

- Department of Internal Medicine, Washington University School of Medicine, St. Louis, MO, USA
IgA is one of the most important molecules in the regulation of intestinal homeostasis. Peyer’s patches have been traditionally recognized as sites for the induction of intestinal IgA responses, however more recent studies demonstrate that isolated lymphoid follicles (ILFs) can perform this function as well. ILF development is dynamic, changing in response to the luminal microbial burden, suggesting that ILFs play an important role providing an expandable reservoir of compensatory IgA inductive sites. However, in situations of immune dysfunction, ILFs can over-develop in response to uncontrollable enteric flora, resulting in ILF hyperplasia. The ability of ILFs to expand and respond to help control the enteric flora makes this dynamic reservoir an important arm of IgA inductive sites in intestinal immunity.
Introduction
IgA is the most abundantly produced antibody, representing 70% of antibody production (Macpherson and Uhr, 2004; Macpherson et al., 2008). The majority of IgA is secreted at mucosal surfaces, and plays a major role in immune homeostasis by protecting these surfaces from bacterial or viral attack. Accordingly, there is significant interest in how and where IgA production is initiated and how this defense mechanism can be enhanced to meet ongoing challenges. In this review we will focus on the role of isolated lymphoid follicles (ILFs) as sites initiating IgA production, highlighting the dynamic nature of ILFs IgA production and the role of environmental stimuli in this process.
Mucosal Lymphoid Tissues in the Gastrointestinal Tract as Sites for the Initiation of IgA Production
It has long been appreciated that the gastrointestinal tract contains well developed lymphoid tissues with a unique environment specialized for IgA production (Fagarasan, 2006; Macpherson, 2006). Instead of mounting a strong systemic Th1, Th2, or IgG response, a role of these lymphoid tissues is to promote homeostatic responses against luminal antigens by inducing IgA, which is secreted into the lumen. Within the mucosal lymphoid tissues of the intestinal tract, made up of Peyer’s patches (PPs), ILFs, and Colonic Patches, TGFβ1 is crucial for IgA production and expressed by several cell types promoting the majority of gut B cells to differentiate into IgA producing plasma cells (Coffman et al., 1989). Mucosal lymphoid tissue dendritic cells (DCs) express the TNF superfamily members a proliferation inducing ligand (APRIL) and B cell activation factor of the TNF family (BAFF) which promote IgA production in B cells expressing the transmembrane activator and calcium modulator and cyclophilin ligand interactor (TACI) and the B cell maturation antigen (BCMA), the receptors for APRIL and BAFF (Fink and Frøkiær, 2008; Massacand et al., 2008; Tsuji et al., 2008). T cells found near the B cells follicles in the intestinal tract express yet another TNF superfamily member, CD40L, which promotes class-switch recombination (CSR) in B cells (Fagarasan et al., 2010). In normal SPF housed mice, the continual induction of IgA producing B cells is evident by the presence of GL7+ germinal centers within PP follicles. Activation-induced cytidine deaminase (AID), necessary for both CSR and somatic hypermutation (SHM), is also present in PP B cells. Currently it is thought PPs utilize mostly T cell dependent routes for IgA production as germinal center formation in PPs is dependent on both CD40–CD40L signaling from T cells (Bergqvist et al., 2006) and the presence of T cells (Tsuji et al., 2008). However mice lacking CD40, CD40L, or T cells still have near normal levels of IgA. This IgA may arise from B-1 B cells and have lower specificity and altered functionality. Still, it is evident there are many compensatory mechanisms for IgA production in the intestine.
The best recognized of the IgA inducing lymphoid tissues are PPs, which are scattered along the anti-mesenteric border throughout the length of the small intestine (Schuurman et al., 1994). The murine colon contains a PP equivalents located in the cecum, or the cecal patch (Owen et al., 1991), and multi-follicle structures throughout the colon, or colonic patches (O’Leary and Sweeney, 1986). Depending on the strain of mouse, there are usually 7–10 PPs in the small intestine, each with multiple follicles containing naïve B-2 B cells (Husband and Gowans, 1978). PPs also contain CD4+ TCRβ+ T cells in between the follicles in T cell zones and different classes of CD11c+ DCs (Iwasaki and Kelsall, 2000). PPs sample luminal antigens through specialized antigen transporting cells known as Microfold cells, or M cells (Gebert et al., 1996). Follicular DCs (FDCs), within the B-cell follicles present the antigen in a T cell dependent manner to B cells, which undergo activation and class-switch from IgM to IgA (Garside et al., 2004).
More recently it was appreciated that the small intestine and colon contained an additional type of lymphoid tissue, the solitary intestinal lymphoid tissues (SILT; Hamada et al., 2002; Lorenz et al., 2003; Pabst et al., 2006). In contrast to PPs, which remain fully developed throughout life, SILT are a spectrum of lymphoid tissues ranging from nascent cryptopatches (CPs) to fully developed mature ILFs (mILFs; Lorenz et al., 2003; Pabst et al., 2006). CPs, aggregates of approximately 1000 cells, are composed of RORγt+ lymphoid tissue inducer (LTi) cells and lymphoid tissue organizer (LTo) cells found around the crypts of the small intestine (Kanamori et al., 1996). By secreting lymphocyte attracting chemokines, CPs are able to develop into ILFs in response to signals originating from the commensal enteric flora (Pabst et al., 2006). Small immature ILFs contain a mixture of T cells and B cells and through further recruitment of B cells develop into mILFs, which have an overlying follicle-associated epithelium (FAE) containing M cells and B cell follicles with germinal centers (Lorenz et al., 2003). Approximately 1000 SILT are scattered throughout the murine small intestine, and in most situations CPs greatly outnumber ILFs. Current evidence shows that de novo development of CPs does not continually occur throughout adulthood (Velaga et al., 2009): as needed, a CP may develop further into an ILF (Taylor et al., 2004), a process that does continue throughout adulthood. In mice aged 2 years, all classes of SILT except CPs, were increased (McDonald et al., 2011), indicating the continual development of ILFs as a site for IgA induction when needed.
Isolated lymphoid follicles were described relatively recently in humans (Moghaddami et al., 1998) and in mice (Hamada et al., 2002) as a small intestinal villi containing a single B cell follicle, but were immediately suggested to be a compensatory mechanism for humoral responses, able to support IgA responses. Similar to PPs, ILFs contain mostly CD19+ B-2 B cells that have yet to undergo CSR (Lorenz and Newberry, 2004). CD4+TCRβ+ T cells and MHCII+ CD11c+ antigen presenting cells make up approximately 25% of the ILFs (Hamada et al., 2002; Lorenz and Newberry, 2004). M cells are found on the FAE of the ILFs, indicating antigen introduction to the follicle also occurs via M cells (Lorenz et al., 2003).
The observation that mice lacking PPs could still produce and secrete IgA in response to luminal antigen led many to believe IgA CSR could occur outside of organized lymphoid tissues (Hamilton et al., 1981; Yamamoto et al., 2000). However following the identification of ILFs, these observations could be reinterpreted to support a role for ILFs in IgA production.
ILFs are a Dynamic Reservoir of IgA Inductive Sites
Though CPs and ILFs require many of the same molecules necessary for PP development, signals from the diet and the enteric flora are required for the development of ILFs, but not PPs. PPs are embryonically imprinted, developing around day 14 of gestation, while SILT is only present postnatally, appearing in the first few weeks of life (Kanamori et al., 1996; McDonald et al., 2010). The initial aggregation of LTi cells in both PPs and CPs require lymphotoxin signaling as neither PPs nor any SILT are found in LTα−/− or LTβR−/− mice (DeTogni et al., 1994; Taylor et al., 2004). However recent work has shown a requirement for the expression of the aryl hydrocarbon receptor (AHR) in the differentiation of LTi cells postnatally, but not during embryogenesis (Kiss et al., 2011; Lee et al., 2011). Knocking out AHR, expressed on RORγt+ LTi cells, unexpectedly resulted in the absence of any CPs or ILFs, while the lymph nodes and PPs were undisturbed (Lee et al., 2011). Keeping mice on diets free of AHR ligands reproduced the lack of CPs and ILFs, while mice fed a diet including indole-3-carbinol, an AHR ligand found in Brassicaceae plants (such as broccoli or cauliflower) developed normal numbers of intestinal lymphoid aggregates (Kiss et al., 2011). Thus diet initiates one of the early steps in ILF development revealing an unappreciated role for nutrition in IgA induction (Figure 1).
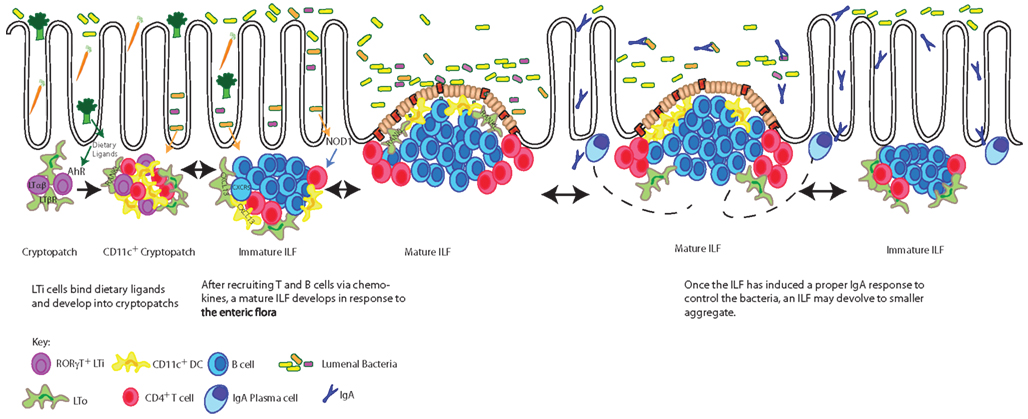
Figure 1. Dietary ligands drive the development of CPs, which expand into mature ILFs in response to bacterial stimuli. The development of CPs requires dietary signals delivered through the aryl hydrocarbon receptor (AHR) expressed by RORγt+ LTi cells. LTi cells in turn express lymphotoxin (LTα1β2) and transmit a signal to lymphotoxin beta receptor (LTβR) expressing LTo (stromal) cells inducing the production of a chemokines resulting in cryptopatch formation. In response to further lymphotoxin signals and enteric flora, chemokines, including CXCL13, recruit lymphocytes to transform CPs into immature ILFs and subsequently mature ILFs. The transition of an immature ILF to a mature ILF is dependent upon further lymphotoxin and RANKL signals driving the development of germinal centers and the development of an FAE. Mature ILFs promote naïve B cells to become IgA plasmablasts responding to luminal antigens delivered via the FAE. These plasmablasts are believed to migrate to the lamina propria by yet undescribed mechanisms to become plasma cells producing IgA specific for luminal antigens and by extension regulating the enteric flora. When challenges from the enteric flora are controlled, mature ILFs can regress to their precursor stages. The pathways presented in this figure are restricted to those discussed in this review.
After the initial development of a CP, the constituent cells secrete chemokines to recruit lymphocytes to the aggregate. As future IgA induction sites, the recruitment of B cells to the SILT is an important step in the development from a CP to an ILF. LTo cells/stromal cells and CD11c+ cells within CPs and/or ILFs express CXCL13, a B cell attractant (Gunn et al., 1998; Ansel et al., 2000; Honda et al., 2001; Cupedo et al., 2004; van de Pavert et al., 2009; McDonald et al., 2010). Knocking out the CXCL13 or its receptor CXCR5 results in normal numbers of CPs, but an absence of mILFs (Velaga et al., 2009; McDonald et al., 2010). Alternatively, SILT can be pushed from CPs to ILFs by increasing the local concentration of CXCL13 through transgenic expression by gut epithelial cells (Marchesi et al., 2009), resulting in an increase in ILFs, but not the absolute number of intestinal aggregates.
Mature ILFs could form as a result of the expansion of a small number of B cells present within the SILT and/or due to the recruitment of B cells from the systemic pool. Work with the AID−/− mice, which have an expanded ILFs compartment, suggested B cells within SILT expand concurrently with ILF development (Fagarasan et al., 2002). However, analysis of individual ILFs from wild type mice demonstrated the ILFs contained a population of polyclonal B cells which reflected the systemic B cell pool (Wang et al., 2006). Further work with naïve mice revealed diverse variable heavy chain usage in ILFs, again reflecting the systemic population of B cells (McDonald et al., 2011). While evidence exists to support both pathways, recruitment of systemic polyclonal B cell population would give ILFs a greater potential to respond to a wide range of antigens.
Along with changes to the cellular components of the ILFs, changes outside the lymphoid structures must occur during the development of the ILFs. The eventual mILF will have moved from the crypt into a villus, displacing the original lamina propria cells. The epithelium overlying the ILF must change from the villus epithelium to FAE, which lacks goblet cells and contains M cells (Lorenz and Newberry, 2004). While this process is not well understood, the cytokine receptor activator of NF-κB ligand (RANKL) appears to be a strong candidate promoting this transition. RANKL is a TNF super family cytokine that is expressed to varying degrees in all GALT (Taylor et al., 2007). RANKL can be found on the stromal cells throughout CPs, but in PPs and ILFs, RANKL staining is confined to the subepithelial dome, the area right beneath the FAE (Taylor et al., 2007). Though initial development of CPs is not dependent on RANKL, there is a significant decrease of CPs in RANKL−/− mice (Knoop et al., 2011). Furthermore RANKL−/− mice fail to develop ILFs due to a lack of CXCL13 expression within CPs (Knoop et al., 2011). As the ILF develops, RANKL expression becomes restricted to the subepithelial dome inducing the development of M cells and FAE from what was once villus epithelium (Knoop et al., 2009) demonstrating multiple roles for RANKL in the development of GALT.
The transition of CP to mILFs can be driven by changes in the enteric flora. While CPs, immature ILFs, and PPs are found in germfree mice (Kanamori et al., 1996), mILFs are rare and increase in response to conventionalization with normal flora (Lorenz et al., 2003). One mechanism appears to be bacterial stimulation of NOD1, an intracellular pattern recognition receptor found in the epithelium, which has been shown to lead to ILF development (Bouskra et al., 2008). Thus two key signals for ILF development are enteric flora and diet, neither of which are required for the development of PPs. It seems likely the purpose of SILT is to adapt to the ever-changing enteric flora and diet of an individual, producing IgA in response to new environmental cues (Figure 1).
Evidence of the adaptability of SILT can be seen in mouse models where PP development is blocked by disrupting lymphotoxin signaling during the embryonic stage; development of PPs is completely blocked and cannot be rescued during adulthood (Rennert et al., 1996; Lorenz et al., 2003). In this model, ILF development still occurs unperturbed throughout adulthood. Furthermore ILF development is increased 10-fold, expanding in response to the lack of PPs (Lorenz et al., 2003). The plasticity of ILF development, seen in the ability of ILFs to expand as needed, aptly compensates for PPs, which are fixed in number and position.
ILF’s Role in IgA Production
There is little doubt that ILFs are able to induce IgA responses. Early studies on ILFs showed the B cells within the follicles are mostly B220+ CD19+CD23+IgMlowIgDhigh cells and relatively few are IgA+ (Hamada et al., 2002), representing a population of B cells poised to undergo activation, CSR, and SHM. Most work showing ILFs contribute to IgA production was completed in mouse models, which lack PPs, yet are still able to develop ILFs. Indeed, LTα−/− mice, which lack any organized lymphoid structures, including PPs and ILFs, make only a small amount of IgA, presumably by B-1 B cells (Thurnheer et al., 2003). LTα−/− mice reconstituted with C57Bl/6 bone marrow develop ILFs, which restore IgA production in the intestine (Lorenz and Newberry, 2004; Hashizume et al., 2007). Similar results were seen with RORγt−/− mice, which lack PPs and ILFs: when reconstituted with RORγt+ bone marrow ILFs develop and IgA production becomes restored (Tsuji et al., 2008).
LTβR blockade by the injection LTβR-Ig fusion protein on or before day 17 of gestation prevents PP development (Rennert et al., 1996) and has been used as a model to infer the role of MLNs in IgA production (Yamamoto et al., 2000). Following in utero LTβR blockade, these mice produce normal levels of IgA and make specific IgA against OVA and cholera toxin (Yamamoto et al., 2000). While these studies attributed this IgA production to the MLN, subsequent work revealed that gestational LTβR-Ig treatment resulted in enhanced ILFs development (Lorenz et al., 2003), and in retrospect ILFs could also contribute to the IgA production in these mice.
To be a useful compensatory mechanism for IgA, an ILF should induce high-affinity IgA specific against antigen epitopes. The specificity of IgA produced in ILFs was called into question when specific IgA was not seen in ILFs against tetanus toxoid following immunization (Hashizume et al., 2008). However other studies provide evidence for ILF generated antigen-specific IgA in response to multiple antigens. Upon oral immunization with the T cell dependent antigen sheep red blood cells, ILFs produced antigen-specific IgA at levels equivalent to PPs (Lorenz and Newberry, 2004). Moreover mice possessing mILFs, but lacking PPs and other secondary lymphoid tissues were found to have antigen-specific IgA in their feces following infection with Salmonella typhimurium (Lorenz and Newberry, 2004). Furthermore, analysis of the B cell repertoire in ILFs demonstrated ILFs contain a pool of B cells similar to the repertoire seen in PPs and spleen (McDonald et al., 2011). Collectively, the literature suggests ILFs have the potential to respond to a wide array of antigens and mount specific responses against enteric bacteria.
Some findings suggest that while PPs are the major source of T cell dependent responses, ILFs contribute a compensatory mechanism by housing T cell independent responses (Tsuji et al., 2008). A T cell independent mechanism for IgA production has been described in TCRβ−/−δ−/−mice, which produce IgA specific for commensal bacteria, and can respond to changes in the gut flora (Macpherson et al., 2000). The T cell independent pathway for IgA has been shown to be dependent on APRIL and suggested to be quite important for the production of IgA specific for commensal bacteria (Castigli et al., 2004). Further work with the TCRβ−/−δ−/− mice suggested ILFs, but not PP, can house T cell independent IgA responses (Tsuji et al., 2008). PPs from these mice formed defective GC and failed to activate AID, while ILFs contained AID+ B cells undergoing CSR to IgA (Tsuji et al., 2008).
During T cell dependent IgA production, B cells become activated by the presentation of antigen from FDCs and by CD40 signals from T cells (MacLennan, 1994). This leads to upregulation of AID, which is responsible for both SHM and CSR (Longerich et al., 2006). TGFβ1, expressed by several cell types in the gut, promotes CSR to IgA (Fagarasan et al., 2010). In the absence of T cells, DCs from ILFs, but not PPs appear to be sufficient for IgA production from B cells (Fagarasan et al., 2010). The DCs are thought to use APRIL, BAFF, and TGFβ1 to promote IgA production. Interestingly, it was found RORγt+ LTi cells are indispensible for the T cell independent production of IgA in this model (Tsuji et al., 2008). The combination of LTi cells, DCs, and naïve B cells are rarely found in the LP outside of ILFs, suggesting this model of T cell independent IgA production can only be induced in organized lymphoid tissue.
Other mouse models indicate that in addition to ILFs, PPs can support T cell independent means of IgA induction. CD40−/− mice were shown to have only rare IgA CSR events in small intestine PPs, though AID was activated (Bergqvist et al., 2006). CD40−/− mice still maintained high levels of IgA, and it was later shown IgA CSR can occur in PPs, ILFs, and colonic patches (Bergqvist et al., 2010). This work concluded though CSR was found in some ILFs, the majority of IgA CSR in the absence of T cells occurs in the PPs. The role for T cells seems to be more important for SHM as the IgA in CD40−/− mice showed few mutations in the variable regions (Bergqvist et al., 2010).
Technology has moved past merely looking for the presence or absence of specific IgA using ELISAs. Now with the genetic analysis options available, the ability to track specific clones of B cells and closely watch the variation in the IgA repertoire during infections can better answer how the majority of IgA is produced in the gut (Spencer et al., 2009). Indeed such genetic analysis of the clonal relationship between B cells and IgA Plasma cells is already being incorporated into reports (Bergqvist et al., 2010) better showing when SHM and CSR has occurred. Not only will this help clarify the field, but also help focus mucosal vaccine work in the attempts to target better IgA responses for protection against gut pathogens.
ILF Hyperplasia as a Sign of Immune Dysfunction
As described in the above sections, multiple studies support that ILFs develop in response to changes or imbalances in the luminal microbial community and function to promote IgA production. Furthermore IgA produced at mucosal surfaces can in turn alter or control the luminal microbial community returning it to homeostasis (Cerutti and Rescigno, 2008). Together these observations suggest that ineffective IgA production would be unable to control the luminal microbiota and result in expansion of ILFs, and thus prolonged ILF hyperplasia could be viewed as a sign of IgA dysfunction.
Though ILF hyperplasia has been known to occur in humans for some time (Webster, 1973), the first mouse model that included ILF hyperplasia was the AID−/− mice (Fagarasan et al., 2002). Since AID is required for both CSR and SHM, these mice can only produce low-affinity IgM. In the absence of high-affinity IgA, the enteric flora quickly becomes altered, with a 100-fold expansion in anaerobic bacteria (Fagarasan et al., 2002). Anaerobes are notable for their opportunistic tendencies to quickly expand without proper IgA control (Suzuki et al., 2004; Ohashi et al., 2010). Intriguingly, AID−/− mice develop massive ILF hyperplasia in response to the increases in the enteric flora (Fagarasan et al., 2002). Similarly, mice with a mutated form of AID that allows for CSR but not SHM still have expanded flora due to low-affinity IgA that is unable to control bacterial growth (Wei et al., 2011) and with the expanded flora, these mice also develop ILF hyperplasia.
The ability for ILFs to rescue an inferior IgA response was seen in recent studies investigating chemokine receptor 10 (CCR10) expression by plasma cells (Hu et al., 2011). IgA+ plasma cells are unable to migrate to the intestine in CCR10-KO/EGFP-knock-in mice, yet the mice maintain normal levels of fecal IgA and have no significant increase in luminal flora due to an increase in IgA producing plasma cells in ILFs. While evidence to support that a large population of IgA producing plasma cells resides in the ILFs in the setting of CCR10 sufficiency is lacking and the migration and fate of ILF generated IgA+ plasmablasts is largely unknown, it is clear in this model that ILF development is increased in response to the commensals, and that IgA is induced within ILFs to control the enteric flora. Furthermore, the number of ILFs decreased upon antibiotic treatment (Hu et al., 2011), illustrating how ILF may devolve once they are no longer needed.
Immune dysfunction does not have to be caused by a genetic mutation; immunosenescence in aged individuals is seen as a dysfunction in protective immunity and can affect gastrointestinal immunity as well as systemic immunity (Schmucker and Daniels, 1986; Schmucker et al., 1996). Mice aged 2 years show many signs of immune dysfunction in the gut (McDonald et al., 2011). There is a threefold increase in all classes of ILFs and these ILFs contain an aberrant cellular population with decreased numbers of B cells and increased numbers of T cells, specifically CD4+ CD8αα+ T cells (McDonald et al., 2011). Along with the greatly elevated amounts of IgA, purportedly from the ILFs, these signs all point to immune dysfunction. Since aged rats have been shown to have increased numbers of anaerobic bacteria (Maczulak et al., 1989). A similar expansion could occur in aged mice when immune dysfunction prevents proper control of the bacteria and drive the development of the ILFs in aged mice.
Clinically an increase of ILFs in the small intestine has long been seen as a symptom of immune dysfunction (Bastlein et al., 1988). In children, a common endoscopic finding is the large number of tiny lymphoid nodules located in the terminal ileum and colon referred to as lymphonodular hyperplasia (LNH), or nodular lymphoid hyperplasia (Laufer and deSa, 1978). It was first described as an age-related finding in children of minimal significance, but further work noted a close association between LNH and the development of food allergies. It was shown that children with LNH were two to three times more likely to have non-IgE mediated allergies to food such as milk or cereal (Kokkonen et al., 1999) including high levels of IgG and IgA against milk proteins (Kokkonen et al., 2002), increased numbers of γδ T cells associated with the lymphoid follicles (Kokkonen et al., 2000), and increased CD4+ T cells with decreased Th1 cytokines (Bellanti et al., 2003). LNH was not found to be associated with Crohn’s disease or NOD2/CARD15 mutations raising speculation that LNH is not related to inflammatory disease, but may be a response to the microbial environment (Shaoul et al., 2006) as children with LNH showed a significant increase in number of bacteria adherent to the intestinal mucosa layer (Conte et al., 2006). Collectively, LNH appears not to be the cause of the food allergies, but rather a symptom of an expanded flora during immune dysfunction.
Lymphoid hyperplasia can also be found in adults, though more rarely than in children (Misra et al., 1998; Carroccio et al., 2009). It is also found alongside food allergies in adults (Krauss et al., 2010) but is generally associated with genetic immune disorders such as common variable immune deficiency (CVID; Webster, 1973; Scharenberg et al., 1993). CVID patients have reduced ability to undergo SHM and as a result have low concentrations of IgG and IgA. LHN develops in about 20% of CVID (Bastlein et al., 1988) and those patients have increased intestinal IgM compared to CVID patients without LHN, consistent with ILFs contributing to the antibody response in these individuals (Webster et al., 1977). Treatment is rarely recommended to stop the development of the follicles, differentiating these lymphoid follicles from more problematic follicles that develop in other intestinal inflammatory diseases. The LHN associated with CVID likely develops in a manner similar to the ILF hyperplasia seen in the AID−/− mice; the lymphoid follicles develop not because of the CVID but due to continual signals from the expanded microflora able to grow uncontrollably without the reins of IgA. Until the flora is controlled, usually by means of antibiotic treatment, the follicles will continue to develop in an attempt to fight the perceived threat to the intestines.
Conclusion
In this review we have illustrated how ILFs develop in response to diet and commensals and how ILFs can compensate for inadequate IgA responses. Though their relative contribution to IgA production has yet to be enumerated compared to PPs, it is clear that induction of IgA responses is a function of an ILF. Further work still needs to examine the specific antigenic targets of the IgA that comes from ILFs, but one would expect most of the targets come from the enteric flora since the development of ILFs is highly responsive to the billions to bacteria that reside in the gut lumen.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ansel, K. M., Ngo, V. N., Hyman, P. L., Luther, S. A., Forster, R., Sedgwick, J. D., Browning, J. L., Lipp, M., and Cyster, J. G. (2000). A chemokine-driven positive feedback loop organizes lymphoid follicles. Nature 406, 309–314.
Bastlein, C., Burlefinger, R., Holzberg, E., Voeth, C., Garbrecht, M., and Ottenjann, R. (1988). Common variable immunodeficiency syndrome and nodular lymphoid hyperplasia in the small intestine. Endoscopy 20, 272–275.
Bellanti, J. A., Zeligs, B. J., Malka-Rais, J., and Sabra, A. (2003). Abnormalities of Th1 function in non-IgE food allergy, celiac disease, and ileal lymphonodular hyperplasia: a new relationship? Ann. Allergy Asthma Immunol. 90, 84–89.
Bergqvist, P., Gardby, E., Stensson, A., Bemark, M., and Lycke, N. Y. (2006). Gut IgA class switch recombination in the absence of CD40 does not occur in the lamina propria and is independent of germinal centers. J. Immunol. 177, 7772–7783.
Bergqvist, P., Stensson, A., Lycke, N. Y., and Bemark, M. (2010). T cell-independent IgA class switch recombination is restricted to the GALT and occurs prior to manifest germinal center formation. J. Immunol. 184, 3545–3553.
Bouskra, D., Brezillon, C., Berard, M., Werts, C., Varona, R., Boneca, I. G., and Eberl, G. (2008). Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature 456, 507–510.
Carroccio, A., Iacono, G., Di Prima, L., Ravelli, A., Pirrone, G., Cefalù, A. B., Florena, A. M., Rini, G. B., and Di Fede, G. (2009). Food hypersensitivity as a cause of rectal bleeding in adults. Clin. Gastroenterol. Hepatol. 7, 120–122.
Castigli, E., Scott, S., Dedeoglu, F., Bryce, P., Jabara, H., Bhan, A. K., Mizoguchi, E., and Geha, R. S. (2004). Impaired IgA class switching in APRIL-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 101, 3903–3908.
Cerutti, A., and Rescigno, M. (2008). The biology of intestinal immunoglobulin A responses. Immunity 28, 740–750.
Coffman, R. L., Lebman, D. A., and Shrader, B. (1989). Transforming growth factor beta specifically enhances IgA production by lipopolysaccharide-stimulated murine B lymphocytes. J. Exp. Med. 170, 1039–1044.
Conte, M. P., Schippa, S., Zamboni, I., Penta, M., Chiarini, F., Seganti, L., Osborn, J., Falconieri, P., Borrelli, O., and Cucchiara, S. (2006). Gut-associated bacterial microbiota in paediatric patients with inflammatory bowel disease. Gut 55, 1760–1767.
Cupedo, T., Vondenhoff, M. F., Heeregrave, E. J., De Weerd, A. E., Jansen, W., Jackson, D. G., Kraal, G., and Mebius, R. E. (2004). Presumptive lymph node organizers are differentially represented in developing mesenteric and peripheral nodes. J. Immunol. 173, 2968–2975.
DeTogni, P., Goellner, J., Ruddle, N. H., Streeter, P. R., Fick, A., Mariathasan, S., Smith, S. C., Carlson, R., Shornick, L. P., Strauss-Schoenberger, J., Russell, J. H., Karr, R., and Chaplin, D. D. (1994). Abnormal development of peripheral lymphoid organs in mice deficient in lymphotoxin. Science 264, 703–707.
Fagarasan, S. (2006). Intestinal IgA synthesis: a primitive form of adaptive immunity that regulates microbial communities in the gut. Curr. Top. Microbiol. Immunol. 308, 137–153.
Fagarasan, S., Kawamoto, S., Kanagawa, O., and Suzuki, K. (2010). Adaptive immune regulation in the gut: T cell-dependent and T cell-independent IgA synthesis. Annu. Rev. Immunol. 28, 243–273.
Fagarasan, S., Muramatsu, M., Suzuki, K., Nagaoka, H., Hiai, H., and Honjo, T. (2002). Critical roles of activation-induced cytidine deaminase in the homeostasis of gut flora. Science 298, 1424–1427.
Fink, L. N., and Frøkiær, H. (2008). Dendritic cells from Peyer’s patches and mesenteric lymph nodes differ from spleen dendritic cells in their response to commensal gut bacteria. Scand. J. Immunol. 68, 270–279.
Garside, P., Millington, O., and Smith, K. M. (2004). The anatomy of mucosal immune responses. Ann. N. Y. Acad. Sci. 1029, 9–15.
Gebert, A., Rothkotter, H. J., and Pabst, R. (1996). M cells in Peyer’s patches of the intestine. Int. Rev. Cytol. 167, 91–159.
Gunn, M. D., Ngo, V. N., Ansel, K. M., Ekland, E. H., Cyster, J. G., and Williams, L. T. (1998). A B-cell-homing chemokine made in lymphoid follicles activates Burkitt’s lymphoma receptor-1. Nature 391, 799–803.
Hamada, H., Hiroi, T., Nishiyama, Y., Takahashi, H., Masunaga, Y., Hachimura, S., Kaminogawa, S., Takahashi-Iwanaga, H., Iwanaga, T., Kiyono, H., Yamamoto, H., and Ishikawa, H. (2002). Identification of multiple isolated lymphoid follicles on the antimesenteric wall of the mouse small intestine. J. Immunol. 168, 57–64.
Hamilton, S. R., Keren, D. F., Yardley, J. H., and Brown, G. (1981). No impairment of local intestinal immune response to keyhole limpet haemocyanin in the absence of Peyer’s patches. Immunology 42, 431–435.
Hashizume, T., Momoi, F., Kurita-Ochiai, T., Kaminogawa, S., Hosono, A., Kataoka, K., Shinozaki-Kuwahara, N., Kweon, M. N., and Yamamoto, M. (2007). Isolated lymphoid follicles are not IgA inductive sites for recombinant Salmonella. Biochem. Biophys. Res. Commun. 360, 388–393.
Hashizume, T., Togawa, A., Nochi, T., Igarashi, O., Kweon, M. N., Kiyono, H., and Yamamoto, M. (2008). Peyer’s patches are required for intestinal immunoglobulin A responses to Salmonella. Infect. Immun. 76, 927–934.
Honda, K., Nakano, H., Yoshida, H., Nishikawa, S., Rennert, P. D., Ikuta, K., Tamechika, M., Yamaguchi, K., Fukumoto, T., Chiba, T., and Nishikawa, S. I. (2001). Molecular basis for hematopoietic/mesenchymal interaction during initiation of peyer's patch organogenesis. J. Exp. Med. 193, 621–630.
Hu, S., Yang, K., Yang, J., Li, M., and Xiong, N. (2011). Critical roles of chemokine receptor CCR10 in regulating memory IgA responses in intestines. Proc. Natl. Acad. Sci. U.S.A. 108, E1035–E1044.
Husband, A. J., and Gowans, J. L. (1978). The origin and antigen-dependent distribution of IgA-containing cells in the intestine. J. Exp. Med. 148, 1146–1160.
Iwasaki, A., and Kelsall, B. L. (2000). Localization of distinct Peyer’s patch dendritic cell subsets and their recruitment by chemokines macrophage inflammatory protein (MIP)-3alpha, MIP-3beta, and secondary lymphoid organ chemokine. J. Exp. Med. 191, 1381–1394.
Kanamori, Y., Ishimaru, K., Nanno, M., Maki, K., Ikuta, K., Nariuchi, H., and Ishikawa, H. (1996). Identification of novel lymphoid tissues in murine intestinal mucosa where clusters of c-kit+ IL-7R+ Thy1+ lympho-hemopoietic progenitors develop. J. Exp. Med. 184, 1449–1459.
Kiss, E. A., Vonarbourg, C., Kopfmann, S., Hobeika, E., Finke, D., Esser, C., and Diefenbach, A. (2011). Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science 334, 1561–1565.
Knoop, K. A., Butler, B. R., Kumar, N., Newberry, R. D., and Williams, I. R. (2011). Distinct developmental requirements for isolated lymphoid follicle formation in the small and large intestine RANKL is essential only in the small intestine. Am. J. Pathol. 179, 1861–1871.
Knoop, K. A., Kumar, N., Butler, B. R., Sakthivel, S. K., Taylor, R. T., Nochi, T., Akiba, H., Yagita, H., Kiyono, H., and Williams, I. R. (2009). RANKL is necessary and sufficient to initiate development of antigen-sampling M cells in the intestinal epithelium. J. Immunol. 183, 5738–5747.
Kokkonen, J., Holm, K., Karttunen, T. J., and Maki, M. (2000). Children with untreated food allergy express a relative increment in the density of duodenal gammadelta+ T cells. Scand. J. Gastroenterol. 35, 1137–1142.
Kokkonen, J., Karttunen, T. J., and Niinimaki, A. (1999). Lymphonodular hyperplasia as a sign of food allergy in children. J. Pediatr. Gastroenterol. Nutr. 29, 57–62.
Kokkonen, J., Tikkanen, S., Karttunen, T. J., and Savilahti, E. (2002). A similar high level of immunoglobulin A and immunoglobulin G class milk antibodies and increment of local lymphoid tissue on the duodenal mucosa in subjects with cow’s milk allergy and recurrent abdominal pains. Pediatr. Allergy Immunol. 13, 129–136.
Krauss, E., Konturek, P., Maiss, J., Kressel, J., Schulz, U., Hahn, E. G., Neurath, M. F., and Raithel, M. (2010). Clinical significance of lymphoid hyperplasia of the lower gastrointestinal tract. Endoscopy 42, 334–337.
Laufer, I., and deSa, D. (1978). Lymphoid follicular pattern: a normal feature of the pediatric colon. AJR Am. J. Roentgenol. 130, 51–55.
Lee, J. S., Cella, M., Mcdonald, K. G., Garlanda, C., Kennedy, G. D., Nukaya, M., Mantovani, A., Kopan, R., Bradfield, C. A., Newberry, R. D., and Colonna, M. (2011). AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat. Immunol. 13, 144–151.
Longerich, S., Basu, U., Alt, F., and Storb, U. (2006). AID in somatic hypermutation and class switch recombination. Curr. Opin. Immunol. 18, 164–174.
Lorenz, R. G., Chaplin, D. D., Mcdonald, K. G., Mcdonough, J. S., and Newberry, R. D. (2003). Isolated lymphoid follicle formation is inducible and dependent upon lymphotoxin-sufficient B lymphocytes, lymphotoxin β receptor, and TNF receptor I function. J. Immunol. 170, 5475–5482.
Lorenz, R. G., and Newberry, R. D. (2004). Isolated lymphoid follicles can function as sites for induction of mucosal immune responses. Ann. N. Y. Acad. Sci. 1029, 44–57.
Macpherson, A. J. (2006). IgA adaptation to the presence of commensal bacteria in the intestine. Curr. Top. Microbiol. Immunol. 308, 117–136.
Macpherson, A. J., Gatto, D., Sainsbury, E., Harriman, G. R., Hengartner, H., and Zinkernagel, R. M. (2000). A primitive T cell-independent mechanism of intestinal mucosal IgA responses to commensal bacteria. Science 288, 2222–2226.
Macpherson, A. J., Mccoy, K. D., Johansen, F. E., and Brandtzaeg, P. (2008). The immune geography of IgA induction and function. Mucosal Immunol. 1, 11–22.
Macpherson, A. J., and Uhr, T. (2004). Induction of protective IgA by intestinal dendritic cells carrying commensal bacteria. Science 303, 1662–1665.
Maczulak, A. E., Wolin, M. J., and Miller, T. L. (1989). Increase in colonic methanogens and total anaerobes in aging rats. Appl. Environ. Microbiol. 55, 2468–2473.
Marchesi, F., Martin, A. P., Thirunarayanan, N., Devany, E., Mayer, L., Grisotto, M. G., Furtado, G. C., and Lira, S. A. (2009). CXCL13 expression in the gut promotes accumulation of IL-22-producing lymphoid tissue-inducer cells, and formation of isolated lymphoid follicles. Mucosal Immunol. 2, 486–494.
Massacand, J. C., Kaiser, P., Ernst, B., Tardivel, A., Burki, K., Schneider, P., and Harris, N. L. (2008). Intestinal bacteria condition dendritic cells to promote IgA production. PLoS ONE 3, e2588. doi:10.1371/journal.pone.0002588
McDonald, K. G., Leach, M. R., Huang, C., Wang, C., and Newberry, R. D. (2011). Aging impacts isolated lymphoid follicle development and function. Immun. Ageing 8, 1.
McDonald, K. G., Mcdonough, J. S., Dieckgraefe, B. K., and Newberry, R. D. (2010). Dendritic cells produce CXCL13 and participate in the development of murine small intestine lymphoid tissues. Am. J. Pathol. 176, 2367–2377.
Misra, S. P., Misra, V., Dwivedi, M., and Singh, P. A. (1998). CASE REPORT: Helicobacter pylori-induced lymphonodular hyperplasia: a new cause of gastric outlet obstruction. J. Gastroenterol. Hepatol. 13, 1191–1194.
Moghaddami, M., Cummins, A., and Mayrhofer, G. (1998). Lymphocyte-filled villi: comparison with other lymphoid aggregations in the mucosa of the human small intestine. Gastroenterology 115, 1414–1425.
Ohashi, Y., Hiraguchi, M., Sunaba, C., Tanaka, C., Fujisawa, T., and Ushida, K. (2010). Colonization of segmented filamentous bacteria and its interaction with the luminal IgA level in conventional mice. Anaerobe 16, 543–546.
O’Leary, A. D., and Sweeney, E. C. (1986). Lymphoglandular complexes of the colon: structure and distribution. Histopathology 10, 267–283.
Owen, R. L., Piazza, A. J., and Ermak, T. H. (1991). Ultrastructural and cytoarchitectural features of lymphoreticular organs in the colon and rectum of adult BALB/c mice. Am. J. Anat. 190, 10–18.
Pabst, O., Herbrand, H., Friedrichsen, M., Velaga, S., Dorsch, M., Berhardt, G., Worbs, T., Macpherson, A. J., and Forster, R. (2006). Adaptation of solitary intestinal lymphoid tissue in response to microbiota and chemokine receptor CCR7 signaling. J. Immunol. 177, 6824–6832.
Rennert, P. D., Browning, J. L., Mebius, R., Mackay, F., and Hochman, P. S. (1996). Surface lymphotoxin α/β complex is required for the development of peripheral lymphoid organs. J. Exp. Med. 184, 1999–2006.
Scharenberg, A. M., Hannibal, M. C., Torgerson, T., Ochs, H.D., and Rawlings, D. J. (1993). “Common variable immune deficiency overview,” in GeneReviews™ [Internet], eds R. A. Pagon, T. D. Bird, C. R. Dolan, K. Stephens, and N. P. Adam (Seattle: University of Washington).
Schmucker, D. L., and Daniels, C. K. (1986). Aging, gastrointestinal infections, and mucosal immunity. J. Am. Geriatr. Soc. 34, 377–384.
Schmucker, D. L., Heyworth, M. F., Owen, R. L., and Daniels, C. K. (1996). Impact of aging on gastrointestinal mucosal immunity. Dig. Dis. Sci. 41, 1183–1193.
Schuurman, H. J., Kuper, C. F., and Vos, J. G. (1994). Histopathology of the immune system as a tool to assess immunotoxicity. Toxicology 86, 187–212.
Shaoul, R., Eliakim, R., Tamir, A., and Karban, A. (2006). Ileal lymphonodular hyperplasia is not associated with NOD2/CARD15 mutations. J. Pediatr. Gastroenterol. Nutr. 43, 30–34.
Spencer, J., Barone, F., and Dunn-Walters, D. (2009). Generation of Immunoglobulin diversity in human gut-associated lymphoid tissue. Semin. Immunol. 21, 139–146.
Suzuki, K., Meek, B., Doi, Y., Muramatsu, M., Chiba, T., Honjo, T., and Fagarasan, S. (2004). Aberrant expansion of segmented filamentous bacteria in IgA-deficient gut. Proc. Natl. Acad. Sci. U.S.A. 101, 1981–1986.
Taylor, R. T., Lugering, A., Newell, K. A., and Williams, I. R. (2004). Intestinal cryptopatch formation in mice requires lymphotoxin α and the lymphotoxin β receptor. J. Immunol. 173, 7183–7189.
Taylor, R. T., Patel, S. R., Lin, E., Butler, B. R., Lake, J. G., Newberry, R. D., and Williams, I. R. (2007). Lymphotoxin-independent expression of TNF-related activation-induced cytokine by stromal cells in cryptopatches, isolated lymphoid follicles, and Peyer’s patches. J. Immunol. 178, 5659–5667.
Thurnheer, M. C., Zuercher, A. W., Cebra, J. J., and Bos, N. A. (2003). B1 cells contribute to serum IgM, but not to intestinal IgA, production in gnotobiotic Ig allotype chimeric mice. J. Immunol. 170, 4564–4571.
Tsuji, M., Suzuki, K., Kitamura, H., Maruya, M., Kinoshita, K., Ivanov, Ii., Itoh, K., Littman, D. R., and Fagarasan, S. (2008). Requirement for lymphoid tissue-inducer cells in isolated follicle formation and T cell-independent immunoglobulin A generation in the gut. Immunity 29, 261–271.
van de Pavert, S. A., Olivier, B. J., Goverse, G., Vondenhoff, M. F., Greuter, M., Beke, P., Kusser, K., Höpken, U. E., Lipp, M., Niederreither, K., Blomhoff, R., Sitnik, K., Agace, W. W., Randall, T. D., De Jonge, W. J., and Mebius, R. E. (2009). Chemokine CXCL13 is essential for lymph node initiation and is induced by retinoic acid and neuronal stimulation. Nat. Immunol. 10, 1193–1199.
Velaga, S., Herbrand, H., Friedrichsen, M., Jiong, T., Dorsch, M., Hoffmann, M. W., Forster, R., and Pabst, O. (2009). Chemokine receptor CXCR5 supports solitary intestinal lymphoid tissue formation, B cell homing, and induction of intestinal IgA responses. J. Immunol. 182, 2610–2619.
Wang, C., Mcdonald, K. G., Mcdonough, J. S., and Newberry, R. D. (2006). Murine isolated lymphoid follicles contain follicular B lymphocytes with a mucosal phenotype. Am. J. Physiol. Gastrointest. Liver Physiol. 291, G595–G604.
Webster, A. D. (1973). Nodular lymphoid hyperplasia with hypogammaglobulinaemia. Proc. R. Soc. Med. 66, 1127–1128.
Webster, A. D., Kenwright, S., Ballard, J., Shiner, M., Slavin, G., Levi, A. J., Loewi, G., and Asherson, G. L. (1977). Nodular lymphoid hyperplasia of the bowel in primary hypogammaglobulinaemia: study of in vivo and in vitro lymphocyte function. Gut 18, 364–372.
Wei, M., Shinkura, R., Doi, Y., Maruya, M., Fagarasan, S., and Honjo, T. (2011). Mice carrying a knock-in mutation of Aicda resulting in a defect in somatic hypermutation have impaired gut homeostasis and compromised mucosal defense. Nat. Immunol. 12, 264–270.
Keywords: isolated lymphoid follicles, IgA, intestine, gut-associated lymphoid tissue
Citation: Knoop KA and Newberry RD (2012) Isolated lymphoid follicles are dynamic reservoirs for the induction of intestinal IgA. Front. Immun. 3:84. doi: 10.3389/fimmu.2012.00084
Received: 27 January 2012; Paper pending published: 15 February 2012;
Accepted: 03 April 2012; Published online: 04 May 2012.
Edited by:
Nils Yngve Lycke, University of Gothenburg, SwedenReviewed by:
J. Rodrigo Mora, Harvard Medical School, USAGlen C. Ulett, Griffith University, Australia
Copyright: © 2012 Knoop and Newberry. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Rodney D. Newberry, Washington University School of Medicine, 660 South Euclid Avenue, Box 8124, St Louis, MO 63110, USA. e-mail:cm5ld2JlcnJ5QHd1c3RsLmVkdQ==