 James Alexander1* and Frank Brombacher2
James Alexander1* and Frank Brombacher2
- 1 Strathclyde Institute of Pharmacy and Biomedical Sciences, University of Strathclyde, Glasgow, UK
- 2 Division of Immunology, Faculty of Health Science, International Center for Genetic Engineering and Biotechnology, Cape Town Component and Institute of Infectious Diseases and Molecular Medicine, University of Cape Town, Cape Town, South Africa
Work in large part on Leishmania major in the 1980s identified two distinct apparently counter-regulatory CD4+ T cell populations, T helper (h)1 and Th2, that controlled resistance/susceptibility to infection respectively. However, the generation of IL-4−/− mice in the 1990s questioned the paramount role of this Th2 archetypal cytokine in the non-healing response to Leishmania infection. The more recent characterization of CD4+ T cell regulatory populations and further effector CD4+ T helper populations, Th17, Th9, and T follicular (f)h cells as well as the acknowledged plasticity in T helper cell function has further added to the complexity of host pathogen interactions. These interactions are complicated by the multiplicity of cells that respond to CD4+ T cell subset signatory cytokines, as well as the diversity of Leishmania species that are often subject to significantly different immune-regulatory controls. In this article we review current knowledge with regard to the role of CD4+ T cells and their products during Leishmania infection. In particular we update on our studies using conditional IL-4Rα gene-deficient mice that have allowed dissection of the cell interplay dictating the disease outcomes of the major Leishmania species infecting humans.
Introduction
The identification, based on their different cytokine profiles, of at least two distinct populations of CD4+ T helper cells was made by Mosmann et al. (1986). Further studies, in large part utilizing the murine model of Leishmania major infection, gave rise to the Th1/Th2 paradigm of resistance/susceptibility to intracellular infection whereby an IL-4 driven Th2 response counter-regulated a protective Th1 response and resulted in non-healing disease (Heinzel et al., 1989). Indeed Leishmania species have long been considered ideal models to study the mechanisms underling persistent intracellular infections. In the first instance protective immunity against all species is by general consensus recognized as being Th1 dependent. However, as the causative agents of both Old World and New World cutaneous leishmaniasis as well as visceral leishmaniasis diverged in evolutionary terms 40–80 million years ago (reviewed McMahon-Pratt and Alexander, 2004) they have had significant time to develop different mechanisms to survive within the mammalian host. As a consequence these various species have provided excellent tools to dissect different pathways of subverting the development of protective Th1 responses. Subsequently, studies using cytokine deficient mice as well as different species and lineages of Leishmania have certainly questioned the simplicity, if not as yet totally undermined the basic premise, of the Th1/Th2 paradigm of resistance/susceptibility to intracellular infection. Part of the re-evaluation of the “Th1/Th2” paradigm results from the identification of further CD4+ T cell populations that can significantly influence disease outcomes (Figure 1). Such populations include CD4+ T cell regulatory populations, as well as further CD4+ T helper populations Th17, Th9, and T follicular helper (fh) cells (Bettelli et al., 2007; Korn et al., 2009; Jäeger and Kuchroo, 2010; Crotty, 2011; Peterson, 2012). There is also increasing evidence of plasticity in function of different CD4+ T cell populations that while adding to the perceived complexity of host pathogen interactions may also clarify previous apparently anomalous reports.
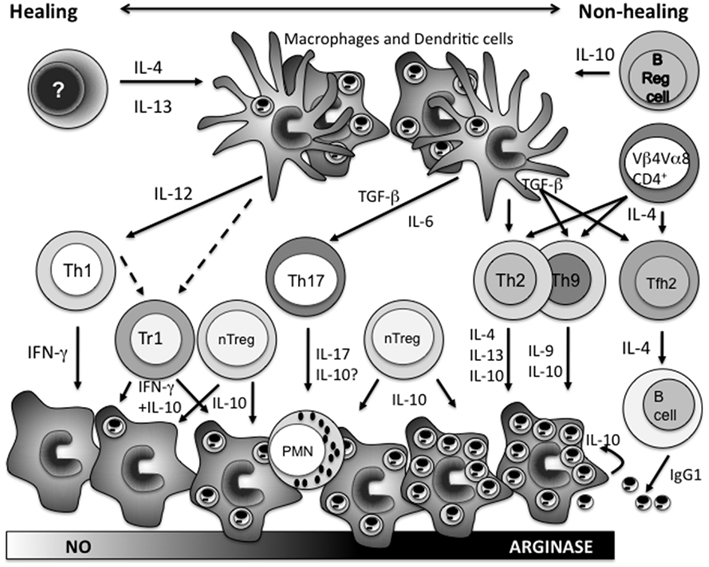
Figure 1. The mechanisms that influence the expansion of different CD4+ T cell populations as part of the adaptive immune response following Leishmania major infection, and their role in determining the outcome of disease. Early IL-4 (IL-13) instructs DCs to produce IL-12 that together with antigen presentation drives the expansion of Th1 cells. IFN-γ production from Th1 cells induces classical macrophage activation, nitric oxide production, and parasite killing. Latent infection, but also in more virulent models, persistent infection is maintained by type-1 regulatory cells producing both IFN-γ and IL-10, and natural T regulatory cells producing IL-10. In the absence of early IL-4, B regulatory cells producing IL-10 instruct the inhibition of DC IL-12 production allowing the expansion of non-Th1 populations. Under the influence of TGF-β, expansion of Th17 cells causes neutrophil influx into lesions which is often associated with increased pathology. Expansion of Th2, Th9, and Tfh2 cells is further facilitated under the influence of IL-4 from Vβ4Vα8 CD4+ T cells responding to the parasite LACK antigen. Type-2 cytokines from Th2/Th9 populations induce alternative macrophage activation, increased arginase expression, and parasite expansion. Tfh2 cells induce B cell IgG1 production and IgG1 opsonized parasites via macrophage FcγR uptake stimulates host cell IL-10 release which together with the influence of cytokines from the other non-Th1 CD4+ T cell populations promotes progressive disease. Overall depending on the virulence of the L. major parasite strains and the animal model examined these various CD4+ T cell populations interact to create a dynamic disease spectrum. Studies on L. donovani infections would suggest that type-2 responses are not disease promoting for visceral leishmaniasis while those with L. mexicana would suggest a significant role.
The traditional counter-regulatory roles for Th1 and Th2 cells and their signatory cytokines IFN-γ and IL-4 are also subject to significant debate as new information has accumulated. For example, the archetypal Th2 cytokines IL-4 and IL-13 need not necessarily counter-regulate a type-1 response as initially proposed, but can also in certain disease models or experimental conditions drive, facilitate, or promote a Th1 response (Alexander et al., 2000; Biedermann et al., 2001; Stager et al., 2003a,b; Murray et al., 2006; McFarlane et al., 2011). Furthermore Th2 responses can be induced independently of the signatory cytokine IL-4 (Mohrs et al., 2000). IL-4/IL-13 mediated Th1 activities include inducing macrophage and dendritic cell IL-12 production (Hochrein et al., 2000; McDonald et al., 2004), enhancing IFN-γ production (Noble and Kemeny, 1995), or synergizing with IFN-γ for enhanced anti-microbial activity (Bogdan et al., 1991; Lean et al., 2003). These studies emphasize the pleiotropic activities of IL-4 and IL-13. Numerous cell types of both the innate and adaptive immune responses not only produce these cytokines but also express their receptors. Thus, many apparently contradictory reports on IL-4/IL-13 influences during infections with different species or strains of Leishmania may result in large part from the hierarchy of importance of different target cell/IL-4 and or IL-13 interactions within the overall global network of IL-4/IL-13 activities in an individual host model system.
In this review we shall outline and discuss, using different species of Leishmania, our current knowledge of how different CD4+ T cells and their cytokine products operate to influence the disease process. In particular we shall update on our studies using tissue specific IL-4Rα−/− mice what information has been discerned with regard to the cell targets for IL-4/IL-13 that promote non-healing or healing disease. Table 1 lists the cell-type specific IL-4Rα deficient mice that have been created to date and indicates which have so far been tested in Leishmania infection models.

Table 1. Global and conditional IL-4Rα gene-deficient mouse models currently available or being characterized.
T Helper 1 CD4+ Cells and Their Role in Leishmaniasis
It is now well established that a protective immune response against both cutaneous leishmaniasis, caused by L. major, L. mexicana, or L. amazonensis, as well as visceral leishmaniasis caused by L. donovani or L. infantum, is dependent on the development of Th1 immunity (reviewed McMahon-Pratt and Alexander, 2004). The general consensus is that IL-12 from antigen-presenting cells (APCs), macrophages, and dendritic cells, drives the differentiation and proliferation of Th1 cells via Stat-4 induced IFN-γ expression which in turn activates Stat-1 to induce T-bet which is recognized as the master regulator of Th1 cells. Stat-1 activation is also essential for the upregulation of CXCR3 and the homing of CD4+ Th1 cells to sites of infection and draining lymph nodes (Barbi et al., 2009). These responses depending on the host/parasite model can be augmented by cytokines such as IL-1β (Von Stebut et al., 2003; Xin et al., 2007), IL-18 (Wei et al., 2004), IL-6 (Stager et al., 2006), and IL-27 (Artis et al., 2004). MHC Class II antigen presentation and cytokine influences alone are not sufficient to stimulate Th1 responses and ligation of co-stimulatory molecules, B7-1/B7-2, and CD40 on the APC with CD28 and CD40L on the T cell respectively have also been described as prerequisites (Bogdan et al., 1996). Ligation of CD40/CD40L enhances APC IL-12 production (Heinzel et al., 1998) although CD40 ligation may not be necessary to promote healing following low level infections (Padigel and Farrell, 2003) and under these circumstances TRANCE-RANK co-stimulation promotes IL-12 production (Padigel et al., 2003b). IFN-γ from Th1 cells, mediates macrophage activation, nitric oxide production, and parasite killing (reviewed Cunningham, 2002). Macrophage leishmanicidal activity induced by IFN-γ may be enhanced by other cytokines such as TNF-α and migration inhibition factor (MIF; Mannheimer et al., 1996; Jüttner et al., 1998; Xu et al., 2003) and type-1 interferons (Diefenbach et al., 1998) as well as CD40/CD40L interactions (Campbell et al., 1996; Kamanaka et al., 1996; Soong et al., 1996; Heinzel et al., 1998). The exquisite susceptibility of BALB/c mice has been attributed to intrinsic defects associated with their Th1 cells such as an inability to upregulate CXCR3 and home to lesions (Barbi et al., 2008). Although the paramount role is for Th1 cells in protection CD8+ T cells are also generally associated with protective immunity following primary infection such as in murine L. major infections (Belkaid et al., 2001, 2002b) or following vaccination against L. amazonensis (Colmenares et al., 2003) or L. donovani (Stager et al., 2003b). Interestingly, BALB/c CD8+T cells also have an intrinsic defect in the ability to upregulate CXCR3 (Barbi et al., 2008).
While a Th1 response and IFN-γ production and inducible nitric oxide synthase expression limits L. mexicana infection in C57BL/6 mice, this is IL-12 independent (Buxbaum et al., 2002). Nevertheless, immunity in this study was dependent on the transcription factor Stat-4, which is required for IL-12 signaling. In this model type-1 interferons have been found not to be the missing activators of Stat-4 (Buxbaum, 2010). This poses the question as to whether L. mexicana can directly activate Stat-4 as has been demonstrated for the virus varicella zoster (Yu et al., 2005). Significantly, a further study on L. mexicana (Aguilar Torrentera et al., 2002) demonstrated early but not late resistance to this parasite in C57BL/6 mice in the absence of IL-12 although the mechanisms involved remain uncharacterized. Alternative mechanisms of Th1 cell induction independently of IL-12 in other Leishmania models have been identified and characterized. Thus, the IL-12 related cytokine IL-27, signaling via WSX-1, has been shown to counter-regulate the early disease exacerbating effects of IL-4 in this mouse strain during L. major infection (Artis et al., 2004) by promoting a Th1 response. IL-27/WSX-1 signaling activates Stat-1 and T-bet (Takeda et al., 2003) which counter-regulates GATA 3 expression and Th2 development (Artis et al., 2004). In the absence of WSX-1 signaling early lesion growth is greatly exacerbated although healing under the influence of IL-12 occurs late in infection.
Th1 cells are not always associated with healing responses but have also been associated with disease exacerbating conditions and playing a regulatory role. For example, a study using a virulent L. major strain that causes non-curing infection in normally resistant C57BL/6 mice has identified a CD4+CD25−FoxP3− Th1 population (Belkaid et al., 2002a; Anderson et al., 2007) that co-expresses IL-10 and IFN-γ and maintains chronic infection. WSX-1 ligation also activates Stat-3 (Yoshimura et al., 2006) that is a transcription factor for IL-10. Consequently, this double expressing Th1 subset is diminished in WSX-1−/− mice (Anderson et al., 2009). The likelihood is that these IFN-γ and IL-10 dual expressing CD4+ T cells are part of a feed-back regulating process to limit pathology in late infection and WSX-1−/− mice have increased pathology following infection though parasite numbers are not increased (Anderson et al., 2009). CD4+CD25−FoxP3− populations producing IL-10, that can be either IFN-γ+ or IFN-γ−, have also been associated with enhanced disease progression in murine L. donovani infections (Stager et al., 2006).
Collectively these studies while highlighting the importance of Th1 responses in protective immunity against Leishmania would suggest potential elements of redundancy in their induction. However, they also suggest an element of plasticity in the Th1 cell population that allows for the induction of a regulatory role in conditions of prolonged inflammation.
T Helper 2 Cells: Are They Responsible for Non-Healing Disease?
Th2 cells were identified at the same time as Th1 cells and characterized as producing IL-4, IL-5, IL-9, and IL-13. They were associated primarily with immunity to helminths and the pathology associated with inflammatory airways diseases. Initially thought to be IL-4 dependent in their generation via Stat-6 activation and GATA 3 induction it is now recognized that GATA 3 is the master regulator of Th2 differentiation and can be induced independently of IL-4/IL-13 and Stat-6 activation (Ouyang et al., 1998, 2000; Brewer et al., 1999; Mohrs et al., 2000).
Early studies in susceptible BALB/c mice demonstrated that a single T cell epitope derived from the parasite LACK antigen (Leishmania homolog of receptors for activated C kinase) induced rapid IL-4 production by Vβ4Vα8CD4+ T cells which apparently rendered T cells unresponsive to IL-12 and correlated with lesion development (Launois et al., 1995). Thus a particularly strong case for the predominant role of IL-4 and a Th2 response in non-healing cutaneous disease was established. Nevertheless, other studies on resistant C57BL/6 mice noted that this strain also produced IL-4 early during infection and yet developed an unimpaired Th1 biased response (Scott et al., 1996). Subsequently studies utilizing IL-4−/− mice seriously questioned the paramount role of IL-4 in the non-healing response of BALB/c mice to L. major as a number of apparently contradictory reports determining the significance of IL-4 were published (Kopf et al., 1996; Noben-Trauth et al., 1996; Mohrs et al., 2000). This posed the question as to whether other cytokines could be regulating Th1 development. As IL-4 and IL-13 share a common signaling pathway through the IL-4 receptor α (IL-4Rα) chain and as IL-4Rα deficient mice were found to be more resistant than IL-4−/− mice in one study this suggested a compensatory disease exacerbating role for IL-13 in the absence of IL-4 (Noben-Trauth et al., 1999). However, in a further study IL-13 was also found to be able to act independently of IL-4 and it was concluded that the effects of IL-4 and IL-13 were also additive (Matthews et al., 2000). Nevertheless, in a further study IL-4Rα−/− BALB/c mice were found to be more susceptible than IL-4−/− animals indicating a protective role for IL-13 in preventing disease dissemination in the absence of IL-4 signaling (Mohrs et al., 1999). While question marks remain over whether IL-4 and IL-13 play a significant role in L. major disease progression in BALB/c mice IL-10 unquestionably does promote parasite persistence in resistant mice and multiplication in susceptible mice (Belkaid et al., 2001; Noben-Trauth et al., 2003). IL-10 could be generated via either Fcγ-mediated uptake of antibody coated amastigotes (Kane and Mosser, 2001), or produced by Th2 cells, or CD4+ CD25+ (Belkaid et al., 2001, 2002a; Sacks and Anderson, 2004) or CD4+CD25−FoxP3+ T regulatory cells (Lages et al., 2008) or indeed CD4+CD25−FoxP3− Th1 population (Anderson et al., 2007).
In the absence of a definitive role for IL-4/IL-13 in non-healing disease it has been proposed that defective T cell responses as a consequence of intrinsic defects in APC function or alternatively in T cell development could be the ultimate underlying problem determining the exquisite susceptibility of BALB/c mice to infection with L. major. While several studies suggest that non-healing disease is a consequence of the failure to produce or respond to IL-12 (Reiner et al., 1994; Himmelrich et al., 1998) this could be irrespective of the presence of a Th2 response and although IL-4 can down-regulate IL-12 production and expression of IL-12Rβ2 on CD4+ T cells (Launois et al., 1995; Himmelrich et al., 1998; Jones et al., 1998), this could also be by IL-4 independent mechanisms (Jones et al., 2000). In this context it is significant that IL-12β2 expression remains impaired in IL-4Rα−/− BALB/c mice compared with resistant C57BL/6 mice infected with L. major (Mohrs et al., 1999). Defects in APC function reported for BALB/c mice include a comparative deficiency in dendritic cell IL-1 production (Von Stebut et al., 2003) compared with L. major-resistant strains. IL-1 has been shown to upregulate dendritic cell IL-12 production as well as MHC Class II and co-stimulatory molecule expression (Eriksson et al., 2003) and to enhance IFN-γ mediated inhibition of Th2 cell proliferation (Oriss et al., 1997). However, as transgenic IL-12Rβ2 BALB/c mice exhibit a non-healing phenotype despite intact IL-12 signaling the importance of an APC defect remains in question (Nishikomori et al., 2001). Irrespective of any potential defect in APC function, BALB/c CD4+ T cells have been demonstrated as having an intrinsic IL-4 independent Th2 bias (Baguet et al., 2004). This may be a consequence of the observed intrinsic weak co-polarization of the T cell receptor and IFN-γ receptor in BALB/c naïve T cells compared with C57BL/6 mice (Maldonado et al., 2004). However, as IL-4 and Stat-6 signaling also suffice to inhibit this polarization immune-regulatory mechanisms as well as intrinsic defects could serve to inhibit Th1 responses.
Global gene-deficient mouse models are complex and more recent studies using cell-type specific IL-4Rα deficient BALB/c mouse models may serve to help reconcile the observations above (Herbert et al., 2004). In contrast to BALB/c mice, which developed ulcerating lesions following infection with L. major, CD4+ T cell-specific IL-4Rα deficient BALB/c mice were resistant and protected against re-infection in a similar manner to healer C57BL/6 mice (Radwanska et al., 2007). Resistance to L. major in CD4+ T cell-specific IL-4Rα−/− mice correlated with reduced numbers of IL-10-secreting cells and early IL-12p35 mRNA induction, leading to increased delayed type hypersensitivity responses, IFN-γ production, and elevated ratios of inducible nitric oxide synthase mRNA/parasite numbers, similar to C57BL/6 mice. These data demonstrated that abrogation of IL-4 signaling in CD4+ T cells is required to transform non-healer BALB/c mice into a healer phenotype. Although global IL-4Rα deficient mice are resistant during acute infection they are unable to control parasite dissemination during chronic disease (Mohrs et al., 1999), despite the deletion of IL-4Rα on T cells. This indicated, that IL-4Rα-responsive cells, other than CD4+ T cells, played a protective role during L. major infection in BALB/c mice. Possible candidates include dendritic cells, as IL-4 treatment of BALB/c mice pre T cell priming has previously been demonstrated to instruct dendritic cells to produce IL-12 and facilitate a protective Th1 response against L. major (Biedermann et al., 2001). To investigate this possibility, we recently generated dendritic cell-specific IL-4Rα deficient BALB/c mice. L. major-infected dendritic cell-specific IL-4Rα−/− mice became hyper-susceptible, providing evidence that IL-4-instructed DC’s are beneficial for host protection (manuscript in preparation). A potential contribution of IL-4/IL-13 responsive non-CD4+cells toward protective immunity may explain why Leishmania vaccines incorporating IFN-γ-promoting adjuvants are relatively ineffective compared with those that also utilize ALUM and subsequently promote an IL-4 response (Kenney et al., 1999; Gicheru et al., 2001). By contrast, L. major-infected macrophage/neutrophil specific IL-4Rα deficient BALB/c mice showed delayed susceptibility compared with control mice, despite the presence of type-2 immune responses (Hoelscher et al., 2006). Presumably impaired alternative macrophage activation with a shift toward classical macrophage activation resulted in enhanced macrophage nitric oxide mediated leishmanicidal activity. Promoting alternative activated macrophage activities by L. major may be an attractive evasion strategy, allowing elevated parasite growth and hijacking host arginase expression for the pathogens own production of polyamines.
New world cutaneous leishmaniasis resulting from infection with parasites of the mexicana complex is under different genetic and immunoregulatory controls to those controlling L. major infection and unlike L. major the majority of mouse strains are susceptible to this parasite. Early studies on L. mexicana complex parasites that examined IL-4 mRNA transcript expression in the draining lymph nodes of resistant female and susceptible male DBA/2 mice indicated that IL-4 played no role in the non-healing response of male mice to this parasite (Satoskar and Alexander, 1995). Nevertheless, follow up experiments using IL-4−/− mice to confirm this observation demonstrated that IL-4 was after all the predominant cytokine responsible for non-healing disease in a number of other mouse strains (Satoskar et al., 1995, 1997). However, these apparently contradictory results are now easy to reconcile as it is now well established that IL-4 transcript expression is not a reliable indicator of IL-4 production (Mohrs et al., 2005). IL-4Rα−/− BALB/c mice were found to be more resistant than IL-4−/− mice to infection with L. mexicana indicating a disease promoting role for IL-13 (Alexander et al., 2002). This was confirmed in studies using IL-13−/− mice that indicate that IL-13 may play a role in preventing disease resolution by inhibiting IL-12Rβ2 expression (Alexander et al., 2002). Inhibiting IL-12Rβ2 would limit Th1 expansion and L. mexicana has indeed been demonstrated to inhibit Th1 differentiation (Hsu and Scott, 2007). Unlike global IL-4Ra−/− mice infected with L. mexicana that display no lesion growth, infected CD4+ T cell specific IL-4Rα−/− mice initially developed lesions indicating that early susceptibility to L. mexicana is dependent on an IL-4 responsive non-CD4+ T cell population (Bryson et al., 2011). However, subsequent lesion growth was significantly curtailed in infected CD4+ T cell specific IL-4Rα−/− mice compared with IL-4Rα intact mice, and a strong Th1 response generated. This clearly demonstrates the paramount importance of an expanding Th2 population in progressive L. mexicana infections. Despite reduced susceptibility in all CD4+ T cell specific IL-4Rα−/− mice, a dichotomy between the sexes was identified during L. mexicana infection and while lesions in female CD4+ T cell specific IL-4Rα−/− mice healed they persisted in male mice associated with elevated IL-4 production in this sex compared with females. This would suggest that an IL-4 responsive non-T cell population is influencing a non-healing response in male mice. In contrast to L. major infection, macrophage/neutrophil signaling via IL-4Rα had minimal effect on the outcome of L. mexicana infection (Bryson et al., 2011), although this latter study was only carried out in female mice.
The paramount role of IL-4 in promoting disease progression is, however, mouse strain and site of infection dependent. Thus the growth of both L. mexicana and L. amazonensis is primarily IL-4 dependent in rump infections in BALB/c and C57BL/6 mice (McMahon-Pratt and Alexander, 2004). By contrast the growth of these parasites in the footpad of C57BL/6 mice, though not BALB/c mice, is IL-4 independent (Jones et al., 2000; McMahon-Pratt and Alexander, 2004) and the contribution of IL-10 to this activity may also depend on whether L. amazonensis or L. mexicana is studied (Padigel et al., 2003a; Buxbaum and Scott, 2005). Overall there is no evidence whatsoever that Th2 cytokines can be beneficial during infection with New World cutaneous leishmaniasis and often they play the classic counter-protective role.
Protective immunity against visceral leishmaniasis, as with species causing cutaneous leishmaniasis, is dependent on an IL-12 driven type-1 response and IFN-γ production (Murray, 1997; Engwerda et al., 1998), which results in the induction of parasite killing by macrophages primarily via the production of reactive nitrogen and oxygen intermediates (Murray and Nathan, 1999). Studies, not only in mice (Kaye et al., 1991), but also humans (Karp et al., 1993; Kemp et al., 1993; Kemp, 2000), suggested that cure was independent of the differential production of Th1 and Th2 cytokines and both IFN-γ and IL-4 producing T cells have been isolated from asymptomatic and cured patients. Indeed in the mouse model it has been shown that the induction of enhanced protection is related to increasing the frequency of cytokine-producing cells rather than altering the IFN-γ/IL-4 balance (Murphy et al., 1998). In addition, while a recent study has shown that the control of L. donovani in the susceptible BALB/c mouse is IL-12 dependent, this cytokine was found to promote the expansion of Th2 as well as Th1 responses (Engwerda et al., 1998). Both human (Kemp et al., 1993; Murphy et al., 1998) and murine studies (Murphy et al., 2001) indicate that IL-10 is the major immunosuppressive cytokine in visceral leishmaniasis. Furthermore it has been demonstrated that the source of this IL-10 is not traditional Th2 cells (Murphy et al., 1998) but more likely a type-1 regulatory T cell population (Bodas et al., 2006) as well as non-T cell regulatory populations.
Thus it would probably be the general consensus of opinion that the Th2 response does not contribute adversely to the immunological control of visceral leishmaniasis. Indeed studies utilizing gene-deficient mice have identified a protective role for IL-4 and IL-4Rα signaling during primary L. donovani infections (Satoskar et al., 1995; Stager et al., 2003a). Furthermore the increased susceptibility of IL-4Rα−/− mice compared with IL-4−/− BALB/c mice as measured by hepatic parasite burdens (Stager et al., 2003a) also suggested a protective role for IL-13 as both IL-4 and IL-13 activities are dependent on the expression of IL-4Rα. Control of parasite growth in the liver was associated with the ability to produce sterile granulomas (Stager et al., 2003a) a mechanism known to be driven by T cell derived IFN-γ (Murray et al., 2000). Enhanced susceptibility of IL-4−/− and IL-4Rα−/− mice was associated with down-regulated type-1 responses and markedly retarded granuloma maturation (Alexander et al., 2000; Stager et al., 2003a,). Decreased IFN-γ and retarded granuloma maturation have also been observed in IL-13−/− mice (Murray et al., 2006; McFarlane et al., 2011); this was associated with an increased parasite burden (McFarlane et al., 2011). The recent availability of cell-type specific IL-4Rα−/− mice has facilitated the dissection of the IL-4 and IL-13 effects during visceral leishmaniasis and so far we have examined the course of L. donovani in macrophage/neutrophil specific IL-4Rα−/− BALB/c mice. The protective activity of IL-4/IL-13 during visceral leishmaniasis is not via these cell populations (McFarlane et al., 2011). Studies using CD4+ T cell specific IL-4Rα−/− BALB/c mice demonstrate it is also not via CD4+ or CD8+ T cells (unpublished).
In addition to playing a significant protective role during primary infection with L. donovani effective sodium stibogluconate (SSG) chemotherapy is dependent on Th2 cytokines (Alexander et al., 2000; McFarlane et al., 2011). Previous studies have demonstrated that successful treatment of visceral leishmaniasis with SSG requires the presence of both CD4+ and CD8+ T cells (Murray et al., 1989) accompanied by the type-1 cytokines IL-12 and IFN-γ (Murray et al., 2000). We have observed that SSG chemotherapy of L. donovani in IL-4−/− (Alexander et al., 2000) or IL-13−/− (McFarlane et al., 2011) BALB/c mice compared with wild-type mice resulted in down-regulated IFN-γ production and increased IL-10 production. This would suggest a requirement for IL-4 and IL-13 in instructing a type-1 response. Significantly, successful immunotherapeutic intervention in the BALB/c mouse using a hybrid cell vaccine which was mediated by a strong CD8+CTL response was associated with downregulation of IL-10 but significantly enhanced IL-4 and IL-13 expression as well as IFN-γ production (Basu et al., 2007) providing further strong circumstantial evidence for these Th2 cytokines facilitating the generation of type-1 immunity. Similarly the protection afforded by a kinetoplast membrane protein protein-11 DNA vaccine which protected hamsters against visceral leishmaniasis correlated with decreased IL-10 levels but with increased IL-4 production and an elevated mixed Th1/Th2 response (Basu et al., 2005). However, vaccination studies utilizing HASPB-1 have provided the most concrete evidence for a Th2 cytokine requirement to instruct a protective type-1 response against visceral leishmaniasis (Stager et al., 2003b) as this vaccine generates a mixed Th1/Th2 response and is totally ineffective in IL-4 or IL-4Rα−/− mice. The primary source of IL-4 has been defined as a CD11b+CD11clo phagocyte (Stager et al., 2003b).
T Regulatory Cells and Their Role in Leishmaniasis
Unlike other CD4+ T cell populations the primary role of T regulatory cells is homeostasis and limiting excessive inflammation via in particular IL-10 production. T regulatory cells include FoxP3+CD4+CD25+ natural T regulatory cells (nTregs) that are thymus derived and inducible Tregs that derive from the periphery and are either FoxP3+ (iTregs) or adaptive FoxP3− type-1 Treg cells (Tr1; reviewed Peterson, 2012).
The role of Tregs during infection with Leishmania is multifaceted and whether their role is disease promoting or protective depends on the experimental model being investigated. Thus in the L. major infection model CD4+CD25+ Tregs have been demonstrated in resistant C57 BL/6 mice to accumulate in the lesions and draining lymph nodes and be responsible for IL-10 dependent parasite persistence and latent infection (Belkaid et al., 2001). In the absence of IL-10 sterile immunity occurs. However, parasite persistence in apparently healed mice is necessary for concomitant immunity and the maintenance of a viable effector Th1 cell population (Belkaid et al., 2002a). Nevertheless not only can latent infection provide a reservoir for potential continued parasite transmission to sandflies but persistent parasites can also be a source of disease reactivation. For example FoxP3+ CD25− T regulatory expand in mice and humans with age and have been demonstrated to be responsible for spontaneous reactivation of L. major infections in aged mice (Lages et al., 2008) as they down-regulate effector Th1 responses. In progressive chronic L. major infection in normally resistant C57BL/6 mice FoxP3−CD25− type-1 regulatory cells would appear to play the dominant role (Anderson et al., 2007). By contrast CD4+CD25+ Treg cells can also play significant disease control roles during L. major infections in susceptible BALB/c mice by regulating the biased Th2 response (Aseffa et al., 2002; Xu et al., 2003). In the absence of CD4+CD25+ Treg cells IL-4 levels were increased and L. major infection was exacerbated (Aseffa et al., 2002). In addition non-healing disease, in CD4+CD25− T cell reconstituted SCID mice, was controlled by addition of CD4+CD25+ regulatory T cells (Aseffa et al., 2002; Xu et al., 2003). Nevertheless impairing the expansion and homing of FoxP3+CD4+CD25+ T cells to sites of infection by inactivating the p110δ isoform of phosphatidylinositol 3 kinase results in increased resistance in both L. major-resistant and susceptible mice (Liu et al., 2009). This implies the potential for therapeutic intervention with pharmacological inhibitors of this enzyme.
In non-healing L. amazonensis infections of C57BL/6 mice CD4+CD25+ regulatory T cell populations can adoptively transfer a degree of resistance to naïve recipients while RAG−/− mice reconstituted with spleen cells depleted of this population have higher parasite burdens and more pathology than those reconstituted with intact spleen cells (Ji et al., 2005). This study indicated a role for nTregs in inducing IFN-γ producing effector cells. In contrast in studies using the related parasite L. mexicana depletion of CD4+CD25+ T cells did not alter the course of infection (Thomas and Buxbaum, 2008).
While CD4+CD25+T cells have been found in L. braziliensis lesions in humans no functional role has been ascribed to them (Campanelli et al., 2006). By contrast a series of elegant studies have highlighted a significant role for Tregs in humans infected with L. guyanensis (Bourreau et al., 2001, 2009). CD4+CD25+ T regulatory cells are found in lesions during early acute as well as chronic infection. While Tregs from both acute and chronic patients inhibited IFN-γ production from CD4+CD25− T cells exposed to parasite antigen, intralesional FoxP3, and IL-10 expression is particularly elevated in those patients with chronic disease.
It is well established that IL-10 is the major influence resulting in non-healing visceral leishmaniasis. However, little role for nTregs has been noted in either human (Nylén et al., 2007) or murine L. donovani infection (Stager et al., 2006). Any disease exacerbatory role for CD4+ Tregs has been attributed to FoxP3−CD25− regulatory (type-1) cells. These may also produce IFN-γ as well as IL-10 suggesting a further stage of Th1 development. Indeed numerous non-T cell populations, including DCs (Svensson et al., 2004) and NK cells (Maroof et al., 2008) have been implicated as playing regulatory roles during visceral leishmaniasis. As with Th1 cells producing IL-10 these non-traditional regulatory populations may come about as a homeostatic mechanism to counteract the effects of prolonged inflammation. However, a recent study has suggested a strong correlation between the parasite burdens in post kala-azar dermal leishmaniasis nodules and FoxP3 and IL-10 levels (Katara et al., 2011).
T Helper 17 Cells and Their Role in Leishmaniasis
T helper 17 cells are primarily associated with protecting mucosal surfaces, but also with excessive pathology, autoimmune diseases, and allergies (reviewed Korn et al., 2009). Their development is committed under the influence of TGF-β and IL-6 and or IL-21 with the retinoic-acid orphan related receptors (ROR-α and ROR-γt) being critical transcription factors in differentiation. Th17 cells are characterized in particular by IL-17 production, and also by IL-22 and IL-21 secretion. IL-17 plays a significant role in the migration, recruitment, and activation of neutrophils. As neutrophils have been demonstrated to have the potential to play important effector and or regulatory functions during Leishmania infections it would be expected that Th17 cells would have significant roles to play during Leishmania infection (reviewed Charmoy et al., 2010). For example while neutrophils contribute to a protective response against L. donovani they promote a non-healing response against L. major in BALB/c mice. Overall the role of Th17 cells in leishmaniasis remains unclear and both disease promoting and protective responses have been attributed to their influence. Undoubtedly, the infecting species as well as the genetic background of the host play a role in Th17 cell activities.
With regard to the “mexicana” complex of parasites there is little by way of published evidence to indicate a role. However, the inadequate immune response against L. amazonensis resulting in chronic infection in mice has been associated with the induction of “pathogenic” CD4+ T cells producing high levels of IL-17 (Xin et al., 2007). A disease exacerbating role for IL-17 following L. major infection has also been observed in studies using IL-17 deficient BALB/c mice which developed smaller lesions with fewer parasites than their wild-type counterparts (Lopez Kostka et al., 2009). While T cell IL-4, IL-10, and IFN-γ production were comparable in IL-17−/− and IL-17+/+ mice, CXCL2 expression, and neutrophil recruitment were significantly reduced in the absence of IL-17.
Th17 cell development, and IL-17 production are down-regulated by IL-27/WSX-1 signaling (Fitzgerald et al., 2007). Prior to the characterization of Th17 cells increased L. major growth in IL-27R−/− (WSX-1−/−) C57 BL/6 mice in the healing C57BL/6 model was attributed to a diminished Th1 response (Artis et al., 2004). Subsequently it has been shown that IL-27R−/− (WSX-1−/−) C57 BL/6 mice infected with a virulent strain of L. major, that induced non-healing disease in these normally resistant mice, had enhanced Th17 development and increased pathology though no difference in parasite numbers compared with wild-type controls (Anderson et al., 2009). However, as the IL-27/WSX-1 signaling pathway has also been associated with promoting Th1 cell development by inhibiting IL-4 production (Artis et al., 2004) and also in enhancing IL-10 production from CD4+ T cells (Anderson et al., 2009) this can often make interpretation of effects utilizing IL-27R−/− (WSX−/−) mice difficult.
While the evidence might suggest a counter-protective role for IL-17 in Old and New world cutaneous leishmaniasis a recent study using a CpG adjuvanted live vaccine protected C57BL/6 mice against L. major associated with the specific induction of Th17 cells (Wu et al., 2010). Similarly a vaccine comprising CpG stimulated bone marrow-derived DCs pulsed with L. infantum KMP-11 peptide induced protection in BALB/c mice against visceral infection associated with overproduction of IL-17 (Agallou et al., 2011). It should be noted that vaccination in both studies also enhanced type-1 responses. However, studies in humans do indeed demonstrate a therapeutic role for Th17 cells as IL-17 and IL-22 have been strongly correlated with protection against visceral leishmaniasis (Pitta et al., 2009). Furthermore studies in IL-27R−/− (WSX-1−/−) C57 BL/6 mice demonstrated them to be significantly more resistant than their wild-type counterparts to infection with L. donovani although this resistance was associated with severe liver pathology (Rosas et al., 2006). While pro-inflammatory cytokine production was significantly up-regulated in IL-27R−/− mice and IL-10 levels down-regulated during chronic disease unfortunately the IL-17 component was not studied at that time.
Finally the uncertainty surrounding the role of IL-17 is emphasized in studies involving the L. (viannia) sub-genus. Healing in a murine model of L. braziliensis infection (Vargas-Inchaustegui et al., 2008) as well as resolution of L. panamensis infections in IL-13−/− mice (Castilho et al., 2010) is associated with elevated IL-17 as well as IFN-γ levels. Similarly elevated levels of IL-17 were identified in sub-clinical L. braziliensis human infections (Novoa et al., 2011) although a significant role for IL-17 mediated neutrophil infiltration has been associated with tissue damage in human mucosal leishmaniasis (Boaventura et al., 2010).
T Helper 9 Cells and Their Role in Leishmaniasis
Th9 cells are often considered a subset of Th2 cells, generated by IL-4, but requiring the additional influence of TGF-β to be reprogrammed, as well as IL-2 (reviewed Jäeger and Kuchroo, 2010; Nowak and Noelle, 2010). Conversely, expansion of the Th9 cell population is inhibited by IFN-γ. Th9 cells express the transcription factor PU.1 but do not express T-bet, GATA 3, ROR-γt, or Foxp3. Th9 cells produce both IL-9 and IL-10 although IL-10 expression is not regulated by PU.1. While Th9 cells are considered the main source of IL-9, this cytokine can also be produced by Th17 and Treg cells. Th9 cells and IL-9 production have been associated with Mast cell recruitment and expansion, the pathology associated with atopic allergy and immunity to nematode infection (Faulkner et al., 1998).
Studies examining the role of IL-9 during leishmaniasis are limited and have only involved L. major. Early studies demonstrated that IL-9 could be induced early during infection in both susceptible BALB/c and resistant DBA/2 and C57BL/6 mice (Gessner et al., 1993; Nashed et al., 2000). However, higher levels of IL-9 were observed in susceptible BALB/c mice and the ability of CD4 T cells to produce IL-9 persisted only in BALB/c mice as infection progressed. The disease promoting activity of IL-9 indicated circumstantially by these early studies was confirmed in BALB/c mice rendered deficient in IL-9 by vaccination with a recombinant IL-9/Freunds Adjuvant complex (Arendse et al., 2005). Following IL-9 depletion BALB/c mice become more resistant to L. major infection and there was a switch in the Th2/Th1 balance in favor of a protective type-1 response. This resulted in a switch from a classically to an alternatively activated macrophage phenotype as characterized by increased microbicidal activity associated with higher NO production and reduced arginase-1 expression.
Follicular Helper CD4+ T Cells and Their Potential Role in Leishmaniasis
Follicular helper (CD4+ T cells) have been recognized as the specialized providers of B cell help (reviewed Crotty, 2011). Although they have been identified as the major producers of IL-4 and IFN-γ in the draining lymph nodes during parasite infection (Reinhardt et al., 2009), they are distinct from Th1 and Th2 cells. They are dependent on expression of the master regulator Bcl6 and the absence of Blimp1, which is expressed by other CD4 T cell sub-populations and counter-regulated by Bcl6. Tfh cells are further characterized by the expression of CXCR5, PD-1, IL-21, and iCOS. They play a major role in lymph node germinal center formation and are critical in the differentiation of B cells into plasma cells and high affinity antibody production. During L. major infection in BALB/c mice Tfh cells and not canonical Th2 cells provided virtually all the IL-4 secreting cells in the draining lymph nodes (Reinhardt et al., 2009). While DCs may be the initial APCs for Tfh cells as they develop from naïve precursors in the T cell zone of lymph nodes activated B cells take over this role at the T cell-B cell zone border (Deenick et al., 2011). Consequently if B cells and or specific antibody production play a significant role in the disease process Tfh cells would comprise a significant population in determining disease outcome.
The role of B cells (as APCs or regulatory cells) and/or antibody in contributing to susceptibility to Leishmania infection appears to depend in large part upon the parasite species examined but also probably to a significant extent upon the host.
Evidence for a role of B cells and/or antibody in the susceptibility to infection with L. major has been variable which may be a result of the particular parasite or mouse strain used (Varkila et al., 1993; Brown and Reiner, 1999). In one study using T cell-reconstituted L. major-resistant C.B-17 SCID mice, B cells were shown to convert resistance into susceptibility. However, other studies have not found B cells to affect the course of infection in T cell reconstituted mice (Varkila et al., 1993). Further, studies employing B cell deficient mice (BALB/c or C57BL/6) and infection with L. major promastigotes or amastigotes have indicated that neither antibodies nor B cells are required for susceptibility to L. major infection (Brown and Reiner, 1999; Colmenares et al., 2002). Nevertheless a further study has shown FcγR−/− BALB/c mice to be resistant to L. major and this resistance was associated with decreased IL-10 production (Padigel and Farrell, 2005). The inference was that FcγR mediated uptake of opsonized parasites and associated IL-10 production mediated non-healing disease in wild-type mice (Kane and Mosser, 2001). However, these apparently contradictory results may now have been reconciled at least in part. Thus Ronet et al. (2008) have demonstrated that while B cell deficient BALB/c mice are more resistant than their wild-type counterparts to infection with the LV39 strain of L. major these mice remained susceptible to two other strains of this parasite. This B cell dependent susceptibility to the LV39 strain of L. major was independent of antibodies and attributed to B cell antigen presentation. A further study determined that regulatory B cells instructed a Th2 response via IL-10 production (Ronet et al., 2010). The lack of evidence for a role for antibody in susceptibility to L. major infection is still somewhat surprising given some of the previous observations (e.g., Padigel and Farrell, 2005). However, it may be that other immunological down-regulatory mechanisms (IL-4 and IL-13) may compensate or that the other mechanisms that induce an IL-10 response are more significant globally. Recently, B cell-specific IL-4Rα deficient BALB/c mice were established (Hoving et al., 2012). As a consequence of impaired IL-4Rα responsiveness, ovalbumin immunization resulted in abrogated B cell-type-2 antibody responses (IgG1 and IgE) with similar low levels as found in global IL-4Rα deficient mice. In contrast, type-1-antibody responses were up-regulated, as shown by elevated ovalbumin-specific IgG2a and IgG2b. L. major infection studies have been initiated and should more definitively define a possible role of IL-4Rα responsive B cells in this experimental model system.
Infection of B cell deficient mice by any species of the L. mexicana complex has been found to be associated with resistance and susceptibility to infection could be passively transferred with antibody but not by B cells trans-genetically reconstituted and capable of antigen presentation (Peters et al., 1995; Kima et al., 2000; Colmenares et al., 2002). The immune responses of CD4+ T cells from the draining lymph nodes of infected B cell deficient and wild-type BALB/c mice were comparable, both in terms of the level of proliferation and the cytokines produced (IL-4, IL-10, IFN-γ). Hence, T cell activation was comparable in both groups of mice and a lack of a T cell response in the B cell deficient mice did not contribute to the diminished pathology. What differed significantly were the responses at the cutaneous site of infection. B cell deficient animals had markedly reduced numbers of monocytes and lymphocytes recruited or retained at the site of cutaneous infection in comparison to wild-type mice, indicating a selective impairment in the local cutaneous immune response. Consistent with this observation and a role for antibody in pathogenesis, FcγR-deficient mice were also relatively refractory to infection (Kima et al., 2000; Buxbaum and Scott, 2005; Thomas and Buxbaum, 2008). C57BL/6 deficient in FcγR or FcγRIII unlike their wild-type counterparts healed following L. mexicana infection and developed a Th1 response while IL-10 levels were reduced in the site of infection in FcγRIII−/− mice (Buxbaum and Scott, 2005; Thomas and Buxbaum, 2008). IL-10 levels in draining lymph nodes were similar from both wild-type and FcγRIII−/− mice and depletion of Treg cells, the main producers of IL-10 in the draining lymph nodes of wild-type mice, did not modify the immune response or disease outcome. In vitro IgG1 and IgG2a/c can both induce similar levels of IL-10 production from macrophages. IgG1 deficient mice have increased resistance against L. mexicana but at the same time have increased IgG2a/c levels. However, while IgG1 induces IL-10 entirely via FcγRIII, IgG2a/c utilizes primarily FcγR1 but also to a lesser degree FcγRIII (Chu et al., 2010). Overall this indicates that a crucial cell for FcγRIII engagement is present in the lesion site to maintain non-healing disease and consequently that Tfh cells excreting IL-4 to induce B cell IgG1 production are likely playing a significant role in pathology. However, although we have found that B cell specific IL-4Rα−/− mice on the BALB/c background did not produce parasite specific IgG1, they remained susceptible to L. mexicana infection (unpublished). Further investigations are currently in progress to better understand this apparent paradox.
In the case of visceral leishmaniasis, both C57BL/6 infected with L. donovani (Smelt et al., 2000) and BALB/c infected with L. infantum (Deak et al., 2010), B cell deficient mice are relatively resistant to infection. However, susceptibility as measured by parasite growth in B cell deficient C57BL/6 mice, unlike BALB/c mice is independent of antibody production, although antibodies do protect C57BL/6 mice from exaggerated pathology during infection. In BALB/c mice infected with L. infantum neither B cell IL-10 production or antigen presentation played a significant role in B cell mediated disease exacerbation. Passive transfer experiments indicated that antibodies, IgM and IgG, induced as a result of parasite induced polyclonal B cell activation promoted parasite growth. While Tfh cells have been reported as triggering hypergammaglobulinemia during chronic Salmonella infection (Ko et al., 2011) whether they can do this early in visceral leishmaniasis remains to be determined.
Conclusion
It has been some quarter of a century since two distinct CD4+ T cell populations, T helper 1 and 2, were identified by their different cytokine profiles. This seminal discovery provided a basic framework around which immunologists could test their various “pet” hypotheses with regard to how immune-regulatory mechanisms controlled infectious diseases on the one hand while on the other hand limiting immune-pathological processes. Early observations, particularly on the murine cutaneous leishmaniasis model, demonstrated that Th1 and Th2 responses via their signatory cytokines, IFNγ, and IL-4 respectively, were counter-regulatory. However, later studies using gene-deficient mice have questioned the precise role of the Th2 response and IL-4 in regulating the Th1 response. Depending on the parasite strain/species/host model studied the Th2 response could either promote infection or at best be irrelevant to disease progression. Furthermore depending on the model IL-4/IL-13 could actually promote a Th1 response as happens in visceral leishmaniasis. Our current studies using cell-type specific IL-4Rα−/− have been making huge inroads into dissecting out and understanding these apparent paradoxes.
Given that both Th1 and Th2 responses are inflammatory, albeit distinct, responses evolved respectively against intracellular parasites and large pathogens, it now seems obvious in retrospect that other distinct regulatory CD4+ T cell populations would operate to control excessive inflammatory processes induced by either population. Subsequently new CD4+ T cell populations, both regulatory and inflammatory, and influencing the outcome of Leishmania infections have been characterized as summarized in the above review. Furthermore the degree of plasticity amongst T cell populations is much greater than previously thought such that type-1 CD4+ T cells can also be regulatory and express IL-10. In the ideal scenario following infection with Leishmania infection Th1 responses are induced sufficient to control infection initially while regulatory mechanisms are necessary laterly to prevent excessive pathology, maintain a latent (sub-clinical) infection and immunity to re-infection. Perturbation of this balance results in either progressive or chronic disease.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
James Alexander is in receipt of funding from the Wellcome Trust and Royal Society (UK). Frank Brombacher is scientific co-ordinator and group leader of ICGEB, a South African Research Chair holder funded by NRF and directs an external MRC Unit within the University of Cape Town.
References
Agallou, M., Margaroni, M., and Karagouni, E. (2011). Cellular vaccination with bone marrow-derived dendritic cells pulsed with a peptide of Leishmania infantum KMP-11 and CpG oligonucleotides induces protection in a murine model of visceral leishmaniasis. Vaccine 29, 5053–5064.
Aguilar Torrentera, F., Laman, J. D., Van Meurs, M., Adorini, L., Muraille, E., and Carlier, Y. (2002). Endogenous interleukin-12 is critical for controlling the late but not the early stage of Leishmania mexicana infection in C57BL/6 mice. Infect. Immun. 70, 5075–5080.
Alexander, J., Brombacher, F., McGachy, H. A., McKenzie, A. N., Walker, W., and Carter, K. C. (2002). An essential role for IL-13 in maintaining a non-healing response following Leishmania mexicana infection. Eur. J. Immunol. 32, 2923–2933.
Alexander, J., Carter, K. C., Al-Fasi, N., Satoskar, A., and Brombacher, F. (2000). Endogenous IL-4 is necessary for effective drug therapy against visceral leishmaniasis. Eur. J. Immunol. 30, 2935–2943.
Anderson, C. F., Oukka, M., Kuchroo, V. J., and Sacks, D. (2007). CD4+CD25-Foxp3-Th1 cells are the source of IL-10-mediated immune suppression in chronic cutaneous leishmaniasis. J. Exp. Med. 204, 285–297.
Anderson, C. F., Stumhofer, J. S., Hunter, C. A., and Sacks, D. (2009). IL-27 regulates IL-10 and IL-17 from CD4+ cells in nonhealing Leishmania major infection. J. Immunol. 183, 4619–4627.
Arendse, B., Van Snick, J., and Brombacher, F. (2005). IL-9 is a susceptibility factor in Leishmania major infection by promoting detrimental Th2/type 2 responses. J. Immunol. 174, 2205–2211.
Artis, D., Johnson, L. M., Joyce, K., Saris, C., Villarino, A., Hunter, C. A., and Scott, P. (2004). Cutting edge: early IL-4 production governs the requirement for IL-27-WSX-1 signaling in the development of protective Th1 cytokine responses following Leishmania major infection. J. Immunol. 172, 4672–4675.
Aseffa, A., Gumy, A., Launois, P., MacDonald, H. R., Louis, J. A., and Tacchini-Cottier, F. (2002). The early IL-4 response to Leishmania major and the resulting Th2 cell maturation steering progressive disease in BALB/c mice are subject to the control of regulatory CD4+CD25+ T cells. J. Immunol. 169, 3232–3241.
Baguet, A., Epler, J., Wen, K. W., and Bix, M. (2004). A Leishmania major response locus identified by interval-specific congenic mapping of a T helper type 2 cell bias-controlling quantitative trait locus. J. Exp. Med. 200, 1605–1612.
Barbi, J., Brombacher, F., and Satoskar, A. R. (2008). T cells from Leishmania major-susceptible BALB/c mice have a defect in efficiently up-regulating CXCR3 upon activation. J. Immunol. 181, 4613–4620.
Barbi, J., Snider, H. M., Bhardwaj, N., Lezama-Dávila, C. M., Durbin, J. E., and Satoskar, A. R. (2009). Signal transducer and activator of transcription 1 in T cells plays an indispensable role in immunity to Leishmania major by mediating Th1 cell homing to the site of infection. FASEB J. 23, 3990–3999.
Basu, R., Bhaumik, S., Basu, J. M., Naskar, K., De, T., and Roy, S. (2005). Kinetoplastid membrane protein-11 DNA vaccination induces complete protection against both pentavalent antimonial-sensitive and -resistant strains of Leishmania donovani that correlates with inducible nitric oxide synthase activity and IL-4 generation: evidence for mixed Th1- and Th2-like responses in visceral leishmaniasis. J. Immunol. 174, 7160–7171.
Basu, R., Bhaumik, S., Haldar, A. K., Naskar, K., De, T., Dana, S. K., Walden, P., and Roy, S. (2007). Hybrid cell vaccination resolves Leishmania donovani infection by eliciting a strong CD8+ cytotoxic T-lymphocyte response with concomitant suppression of interleukin-10 (IL-10) but not IL-4 or IL-13. Infect. Immun. 75, 5956–5966.
Belkaid, Y., Hoffmann, K. F., Mendez, S., Kamhawi, S., Udey, M. C., Wynn, T. A., and Sacks, D. L. (2001). The role of interleukin (IL)-10 in the persistence of Leishmania major in the skin after healing and the therapeutic potential of anti-IL-10 receptor antibody for sterile cure. J. Exp. Med. 194, 1497–1506.
Belkaid, Y., Piccirillo, C. A., Mendez, S., Shevach, E. M., and Sacks, D. L. (2002a). CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature 420, 502–507.
Belkaid, Y., Von Stebut, E., Mendez, S., Lira, R., Caler, E., Bertholet, S., Udey, M. C., and Sacks, D. (2002b). CD8+ T cells are required for primary immunity in C57BL/6 mice following low-dose, intradermal challenge with Leishmania major. J. Immunol. 168, 3992–4000.
Bettelli, E., Oukka, M., and Kuchroo, V. K. (2007). T(H)-17 cells in the circle of immunity and autoimmunity. Nat. Immunol. 8, 345–350.
Biedermann, T., Zimmermann, S., Himmelrich, H., Gumy, A., Egeter, O., Sakrauski, A. K., Seegmuller, I., Voigt, H., Launois, P., Levine, A. D., Wagner, H., Heeg, K., Louis, J. A., and Rocken, M. (2001). IL-4 instructs TH1 responses and resistance to Leishmania major in susceptible BALB/c mice. Nat. Immunol. 2, 1054–1060.
Boaventura, V. S., Santos, C. S., Cardoso, C. R., de Andrade, J., Dos Santos, W. L., Clarêncio, J., Silva, J. S., Borges, V. M., Barral-Netto, M., Brodskyn, C. I., and Barral, A. (2010). Human mucosal leishmaniasis: neutrophils infiltrate areas of tissue damage that express high levels of Th17-related cytokines. Eur. J. Immunol. 40, 2830–2836.
Bodas, M., Jain, N., Awasthi, A., Martin, S., Penke Loka, R. K., Dandekar, D., Mitra, D., and Saha, B. (2006). Inhibition of IL-2 induced IL-10 production as a principle of phase-specific immunotherapy. J. Immunol. 177, 4636–4643.
Bogdan, C., Gessner, A., Solbach, W., and Röllinghoff, M. (1996). Invasion, control and persistence of Leishmania parasites. Curr. Opin. Immunol. 8, 517–525.
Bogdan, C., Stenger, S., Rollinghoff, M., and Solbach, W. (1991). Cytokine interactions in experimental cutaneous leishmaniasis. Interleukin 4 synergizes with interferon-gamma to activate murine macrophages for killing of Leishmania major amastigotes. Eur. J. Immunol. 21, 327–333.
Bourreau, E., Prévot, G., Gardon, J., Pradinaud, R., and Launois, P. (2001). High intralesional interleukin-10 messenger RNA expression in localized cutaneous leishmaniasis is associated with unresponsiveness to treatment. J. Infect. Dis. 184, 1628–1630.
Bourreau, E., Ronet, C., Darcissac, E., Lise, M. C., Sainte Marie, D., Clity, E., Tacchini-Cottier, F., Couppie, P., and Launois, P. (2009). Intralesional regulatory T-cell suppressive function during human acute and chronic cutaneous leishmaniasis due to Leishmania guyanensis. Infect. Immun. 77, 1465–1474.
Brewer, J. M., Conacher, M., Hunter, C. A., Mohrs, M., Brombacher, F., and Alexander, J. (1999). Aluminium hydroxide adjuvant initiates strong antigen-specific Th2 responses in the absence of IL-4- or IL-13-mediated signalling. J. Immunol. 163, 6448–6454.
Brown, D. R., and Reiner, S. L. (1999). Polarized helper-T-cell responses against Leishmania major in the absence of B cells. Infect. Immun. 67, 266–270.
Bryson, K. J., Millington, O. R., Mokgethi, T., McGachy, H. A., Brombacher, F., and Alexander, J. (2011). BALB/c mice deficient in CD4 T cell IL-4Rα expression control Leishmania mexicana Load although female but not male mice develop a healer phenotype. PLoS Negl. Trop. Dis. 5, e930. doi:10.1371/journal.pntd.0000930
Buxbaum, L. U. (2010). Type I IFNs promote the early IFN-gamma response and the IL-10 response in Leishmania mexicana infection. Parasite Immunol. 32, 153–160.
Buxbaum, L. U., and Scott, P. (2005). Interleukin 10- and Fcgamma receptor-deficient mice resolve Leishmania mexicana lesions. Infect. Immun. 73, 2101–2108.
Buxbaum, L. U., Uzonna, J. E., Goldschmidt, M. H., and Scott, P. (2002). Control of new world cutaneous leishmaniasis is IL-12 independent but STAT4 dependent. Eur. J. Immunol. 32, 3206–3215.
Campanelli, A. P., Roselino, A. M., Cavassani, K. A., Pereira, M. S., Mortara, R. A., Brodskyn, C. I., Goncalves, H. S., Belkaid, Y., Barral-Netto, M., Barral, A., and Silva, J. S. (2006). CD4+CD25+ T cells in skin lesions of patients with cutaneous leishmaniasis exhibit phenotypic and functional characteristics of natural regulatory T cells. J. Infect. Dis. 193, 1313–1322.
Campbell, K. A., Ovendale, P. J., Kennedy, M. K., Fanslow, W. C., Reed, S. G., and Maliszewski, C. R. (1996). CD40 ligand is required for protective cell-mediated immunity to Leishmania major. Immunity 4, 283–289.
Castilho, T. M., Goldsmith-Pestana, K., Lozano, C., Valderrama, L., Saravia, N. G., and McMahon-Pratt, D. (2010). Murine model of chronic L. (Viannia) panamensis infection: role of IL-13 in disease. Eur. J. Immunol. 40, 2816–2829.
Charmoy, M., Auderset, F., Allenbach, C., and Tacchini-Cottier, F. (2010). The prominent role of neutrophils during the initial phase of infection by Leishmania parasites. J. Biomed. Biotechnol. 2010, 719361. doi:10.1155/2010/719361
Chu, N., Thomas, B. N., Patel, S. R., and Buxbaum, L. U. (2010). IgG1 is pathogenic in Leishmania mexicana infection. J. Immunol. 185, 6939–6946.
Colmenares, M., Constant, S. L., Kima, P. E., and McMahon-Pratt, D. (2002). Leishmania pifanoi pathogenesis: selective lack of a local cutaneous response in the absence of circulating antibody. Infect. Immun. 70, 6597–6605.
Colmenares, M., Kima, P. E., Samoff, E., Soong, L., and McMahon-Pratt, D. (2003). Perforin and gamma interferon are critical CD8+ T-cell-mediated responses in vaccine-induced immunity against Leishmania amazonensis infection. Infect. Immun. 71, 3172–3182.
Cunningham, A. C. (2002). Parasitic adaptive mechanisms in infection by Leishmania. Exp. Mol. Pathol. 72, 132–141.
Deak, E., Jayakumar, A., Cho, K. W., Goldsmith-Pestana, K., Dondji, B., Lambris, J. D., and McMahon-Pratt, D. (2010). Murine visceral leishmaniasis: IgM and polyclonal B-cell activation lead to disease exacerbation. Eur. J. Immunol. 40, 1355–1368.
Deenick, E. K., Ma, C. S., Brink, R., and Tangye, S. G. (2011). Regulation of T follicular helper cell formation and function by antigen presenting cells. Curr. Opin. Immunol. 23, 111–118.
Dewals, B., Hoving, J. C., Leeto, M., Marillier, R. G., Govender, U., Cutler, A. J., Horsnell, W. G., and Brombacher, F. (2009). IL-4Ralpha responsiveness of non-CD4 T cells contributes to resistance in Schistosoma mansoni infection in pan-T cell-specific IL-4Ralpha-deficient mice. Am. J. Pathol. 175, 706–716.
Diefenbach, A., Schindler, H., Donhauser, N., Lorenz, E., Laskay, T., MacMicking, J., Röllinghoff, M., Gresser, I., and Bogdan, C. (1998). Type 1 interferon (IFNalpha/beta) and type 2 nitric oxide synthase regulate the innate immune response to a protozoan parasite. Immunity 8, 77–87.
Engwerda, C. R., Murphy, M. L., Cotterell, S. E., Smelt, S. C., and Kaye, P. M. (1998). Neutralization of IL-12 demonstrates the existence of discrete organ-specific phases in the control of Leishmania donovani. Eur. J. Immunol. 28, 669–680.
Eriksson, U., Kurrer, M. O., Sonderegger, I., Iezzi, G., Tafuri, A., Hunziker, L., Suzuki, S., Bachmaier, K., Bingisser, R. M., Penninger, J. M., and Kopf, M. (2003). Activation of dendritic cells through the interleukin 1 receptor 1 is critical for the induction of autoimmune myocarditis. J. Exp. Med. 197, 323–331.
Faulkner, H., Renauld, J. C., Van Snick, J., and Grencis, R. K. (1998). Interleukin-9 enhances resistance to the intestinal nematode Trichuris muris. Infect. Immun. 66, 3832–3840.
Fitzgerald, D. C., Ciric, B., Touil, T., Harle, H., Grammatikopolou, J., Das Sarma, J., Gran, B., Zhang, G. X., and Rostami, A. (2007). Suppressive effect of IL-27 on encephalitogenic Th17 cells and the effector phase of experimental autoimmune encephalomyelitis. J. Immunol. 179, 3268–3275.
Gessner, A., Blum, H., and Röllinghoff, M. (1993). Differential regulation of IL-9-expression after infection with Leishmania major in susceptible and resistant mice. Immunobiology 189, 419–435.
Gicheru, M. M., Olobo, J. O., Anjili, C. O., Orago, A. S., Modabber, F., and Scott, P. (2001). Vervet monkeys vaccinated with killed Leishmania major parasites and interleukin-12 develop a type 1 immune response but are not protected against challenge infection. Infect. Immun. 69, 245–251.
Heinzel, F. P., Rerko, R. M., and Hujer, A. M. (1998). Underproduction of interleukin-12 in susceptible mice during progressive leishmaniasis is due to decreased CD40 activity. Cell. Immunol. 184, 129–142.
Heinzel, F. P., Sadick, M. D., Holaday, B. J., Coffman, R. L., and Locksley, R. M. (1989). Reciprocal expression of interferon gamma or interleukin 4 during the resolution or progression of murine leishmaniasis. Evidence for expansion of distinct helper T cell subsets. J. Exp. Med. 169, 59–72.
Herbert, D. R., Hölscher, C., Mohrs, M., Arendse, B., Schwegmann, A., Radwanska, M., Leeto, M., Kirsch, R., Hall, P., Mossmann, H., Claussen, B., Förster, I., and Brombacher, F. (2004). Alternative macrophage activation is essential for survival during schistosomiasis and downmodulates T helper 1 responses and immunopathology. Immunity 20, 623–635.
Himmelrich, H., Parra-Lopez, C., Tacchini-Cottier, F., Louis, J. A., and Launois, P. (1998). The IL-4 rapidly produced in BALB/c mice after infection with Leishmania major down-regulates IL-12 receptor beta 2-chain expression on CD4+ T cells resulting in a state of unresponsiveness to IL-12. J. Immunol. 161, 6156–6163.
Hochrein, H., O’Keeffe, M., Luft, T., Vandenabeele, S., Grumont, R. J., Maraskovsky, E., and Shortman, K. (2000). Interleukin (IL-4) is a major regulatory cytokine governing bioactive IL-12 production by mouse and human dendritic cells. J. Exp. Med. 192, 823–833.
Hoelscher, C., Arendse, B., Schwegmann, A., Myburgh, E., and Brombacher, F. (2006). Impairment of alternative macrophage activation delays cutaneous leishmaniasis in nonhealing BALB/c mice. J. Immunol. 176, 1115–1121.
Horsnell, W. G., Cutler, A. J., Hoving, J. C., Mearns, H., Myburgh, E., Arendse, B., Finkelman, F. D., Owens, G. K., Erle, D., and Brombacher, F. (2007). Delayed goblet cell hyperplasia, acetylcholine receptor expression, and worm expulsion in SMC-specific IL-4Ralpha-deficient mice. PLoS Pathog. 3, e1. doi:10.1371/journal.ppat.0030001
Hoving, J. C., Kirstein, F., Nieuwenhuizen, N. E., Fick, L. C., Hobeika, E., Reth, M., and Brombacher, F. (2012). B cells that produce immunoglobulin e mediate colitis in BALB/c mice. Gastroenterology 142, 96–108.
Hsu, A. C., and Scott, P. (2007). Leishmania mexicana infection induces impaired lymph node expansion and Th1 cell differentiation despite normal T cell proliferation. J. Immunol. 179, 8200–8207.
Jäeger, A., and Kuchroo, V. K. (2010). Effector and regulatory T-cell subsets in autoimmunity and tissue inflammation. Scand. J. Immunol. 72, 173–184.
Ji, J., Masterson, J., Sun, J., and Soong, L. (2005). CD4+CD25+ regulatory T cells restrain pathogenic responses during Leishmania amazonensis infection. J. Immunol. 174, 7147–7153.
Jones, D., Elloso, M. M., Showe, L., Williams, D., Trinchieri, G., and Scott, P. (1998). Differential regulation of the interleukin-12 receptor during the innate immune response to Leishmania major. Infect. Immun. 66, 3818–3824.
Jones, D. E., Buxbaum, L. U., and Scott, P. (2000). IL-4-independent inhibition of IL-12 responsiveness during Leishmania amazonensis infection. J. Immunol. 165, 364–372.
Jüttner, S., Bernhagen, J., Metz, C. N., Röllinghoff, M., Bucala, R., and Gessner, A. (1998). Migration inhibitory factor induces killing of Leishmania major by macrophages: dependence on reactive nitrogen intermediates and endogenous TNF-alpha. J. Immunol. 161, 2383–2390.
Kamanaka, M., Yu, P., Yasui, T., Yoshida, K., Kawabe, T., Horii, T., Kishimoto, T., and Kikutani, H. (1996). Protective role of CD40 in Leishmania major infection at two distinct phases of cell-mediated immunity. Immunity 4, 275–281.
Kane, M. M., and Mosser, D. M. (2001). The role of IL-10 in promoting disease progression in leishmaniasis. J. Immunol. 166, 1141–1147.
Karp, C. L., el-Safi, S. H., Wynn, T. A., Satti, M. M., Kordofani, A. M., Hashim, F. A., Hag-Ali, M., Neva, F. A., Nutman, T. B., and Sacks, D. L. (1993). In vivo cytokine profiles in patients with kala-azar. Marked elevation of both interleukin-10 and interferon-gamma. J. Clin. Invest. 91, 1644–1648.
Katara, G. K., Ansari, N. A., Verma, S., Ramesh, V., and Salotra, P. (2011). Foxp3 and IL-10 expression correlates with parasite burden in lesional tissues of post kala azar dermal leishmaniasis (PKDL) patients. PLoS Negl. Trop. Dis. 5, e1171. doi:10.1371/journal.pntd.0001171
Kaye, P. M., Curry, A. J., and Blackwell, J. M. (1991). Differential production of Th1- and Th2-derived cytokines does not determine the genetically controlled or vaccine-induced rate of cure in murine visceral leishmaniasis. J. Immunol. 146, 2763–2770.
Kemp, K. (2000). Cytokine-producing T cell subsets in human leishmaniasis. Arch. Immunol. Ther. Exp. 48, 173–177.
Kemp, M., Kurtzhals, J. A., Bendtzen, K., Poulsen, L. K., Hansen, M. B., Koech, D. K., Kharazmi, A., and Theander, T. G. (1993). Leishmania donovani-reactive Th1- and Th2-like T-cell clones from individuals who have recovered from visceral leishmaniasis. Infect. Immun. 61, 1069–1073.
Kenney, R. T., Sacks, D. L., Sypek, J. P., Vilela, L., Gam, A. A., and Evans-Davis, K. (1999). Protective immunity using recombinant human IL-12 and alum as adjuvants in a primate model of cutaneous leishmaniasis. J. Immunol. 163, 4481–4488.
Kima, P. E., Constant, S. L., Hannum, L., Colmenares, M., Lee, K. S., Haberman, A. M., Shlomchik, M. J., and McMahon-Pratt, D. (2000). Internalization of Leishmania mexicana complex amastigotes via the Fc receptor is required to sustain infection in murine cutaneous leishmaniasis. J. Exp. Med. 191, 1063–1068.
Ko, H. J., Yang, H., Yang, J. Y., Seo, S. U., Chang, S. Y., Seong, J. K., and Kweon, M. N. (2011). Expansion of Tfh-like cells during chronic Salmonella exposure mediates the generation of autoimmune hypergammaglobulinemia in MyD88-deficient mice. Eur. J. Immunol. doi: 10.1002/eji.201141748. [Epub ahead of print].
Kopf, M., Brombacher, F., Kohler, G., Kienzle, G., Widmann, K. H., Lefrang, K., Humborg, C., Ledermann, B., and Solbach, W. (1996). IL-4-deficient Balb/c mice resist infection with Leishmania major. J. Exp. Med. 184, 1127–1136.
Korn, T., Bettelli, E., Oukka, M., and Kuchroo, V. K. (2009). IL-17 and Th17 Cells. Annu. Rev. Immunol. 27, 485–517.
Lages, C. S., Suffia, I., Velilla, P. A., Huang, B., Warshaw, G., Hildeman, D. A., Belkaid, Y., and Chougnet, C. (2008). Functional regulatory T cells accumulate in aged hosts and promote chronic infectious disease reactivation. J. Immunol. 181, 1835–1848.
Launois, P., Ohteki, T., Swihart, K., MacDonald, H. R., and Louis, J. A. (1995). In susceptible mice, Leishmania major induce very rapid interleukin-4 production by CD4+ T cells which are NK1.1 . Eur. J. Immunol. 25, 3298–3307.
Lean, I. S., McDonald, S. A., Bajaj-Elliott, M., Pollok, R. C., Farthing, M. J., and McDonald, V. (2003). Interleukin-4 and transforming growth factor-beta have opposing regulatory effects on gamma interferon-mediated inhibition of Cryptosporidium parvum reproduction. Infect. Immun. 71, 4580–4585.
Liu, D., Zhang, T., Marshall, A. J., Okkenhaug, K., Vanhaesebroeck, B., and Uzonna, J. E. (2009). The p110delta isoform of phosphatidylinositol 3-kinase controls susceptibility to Leishmania major by regulating expansion and tissue homing of regulatory T cells. J. Immunol. 183, 1921–1933.
Lopez Kostka, S., Dinges, S., Griewank, K., Iwakura, Y., Udey, M. C., and von Stebut, E. (2009). IL-17 promotes progression of cutaneous leishmaniasis in susceptible mice. J. Immunol. 182, 3039–3046.
Maldonado, R. A., Irvine, D. J., Schreiber, R., and Glimcher, L. H. (2004). A role for the immunological synapse in lineage commitment of CD4 lymphocytes. Nature 431, 527–532.
Mannheimer, S. B., Hariprashad, J., Stoeckle, M. Y., and Murray, H. W. (1996). Induction of macrophage antiprotozoal activity by monocyte chemotactic and activating factor. FEMS Immunol. Med. Microbiol. 14, 59–61.
Maroof, A., Beattie, L., Zubairi, S., Svensson, M., Stager, S., and Kaye, P. M. (2008). Posttranscriptional regulation of II10 gene expression allows natural killer cells to express immunoregulatory function. Immunity 29, 295–305.
Matthews, D. J., Emson, C. L., McKenzie, G. J., Jolin, H. E., Blackwell, J. M., and McKenzie, A. N. (2000). IL-13 is a susceptibility factor for Leishmania major infection. J. Immunol. 164, 1458–1462.
McDonald, S. A., O’Grady, J. E., Bajaj-Elliott, M., Notley, C. A., Alexander, J., Brombacher, F., and McDonald, V. (2004). Protection against the early acute phase of Cryptosporidium parvum infection conferred by interleukin-4-induced expression of T helper 1 cytokines. J. Infect. Dis. 190, 1019–1025.
McFarlane, E., Carter, K. C., McKenzie, A. N., Kaye, P. M., Brombacher, F., and Alexander, J. (2011). Endogenous IL-13 plays a crucial role in liver granuloma maturation during Leishmania donovani infection, independent of IL-4Rα-responsive macrophages and neutrophils. J. Infect. Dis. 204, 36–43.
McMahon-Pratt, D., and Alexander, J. (2004). Does the Leishmania major paradigm of pathogenesis and protection hold for New World cutaneous leishmaniasis or the visceral disease? Immunol. Rev. 201, 206–224.
Mohrs, K., Wakil, A. E., Killeen, N., Locksley, R. M., and Mohrs, M. (2005). A two-step process for cytokine production revealed by IL-4 dual-reporter mice. Immunity 23, 419–429.
Mohrs, M., Holscher, C., and Brombacher, F. (2000). Interleukin-4 receptor alpha-deficient BALB/c mice show an unimpaired T helper 2 polarization in response to Leishmania major infection. Infect. Immun. 68, 1773–1780.
Mohrs, M., Ledermann, B., Kohler, G., Dorfmuller, A., Gessner, A., and Brombacher, F. (1999). Differences between IL-4- and IL-4 receptor alpha-deficient mice in chronic leishmaniasis reveal a protective role for IL-13 receptor signaling. J. Immunol. 162, 7302–7308.
Mosmann, T. R., Cherwinski, H., Bond, M. W., Giedlin, M. A., and Coffman, R. L. (1986). Two types of murine helper T cell clone. 1. Definition according to profiles of lymphokine activities and secreted proteins. J. Immunol. 136, 2348–2357.
Murphy, M. L., Cotterell, S. E. J., Gorak, P. M. A., Engwerda, C. R., and Kaye, P. M. (1998). Blockade of CTLA4 enhances host resistance to the intracellular pathogen, Leishmania donovani. J. Immunol. 166, 4153–4160.
Murphy, M. L., Wille, U., Villegas, E. N., Hunter, C. A., and Farrell, J. P. (2001). IL-10 mediates susceptibility to Leishmania donovani infection. Eur. J. Immunol. 31, 2848–2856.
Murray, H. W. (1997). Endogenous interleukin-12 regulates acquired resistance in experimental visceral leishmaniasis. J. Infect. Dis. 175, 1477–1479.
Murray, H. W., and Nathan, C. F. (1999). Macrophage microbicidal mechanisms in vivo: reactive nitrogen versus oxygen intermediates in the killing of intracellular visceral Leishmania donovani. J. Exp. Med. 189, 741–746.
Murray, H. W., Montelibano, C., Peterson, R., and Sypek, J. P. (2000). Interleukin-12 regulates the response to chemotherapy in experimental visceral leishmaniasis. J. Infect. Dis. 182, 1497–1502.
Murray, H. W., Oca, M. J., Granger, A. M., and Schreiber, R. D. (1989). Requirement for T cells and effect of lymphokines in successful chemotherapy for an intracellular infection. J. Clin. Invest. 83, 1253–1257.
Murray, H. W., Tsai, C. W., Liu, J., and Ma, X. (2006). Visceral Leishmania donovani infection in interleukin-13-/- mice. Infect. Immun. 74, 2487–2490.
Nashed, B. F., Maekawa, Y., Takashima, M., Zhang, T., Ishii, K., Dainichi, T., Ishikawa, H., Sakai, T., Hisaeda, H., and Himeno, K. (2000). Different cytokines are required for induction and maintenance of the Th2 type response in DBA/2 mice resistant to infection with Leishmania major. Microbes Infect. 2, 1435–1443.
Nishikomori, R., Gurunathan, S., Nishikomori, K., and Strober, W. (2001). BALB/c mice bearing a transgenic IL-12 receptor beta 2 gene exhibit a nonhealing phenotype to Leishmania major infection despite intact IL-12 signaling. J. Immunol. 166, 6776–6783.
Noben-Trauth, N., Kropf, P., and Muller, I. (1996). Susceptibility to Leishmania major infection in interleukin-4-deficient mice. Science 271, 987–990.
Noben-Trauth, N., Lira, R., Nagase, H., Paul, W. E., and Sacks, D. L. (2003). The relative contribution of IL-4 receptor signaling and IL-10 to susceptibility to Leishmania major. J. Immunol. 170, 5152–5158.
Noben-Trauth, N., Paul, W. E., and Sacks, D. L. (1999). IL-4- and IL-4 receptor-deficient BALB/c mice reveal differences in susceptibility to Leishmania major parasite substrains. J. Immunol. 162, 6132–6140.
Noble, A., and Kemeny, D. M. (1995). Interleukin-4 enhances interferon-g synthesis but inhibits development of interferon-g-producing cells. Immunology 85, 357–363.
Novoa, R., Bacellar, O., Nascimento, M., Cardoso, T. M., Ramasawmy, R., Oliveira, W. N., Schriefer, A., and Carvalho, E. M. (2011). IL-17 and regulatory cytokines (IL-10 and IL-27) in L. braziliensis Infection. Parasite Immunol. 33, 132–136.
Nowak, E. C., and Noelle, R. J. (2010). Interleukin-9 as a T helper type 17 cytokine. Immunology 131, 169–173.
Nylén, S., Maurya, R., Eidsmo, L., Manandhar, K. D., Sundar, S., and Sacks, D. (2007). Splenic accumulation of IL-10 mRNA in T cells distinct from CD4+CD25+ (Foxp3) regulatory T cells in human visceral leishmaniasis. J. Exp. Med. 204, 805–817.
Oriss, T. B., McCarthy, S. A., Morel, B. F., Campana, M. A., and Morel, P. A. (1997). Cross-regulation between T helper cell (Th)1 and Th2: inhibition of Th2 proliferation by IFN-γ involves interference with IL-1. J. Immunol. 158, 3666–3672.
Ouyang, W., Löhning, M., Gao, Z., Assenmacher, M., Ranganath, S., Radbruch, A., and Murphy, K. M. (2000). Stat6-independent GATA-3 autoactivation directs IL-4-independent Th2 development and commitment. Immunity 12, 27–37.
Ouyang, W., Ranganath, S. H., Weindel, K., Bhattacharya, D., Murphy, T. L., Sha, W. C., and Murphy, K. M. (1998). Inhibition of Th1 development mediated by GATA-3 through an IL-4-independent mechanism. Immunity 9, 745–755.
Padigel, U. M., Alexander, J., and Farrell, J. P. (2003a). The role of interleukin-10 in susceptibility of BALB/c mice to infection with Leishmania mexicana and Leishmania amazonensis. J. Immunol. 171, 3705–3710.
Padigel, U. M., Kim, N., Choi, Y., and Farrell, J. P. (2003b). TRANCE-RANK costimulation is required for IL-12 production and the initiation of a Th1-type response to Leishmania major infection in CD40L-deficient mice. J. Immunol. 171, 5437–5441.
Padigel, U. M., and Farrell, J. P. (2003). CD40-CD40 ligand costimulation is not required for initiation and maintenance of a Th1-type response to Leishmania major infection. Infect. Immun. 71, 1389–1395.
Padigel, U. M., and Farrell, J. P. (2005). Control of infection with Leishmania major in susceptible BALB/c mice lacking the common gamma-chain for FcR is associated with reduced production of IL-10 and TGF-beta by parasitized cells. J. Immunol. 174, 6340–6345.
Peters, C., Aebischer, T., Stierhof, Y. D., Fuchs, M., and Overath, P. (1995). The role of macrophage receptors in adhesion and uptake of Leishmania mexicana amastigotes. J. Cell. Sci. 108, 3715–3724.
Peterson, R. A. (2012). Regulatory T-cells: diverse phenotypes integral to immune homeostasis and suppression. Toxicol. Pathol. 40, 186–204.
Pitta, M. G., Romano, A., Cabantous, S., Henri, S., Hammad, A., Kouriba, B., Argiro, L., el Kheir, M., Bucheton, B., Mary, C., El-Safi, S. H., and Dessein, A. (2009). IL-17 and IL-22 are associated with protection against human kala azar caused by Leishmania donovani. J. Clin. Invest. 119, 2379–2387.
Radwanska, M., Cutler, A. J., Hoving, J. C., Magez, S., Holscher, C., Bohms, A., Arendse, B., Kirsch, R., Hunig, T., Alexander, J., Kaye, P., and Brombacher, F. (2007). Deletion of IL-4Ralpha on CD4 T cells renders BALB/c mice resistant to Leishmania major infection. PLoS Pathog. 3, e68. doi:10.1371/journal.ppat.0030068
Reiner, S. L., Zheng, S., Wang, Z. E., Stowring, L., and Locksley, R. M. (1994). Leishmania promastigotes evade interleukin 12 (IL-12) induction by macrophages and stimulate a broad range of cytokines from CD4+ T cells during initiation of infection. J. Exp. Med. 179, 447–456.
Reinhardt, R. L., Liang, H. E., and Locksley, R. M. (2009). Cytokine-secreting follicular T cells shape the antibody repertoire. Nat. Immunol. 10, 385–393.
Ronet, C., Hauyon-La Torre, Y., Revaz-Breton, M., Mastelic, B., Tacchini-Cottier, F., Louis, J., and Launois, P. (2010). Regulatory B cells shape the development of Th2 immune responses in BALB/c mice infected with Leishmania major through IL-10 production. J. Immunol. 184, 886–894.
Ronet, C., Voigt, H., Himmelrich, H., Doucey, M. A., Hauyon-La Torre, Y., Revaz-Breton, M., Tacchini-Cottier, F., Bron, C., Louis, J., and Launois, P. (2008). Leishmania major-specific B cells are necessary for Th2 cell development and susceptibility to L. major LV39 in BALB/c mice. J. Immunol. 180, 4825–4835.
Rosas, L. E., Satoskar, A. A., Roth, K. M., Keiser, T. L., Barbi, J., Hunter, C., de Sauvage, F. J., and Satoskar, A. R. (2006). Interleukin-27R (WSX-1/T-cell cytokine receptor) gene-deficient mice display enhanced resistance to Leishmania donovani infection but develop severe liver immunopathology. Am. J. Pathol. 168, 158–169.
Sacks, D., and Anderson, C. (2004). Re-examination of the immunosuppressive mechanisms mediating non-cure of Leishmania infection in mice. Immunol. Rev. 201, 225–238.
Satoskar, A., and Alexander, J. (1995). Sex-determined susceptibility and differential IFN-gamma and TNF-alpha mRNA expression in DBA/2 mice infected with Leishmania mexicana. Immunology 84, 1–4.
Satoskar, A., Bluethmann, H., and Alexander, J. (1995). Disruption of the murine interleukin 4 gene inhibits disease progression during Leishmania mexicana infection but does not increase control of Leishmania donovani infection. Infect. Immun. 63, 4894–4899.
Satoskar, A., Brombacher, F., Dai, W. J., McInnes, I., Liew, F. Y., Alexander, J., and Walker, W. (1997). SCID mice reconstituted with IL-4-deficient lymphocytes, but not immunocompetent lymphocytes, are resistant to cutaneous leishmaniasis. J. Immunol. 159, 5005–5013.
Scott, P., Eaton, A., Gause, W. C., di Zhou, X., and Hondowicz, B. (1996). Early IL-4 production does not predict susceptibility to Leishmania major. Exp. Parasitol. 84, 178–187.
Smelt, S. C., Cotterell, S. E., Engwerda, C. R., and Kaye, P. M. (2000). B cell-deficient mice are highly resistant to Leishmania donovani infection, but develop neutrophil-mediated tissue pathology. J. Immunol. 164, 3681–3688.
Soong, L., Xu, J. C., Grewal, I. S., Kima, P., Sun, J., Longley, B. J. Jr., Ruddle, N. H., McMahon-Pratt, D., and Flavell, R. A. (1996). Disruption of CD40-CD40 ligand interactions results in an enhanced susceptibility to Leishmania amazonensis infection. Immunity 4, 263–273.
Stager, S., Alexander, J., Carter, K. C., Brombacher, F., and Kaye, P. M. (2003a). Both interleukin-4 (IL-4) and IL-4 receptor alpha signaling contribute to the development of hepatic granulomas with optimal antileishmanial activity. Infect. Immun. 71, 4804–4807.
Stager, S., Alexander, J., Kirby, A. C., Botto, M., Rooijen, N. V., Smith, D. F., Brombacher, F., and Kaye, P. M. (2003b). Natural antibodies and complement are endogenous adjuvants for vaccine-induced CD8+ T-cell responses. Nat. Med. 9, 1287–1292.
Stager, S., Maroof, A., Zubairi, S., Sanos, S. L., Kopf, M., and Kaye, P. M. (2006). Distinct roles for IL-6 and IL-12p40 in mediating protection against Leishmania donovani and the expansion of IL-10+ CD4+ T cells. Eur. J. Immunol. 36, 1764–1771.
Svensson, M., Maroof, A., Ato, M., and Kaye, P. M. (2004). Stromal cells direct local differentiation of regulatory dendritic cells. Immunity 21, 805–816.
Takeda, A., Hamano, S., Yamanaka, A., Hanada, T., Ishibashi, T., Mak, T. W., Yoshimura, A., and Yoshida, H. (2003). Cutting edge: role of IL-27/WSX-1 signaling for induction of T-bet through activation of STAT1 during initial Th1 commitment. J. Immunol. 170, 4886–4890.
Thomas, B. N., and Buxbaum, L. U. (2008). FcgammaRIII mediates immunoglobulin G-induced interleukin-10 and is required for chronic Leishmania mexicana lesions. Infect. Immun. 76, 623–631.
Vargas-Inchaustegui, D. A., Xin, L., and Soong, L. (2008). Leishmania braziliensis infection induces dendritic cell activation, ISG15 transcription, and the generation of protective immune responses. J. Immunol. 180, 7537–7545.
Varkila, K., Chatelain, R., Leal, L. M., and Coffman, R. L. (1993). Reconstitution of C.B-17 scid mice with BALB/c T cells initiates a T helper type-1 response and renders them capable of healing Leishmania major infection. Eur. J. Immunol. 23, 262–268.
Von Stebut, E., Ehrchen, J. M., Belkaid, Y., Kostka, S. L., Molle, K., Knop, J., Sunderkotter, C., and Udey, M. C. (2003). Interleukin 1alpha promotes Th1 differentiation and inhibits disease progression in Leishmania major-susceptible BALB/c mice. J. Exp. Med. 198, 191–199.
Wei, X. Q., Niedbala, W., Xu, D., Luo, Z. X., Pollock, K. G., and Brewer, J. M. (2004). Host genetic background determines whether IL-18 deficiency results in increased susceptibility or resistance to murine Leishmania major infection. Immunol. Lett. 94, 35–37.
Wu, W., Huang, L., and Mendez, S. (2010). A live Leishmania major vaccine containing CpG motifs induces the de novo generation of Th17 cells in C57BL/6 mice. Eur. J. Immunol. 40, 2517–2527.
Xin, L., Li, Y., and Soong, L. (2007). Role of interleukin-1beta in activating the CD11c(high) CD45RB-dendritic cell subset and priming Leishmania amazonensis-specific CD4+ T cells in vitro and in vivo. Infect. Immun. 75, 5018–5026.
Xu, D., Liu, H., Komai-Koma, M., Campbell, C., McSharry, C., Alexander, J., and Liew, F. Y. (2003). CD4+CD25+ regulatory T cells suppress differentiation and functions of Th1 and Th2 cells, Leishmania major infection, and colitis in mice. J. Immunol. 170, 394–399.
Yoshimura, T., Takeda, A., Hamano, S., Miyazaki, Y., Kinjyo, I., Ishibashi, T., Yoshimura, A., and Yoshida, H. (2006). Two-sided roles of IL-27: induction of Th1 differentiation on naive CD4+ T cells versus suppression of proinflammatory cytokine production including IL-23-induced IL-17 on activated CD4+ T cells partially through STAT3-dependent mechanism. J. Immunol. 177, 5377–5385.
Keywords: Leishmania, T helper 1, T helper 2, T regulatory cells, T helper 17, T follicular helper cells, interleukin-4, interferon-gamma
Citation: Alexander J and Brombacher F (2012) T helper1/T helper2 cells and resistance/susceptibility to Leishmania infection: is this paradigm still relevant? Front. Immun. 3:80. doi: 10.3389/fimmu.2012.00080
Received: 06 February 2012; Accepted: 28 March 2012;
Published online: 17 April 2012.
Edited by:
Nathan Peters, National Institute of Allergy and Infectious Diseases, USAReviewed by:
Emilio Luis Malchiodi, University of Buenos Aires, ArgentinaDavid Sacks, National Institutes of Health, USA
Copyright: © 2012 Alexander and Brombacher. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: James Alexander, Strathclyde Institute of Pharmacy and Biomedical Sciences, University of Strathclyde, Glasgow, G4 ORE, UK. e-mail:ai5hbGV4YW5kZXJAc3RyYXRoLmFjLnVr