
 Jiandong Huo
Jiandong Huo
- MRC Human Immunology Unit, Nuffield Department of Clinical Medicine, John Radcliffe Hospital, University of Oxford, Headington, Oxford, UK
Introduction
The T-cell receptor (TCR) is responsible first of all for recognizing small peptides embedded in major histocompatibility complex molecules (pMHC). The information that a complex has thus formed is then transduced across the membrane via “triggering” of the TCR. This results in a net increase in the number and/or half-life of phosphorylated immunoreceptor tyrosine-based activation motifs (ITAMs) present in the CD3 subunits of the TCR (Smith-Garvin et al., 2009). Signaling via tyrosine-containing motifs phosphorylated by Src tyrosine kinases is not unique to the TCR complex; in fact, a large number of receptors present in lymphocytes likely signal in the same manner (Davis and van der Merwe, 2006). Almost 30 years after the TCR was discovered, however, the regulation of its phosphorylation remains poorly understood. An important recent development is that relatively high levels of active forms of the Src kinase Lck are present in resting T-cells (Nika et al., 2010), prompting the question: why are T cells not constitutively activated?
Drawing on recently published and unpublished work on the TCR and, in particular, the extensive literature on the regulation of Src kinases, in this Opinion article we discuss how TCR phosphorylation might be controlled in T cells. We consider how, at a global level, the homeostatic balancing of tyrosine kinase and phosphatase expression levels might keep TCR phosphorylation under tonic control, and how a previously overlooked mechanism of Src kinase activation might contribute to ultra-sensitive signaling in T cells, helping to locally counteract these global homeostatic effects. Rather than emphasizing its unconventionality, we highlight ways in which TCR behavior likely resonates with known molecular and cellular processes. As discussed previously (James et al., 2007, 2011), we assume that the “resting” TCR is monovalent. TCR triggering per se is also considered elsewhere (Davis and van der Merwe, 1996, 2006).
Global Control of TCR Phosphorylation in Resting Cells
TCR-based signaling occurs in seconds (<10 s) and is sensitive enough to detect single agonist pMHC among large numbers of non-agonist pMHC (Irvine et al., 2002; Huse et al., 2007). It was long assumed that the TCR would activate Src kinases, leading to receptor phosphorylation (Smith-Garvin et al., 2009). Indeed, the levels of TCR phosphorylation in resting cells are low (see e.g., Patel et al., 1987), consistent with most if not all of the tyrosine kinase being inactive. However, recent analyses suggest that a significant pool of Lck is constitutively active (Nika et al., 2010), raising an interesting question: how is the TCR kept largely unphosphorylated in a resting T-cell in the presence of active kinase?
The possibility that positively charged residues present in the CD3ε and CD3ζ cytoplasmic domains interact with the cell membrane offered a novel explanation: that membrane association prevents kinase access to the TCR (Xu et al., 2008). However, mutation of these positively charged residues does not lead to receptor phosphorylation and has little or no effect on T-cell development (Deford-Watts et al., 2009; Fernandes et al., 2010). Instead, mutations that “free” the cytoplasmic domains of CD3ε or CD3ζ slow ITAM phosphorylation and reduce T-cell activation (Fernandes et al., 2010; Gagnon et al., 2010; Zhang et al., 2011). One possible explanation for this is that, if they occur at all, transient interactions of the relatively long and flexible CD3 cytoplasmic domains with the cell membrane could increase the frequency of productive encounters between ITAMs and the active sites of tyrosine kinases, whose positions are largely fixed by N-terminal membrane attachment.
Another proposed mechanism protecting the TCR from random phosphorylation is based on the physical segregation of the complex from Src kinases, predicated on the existence of “lipid rafts” (He and Marguet, 2008). Recently, in situ ultra high-resolution approaches, i.e., stimulated emission depletion far-field fluorescence microscopy and fluctuation correlation spectroscopy, were used to study the “nano-scale” organization of membrane lipids in situ (Eggeling et al., 2009). This showed that domains containing sphingolipids and glycosylphosphatidylinositol-anchored proteins, i.e., lipid rafts, might be as small as <20 nm diameter and very short-lived (∼10–20 ms; Eggeling et al., 2009). Whether such structures nevertheless prevent the interaction in resting cells of, e.g., receptors and Src kinases, could be probed using Förster resonance energy transfer (RET; Figure 1A), which is highly sensitive to random and non-random interactions of proteins within membranes, at roughly this length-scale (<10 nm; James et al., 2006). For now, whether or not receptors and kinases are pre-segregated within the membrane prior to triggering remains an open question.
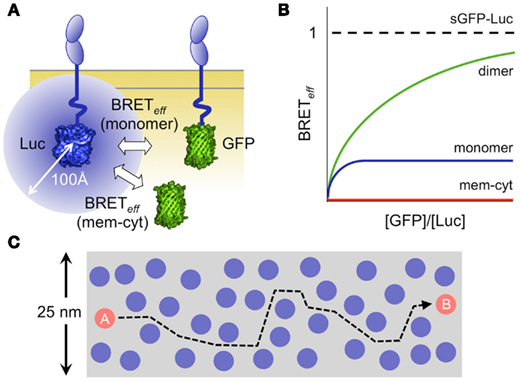
Figure 1. Analysis of protein interactions using bioluminescence resonance energy transfer (BRET). (A) Schematic showing a monomeric protein expressed at the cell surface as chimeras with luciferase (Luc) and a green fluorescent protein (GFP) acting as donors and acceptors, respectively. The blue circle represents the 100 Å sphere within which random or stable co-association of Luc and GFP allows energy transfer. (B) Graphical representation of the relationship of energy transfer efficiency (BRETeff) to the acceptor/donor ratio for dimeric (green) and monomeric (blue) protein pairs expressed at the membrane, versus that measured for a pair of proteins one of which is expressed at the membrane (mem) and the other in the cytoplasm (cyt; James et al., 2006, 2011). In practice, the transfer efficiency is normalized against that measured for a soluble GFP/Luc fusion protein (sGFP–Luc) expressed in the cytoplasm. (C) The efficiency of energy transfer arising via random interactions is explained by the high density of membrane surface proteins and by their high mobility. Protein density is illustrated to scale, assuming that there are 20,000 molecules/μm2, and that each protein is 4 nm in diameter (Grasberger et al., 1986). It takes 0.2–0.3 s for a protein to move from position A to position B, based on measurements of the TCR diffusion rate (James et al., 2007). Since many of the proteins are likely diffusing at comparable rates, numerous random interactions seem unavoidable. Some estimates for the expression levels of cell surface proteins are substantially higher (Quinn et al., 1984).
A striking outcome of the few RET experiments done on leukocyte surface proteins, however, is the extent to which non-oligomeric proteins interact randomly within the membrane (James et al., 2006). The RET efficiency for non-associated proteins interacting via random collisions is only four- to fivefold lower than that for covalent dimers (Figure 1B; James et al., 2006, 2011). The scale of the effect of membrane association is illustrated by the observation that the efficiency of energy transfer between membrane and cytoplasmic proteins is virtually zero (James et al., 2011), and explained by both the density at which proteins are packed in the membrane (Grasberger et al., 1986), and the rates of their diffusion (James et al., 2007; Figure 1C). This suggests that random contacts of the TCR with receptor-type protein tyrosine phosphatases (PTPs) might explain the low levels of TCR phosphorylation in resting T-cells. Further supporting this idea, receptor-type PTPs such as CD45 account for ∼10% of the cell surface protein content in a T-cell (Williams and Barclay, 1986), and have broad specificity (Barr et al., 2009) and very high catalytic activity (Fischer et al., 1992).
Such effects might not be restricted to antigen receptors, or lymphocytes. CD45 directly dephosphorylates and inactivates JAK-family kinases, and inhibits JAK/STAT dependent signaling by interleukin receptors in response to IL-3, IL-4, erythropoietin, and IFNα (Irie-Sasaki et al., 2001; Yamada et al., 2002). Like the interleukin receptors, the epidermal growth factor receptor (EGFR) was also assumed to rely only on dimerization-induced kinase activation (Ullrich and Schlessinger, 1990). Recent work, however, suggests that unliganded EGFR monomers and dimers exist in equilibrium with the receptor cycling between phosphorylated and unphosphorylated states (Chung et al., 2010; Kleiman et al., 2011). EGFR phosphorylation thus seems to be under tonic control by membrane PTPs, with ligand binding perturbing the steady state via the induction of kinase activity and/or phosphatase inhibition (Reynolds et al., 2003). This mirrors the control of information transfer by phosphorylation–dephosphorylation cycles established for cytosolic proteins, such as ERK, Akt, Jnk, p38, and cyclins (Domingo-Sananes et al., 2011).
To preserve responsiveness in such a setting, the relative levels of kinase and phosphatase activity in resting cells would need to be tightly controlled. We examined whether TCR phosphorylation is controlled as part of a global homeostatic system involving the TCR, Lck, and CD45. Such a system would be highly sensitive to changes in the levels of expression of each individual component, and should respond with compensatory changes that return the status quo. We found that shRNAs that acutely down-regulate CD45 in Jurkat T-cells rapidly induce strong, Src kinase-dependent TCR phosphorylation and T-cell activation. Soon after, however, the cells establish a new steady state in which a “resting” phenotype is restored (R. A. Fernandes et al., unpublished observations). Importantly, in the new state, the cells express less TCR and Lck, directing the resting equilibrium “back” toward low levels of TCR phosphorylation. Interestingly, the new “ground state” was reached over several days, suggesting that a form of feedback control is operating that might be transcription dependent. We speculate that such effects may have contributed to some of the phenotypes observed in CD45 knockdown cells, and masked the role of CD45 in TCR triggering (Hermiston et al., 2009).
Local Regulation of Lck Activity
The key Src kinase involved in TCR triggering is Lck (Smith-Garvin et al., 2009). The present view of Lck regulation is based on biochemical analysis and crystal structures of several Src family kinases, including Lck (Boggon and Eck, 2004). Phosphorylation of the C-terminal Tyr505 residue of Lck by Csk is believed to inhibit the kinase whereas trans auto-phosphorylation of activation-loop Tyr394 promotes its activity (Figure 2A). CD45 dephosphorylates both Tyr sites, complicating the interpretation of its role in receptor signaling. Crystal structures of Src and Hck have revealed how Tyr505 phosphorylation promotes binding of the SH2 domain to the C-terminal tail, stabilizing an association of the SH3 domain with a PxxP motif present in the linker segment connecting the SH2 and kinase domain and “closing” the kinase (Boggon and Eck, 2004).
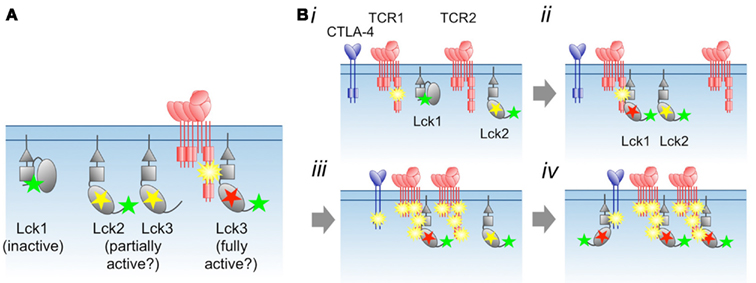
Figure 2. Proposal for regulation of Lck by phosphorylation and substrate binding. (A) The distinct states of phosphorylated Lck, and their proposed levels of activity. Yellow and green stars correspond to phosphorylated Tyr394 and Tyr505, respectively, whereas the red star marks Lck allosterically and/or stably activated by substrate binding. (B) Local, switch-like activation of Lck. (i) Immediately following incipient receptor triggering, the TCR is weakly phosphorylated. Via random collisions with other kinases or phosphatases in the membrane (see Figure 1), Lck transits between inactive (Lck1) and partially active (Lck2) states. (ii) The phosphorylated receptor competes with phosphorylated Lck1 Tyr505 for binding to the SH2 domain of Lck1, which activates the kinase allosterically and/or by stabilizing an open, activated state for the enzyme. The fully activated state also requires phosphorylation of Lck1 by Lck2. (iii) As the result of more random collisions, receptor-bound, fully activated Lck1 phosphorylates adjacent locally engaged (and non-engaged?) receptors, including inhibitory ones such as CTLA-4, which each then recruit and activate more Lck (iv). Differences in the degree of initial phosphorylation of the TCR in step (i), and, consequently, the amount of Lck activation in step (ii) allows signal discrimination. For the present purposes we do not draw distinctions between free and co-receptor associated Lck, although in step (ii) Lck/co-receptor recruitment to the TCR would be expected to be favored over free Lck recruitment.
The dynamics of the inter-conversion of the closed/inactive and open/active states of Lck are poorly understood but likely to be important given that SH2/Tyr505 and SH3/PxxP interactions are of modest affinity and that increasing the SH2/Tyr505 affinity substantially reduces Lck activity (Nika et al., 2007). Small-angle X-ray solution scattering and molecular dynamic simulations have revealed that unphosphorylated forms of Src kinases mainly adopt two distinct states in solution, with around 84% of Src or Hck assuming a closed/inactive conformation and 16% an open/active state; phosphorylated Tyr505 Src forms a single, closed conformation (Bernadó et al., 2008; Yang et al., 2010). Nika et al. (2010) have used the levels of phosphorylation of Tyr505 and Tyr394 to make inferences about the distribution of Lck states in vivo, and concluded that up to 40% of Lck is constitutively active. An additional, striking outcome was that upon T-cell activation there was no apparent increase in the levels of activated, i.e., Tyr394-phosphorylated Lck. However, an important caveat is that local changes in Lck phosphorylation might have gone undetected in their global analyses. But what local processes might occur?
Among several possibilities (Davis and van der Merwe, 2011), the binding of kinase SH2 domains to their substrates, which is itself phosphorylation dependent, would stabilize the kinase in an active state, favoring multi-site phosphorylation of receptors with more than one tyrosine. Classical “feed-forward” regulation of this type is observed for c-Src, Hck, and the closely related Fes and Abl kinases (see e.g., Alexandropoulos and Baltimore, 1996), but does it apply to Lck? We have found that phosphorylated CD3 ITAM-derived peptides strongly activate Lck measured with enolase as substrate (R. A. Fernandes et al., unpublished data). This suggests that receptor phosphorylation could, in principle, be regulated in a local, ultra-sensitive, switch-like manner via Lck/substrate encounters (Figure 2B). The extent to which this occurs in vivo would likely depend on: (1) the effective relative affinities of the SH2 domain of Lck for phosphorylated Tyr505 versus tyrosine-phosphorylated substrates; (2) the competing activities of Csk and other kinases for Tyr505 and Tyr394 on Lck; (3) the degree to which re-binding is favored following encounter complex formation (Dushek et al., 2011); and (4) the time TCR and Lck spend in regions lacking phosphatase activity (Davis and van der Merwe, 2006, 2011).
If Lck is only fully active after it binds phosphorylated substrates, and receptor phosphorylation levels are low, then the levels of active Lck in resting cells may in fact be low, irrespective of the phosphorylation status of Tyr394. Moreover, Lck activation per se will not be observable as changes in Tyr394 phosphorylation, which might remain unchanged during T-cell activation, as observed (Nika et al., 2007). Subtle, local regulation of this type would also explain why the allosteric regulation of Lck is so complex (Davis and van der Merwe, 2011). A final point is that Src kinase SH2 domains seem unique among the SH2 domains expressed by T-cells insofar as the purified SH2 domains bind the tyrosine-phosphorylated motifs of most receptors we have tested in surface plasmon resonance-based assays (J. Huo et al., unpublished data). This suggests the possibility that, should the dynamics allow it, many phosphorylated receptors might activate Lck, including inhibitory ones (e.g., CTLA-4; Figure 2B).
Concluding Remarks
The solution to the TCR “triggering problem” continues to elude us, but there is also much still to be learned about the regulation of the “resting” state of the TCR. An important challenge is to identify feedback control mechanisms underpinning the homeostatic control of TCR phosphorylation in resting cells, if this occurs. A much better understanding of the intrinsic and extrinsic factors driving the inter-conversion of the active and inactive states of the Src kinases is also required, in resting cells and under conditions of receptor triggering. It will also be helpful to know the effective affinities of the SH2 domain of Lck for phosphorylated substrate and Tyr505 residues, so that the extent to which, if at all, local substrate interactions impose “switch-like” behavior on Lck can be established.
The coupling of SH2 and kinase domains appeared early in evolution, soon after the appearance of tyrosine phosphorylation (Pawson, 1995; Filippakopoulos et al., 2009). This may have provided a basis for targeting kinases to their substrates and, later, an allosteric mechanism for controlling their local activity, increasing signaling complexity in multicellular organisms (Pawson, 2004; Kuriyan and Eisenberg, 2007). We have found that the expression of Src kinases is biased toward leukocytes, which also express the largest number of receptors with tyrosine phosphorylation motifs (Y. Lui et al., unpublished data). During metazoan evolution, the expansion of this family of kinases might have “solved” a specialized triggering problem, perhaps one involving the transient interactions of functionally interactive cells, such as leukocytes. This may in turn have allowed rich and varied signaling by an expanded set of receptors structurally related to a precursor of the TCR (Yu et al., 2011). When the receptor triggering “domino” eventually falls for the TCR, the triggering mechanisms for these other receptors are expected to quickly follow.
Acknowledgments
The work described in this article was funded by The Wellcome Trust, the UK Medical Research Council and the Fundação para a Ciência e a Tecnologia of Portugal.
References
Alexandropoulos, K., and Baltimore, D. (1996). Coordinate activation of c-Src by SH3- and SH2-binding sites on a novel, p130(Cas)-related protein, Sin. Genes Dev. 10, 1341–1355.
Barr, A. J., Ugochukwu, E., Lee, W. H., King, O. N., Filippakopoulos, P., Alfano, I., Savitsky, P., Burgess-Brown, N. A., Müller, S., and Knapp, S. (2009). Large-scale structural analysis of the classical human protein tyrosine phosphatome. Cell 136, 352–353.
Bernadó, P., Pérez, Y., Svergun, D. I., and Pons, M. (2008). Structural characterization of the active and inactive states of Src kinase in solution by small-angle X-ray scattering. J. Mol. Biol. 376, 492–505.
Boggon, T. J., and Eck, M. J. (2004). Structure and regulation of Src family kinases. Oncogene 23, 7918–7927.
Chung, I., Akita, R., Vandlen, R., Toomre, D., Schlessinger, J., and Mellman, I. (2010). Spatial control of EGF receptor activation by reversible dimerization on living cells. Nature 464, 783–787.
Davis, S. J., and van der Merwe, P. A. (1996). The structure and ligand interactions of CD2: implications for T-cell function. Immunol. Today 17, 177–187.
Davis, S. J., and van der Merwe, P. A. (2006). The kinetic-segregation model: TCR triggering and beyond. Nat. Immunol. 7, 803–809.
Davis, S. J., and van der Merwe, P. A. (2011). Lck and the nature of the T cell receptor trigger. Trends Immunol. 32, 1–5.
Deford-Watts, L. M., Tassin, T. C., Becker, A. M., Medeiros, J. J., Albanesi, J. P., Love, P. E., Wülfing, C., and van Oers, N. S. (2009). The cytoplasmic tail of the T cell receptor CD3 epsilon subunit contains a phospholipid-binding motif that regulates T cell functions. J. Immunol. 183, 1055–1064.
Domingo-Sananes, M. R., Kapuy, O., Hunt, T., and Novak, B. (2011). Switches and latches: a biochemical tug-of-war between the kinases and phosphatases that control mitosis. Philos. Trans. R. Soc. Lond. B Biol. Sci. 366, 3584–3594.
Dushek, O., van der Merwe, P. A., and Shahrezaei, V. (2011). Ultrasensitivity in multisite phosphorylation of membrane-anchored proteins. Biophys. J. 100, 1189–1197.
Eggeling, C., Ringemann, C., Medda, R., Schwarzmann, G., Sandhoff, K., Polyakova, S., Belov, V. N., Hein, B., von Middendorff, C., Schönle, A., and Hell, S. W. (2009). Direct observation of the nanoscale dynamics of membrane lipids in a living cell. Nature 457, 1159–1162.
Fernandes, R. A., Yu, C., Carmo, A. M., Evans, E. J., van der Merwe, P. A., and Davis, S. J. (2010). What controls T cell receptor phosphorylation? Cell 142, 668–669.
Filippakopoulos, P., Muller, S., and Knapp, S. (2009). SH2 domains: modulators of nonreceptor tyrosine kinase activity. Curr. Opin. Struct. Biol. 19, 643–649.
Fischer, E. H., Charbonneau, H., Cool, D. E., and Tonks, N. K. (1992). Tyrosine phosphatases and their possible interplay with tyrosine kinases. Ciba Found. Symp. 164, 132–144.
Gagnon, E., Xu, C., Yang, W., Chu, H. H., Call, M. E., Chou, J. J., and Wucherpfennig, K. W. (2010). Response: multilayered control of T cell receptor phosphorylation. Cell 142, 669–671.
Grasberger, B., Minton, A. P., DeLisi, C., and Metzger, H. (1986). Interaction between proteins localized in membranes. Proc. Natl. Acad. Sci. U.S.A. 83, 6258–6262.
He, H. T., and Marguet, D. (2008). T-cell antigen receptor triggering and lipid rafts: a matter of space and time scales. EMBO Rep. 9, 525–530.
Hermiston, M. L., Zikherman, J., and Zhu, J. W. (2009). CD45, CD148, and Lyp/Pep: critical phosphatases regulating Src family kinase signaling networks in immune cells. Immunol. Rev. 228, 288–311.
Huse, M., Klein, L. O., Girvin, A. T., Faraj, J. M., Li, Q. J., Kuhns, M. S., and Davis, M. M. (2007). Spatial and temporal dynamics of T cell receptor signaling with a photoactivatable agonist. Immunity 27, 76–88.
Irie-Sasaki, J., Sasaki, T., Matsumoto, W., Opavsky, A., Cheng, M., Welstead, G., Griffiths, E., Krawczyk, C., Richardson, C. D., Aitken, K., Iscove, N., Koretzky, G., Johnson, P., Liu, P., Rothstein, D. M., and Penninger, J. M. (2001). CD45 is a JAK phosphatase and negatively regulates cytokine receptor signalling. Nature 409, 349–354.
Irvine, D. J., Purbhoo, M. A., Krogsgaard, M., and Davis, M. M. (2002). Direct observation of ligand recognition by T cells. Nature 419, 845–849.
James, J. R., McColl, J., Oliveira, M. I., Dunne, P. D., Huang, E., Jansson, A., Nilsson, P., Sleep, D. L., Gonçalves, C. M., Morgan, S. H., Felce, J. H., Mahen, R., Fernandes, R. A., Carmo, A. M., Klenerman, D., and Davis, S. J. (2011). The T cell receptor triggering apparatus is composed of monovalent or monomeric proteins. J. Biol. Chem. 286, 31993–32001.
James, J. R., Oliveira, M. I., Carmo, A. M., Iaboni, A., and Davis, S. J. (2006). A rigorous experimental framework for detecting protein oligomerization using bioluminescence resonance energy transfer. Nat. Methods 3, 1001–1006.
James, J. R., White, S. S., Clarke, R. W., Johansen, A. M., Dunne, P. D., Sleep, D. L., Fitzgerald, W. J., Davis, S. J., and Klenerman, D. (2007). Single-molecule level analysis of the subunit composition of the T cell receptor on live T cells. Proc. Natl. Acad. Sci. U.S.A. 104 17662–17667.
Kleiman, L. B., Maiwald, T., Conzelmann, H., Lauffenburger, D. A., and Sorger, P. K. (2011). Rapid phospho-turnover by receptor tyrosine kinases impacts downstream signaling and drug binding. Mol. Cell 43, 723–737.
Kuriyan, J., and Eisenberg, D. (2007). The origin of protein interactions and allostery in colocalization. Nature 450, 983–990.
Nika, K., Soldani, C., Salek, M., Paster, W., Gray, A., Etzensperger, R., Fugger, L., Polzella, P., Cerundolo, V., Dushek, O., Höfer, T., Viola, A., and Acuto, O. (2010). Constitutively active Lck kinase in T cells drives antigen receptor signal transduction. Immunity 32, 766–777.
Nika, K., Tautz, L., Arimura, Y., Vang, T., Williams, S., and Mustelin, T. (2007). A weak Lck tail bite is necessary for Lck function in T cell antigen receptor signaling. J. Biol. Chem. 282, 36000–36009.
Patel, M. D., Samelson, L. E., and Klausner, R. D. (1987). Multiple kinases and signal transduction. Phosphorylation of the T cell antigen receptor complex. J. Biol. Chem. 262, 5831–5838.
Pawson, T. (2004). Specificity in signal transduction: from phosphotyrosine-SH2 domain interactions to complex cellular systems. Cell 116, 191–203.
Quinn, P., Griffiths, G., and Warren, G. (1984). Density of newly synthesized plasma membrane proteins in intracellular membranes II. Biochemical studies. J. Cell Biol. 98, 2142–2147.
Reynolds, A. R., Tischer, C., Verveer, P. J., Rocks, O., and Bastiaens, P. I. (2003). EGFR activation coupled to inhibition of tyrosine phosphatases causes lateral signal propagation. Nat. Cell Biol. 5, 447–453.
Smith-Garvin, J. E., Koretzky, G. A., and Jordan, M. S. (2009). T cell activation. Annu. Rev. Immunol. 27, 591–619.
Ullrich, A., and Schlessinger, J. (1990). Signal transduction by receptors with tyrosine kinase activity. Cell 61, 203–212.
Williams, A. F., and Barclay, A. N. (1986). “Glycoprotein antigens of the lymphocyte surface and their purification by antibody affinity chromatography,” in Handbook of Experimental Immunology, eds D. M. Weir and L. A. Herzenberg (Oxford: Blackwell Scientific Publications), 22.1–22.24.
Xu, C., Gagnon, E., Call, M. E., Schnell, J. R., Schwieters, C. D., Carman, C. V., Chou, J. J., and Wucherpfennig, K. W. (2008). Regulation of T cell receptor activation by dynamic membrane binding of the CD3 epsilon cytoplasmic tyrosine-based motif. Cell 135, 702–713.
Yamada, T., Zhu, D., Saxon, A., and Zhang, K. (2002). CD45 controls interleukin-4-mediated IgE class switch recombination in human B cells through its function as a Janus kinase phosphatase. J. Biol. Chem. 277, 28830–28835.
Yang, S., Blachowicz, L., Makowski, L., and Roux, B. (2010). Multidomain assembled states of Hck tyrosine kinase in solution. Proc. Natl. Acad. Sci. U.S.A. 107, 15757–15762.
Yu, C., Sonnen, A. F., George, R., Dessailly, B. H., Stagg, L. J., Evans, E. J., Orengo, C. A., Stuart, D. I., Ladbury, J. E., Ikemizu, S., Gilbert, R. J., and Davis, S. J. (2011). Rigid-body ligand recognition drives cytotoxic T-lymphocyte antigen 4 (CTLA-4) receptor triggering. J. Biol. Chem. 286, 6685–6696.
Citation: Fernandes RA, Huo J, Lui Y, Felce JH and Davis SJ (2012) On the control of TCR phosphorylation. Front. Immun. 3:92. doi: 10.3389/fimmu.2012.00092
Received: 01 February 2012; Accepted: 10 April 2012;
Published online: 07 May 2012.
Copyright: © 2012 Fernandes, Huo, Lui, Felce and Davis. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence:c2ltb24uZGF2aXNAbmRtLm94LmFjLnVr