-1CA047407, R01CA119045, and RO-1CA094397.
References
Aalberse, J. A., Kapitein, B., de Roock, S., Klein, M. R., de Jager, W., van der Zee, R., Hoekstra, M. O., van Wijk, F., and Prakken, B. J. (2011). Cord blood CD4+ T cells respond to self heat shock protein 60 (HSP60). PLoS ONE 6, e24119. doi:10.1371/jour-nal.pone.0024119
Amigorena, S., and Savina, A. (2010). Intracellular mechanisms of antigen cross presentation in dendritic cells. Curr. Opin. Immunol. 22, 109–117.
Asea, A., Kraeft, S. K., Kurt-Jones, E. A., Stevenson, M. A., Chen, L. B., Finberg, R. W., Koo, G. C., and Calderwood, S. K. (2000). HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat. Med. 6, 435–442.
Asea, A., Rehli, M., Kabingu, E., Boch, J. A., Bare, O., Auron, P. E., Stevenson, M. A., and Calderwood, S. K. (2002). Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J. Biol. Chem. 277, 15028–15034.
Basu, S., Binder, R. J., Ramalingam, T., and Srivastava, P. K. (2001). CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity 14, 303–313.
Bennett, S. R., Carbone, F. R., Karamalis, F., Flavell, R. A., Miller, J. F., and Heath, W. R. (1998). Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature 393, 478–480.
Bennett, S. R., Carbone, F. R., Karamalis, F., Miller, J. F., and Heath, W. R. (1997). Induction of a CD8+ cytotoxic T lymphocyte response by cross-priming requires cognate CD4+ T cell help. J. Exp. Med. 186, 65–70.
Burgdorf, S., and Kurts, C. (2008). Endocytosis mechanisms and the cell biology of antigen presentation. Curr. Opin. Immunol. 20, 89–95.
Calderwood, S. K., Gong, J., Theriault, J. R., Mambula, S. S., and Gray, P. J. Jr. (2008). Cell stress proteins: novel immunotherapeutics. Novartis Found. Symp. 291, 115–131; discussion 131–140.
Castellino, F., and Germain, R. N. (2006). Cooperation between CD4+ and CD8+ T cells: when, where, and how. Annu. Rev. Immunol. 24, 519–540.
Chalmin, F., Ladoire, S., Mignot, G., Vincent, J., Bruchard, M., Remy-Martin, J. P., Boireau, W., Rouleau, A., Simon, B., Lanneau, D., De Thonel, A., Multhoff, G., Hamman, A., Martin, F., Chauffert, B., Solary, E., Zitvogel, L., Garrido, C., Ryffel, B., Borg, C., Apetoh, L., Rébé, C., and Ghiringhelli, F. (2010). Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J. Clin. Invest. 120, 457–471.
Chandawarkar, R. Y., Wagh, M. S., Kovalchin, J. T., and Srivastava, P. (2004). Immune modulation with high-dose heat-shock protein gp96: therapy of murine autoimmune diabetes and encephalomyelitis. Int. Immunol. 16, 615–624.
Chase, M. A., Wheeler, D. S., Lierl, K. M., Hughes, V. S., Wong, H. R., and Page, K. (2007). Hsp72 induces inflammation and regulates cytokine production in airway epithelium through a TLR4- and NF-kappaB-dependent mechanism. J. Immunol. 179, 6318–6324.
Chen, T., Guo, J., Han, C., Yang, M., and Cao, X. (2009). Heat shock protein 70, released from heat-stressed tumor cells, initiates antitumor immunity by inducing tumor cell chemokine production and activating dendritic cells via TLR4 pathway. J. Immunol. 182, 1449–1459.
Chouaib, S., Thiery, J., Gati, A., Guerra, N., El Behi, M., Dorothée, G., Mami-Chouaib, F., Bellet, D., and Caignard, A. (2002). Tumor escape from killing: role of killer inhibitory receptors and acquisition of tumor resistance to cell death. Tissue Antigens 60, 273–281.
Ciocca, D. R., and Calderwood, S. K. (2005). Heat shock proteins in cancer: diagnostic, prognostic, predictive, and treatment implications. Cell Stress Chaperones 10, 86–103.
Daniels, G. A., Sanchez-Perez, L., Diaz, R. M., Kottke, T., Thompson, J., Lai, M., Gough, M., Karim, M., Bushell, A., Chong, H., Melcher, A., Harrington, K., and Vile, R. G. (2004). A simple method to cure established tumors by inflammatory killing of normal cells. Nat. Biotechnol. 22, 1125–1132.
de Kleer, I., Vercoulen, Y., Klein, M., Meerding, J., Albani, S., van der Zee, R., Sawitzki, B., Hamann, A., Kuis, W., and Prakken, B. (2010). CD30 discriminates heat shock protein 60-induced FOXP3+ CD4+ T cells with a regulatory phenotype. J. Immunol. 185, 2071–2079.
Delneste, Y., Magistrelli, G., Gauchat, J., Haeuw, J., Aubry, J., Nakamura, K., Kawakami-Honda, N., Goetsch, L., Sawamura, T., Bonnefoy, J., and Jeannin, P. (2002). Involvement of LOX-1 in dendritic cell-mediated antigen cross-presentation. Immunity 17, 353–362.
Enomoto, Y., Bharti, A., Khaleque, A. A., Song, B., Liu, C., Apostolopoulos, V., Xing, P. X., Calderwood, S. K., and Gong, J. (2006). Enhanced immunogenicity of heat shock protein 70 peptide complexes from dendritic cell-tumor fusion cells. J. Immunol. 177, 5946–5955.
Evans, S. S., Wang, W. C., Bain, M. D., Burd, R., Ostberg, J. R., and Repasky, E. A. (2001). Fever-range hyperthermia dynamically regulates lymphocyte delivery to high endothelial venules. Blood 97, 2727–2733.
Figueiredo, C., Wittmann, M., Wang, D., Dressel, R., Seltsam, A., Blasczyk, R., and Eiz-Vesper, B. (2009). Heat shock protein 70 (HSP70) induces cytotoxicity of T-helper cells. Blood 113, 3008–3016.
Gong, J., Zhang, Y., Durfee, J., Weng, D., Liu, C., Koido, S., Song, B., Apostolopoulos, V., and Calderwood, S. K. (2010). A heat shock protein 70-based vaccine with enhanced immunogenicity for clinical use. J. Immunol. 184, 488–496.
Gong, J., Zhu, B., Murshid, A., Adachi, H., Song, B., Lee, A., Liu, C., and Calderwood, S. K. (2009). T cell activation by heat shock protein 70 vaccine requires TLR signaling and scavenger receptor expressed by endothelial cells-1. J. Immunol. 183, 3092–3098.
Heath, W. R., and Carbone, F. R. (2009). Dendritic cell subsets in primary and secondary T cell responses at body surfaces. Nat. Immunol. 10, 1237–1244.
Jeannin, P., Bottazzi, B., Sironi, M., Doni, A., Rusnati, M., Presta, M., Maina, V., Magistrelli, G., Haeuw, J. F., Hoeffel, G., Thieblemont, N., Corvaia, N., Garlanda, C., Delneste, Y., and Mantovani, A. (2005). Complexity and complementarity of outer membrane protein A recognition by cellular and humoral innate immunity receptors. Immunity 22, 551–560.
Kamal, A., Thao, L., Sensintaffar, J., Zhang, L., Boehm, M. F., Fritz, L. C., and Burrows, F. J. (2003). A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 425, 407–410.
Kampinga, H. H., Hageman, J., Vos, M. J., Kubota, H., Tanguay, R. M., Bruford, E. A., Cheetham, M. E., Chen, B., and Hightower, L. E. (2009). Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 14, 105–111.
Kono, H., and Rock, K. L. (2008). How dying cells alert the immune system to danger. Nat. Rev. Immunol. 8, 279–289.
Kottke, T., Pulido, J., Thompson, J., Sanchez-Perez, L., Chong, H., Calderwood, S. K., Selby, P., Harrington, K., Strome, S. E., Melcher, A., and Vile, R. G. (2009). Antitumor immunity can be uncoupled from autoimmunity following heat shock protein 70-mediated inflammatory killing of normal pancreas. Cancer Res. 69, 7767–7774.
Kottke, T., Sanchez-Perez, L., Diaz, R. M., Thompson, J., Chong, H., Harrington, K., Calderwood, S. K., Pulido, J., Georgopoulos, N., Selby, P., Melcher, A., and Vile, R. (2007). Induction of hsp70-mediated Th17 autoimmunity can be exploited as immunotherapy for metastatic prostate cancer. Cancer Res. 67, 11970–11979.
Kraman, M., Bambrough, P. J., Arnold, J. N., Roberts, E. W., Magiera, L., Jones, J. O., Gopinathan, A., Tuveson, D. A., and Fearon, D. T. (2010). Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science 330, 827–830.
Kurts, C., Robinson, B. W., and Knolle, P. A. (2010). Cross-priming in health and disease. Nat. Rev. Immunol. 10, 403–414.
Li, G. C., and Werb, Z. (1982). Correlation between the synthesis of heat shock proteins and the development of thermotolerance in CHO fibroblasts. Proc. Natl. Acad. Sci. U.S.A. 79, 3218–3222.
Liu, Z., Li, X., Qiu, L., Zhang, X., Chen, L., Cao, S., Wang, F., and Meng, S. (2009). Treg suppress CTL responses upon immunization with HSP gp96. Eur. J. Immunol. 39, 3110–3120.
Ma, D. Y., and Clark, E. A. (2009). The role of CD40 and CD154/CD40L in dendritic cells. Semin. Immunol. 21, 265–272.
Mambula, S. S., and Calderwood, S. K. (2006a). Heat induced release of Hsp70 from prostate carcinoma cells involves both active secretion and passive release from necrotic cells. Int. J. Hyperthermia 22, 575–585.
Mambula, S. S., and Calderwood, S. K. (2006b). Heat shock protein 70 is secreted from tumor cells by a nonclassical pathway involving lysosomal endosomes. J. Immunol. 177, 7849–7857.
Manjili, M. H., Henderson, R., Wang, X. Y., Chen, X., Li, Y., Repasky, E., Kazim, L., and Subjeck, J. R. (2002). Development of a recombinant HSP110-HER-2/neu vaccine using the chaperoning properties of HSP110. Cancer Res. 62, 1737–1742.
Manjili, M. H., Wang, X. Y., Chen, X., Martin, T., Repasky, E. A., Henderson, R., and Subjeck, J. R. (2003). HSP110-HER2/neu chaperone complex vaccine induces protective immunity against spontaneous mammary tumors in HER-2/neu transgenic mice. J. Immunol. 171, 4054–4061.
Mantovani, A., Allavena, P., Sica, A., and Balkwill, F. (2008). Cancer-related inflammation. Nature 454, 436–444.
Marigo, I., Dolcetti, L., Serafini, P., Zanovello, P., and Bronte, V. (2008). Tumor-induced tolerance and immune suppression by myeloid derived suppressor cells. Immunol. Rev. 222, 162–179.
Moller, P., and Hammerling, G. (1992). The role of surface HLA A, B, C molecules in tumor immunity. Cancer Surv. 13, 101–127.
Multhoff, G., and Hightower, L. E. (2011). Distinguishing integral and receptor-bound heat shock protein 70 (Hsp70) on the cell surface by Hsp70-specific antibodies. Cell Stress Chaperones 16, 251–255.
Murshid, A., and Calderwood, S. K. (2012). “Pathways of Hsp70 release: lessons from cytokines,” in Cellular Trafficking of Molecular Chaperones in Health and Disease, eds B. Henderson, and A. G. Pockley.
Murshid, A., Gong, J., and Calderwood, S. K. (2010). Heat shock protein 90 mediates efficient antigen cross presentation through the scavenger receptor expressed by endothelial cells-I. J. Immunol. 185, 2903–2917.
Murshid, A., Gong, J., and Calderwood, S. K. (2012). “Molecular chaperone receptors: binding and trafficking of molecular chaperones by class F and class G scavenger receptors,” in Cellular Trafficking of Molecular Chaperones in Health and Disease, eds B. Henderson, and A. G. Pockley (in press).
Murshid, A., Gong, J., Stevenson, M. A., and Calderwood, S. K. (2011a). Heat shock proteins and cancer vaccines: developments in the past decade and chaperoning in the decade to come. Expert Rev. Vaccines 10, 1553–1568.
Murshid, A., Theriault, J., Gong, J., and Calderwood, S. K. (2011b). Investigating receptors for extracellular heat shock proteins. Methods Mol. Biol. 787, 289–302.
Nicchitta, C. V. (2003). Re-evaluating the role of heat-shock protein-peptide interactions in tumour immunity. Nat. Rev. Immunol. 3, 427–432.
Nicchitta, C. V., Carrick, D. M., and Baker-Lepain, J. C. (2004). The messenger and the message: gp96 (GRP94)-peptide interactions in cellular immunity. Cell Stress Chaperones 9, 325–331.
Noessner, E., Gastpar, R., Milani, V., Brandl, A., Hutzler, P. J., Kuppner, M. C., Roos, M., Kremmer, E., Asea, A., Calderwood, S. K., and Issels, R. D. (2002). Tumor-derived heat shock protein 70 peptide complexes are cross-presented by human dendritic cells. J. Immunol. 169, 5424–5432.
Oppermann, M. (2004). Chemokine receptor CCR5: insights into structure, function, and regulation. Cell. Signal. 16, 1201–1210.
Pardoll, D. (2003). Does the immune system see tumors as foreign or self? Annu. Rev. Immunol. 21, 807–839.
Parra, E., Varga, M., Sigvardsson, M., Hedlund, G., Kalland, T., Leanderson, T., Sjögren, H. O., and Dohlsten, M. (1995). Costimulation of human CD4+ T cells with LFA-3 and B7 induce distinct effects on AP-1 and NF-kappa B transcription factors. J. Immunol. 155, 1132–1140.
Pekarek, L. A., Starr, B. A., Toledano, A. Y., and Schreiber, H. (1995). Inhibition of tumor growth by elimination of granulocytes. J. Exp. Med. 181, 435–440.
Qin, Z., DeFee, M., Isaacs, J. S., and Parsons, C. (2010). Extracellular Hsp90 serves as a co-factor for MAPK activation and latent viral gene expression during de novo infection by KSHV. Virology 403, 92–102.
Rerole, A. L., Jego, G., and Garrido, C. (2011). Hsp70: anti-apoptotic and tumorigenic protein. Methods Mol. Biol. 787, 205–230.
Rudd, C. E., Taylor, A., and Schneider, H. (2009). CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol. Rev. 229, 12–26.
Sanchez-Perez, L., Kottke, T., Daniels, G. A., Diaz, R. M., Thompson, J., Pulido, J., Melcher, A., and Vile, R. G. (2006). Killing of normal melanocytes, combined with heat shock protein 70 and CD40L expression, cures large established melanomas. J. Immunol. 177, 4168–4177.
Schurich, A., Böttcher, J. P., Burgdorf, S., Penzler, P., Hegenbarth, S., Kern, M., Dolf, A., Endl, E., Schultze, J., Wiertz, E., Stabenow, D., Kurts, C., and Knolle, P. (2009). Distinct kinetics and dynamics of cross-presentation in liver sinusoidal endothelial cells compared to dendritic cells. Hepatology 50, 909–919.
Sidera, K., Gaitanou, M., Stellas, D., Matsas, R., and Patsavoudi, E. (2008). A critical role for HSP90 in cancer cell invasion involves interaction with the extracellular domain of HER-2. J. Biol. Chem. 283, 2031–2041.
Singh-Jasuja, H., Scherer, H. U., Hilf, N., Arnold-Schild, D., Rammensee, H. G., Toes, R. E., and Schild, H. (2000a). The heat shock protein gp96 induces maturation of dendritic cells and down-regulation of its receptor. Eur. J. Immunol. 30, 2211–2215.
Singh-Jasuja, H., Toes, R. E., Spee, P., Münz, C., Hilf, N., Schoenberger, S. P., Ricciardi-Castagnoli, P., Neefjes, J., Rammensee, H. G., Arnold-Schild, D., and Schild, H. (2000b). Cross-presentation of glycoprotein 96-associated antigens on major histocompatibility complex class I molecules requires receptor-mediated endocytosis. J. Exp. Med. 191, 1965–1974.
Srivastava, P. (2002). Interaction of heat shock proteins with peptides and antigen presenting cells: chaperoning of the innate and adaptive immune responses. Annu. Rev. Immunol. 20, 395–425.
Srivastava, P. (2003). Hypothesis: controlled necrosis as a tool for immunotherapy of human cancer. Cancer Immun. 3, 4.
Srivastava, P. K. (1996). Do human cancers express shared protective antigens? or the necessity of remembrance of things past. Semin. Immunol. 8, 295–302.
Srivastava, P. K. (2000). Heat shock protein-based novel immunotherapies. Drug News Perspect. 13, 517–522.
Srivastava, P. K., and Amato, R. J. (2001). Heat shock proteins: the “Swiss Army Knife” vaccines against cancers and infectious agents. Vaccine 19, 2590–2597.
Srivastava, P. K., and Old, L. J. (1988). Individually distinct transplantation antigens of chemically induced mouse tumors. Immunol. Today 9, 78–83.
Suzue, K., Zhou, X., Eisen, H. N., and Young, R. A. (1997). Heat shock fusion proteins as vehicles for antigen delivery into the major histocompatibility complex class I presentation pathway. Proc. Natl. Acad. Sci. U.S.A. 94, 13146–13151.
Tamura, Y., Peng, P., Liu, K., Daou, M., and Srivastava, P. K. (1997). Immunotherapy of tumors with autologous tumor-derived heat shock protein preparations. Science 278, 117–120.
Terabe, M., Swann, J., Ambrosino, E., Sinha, P., Takaku, S., Hayakawa, Y., Godfrey, D. I., Ostrand-Rosenberg, S., Smyth, M. J., and Berzofsky, J. A. (2005). A nonclassical non-Valpha14Jalpha18 CD1d-restricted (type II) NKT cell is sufficient for down-regulation of tumor immunosurveillance. J. Exp. Med. 202, 1627–1633.
Theriault, J. R., Adachi, H., and Calderwood, S. K. (2006). Role of scavenger receptors in the binding and internalization of heat shock protein 70. J. Immunol. 177, 8604–8611.
Tischer, S., Basila, M., Maecker-Kolhoff, B., Immenschuh, S., Oelke, M., Blasczyk, R., and Eiz-Vesper, B. (2011). Heat shock protein 70/peptide complexes: potent mediators for the generation of antiviral T cells particularly with regard to low precursor frequencies. J. Transl. Med. 9, 175.
Trepel, J., Mollapour, M., Giaccone, G., and Neckers, L. (2010). Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 10, 537–549.
Triantafilou, M., Miyake, K., Golenbock, D. T., and Triantafilou, K. (2002). Mediators of innate immune recognition of bacteria concentrate in lipid rafts and facilitate lipopolysaccharide-induced cell activation. J. Cell. Sci. 115, 2603–2611.
Tsan, M. F., and Gao, B. (2004). Cytokine function of heat shock proteins. Am. J. Physiol. Cell Physiol. 286, C739–C744.
Udono, H., and Srivastava, P. K. (1993). Heat shock protein 70-associated peptides elicit specific cancer immunity. J. Exp. Med. 178, 1391–1396.
Vabulas, R. M., Ahmad-Nejad, P., Ghose, S., Kirschning, C. J., Issels, R. D., and Wagner, H. (2002). HSP70 as endogenous stimulus of the toll/interleukin-1 receptor signal pathway. J. Biol. Chem. 277, 15107–15112.
van der Bruggen, P., Traversari, C., Chomez, P., Lurquin, C., De Plaen, E., Van den Eynde, B., Knuth, A., and Boon, T. (1991). A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 254, 1643–1647.
van Eden, W., Spiering, R., Broere, F., and van der Zee, R. (2012). A case of mistaken identity: HSPs are no DAMPs but DAMPERs. Cell Stress Chaperones 17, 281–292.
Wang, L., Rollins, L., Gu, Q., Chen, S. Y., and Huang, X. F. (2009). A mage3/heat shock protein70 DNA vaccine induces both innate and adaptive immune responses for the antitumor activity. Vaccine 28, 561–570.
Wang, R., Kovalchin, J. T., Muhlenkamp, P., and Chandawarkar, R. Y. (2006a). Exogenous heat shock protein 70 binds macrophage lipid raft microdomain and stimulates phagocytosis, processing, and MHC-II presentation of antigens. Blood 107, 1636–1642.
Wang, R., Town, T., Gokarn, V., Flavell, R. A., and Chandawarkar, R. Y. (2006b). HSP70 enhances macrophage phagocytosis by interaction with lipid raft-associated TLR-7 and upregulating p38 MAPK and PI3K pathways. J. Surg. Res. 136, 58–69.
Wang, X. Y., Kazim, L., Repasky, E. A., and Subjeck, J. R. (2003). Immunization with tumor-derived ER chaperone grp170 elicits tumor-specific CD8+ T-cell responses and reduces pulmonary metastatic disease. Int. J. Cancer 105, 226–231.
Wheeler, D. S., Chase, M. A., Senft, A. P., Poynter, S. E., Wong, H. R., and Page, K. (2009). Extracellular Hsp72, an endogenous DAMP, is released by virally infected airway epithelial cells and activates neutrophils via toll-like receptor (TLR)-4. Respir. Res. 10, 31.
Whitesell, L., and Lindquist, S. L. (2005). HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 5, 761–772.
Whittall, T., Wang, Y., Younson, J., Kelly, C., Bergmeier, L., Peters, B., Singh, M., and Lehner, T. (2006). Interaction between the CCR5 chemokine receptors and microbial HSP70. Eur. J. Immunol. 36, 2304–2314.
Yamamoto, M., and Takeda, K. (2010). Current views of toll-like receptor signaling pathways. Gastroenterol. Res. Pract. 2010, 240365.
Aosai, F., Rodriguez Pena, M. S., Mun, H. S., Fang, H., Mitsunaga, T., Norose, K., Kang, H. K., Bae, Y. S., and Yano, A. (2006). Toxoplasma gondii-derived heat shock protein 70 stimulates maturation of murine bone marrow-derived dendritic cells via toll-like receptor 4. Cell Stress Chaperones 11, 13–22.
Appendix
Evidence for and Against a Role for Hsp70–TLR4 Interactions in a Range of Pathophysiological Processes
The following reports indicate that HSP70 and TLR are involved in pro-inflammatory signaling in tissue transplantation, lung pathology, stroke, hyperthermia, surgical stress, infection and pancreatitis, anaphylactic shock, DC maturation, liver metabolism, atherosclerotic lesions, neurodegeneration, and traumatic brain injury in studies carried out largely in vivo (Dybdahl et al., 2002; Kakimura et al., 2002; Vabulas et al., 2002; Curry et al., 2003; Becker et al., 2005; Chen et al., 2005; Mun et al., 2005; Andrade et al., 2006; Aosai et al., 2006; Fekete et al., 2006; Mortaz et al., 2006; Satoh et al., 2006; Chase et al., 2007; Karoly et al., 2007; Fang et al., 2008; Galloway et al., 2008; Ishikawa et al., 2008; Khan et al., 2008; Luo et al., 2008; Marincek et al., 2008; Song et al., 2008; Zou et al., 2008; Chen et al., 2009; de Jong et al., 2009; Wheeler et al., 2009; Su et al., 2010; Suzuki et al., 2010; Brea et al., 2011; Fang et al., 2011; Makino et al., 2011; Mathur et al., 2011; Zhang et al., 2011).
These reports indicate that it is difficult to demonstrate a direct role for HSP–TLR interaction in tissue culture cells in vitro and that contamination of purified HSPs with PAMPs may be a problem in interpreting in vitro studies (Tsan and Gao, 2004; Ye and Gan, 2007).
References
Andrade, C. F., Kaneda, H., Der, S., Tsang, M., Lodyga, M., Chimisso Dos Santos, C., Keshavjee, S., and Liu, M. (2006). Toll-like receptor and cytokine gene expression in the early phase of human lung transplantation. J. Heart Lung Transplant. 25, 1317–1323.
Becker, S., Dailey, L., Soukup, J. M., Silbajoris, R., and Devlin, R. B. (2005). TLR-2 is involved in airway epithelial cell response to air pollution particles. Toxicol. Appl. Pharmacol. 203, 45–52.
Brea, D., Blanco, M., Ramos-Cabrer, P., Moldes, O., Arias, S., Perez-Mato, M., Leira, R., Sobrino, T., and Castillo, J. (2011). Toll-like receptors 2 and 4 in ischemic stroke: outcome and therapeutic values. J. Cereb. Blood Flow Metab. 31, 1424–1431.
Chen, X. Z., Sun, Z. Q., Du, X. L., Liu, Y., Wu, L., Liu, C., and Chen, J. J. (2005). Heat-shock protein 70 may be a putative endogenous ligand of toll-like receptor-4 of human monocytes. Zhonghua Yi Xue Za Zhi 85, 483–486.
Curry, J. L., Qin, J. Z., Bonish, B., Carrick, R., Bacon, P., Panella, J., Robinson, J., and Nickoloff, B. J. (2003). Innate immune-related receptors in normal and psoriatic skin. Arch. Pathol. Lab. Med. 127, 178–186.
de Jong, P. R., Schadenberg, A. W., Jansen, N. J., and Prakken, B. J. (2009). Hsp70 and cardiac surgery: molecular chaperone and inflammatory regulator with compartmentalized effects. Cell Stress Chaperones 14, 117–131.
Dybdahl, B., Wahba, A., Lien, E., Flo, T. H., Waage, A., Qureshi, N., Sellevold, O. F., Espevik, T., and Sundan, A. (2002). Inflammatory response after open heart surgery: release of heat-shock protein 70 and signaling through toll-like receptor-4. Circulation 105, 685–690.
Fang, H., Mun, H. S., Kikumura, A., Sayama, Y., Norose, K., Yano, A., and Aosai, F. (2008). Toxoplasma gondii-derived heat shock protein 70 induces lethal anaphylactic reaction through activation of cytosolic phospholipase A2 and platelet-activating factor via toll-like receptor 4/myeloid differentiation factor 88. Microbiol. Immunol. 52, 366–374.
Fang, H., Wu, Y., Huang, X., Wang, W., Ang, B., Cao, X., and Wan, T. (2011). Toll-like receptor 4 (TLR4) is essential for Hsp70-like protein 1 (HSP70L1) to activate dendritic cells and induce Th1 response. J. Biol. Chem. 286, 30393–30400.
Fekete, A., Viklicky, O., Hubacek, J. A., Rusai, K., Erdei, G., Treszl, A., Vitko, S., Tulassay, T., Heemann, U., Reusz, G., and Szabo, A. J. (2006). Association between heat shock protein 70s and toll-like receptor polymorphisms with long-term renal allograft survival. Transpl. Int. 19, 190–196.
Galloway, E., Shin, T., Huber, N., Eismann, T., Kuboki, S., Schuster, R., Blanchard, J., Wong, H. R., and Lentsch, A. B. (2008). Activation of hepatocytes by extracellular heat shock protein 72. Am. J. Physiol. Cell Physiol. 295, C514–C520.
Ishikawa, Y., Satoh, M., Itoh, T., Minami, Y., Takahashi, Y., and Akamura, M. (2008). Local expression of toll-like receptor 4 at the site of ruptured plaques in patients with acute myocardial infarction. Clin. Sci. 115, 133–140.
Kakimura, J., Kitamura, Y., Takata, K., Umeki, M., Suzuki, S., Shibagaki, K., Taniguchi, T., Nomura, Y., Gebicke-Haerter, P. J., Smith, M. A., Perry, G., and Shimohama, S. (2002). Microglial activation and amyloid-beta clearance induced by exogenous heat-shock proteins. FASEB J. 16, 601–603.
Karoly, E., Fekete, A., Banki, N. F., Szebeni, B., Vannay, A., Szabo, A. J., Tulassay, T., and Reusz, G. S. (2007). Heat shock protein 72 (HSPA1B) gene polymorphism and toll-like receptor (TLR) 4 mutation are associated with increased risk of urinary tract infection in children. Pediatr. Res. 61, 371–374.
Khan, K. N., Kitajima, M., Imamura, T., Hiraki, K., Fujishita, A., Sekine, I., Ishimaru, T., and Masuzaki, H. (2008). Toll-like receptor 4-mediated growth of endometriosis by human heat-shock protein 70. Hum. Reprod. 23, 2210–2219.
Luo, X., Zuo, X., Zhang, B., Song, L., Wei, X., Zhou, Y., and Xiao, X. (2008). Release of heat shock protein 70 and the effects of extracellular heat shock protein 70 on the production of IL-10 in fibroblast-like synoviocytes. Cell Stress Chaperones 13, 365–373.
Makino, M., Uemura, N., Moroda, M., Kikumura, A., Piao, L. X., Mohamed, R. M., and Aosai, F. (2011). Innate immunity in DNA vaccine with Toxoplasma gondii-heat shock protein 70 gene that induces DC activation and Th1 polarization. Vaccine 29, 1899–1905.
Marincek, B. C., Kuhnle, M. C., Srokowski, C., Schild, H., Hammerling, G., and Momburg, F. (2008). Heat shock protein-antigen fusions lose their enhanced immunostimulatory capacity after endotoxin depletion. Mol. Immunol. 46, 181–191.
Mathur, S., Walley, K. R., Wang, Y., Indrambarya, T., and Boyd, J. H. (2011). Extracellular heat shock protein 70 induces cardiomyocyte inflammation and contractile dysfunction via TLR2. Circ. J. 75, 2445–2452.
Mortaz, E., Redegeld, F. A., Nijkamp, F. P., Wong, H. R., and Engels, F. (2006). Acetylsalicylic acid-induced release of HSP70 from mast cells results in cell activation through TLR pathway. Exp. Hematol. 34, 8–18.
Mun, H. S., Aosai, F., Norose, K., Piao, L. X., Fang, H., Akira, S., and Yano, A. (2005). Toll-like receptor 4 mediates tolerance in macrophages stimulated with Toxoplasma gondii-derived heat shock protein 70. Infect. Immun. 73, 4634–4642.
Satoh, M., Shimoda, Y., Akatsu, T., Ishikawa, Y., Minami, Y., and Nakamura, M. (2006). Elevated circulating levels of heat shock protein 70 are related to systemic inflammatory reaction through monocyte toll signal in patients with heart failure after acute myocardial infarction. Eur. J. Heart Fail. 8, 810–815.
Song, J. M., Liu, H. X., Li, Y., Zeng, Y. J., Zhou, Z. G., Liu, H. Y., Xu, B., Wang, L., Zhou, B., and Wang, R. (2008). Extracellular heat-shock protein 70 aggravates cerulein-induced pancreatitis through toll-like receptor-4 in mice. Chin. Med. J. 121, 1420–1425.
Su, X., Sykes, J. B., Ao, L., Raeburn, C. D., Fullerton, D. A., and Meng, X. (2010). Extracellular heat shock cognate protein 70 induces cardiac functional tolerance to endotoxin: differential effect on TNF-alpha and ICAM-1 levels in heart tissue. Cytokine 51, 60–66.
Suzuki, S., Morimatsu, H., Omori, E., Shimizu, H., Takahashi, T., Yamatsuji, T., Naomoto, Y., and Morita, K. (2010). Responses to surgical stress after esophagectomy: gene expression of heat shock protein 70, toll-like receptor 4, tumor necrosis factor-alpha and inducible nitric oxide synthase. Mol. Med. Report. 3, 765–769.
Ye, Z., and Gan, Y. H. (2007). Flagellin contamination of recombinant heat shock protein 70 is responsible for its activity on T cells. J. Biol. Chem. 282, 4479–4484.
Zhang, Z., Zhang, Z. Y., Wu, Y., and Schluesener, H. J. (2011). Immuno-localization of toll-like receptors 2 and 4 as well as their endogenous ligand, heat shock protein 70, in rat traumatic brain injury. Neuroimmunomodulation 19, 10–19.
Zou, N., Ao, L., Cleveland, J. C. Jr., Yang, X., Su, X., Cai, G. Y., Banerjee, A., Fullerton, D. A., and Meng, X. (2008). Critical role of extracellular heat shock cognate protein 70 in the myocardial inflammatory response and cardiac dysfunction after global ischemia-reperfusion. Am. J. Physiol. Heart Circ. Physiol. 294, H2805–H2813.
Keywords: heat shock protein, vaccine, inflammation, antigen presentation
Citation: Calderwood SK, Murshid A and Gong J (2012) Heat shock proteins: conditional mediators of inflammation in tumor immunity. Front. Immun. 3:75. doi: 10.3389/fimmu.2012.00075
Received: 25 November 2011; Paper pending published: 15 February 2012;
Accepted: 23 March 2012; Published online: 09 April 2012.
Edited by:
Willem Van Eden, Utrecht University, NetherlandsReviewed by:
Miriam Wittmann, University of Leeds, UKWillem Van Eden, Utrecht University, Netherlands
Copyright: © 2012 Calderwood, Murshid and Gong. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Stuart K. Calderwood, Department of Radiation Oncology, Beth Israel Medical Center, Harvard Medical School, 99 Brookline Avenue, Boston, MA 02215, USA. e-mail:c2NhbGRlcndAYmlkbWMuaGFydmFyZC5lZHU=