

 Roy Cohen
Roy Cohen

- Department of Chemistry and Chemical Biology, Cornell University, Ithaca, NY, USA
Mobilization of Ca2+ in response to IgE receptor-mediated signaling is a key process in many aspects of mast cell function. Here we summarize our current understanding of the molecular bases for this process and the roles that it plays in physiologically relevant mast cell biology. Activation of IgE receptor signaling by antigen that crosslinks these complexes initiates Ca2+ mobilization as a fast wave that is frequently followed by a series of Ca2+ oscillations which are dependent on Ca2+ influx-mediated by coupling of the endoplasmic reticulum luminal Ca2+ sensor STIM1 to the calcium release activated calcium channel protein Orai1. Granule exocytosis depends on this process, together with the activation of protein kinase C isoforms, and specific roles for these signaling steps are beginning to be understood. Ca2+ mobilization also plays important roles in stimulated exocytosis of recycling endosomes and newly synthesized cytokines, as well as in antigen-mediated chemotaxis of rat mucosal mast cells. Phosphoinositide metabolism plays key roles in all of these processes, and we highlight these roles in several cases.
Introduction
Mast cell degranulation represents one of the earliest described examples of a Ca2+-dependent biological process in non-excitable cells (Foreman et al., 1973; Kanno et al., 1973). The capacity for IgE-mediated activation of this process via crosslinking of IgE receptor (FcεRI) complexes at the mast cell surface was established in the 1970s (Metzger, 1978). Since biochemical identification (Metzger et al., 1986) and cloning (Ravetch and Kinet, 1991) of the three different subunits of FcεRI in the 1980s, much has been learned about the signaling pathways activated via this prototypic multichain immune recognition receptor and their roles in stimulated Ca2+ responses leading to granule exocytosis (Siraganian et al., 2002; Kraft and Kinet, 2007). In the present review we focus on the mechanisms by which FcεRI signaling mediates elevation of cytoplasmic Ca2+ and the consequences of this process in mast cell functional responses, including granule exocytosis, chemotaxis to antigen, and stimulated endosomal trafficking, including cytokine secretion and recycling endosomal exocytosis. We will emphasize contributions to this area from our own laboratory, and we will focus on subcellular spatiotemporal relationships where this information is available.
Mechanisms of Ca2+ Mobilization in Mast Cells
Although the importance of stimulated Ca2+ influx for mast cell degranulation has been known for decades, the mechanisms by which FcεRI-mediated Ca2+ entry occurs and the role of this process in stimulating granule exocytosis are incompletely understood. Mast cells do not exhibit voltage gated Ca2+ influx, a property of excitable cells (Jarvis and Zamponi, 2007). However, release of Ca2+ from endoplasmic reticulum (ER) stores by antigen or thapsigargin (an irreversible inhibitor of the SERCA ATPase, which normally maintains high Ca2+ levels in the ER) is sufficient to activate Ca2+ influx necessary for degranulation from these cells (Wolfe et al., 1996). This Ca2+ influx mechanism is commonly referred to as store-operated Ca2+ entry (SOCE), and it was originally shown by Putney and colleagues to depend critically on depletion of luminal Ca2+ from the ER (Takemura et al., 1989). The Ca2+ channel that mediates SOCE, known as the calcium release activated calcium (CRAC) channel, was defined electrophysiologically about two decades ago by Hoth and Penner (1992) in mast cells and by Lewis and Cahalan (1989) in T cells. However, its molecular identity remained unknown until 2006, when three papers from independent groups described cloning and functional reconstitution of the tetraspan channel protein Orai1/CRACM1 (Feske et al., 2006; Vig et al., 2006; Zhang et al., 2006).
In 2005, studies using large-scale siRNA screens to identify proteins important for SOCE discovered another protein, STIM1, that is critical for activation of Orai1 (Liou et al., 2005; Zhang et al., 2005). Unlike the CRAC channel protein, which is localized to the plasma membrane, STIM1 is an ER-localized Type I membrane protein that redistributes to discrete regions of the ER proximal to the plasma membrane upon Ca2+ depletion from ER stores (Liou et al., 2005; Wu et al., 2006). The mechanism of this redistribution, which appears to be critical for activation of Orai1/CRACM1, involves oligomerization of STIM1 that results from the loss of Ca2+ binding to EF-hand motifs in a luminal domain of this protein (Liou et al., 2007). Recent knockout studies demonstrated essential roles for STIM1 (Baba et al., 2008) and Orai1/CRACM1 (Vig et al., 2008) in mast cell degranulation and other processes in the allergic response (reviewed in Di Capite et al., 2011).
Our laboratory developed an imaging-based method to monitor the time-dependence of stimulated association of Orai1 with STIM1 that utilizes fluorescence resonance energy transfer (FRET) from monomeric AcGFP-Orai1 to STIM1-mRFP co-expressed in RBL mast cells (Calloway et al., 2009). Using this method, we found that thapsigargin stimulates much more extensive association of Orai1 with STIM1 than does antigen-mediated crosslinking of IgE receptors in the presence of extracellular Ca2+. However, removal of extracellular Ca2+ results in antigen-stimulated FRET that is comparable to that stimulated by thapsigargin in the presence of Ca2+, indicating that antigen-stimulated association of Orai1 with STIM1 is highly regulated in these cells (Calloway et al., 2009). Never the less, this more highly regulated coupling can still achieve robust SOCE in response to antigen (Parekh et al., 1997; Vasudevan et al., 2009). Interestingly, G protein-coupled receptors typically activate transient Ca2+ responses in mast cells in response to ligands such as adenosine or prostaglandin E2, but they fail to activate SOCE (Gilfillan and Beaven, 2011).
In our initial study, we defined an acidic amino acid segment in a putative coiled-coil sequence at the C-terminus of Orai1 that is important for stimulated SOCE (Calloway et al., 2009; Figure 1). We subsequently showed that this acidic segment (E272–D291) couples to a short basic sequence (K382–R387) in the C-terminal segment of STIM1 to activate Ca2+ entry under conditions in which STIM1/Orai1 association is promoted (Calloway et al., 2010). This apparent electrostatic interaction, while necessary for Ca2+ entry, is not necessary for stimulated association of STIM1 with Orai1, which depends on a larger, ∼110 amino acid sequence called the “CAD domain” that can constitutively activate CRAC channels when expressed as a cytoplasmic protein (Kawasaki et al., 2009; Muik et al., 2009; Park et al., 2009; Yuan et al., 2009). More recent studies have shown that the sequence comprising five basic amino acids within the CAD domain is inaccessible to Orai1 prior to ER store depletion because of a conformational restriction in STIM1 that sequesters it by intramolecular association with an acidic sequence (E302–D322) that is N-terminal to the CAD domain in the STIM1 cytoplasmic tail (Korzeniowski et al., 2010; Muik et al., 2011). By a mechanism that is still unclear, homo-clustering of STIM1 following depletion of ER luminal Ca2+ results in a conformational transition to a more extended C-terminus that readily couples to Orai1 to activate Ca2+ influx into the cytoplasm through a productive Orai1 channel.
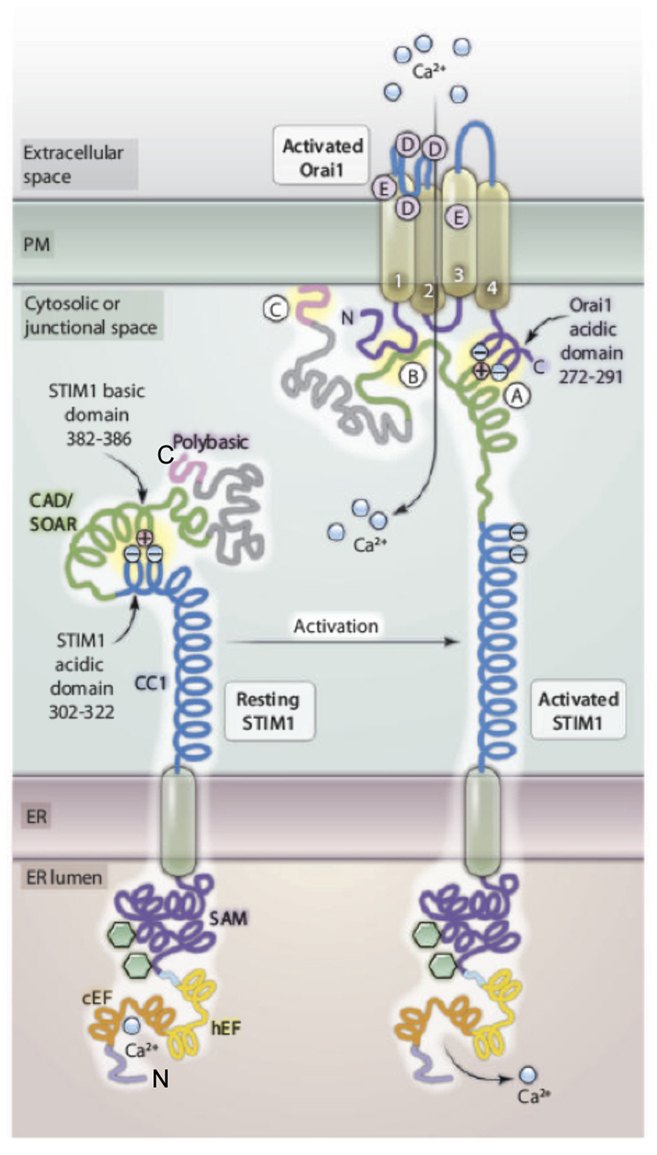
Figure 1. Molecular features of STIM1–Orai1 interactions. Prior to activation, acidic residues of the cytosolic coiled-coil domain 1 (CC1) of STIM1 electrostatically bind to and mask the basic residues in the CAD domain that are critical for CRAC channel gating. Activation by depletion of luminal ER Ca2+ causes conformational changes in both luminal and cytosolic segments of STIM1. Changes in the luminal EF-hand motifs and the sterile A motif (SAM) results in clustering of STIM1 molecules (not shown) that leads to alterations in the cytosolic region: Intramolecular electrostatic interaction between CC1 and CAD are broken, and the positively charged sequence in the CAD domain is free to interact with the acidic domain within the C-terminal domain of Orai1 (A) to activate the CRAC channel formed by Orai1. STIM1 also interacts with the N-terminus of Orai1 (B), and the C-terminal polybasic sequence of STIM1 associates with PIPs at the plasma membrane (C). STIM1 and Orai1 monomers are depicted for simplicity; interactions involve oligomers of these proteins in a multimeric complex. Modified from Wang et al. (2010).
Many ion channels are regulated by phosphoinositides in the plasma membrane, but the roles for these lipids in SOCE are controversial (Huang, 2007). Korzeniowski et al. (2009) demonstrated that activation of SOCE is sensitive to inhibition of phosphatidylinositol 4-kinase, but is not prevented by acute depletion of phosphatidylinositol 4,5-bisphosphate (PIP2) at the plasma membrane of COS-7 cells, suggesting a role for PI4P but not for PIP2. Walsh et al. (2010) showed that inhibition of multiple pathways of PIP2 generation is necessary to prevent thapsigargin-mediated translocation of STIM1 to the plasma membrane in HeLa cells, but that co-expression of Orai1 permits STIM1 to concentrate at ER–PM junctions even in the absence of PIP2. A previous study in our laboratory provided evidence that different PI5-kinase family members synthesize functionally different pools of PIP2 (Vasudevan et al., 2009). To explore whether these functionally different pools of PIP2 might be synthesized in detergent-resistant ordered lipid domains and detergent-sensitive disordered lipid domains (Lingwood and Simons, 2010), we used transient expression of an inositol 5′-phosphatase that is targeted to either ordered or disordered lipids in RBL mast cells, and we found that the thapsigargin-stimulated association of STIM1 with Orai1 is regulated by the balance between PIP2 associated with ordered lipids and PIP2 associated with disordered lipids, such that PIP2 in ordered lipid domains promotes this association and PIP2 in disordered lipid domains reduces it, with corresponding alterations in SOCE (Calloway et al., 2011). Furthermore, we found that over-expression of PIP5-kinase Iβ, which enhances PIP2 levels in both ordered and disordered lipid domains, enhances the stimulated association of STIM1 and Orai1, whereas over-expression of PIP5-kinase Iγ, which enhances PIP2 levels only in the disordered lipid domains, has an inhibitory effect on this association. This regulation of STIM1/Orai1 coupling by PIP2 could be attributed to a basic sequence in the N-terminal segment of Orai1 and a basic sequence at the C-terminus of STIM1 because deletion of either of these prevents the effects of both PI5-kinase over-expression and targeted inositol 5-phosphatase expression on stimulated FRET (Calloway et al., 2011). A simple model for these effects is illustrated in Figure 2, in which ER store depletion causes STIM1 to bind initially to PIP2 in ordered domains at the plasma membrane, followed by redistribution of Orai1 from disordered regions of the plasma membrane to its association with STIM1 in ordered lipid domains. We are currently testing this hypothesis.
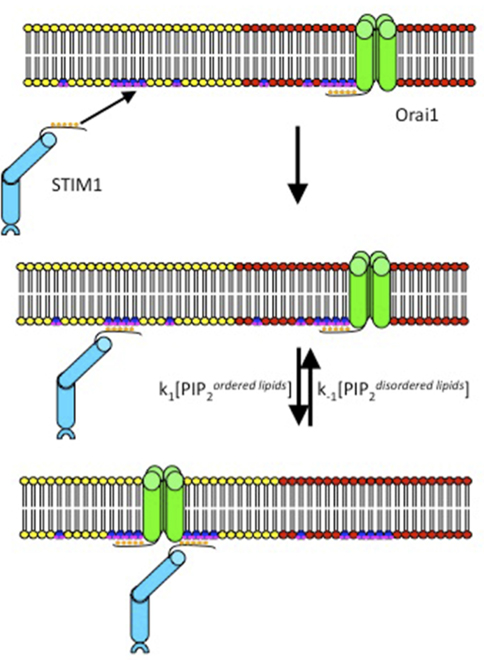
Figure 2. Model for the sequence of events that initiate coupling of the ER sensor protein STIM1 to the CRAC channel protein Orai1. Activated STIM1 in the ER (not shown) binds initially via its C-terminal polylysine sequence to PIP2 (blue/violet) in ordered lipids (yellow) at the inner leaflet of the plasma membrane. Concentration of STIM1 at this site facilitates its coupling to Orai1 that redistributes from disordered lipids (red) to ordered lipids. A basic polyarginine sequence in the N-terminus of Orai1 mediates its electrostatic interactions with PIPs and STIM1.
It is well-established that antigen-stimulated Ca2+ mobilization in mast cells is initiated by activation of phospholipase C (PLC) γ-1 and γ-2, which depends on tyrosine phosphorylation of specific residues in these proteins by Syk kinase and/or by the Tec kinase BTK (Gilfillan and Rivera, 2009; Ma and Beaven, 2009). These lipases hydrolyze PIP2 to produce inositol 1,4,5-trisphosphate (IP3) and diacylglycerol, and it is IP3-mediated release of Ca2+ from ER stores that activates the association of STIM1 with Orai1 to form productive CRAC channels as discussed above. Using fast confocal imaging of the genetically encoded Ca2+ sensor, GCaMP2, and proximal antigen delivery via a micropipette, we found that antigen-stimulated Ca2+ responses usually initiate as a fast Ca2+ wave that begins at the tips of extended membrane protrusions, which are common on well-attached cells (Figure 3; Cohen et al., 2009). Not surprisingly, this Ca2+ wave initiation depends on PLC activity, but it does not utilize CRAC channels because it is not inhibited by Gd3+, which selectively blocks these channels in RBL mast cells (Cohen et al., 2009; Calloway et al., 2011). In the absence of extracellular Ca2+, wave initiation occurs more frequently at other sites along the cell body, suggesting that Ca2+ entry is important for its initiation from protrusions. A previous study provided evidence that certain six transmembrane-structured TRPC cation channel proteins can contribute to antigen-stimulated Ca2+ entry in RBL mast cells (Ma et al., 2008). Our results from shRNA knockdown experiments indicated that wave initiation from protrusions depends on Ca2+ entry via two particular TRPC channels, TRPC1 and TRPC3, particularly at limiting concentrations of antigen (Cohen et al., 2009).
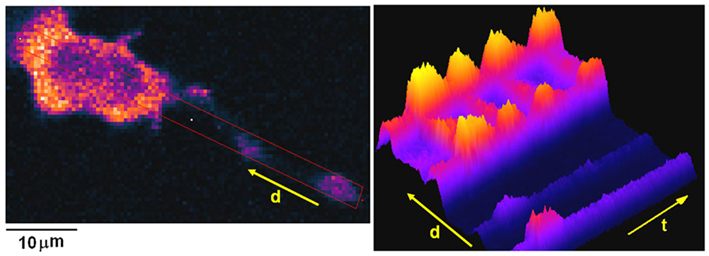
Figure 3. Ca2+ wave initiation from an extended protrusion in RBL mast cells. Left panel: image of GCaMP2 expressed in an IgE-sensitized RBL cell. Note protrusion terminus in lower right hand corner. Red rectangle shows region of interest that is monitored as a function of distance (originating from protrusion) and of time after stimulation by antigen in right panel. Warmer colors correspond to larger increases in intracellular [Ca2+]. Right panel: Time vs distance plot of [Ca2+] shows initial response from protrusion (lower left), followed by a series of global Ca2+ oscillations over the course of several hundred seconds.
Consequences of Ca2+ Mobilization
Degranulation
In recent experiments, we found that Ca2+ wave initiation from protrusions confers a spatial preference for granule exocytosis from sites along these protrusions, particularly at the same low doses of antigen that enhance wave initiation from the tips of these structures (Cohen et al., 2012). This spatial and temporal comparison of granule exocytosis and Ca2+ mobilization is possible because of the implementation of fast confocal imaging of fluorescein isothiocyanate (FITC)–dextran-labeled granules, together with Fura Red-indicated Ca2+ responses. These methods allow us to detect individual exocytosis events as bursts of FITC fluorescence and to compare the spatial and temporal relationships of these events to stimulated Ca2+ oscillations in these cells. Consistent with an earlier study that used amperometry to compare the temporal relationship between granule exocytosis and Ca2+ oscillations (Kim et al., 1997), we find that exocytosis events monitored by FITC–dextran bursts occur primarily at the peaks, or just following the peaks, of sustained Ca2+ oscillations that depend on SOCE (Cohen et al., 2009). We also find that granule exocytosis under these conditions is sensitive to knockdown of the TRPC1 Ca2+ channel protein, further supporting a role for Ca2+-dependent wave initiation via this channel in spatially directed granule exocytosis (Cohen et al., 2012).
Multiple lines of evidence point to the participation of protein kinase C (PKC) isoforms in mast cell granule exocytosis, but the mechanism has been unclear (Rivera and Beaven, 1997). Molecular genetic studies highlight a specific role for PKC β (PKCβ) in this process (Nechushtan et al., 2000). Our studies to understand PKC in stimulated granule exocytosis have utilized the effector domain (ED) of the ubiquitous MARCKS protein. MARCKS-ED is a 25-amino acid segment that contains 13 basic residues that mediate its very tight binding to polyphosphoinositides (PIPs) at the inner leaflet of the plasma membrane (Gambhir et al., 2004; Heo et al., 2006). Protein kinase C normally causes dissociation of MARCKS by phosphorylating three serine residues in the ED sequence, thus adding negative charges to reduce the electrostatic interaction of the ED and thereby increasing access to other PIP-binding proteins in processes such as granule exocytosis (Figure 4).
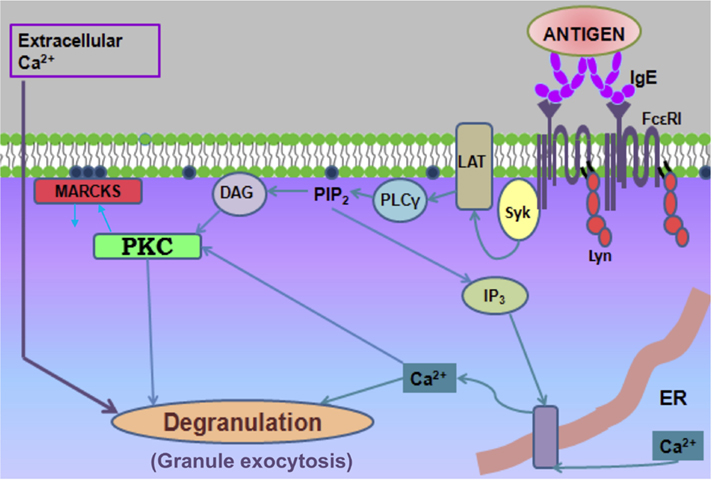
Figure 4. Cartoon depicting key steps in FcεRI signaling leading to degranulation, including PKC-dependent dissociation of the MARCKS protein from the plasma membrane. Crosslinking of IgE–FcεRI complexes by antigen results in receptor phosphorylation by Lyn, recruitment and activation of Syk, and downstream activation of Ca2+ mobilization and PKC activation via adaptor protein LAT and PLCγ, which produces IP3 and diacylglycerol in this process. Not shown is the activation of SOCE depicted in Figure 1.
To explore the role of this PIP sequestration by MARCKS-ED in stimulated mast cell granule exocytosis, we over-expressed a ser → ala mutated form of this polypeptide, which lacks the capacity to be displaced by PKC phosphorylation. We found that this polypeptide construct effectively delays antigen-stimulated Ca2+ mobilization, including release of Ca2+ from intracellular stores, most likely because it reduces access to and hydrolysis of PIP2 by PLC. Despite this delay, the sustained phase of SOCE stimulated by antigen, and the Ca2+ response to thapsigargin, are not inhibited. However, the degranulation response to each of these stimuli is substantially delayed and reduced (Figure 5; Gadi et al., 2011). These results are consistent with a model in which the availability of PIPs is important for stimulated exocytosis at a step that is downstream of Ca2+ mobilization. Synaptotagmins are multidomain proteins known to play key roles in Ca2+-dependent exocytosis in neuronal and other cell types (Chapman, 2008). We hypothesize that PIP2-dependent binding of synaptotagmin to Ca2+ (Bai et al., 2004) is critically inhibited by mutated MARCKS-ED, although we cannot exclude a role for PIP2 in other SNARE-dependent steps (James et al., 2010). In general, the ser → ala mutated MARCKS-ED has proven to be a useful reagent for evaluating PIP participation in signaling processes based on the capacity of this polypeptide to sequester these key lipids in a manner that is not reversed by PKC phosphorylation.
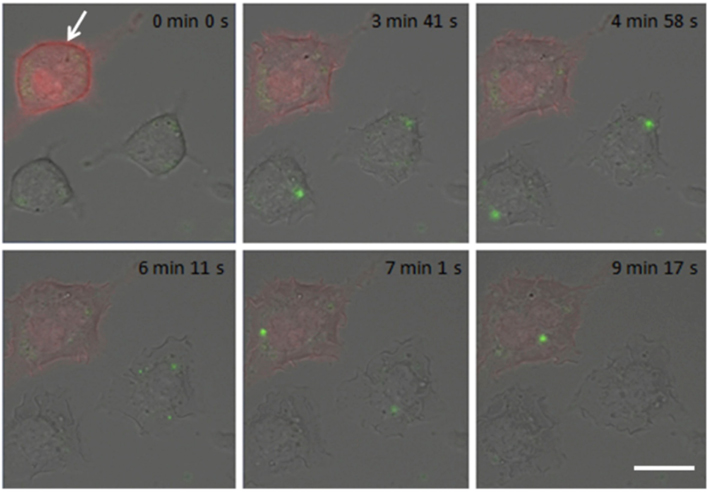
Figure 5. Mutated (ser → ala) MARCKS-ED inhibits antigen-stimulated degranulation. In real-time imaging, degranulation is monitored as FITC–dextran bursts (green puncta) from antigen-stimulated RBL cells. Representative cell expressing mutated mRFP-MARCKS-ED (red; white arrow) shows delayed and reduced response compared to cell that is not transfected with this construct. Times in images specify intervals after antigen addition. Scale bar is 10 μm. From Gadi et al. (2011).
Does PKC phosphorylation of endogenous MARCKS play a physiological role in the regulation of stimulated granule exocytosis? One indication of this possibility comes from a comparison that we made between the effects of over-expression of the wild type sequence of MARCKS-ED and the effects of the ser → ala mutated MARCKS-ED summarized above. In contrast to the inhibitory effects of mutated MARCKS-ED on stimulated granule exocytosis, we found that the wild type sequence does not inhibit this process, confirming that serine phosphorylation by PKC facilitates the dissociation of this sequence during cell signaling to facilitate subsequent events (Gadi et al., 2011).
MARCKS has long been known as a substrate for PKCs, but the physiological role of the full-length protein in cells has been elusive (Stumpo et al., 1995; Trifaro et al., 2008). To evaluate more directly the role of endogenous MARCKS in mast cell functional responses, we knocked down this protein expression using two different siRNA sequences. We were initially surprised by our results indicating that endogenous MARCKS exhibits a positive effect on antigen-stimulated Ca2+ mobilization: Both siRNA constructs cause a significant reduction in SOCE (31 ± 7% inhibition, n = 5), without inhibiting antigen-stimulated Ca2+ release from ER stores (Figure 6A). Consistent with this effect, we also found that these siRNA sequences inhibit antigen-stimulated granule exocytosis measured in suspended RBL cells with granule-incorporated FITC–dextran (Figure 6B; 30 ± 3% inhibition, n = 2). These results indicate that endogenous MARCKS normally facilitates antigen-stimulated Ca2+ and degranulation responses.
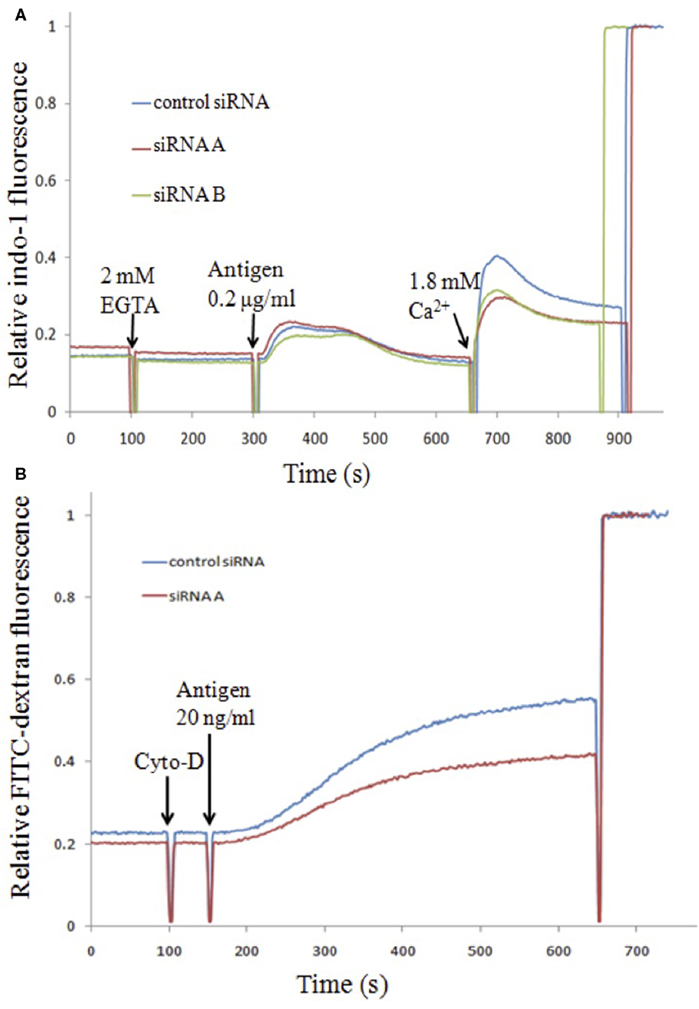
Figure 6. Effects of siRNA knockdown of endogenous MARCKS protein on Ca2+ (A) and degranulation (B) responses in RBL cells. (A) RBL cells transfected with irrelevant control siRNA or two different siRNA sequences specific for rat MARCKS (Gadi, 2012) were loaded with the [Ca2+] indicator indo-1 before stimulating with antigen in the absence of extracellular Ca2+. Then millimolar Ca2+ was added to enable SOCE. (B) RBL cells, transfected as in (A), and loaded with FITC–dextran (Gadi et al., 2011), were treated with 1 mM cytochalasin D and then stimulated with antigen. FITC fluorescence increases due to stimulated degranulation were monitored by steady state fluorimetry.
How might MARCKS contribute to enhancement of stimulated SOCE that supports enhanced exocytosis? One possibility is that MARCKS dissociates from the plasma membrane and associates with Ca2+-bound calmodulin under stimulating conditions. This would occur upon elevation of intracellular Ca2+, and it could provide a mechanism by which Ca2+/calmodulin-mediated processes are regulated (McLaughlin and Murray, 2005). One of these well-studied processes is the rapid inactivation of STIM1-mediated SOCE, which has recently been shown to depend on the binding of Ca2+/calmodulin to a specific sequence in STIM1 (Mullins et al., 2009). Inhibition of this interaction by MARCKS association with Ca2+/calmodulin would provide a plausible mechanism for the enhancing effects of PKC-phosphorylated MARCKS on SOCE and consequent enhancement of granule exocytosis.
Cell Motility and Chemotaxis
Cell migration is critical for various biological functions, such as embryogenesis, wound healing, and immune responses, and it can also contribute to the pathogenesis of diseases including cancer and transplant rejection. Understanding basic mechanisms of cell motility has been a goal of scientific investigations since the emergence of optical microscopy. Cell motility requires the actin cytoskeleton, asymmetric morphology of the cell, and polarized intracellular signaling (Mitchison and Cramer, 1996; Petrie et al., 2009). Polarization and development of leading and trailing edges of the cell mediate cell locomotion by dynamic extension and retraction of cellular protrusions, including pseudopods, filopodia, and lamellipodia (Gupton and Gertler, 2007).
Cells often respond to a gradient of external factors or asymmetric environmental cues by means of a compass or steering mechanism coupled to basal motility machinery, resulting in directed migration. Similarly, chemotaxis occurs in response to soluble cues (Bourne and Weiner, 2002; Arrieumerlou and Meyer, 2005). Mast cells accumulate at sites of inflammation in response to parasite and bacterial infections (Madden et al., 1991; Echtenacher et al., 1996). Differentiated mucosal mast cells are known to redistribute from the submucosa or crypt area to the lamina propria and intraepithelial regions of jejunal villi during the course of an immune response to certain parasitic infections (Friend et al., 1996). Mast cell chemotaxis toward IgE-specific antigen was first described for RBL cells (Orida et al., 1983) and has also been characterized in bone marrow-derived mouse mast cells (Kitaura et al., 2005; Olivera et al., 2006). Recently, we observed with real-time imaging that Ca2+ influx via Orai1 plays an important role in regulating both spontaneous, random motility, as well as chemotaxis toward antigen for both RBL mast cells and bone marrow-derived rat mast cells (J. Lee et al., submitted). Inhibition of Ca2+ influx, or knockdown of the Ca2+ entry channel protein, Orai1, by shRNA in RBL cells reduces both of these processes. RBL cells expressing the Ca2+ sensor, GCaMP3, exhibit spontaneous Ca2+ transients that depend on Ca2+ influx, and their appearance correlates with cell motility. Our results thus identify a novel Ca2+ influx-mediated, Orai1-dependent mechanism for mast cell migration.
Other Ca2+-Dependent Processes
In addition to degranulation and chemotaxic responses to antigen-mediated crosslinking of IgE receptor complexes, rat mast cells undergo additional exocytotic processes that depend on Ca2+ mobilization. One of these is the stimulated outward trafficking of recycling endosomes that are labeled with FITC–cholera toxin B (CTB) bound to ganglioside GM1, which we previously characterized (Naal et al., 2003; Smith et al., 2010). We found that this process depends on Ca2+ mobilization and normal cholesterol levels, but not on PKC activity. In the absence of extracellular Ca2+, this process is transient, indicating that Ca2+ influx is necessary for a sustained response. This stimulated trafficking targets delivery of CTB/GM1 to spatially restricted sites where IgE receptors are clustered and activated (Wu et al., 2007), and this is reminiscent of the cholesterol-dependent delivery of TNFα to phagocytic cups in murine macrophages (Murray et al., 2005; Kay et al., 2006). The macrophage studies provided evidence that recycling endosomes participate in cytokine secretion in those cells. TNFα, as well as other cytokines, are synthesized de novo in rodent mast cells in response to activation via IgE receptors (Baumgartner et al., 1994). Stimulated secretion of TNFα does not involve trafficking to secretory lysosomes as it does for pre-stored TNFα in human mast cells (Gordon and Galli, 1991). Both the initial phase of stimulated transcriptional activation and the secretion of TNFα require Ca2+ mobilization (Baumgartner et al., 1994). Activation of NFAT transcriptional factors via Ca2+/calcineurin-mediated dephosphorylation of these proteins is an important part of this process (Andrade et al., 2011). In contrast, constitutive synthesis and trafficking of another cytokine, TGF-β, does not require elevated cytoplasmic Ca2+ (Baumgartner et al., 1996). Thus, the requirement for elevated cytoplasmic Ca2+ for stimulated cytokine production suggests a different mechanism for their exocytotic release than for constitutive biosynthetic protein trafficking. An interesting question to be pursued from these results is whether antigen-stimulated cytokine secretion in mast cells utilizes trafficking through recycling endosomes en route to the plasma membrane.
Stimulated outward trafficking of recycling endosomes labeled with FITC–CTB is inhibited by basic sphingosine derivatives that spontaneously flip to the inner leaflet of the plasma membrane. Sphingosine derivatives that do not readily flip, including N,N′,N′′-trimethylsphingosine and glycosylated sphingosine (psychosine), do not inhibit this process (Smith et al., 2010). Using the ser → ala mutated MARCKS-ED construct, we further showed that inhibition by these long chain bases correlates with electrostatic neutralization of negatively charged PIPs at the cytoplasmic face of the plasma membrane. Similarly, the sphingosine derivatives that readily flip across the membrane inhibit antigen-stimulated Ca2+ mobilization and granule exocytosis, providing evidence for a general mechanism of phosphoinositide sequestration by these compounds (Smith et al., 2010). This consistent set of results lends new insight into the interactions by which sphingosines inhibit downstream signaling in many different published studies.
An additional Ca2+-dependent process that generates important mediators in stimulated mast cell responses is the activation of phospholipase A2 to produce arachidonic acid, which is the precursor for leukotrienes and prostaglandins, two different family of eicosanoids that act as ligands for specific G protein-coupled receptors (Beaven, 2009). Cytoplasmic phospholipase A2 is activated by Ca2+ binding together with Erk-dependent phosphorylation, which in turn is activated by the MAP kinase cascade (Hirasawa et al., 1995). These eicosanoid mediators can act in an autocrine fashion to synergize with antigen stimulation of mast cells, as well as to activate other cells in processes such as anaphylaxis (Beaven, 2009).
Outstanding Questions
Mast cells are receiving increasing attention as mediators of innate immune responses in addition to their better-known role in the adaptive allergic response (Metz et al., 2007; Brown et al., 2008). Although mast cells are recruited to the gut where they undergo dramatic proliferation in response to certain parasitic infections such as with Trichinella spiralis (Friend et al., 1996), a recent study has shown that they do not contribute significantly to parasite clearance during the secondary immune response (Blum et al., 2009). This suggests that mast cells participate in the initiation of the Th2 immune response, rather than in the effector phase. Our studies summarized above suggest that antigen-specific recruitment of mast cells to small intestinal villi during such immune responses may be driven by Ca2+ mobilization in a process that utilizes STIM1-mediated activation of CRAC channels that are formed by Orai1. Understanding the mechanism by which these proteins and the Ca2+ response they elicit provide a cell polarization cue in response to antigen will be a challenging goal for future studies.
Mast cells also respond to other chemotactic cues, such as the lipophilic ligand for G protein-coupled receptors, sphingosine-1-phosphate (Jolly et al., 2004; Olivera et al., 2007). We found that RBL cells chemotax toward this ligand with a robust response, but in this case, chemotaxis is not dependent on extracellular Ca2+ (J. Lee et al., submitted). The mechanism of signaling involved in this process remains to be determined.
RBL mast cells undergo robust phagocytosis of beads coated with ligands for IgE receptors, and, under these conditions, granule exocytosis is severely restricted (Pierini et al., 1996). The mechanism for this differential regulation is unclear, and recent experiments indicate that Ca2+ mobilization and PKC recruitment to the plasma membrane are not limiting under these conditions (K. Corwith, unpublished results). In macrophages, trafficking of recycling endsomes to the forming phagosome has been shown to contribute to this process (Cox et al., 2000), and we are interested in the potential role of this trafficking in mast cell phagocytosis and in the signaling consequences. Recent studies have revealed a role for recycling endosomal trafficking in the maintenance of the parasitic vacuole that is formed by invading Toxoplasma gondii in mast cells, as well as disruption of normal FcεRI signaling by this process (N. Smith et al., in preparation). Together, these studies reveal dynamic alterations of IgE receptor signaling by phagocytosis or parasite invasion, but the molecular mechanisms of these alterations remain to be characterized.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Andrade, M. V., Iwaki, S., Ropert, C., Gazzinelli, R. T., Cunha-Melo, J. R., and Beaven, M. A. (2011). Amplification of cytokine production through synergistic activation of NFAT and AP-1 following stimulation of mast cells with antigen and IL-33. Eur. J. Immunol. 41, 760–772.
Arrieumerlou, C., and Meyer, T. (2005). A local coupling model and compass parameter for eukaryotic chemotaxis. Dev. Cell 8, 215–227.
Baba, Y., Nishida, K., Fujii, Y., Hirano, T., Hikida, M., and Kurosaki, T. (2008). Essential function for the calcium sensor STIM1 in mast cell activation and anaphylactic responses. Nat. Immunol. 9, 81–88.
Bai, J., Tucker, W. C., and Chapman, E. R. (2004). PIP2 increases the speed of response of synaptotagmin and steers its membrane-penetration activity toward the plasma membrane. Nat. Struct. Mol. Biol. 11, 36–44.
Baumgartner, R. A., Deramo, V. A., and Beaven, M. A. (1996). Constitutive and inducible mechanisms for synthesis and release of cytokines in immune cell lines. J. Immunol. 157, 4087–4093.
Baumgartner, R. A., Yamada, K., Deramo, V. A., and Beaven, M. A. (1994). Secretion of TNF from a rat mast cell line is a brefeldin A-sensitive and a calcium/protein kinase C-regulated process. J. Immunol. 153, 2609–2617.
Beaven, M. A. (2009). Our perception of the mast cell from Paul Erlich until now. Eur. J. Immunol. 39, 11–25.
Blum, L. K., Thrasher, S. M., Gagliardo, L. F., Fabre, V., and Appleton, J. A. (2009). Expulsion of secondary Trichinella spiralis infection in rats occurs independently of mucosal mast cell release of mast cell protease II. J. Immunol. 183, 5816–5822.
Brown, M. A., Sayed, B. A., and Christy, A. (2008). Mast cells and the adaptive immune response. J. Clin. Immunol. 28, 671–676.
Calloway, N., Holowka, D., and Baird, B. (2010). A basic sequence in STIM1 promotes Ca2+ influx by interacting with the C-terminal acidic coiled coil of Orai1. Biochemistry 49, 1067–1071.
Calloway, N., Owens, T., Corwith, K., Rodgers, W., Holowka, D., and Baird, B. (2011). Stimulated association of STIM1 and Orai1 is regulated by the balance of PtdIns(4,5)P2 between distinct membrane pools. J. Cell. Sci. 124, 2602–2610.
Calloway, N., Vig, M., Kinet, J. P., Holowka, D., and Baird, B. (2009). Molecular clustering of STIM1 with Orai1/CRACM1 at the plasma membrane depends dynamically on depletion of Ca2+ stores and on electrostatic interactions. Mol. Biol. Cell 20, 389–399.
Chapman, E. R. (2008). How does synaptotagmin triggers neurotransmitter release? Annu. Rev. Biochem. 77, 615–641.
Cohen, R., Corwith, K., Holowka, D., and Baird, B. (2012). Spatiotemporal resolution of mast cell granule exocytosis reveals correlation with Ca2+ wave initiation. J. Cell. Sci. (in press).
Cohen, R., Torres, A., Ma, H. T., Holowka, D., and Baird, B. (2009). Ca2+ waves initiate antigen-stimulated Ca2+ responses in mast cells. J. Immunol. 183, 6478–6488.
Cox, D., Lee, D. J., Dale, B. M., Calafat, J., and Greenberg, S. (2000). A Rab11-containing rapidly recycling compartment in macrophages that promotes phagocytosis. Proc. Natl. Acad. Sci. U.S.A. 97, 680–685.
Di Capite, J. L., Bates, G. J., and Parekh, A. B. (2011). Mast cell CRAC channel as a novel therapeutic target in allergy. Curr. Opin. Allergy Clin. Immunol. 11, 33–38.
Echtenacher, B., Mannel, D. N., and Hultner, L. (1996). Critical protective role of mast cells in a model of acute septic peritonitis. Nature 381, 75–77.
Feske, S., Gwack, Y., Prakriya, M., Srikanth, S., Puppel, S. H., Tanasa, B., Hogan, P. G., Lewis, R. S., Daly, M., and Rao, A. (2006). A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 441, 179–185.
Foreman, J. C., Mongar, J. L., and Gomperts, B. D. (1973). Calcium ionophores and movement of calcium ions following the physiological stimulus to a secretory process. Nature 245, 249–251.
Friend, D. S., Ghildyal, N., Austen, K. F., Gurish, M. F., Matsumoto, R., and Stevens, R. L. (1996). Mast cells that reside at different locations in the jejunum of mice infected with Trichinella spiralis exhibit sequential changes in their granule ultrastructure and chymase phenotype. J. Cell Biol. 135, 279–290.
Gadi, D. (2012). Interplay of Protein Kinase C, the Marcks Protein, and Phosphoinositides in Regulating Mast Cell Signaling. Ph.D. thesis, Cornell University, Ithaca, NY.
Gadi, D., Wagenknecht-Wiesner, A., Holowka, D., and Baird, B. (2011). Sequestration of phosphoinositides by mutated marcks effector domain inhibits stimulated Ca2+ mobilization and degranulation in mast cells. Mol. Biol. Cell 22, 4908–4917.
Gambhir, A., Hangyas-Mihalyne, G., Zaitseva, I., Cafiso, D. S., Wang, J., Murray, D., Pentyala, S. N., Smith, S. O., and McLaughlin, S. (2004). Electrostatic sequestration of PIP2 on phospholipid membranes by basic/aromatic regions of proteins. Biophys. J. 86, 2188–2207.
Gilfillan, A. M., and Beaven, M. A. (2011). Regulation of mast cell responses in health and disease. Crit. Rev. Immunol. 31, 475–530.
Gilfillan, A. M., and Rivera, J. (2009). The tyrosine kinase network regulating mast cell activation. Immunol. Rev. 228, 149–169.
Gordon, J. R., and Galli, S. J. (1991). Release of both preformed and newly synthesized tumor necrosis factor alpha (TNF-alpha)/cachectin by mouse mast cells stimulated via the Fc epsilon RI. A mechanism for the sustained action of mast cell-derived TNF-alpha during IgE-dependent biological responses. J. Exp. Med. 174, 103–107.
Gupton, S. L., and Gertler, F. B. (2007). Filopodia: the fingers that do the walking. Sci. STKE 2007, re5.
Heo, W. D., Inoue, T., Park, W. S., Kim, M. L., Park, B. O., Wandless, T. J., and Meyer, T. (2006). PI(3,4,5)P3 and PI(4,5)P2 lipids target proteins with polybasic clusters to the plasma membrane. Science 314, 1458–1461.
Hirasawa, N., Santini, F., and Beaven, M. A. (1995). Activation of the mitogen-activated protein kinase/cytosolic phospholipase A2 pathway in a rat mast cell line. Indications of different pathways for release of arachidonic acid and secretory granules. J. Immunol. 154, 5391–5402.
Hoth, M., and Penner, R. (1992). Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature 355, 353–356.
Huang, C. L. (2007). Complex roles of PIP2 in the regulation of ion channels and transporters. Am. J. Physiol. Renal Physiol. 293, F1761–F1765.
James, D. J., Khodthong, C., Kowalchyk, J. A., and Martin, T. F. (2010). Phosphatidylinositol 4,5-bisphosphate regulation of SNARE function in membrane fusion mediated by CAPS. Adv. Enzyme Regul. 50, 62–70.
Jarvis, S. E., and Zamponi, G. W. (2007). Trafficking and regulation of neuronal voltage-gated calcium channels. Curr. Opin. Cell Biol. 19, 474–482.
Jolly, P. S., Bektas, M., Olivera, A., Gonzalez-Espinosa, C., Proia, R. L., Rivera, J., Milstien, S., and Spiegel, S. (2004). Transactivation of sphingosine-1-phosphate receptors by FcepsilonRI triggering is required for normal mast cell degranulation and chemotaxis. J. Exp. Med. 199, 959–970.
Kanno, T., Cochrane, D. E., and Douglas, W. W. (1973). Exocytosis (secretory granule extrusion) induced by injection of calcium into mast cells. Can. J. Physiol. Pharmacol. 51, 1001–1004.
Kawasaki, T., Lange, I., and Feske, S. (2009). A minimal regulatory domain in the C terminus of STIM1 binds to and activates ORAI1 CRAC channels. Biochem. Biophys. Res. Commun. 385, 49–54.
Kay, J. G., Murray, R. Z., Pagan, J. K., and Stow, J. L. (2006). Cytokine secretion via cholesterol-rich lipid raft-associated SNAREs at the phagocytic cup. J. Biol. Chem. 281, 11949–11954.
Kim, T. D., Eddlestone, G. T., Mahmoud, S. F., Kuchtey, J., and Fewtrell, C. (1997). Correlating Ca2+ responses and secretion in individual RBL-2H3 mucosal mast cells. J. Biol. Chem. 272, 31225–31229.
Kitaura, J., Kinoshita, T., Matsumoto, M., Chung, S., Kawakami, Y., Leitges, M., Wu, D., Lowell, C. A., and Kawakami, T. (2005). IgE- and IgE+ Ag-mediated mast cell migration in an autocrine/paracrine fashion. Blood 105, 3222–3229.
Korzeniowski, M. K., Manjarres, I. M., Varnai, P., and Balla, T. (2010). Activation of STIM1-Orai1 involves an intramolecular switching mechanism. Sci. Signal. 3, ra82.
Korzeniowski, M. K., Popovic, M. A., Szentpetery, Z., Varnai, P., Stojilkovic, S. S., and Balla, T. (2009). Dependence of STIM1/Orai1-mediated calcium entry on plasma membrane phosphoinositides. J. Biol. Chem. 284, 21027–21035.
Kraft, S., and Kinet, J. P. (2007). New developments in FcepsilonRI regulation, function and inhibition. Nat. Rev. Immunol. 7, 365–378.
Lewis, R. S., and Cahalan, M. D. (1989). Mitogen-induced oscillations of cytosolic Ca2+ and transmembrane Ca2+ current in human leukemic T cells. Cell Regul. 1, 99–112.
Lingwood, D., and Simons, K. (2010). Lipid rafts as a membrane organizing principle. Science 327, 46–50.
Liou, J., Fivaz, M., Inoue, T., and Meyer, T. (2007). Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc. Natl. Acad. Sci. U.S.A. 104, 9301–9306.
Liou, J., Kim, M. L., Heo, W. D., Jones, J. T., Myers, J. W., Ferrell, J. E. Jr., and Meyer, T. (2005). STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 15, 1235–1241.
Ma, H. T., and Beaven, M. A. (2009). Regulation of Ca2+ signaling with particular focus on mast cells. Crit. Rev. Immunol. 29, 155–186.
Ma, H. T., Peng, Z., Hiragun, T., Iwaki, S., Gilfillan, A. M., and Beaven, M. A. (2008). Canonical transient receptor potential 5 channel in conjunction with Orai1 and STIM1 allows Sr2+ entry, optimal influx of Ca2+, and degranulation in a rat mast cell line. J. Immunol. 180, 2233–2239.
Madden, K. B., Urban, J. F. Jr., Ziltener, H. J., Schrader, J. W., Finkelman, F. D., and Katona, I. M. (1991). Antibodies to IL-3 and IL-4 suppress helminth-induced intestinal mastocytosis. J. Immunol. 147, 1387–1391.
McLaughlin, S., and Murray, D. (2005). Plasma membrane phosphoinositide organization by protein electrostatics. Nature 438, 605–611.
Metz, M., Grimbaldeston, M. A., Nakae, S., Piliponsky, A. M., Tsai, M., and Galli, S. J. (2007). Mast cells in the promotion and limitation of chronic inflammation. Immunol. Rev. 217, 304–328.
Metzger, H. (1978). The IgE-mast cell system as a paradigm for the study of antibody mechanisms. Immunol. Rev. 41, 186–199.
Metzger, H., Alcaraz, G., Hohman, R., Kinet, J. P., Pribluda, V., and Quarto, R. (1986). The receptor with high affinity for immunoglobulin E. Annu. Rev. Immunol. 4, 419–470.
Mitchison, T. J., and Cramer, L. P. (1996). Actin-based cell motility and cell locomotion. Cell 84, 371–379.
Muik, M., Fahrner, M., Derler, I., Schindl, I., Bergsmann, J., Frischauf, J., Groschner, K., and Romanin, C. (2009). A cytosolic homomerization and a modulatory domain within STIM1 C terminus determine coupling to ORAI1 channels. J. Biol. Chem. 284, 8421–8426.
Muik, M., Fahrner, M., Schindl, R., Stathopulos, P., Frischauf, I., Derler, I., Plenk, P., Lackner, B., Groschner, K., Ikura, M., and Romanin, C. (2011). STIM1 couples to ORAI1 via an intramolecular transition into an extended conformation. EMBO J. 30, 1678–1689.
Mullins, F. M., Park, C. Y., Dolmetsch, R. E., and Lewis, R. S. (2009). STIM1 and calmodulin interact with Orai1 to induce Ca2+-dependent inactivation of CRAC channels. Proc. Natl. Acad. Sci. U.S.A. 106, 15495–15500.
Murray, R. Z., Kay, J. G., Sangermani, D. G., and Stow, J. L. (2005). A role for the phagosome in cytokine secretion. Science 310, 1492–1495.
Naal, R. M., Holowka, E. P., Baird, B., and Holowka, D. (2003). Antigen-stimulated trafficking from the recycling compartment to the plasma membrane in RBL mast cells. Traffic 4, 190–200.
Nechushtan, H., Leitges, M., Cohen, C., Kay, G., and Razin, E. (2000). Inhibition of degranulation and interleukin-6 production in mast cells derived from mice deficient in protein kinase Cbeta. Blood 95, 1752–1757.
Olivera, A., Mizugishi, K., Tikhonova, A., Ciaccia, L., Odom, S., Proia, R. L., and Rivera, J. (2007). The sphingosine kinase-sphingosine-1-phosphate axis is a determinant of mast cell function and anaphylaxis. Immunity 26, 287–297.
Olivera, A., Urtz, N., Mizugishi, K., Yamashita, Y., Gilfillan, A. M., Furumoto, Y., Gu, H., Proia, R. L., Baumruker, T., and Rivera, J. (2006). IgE-dependent activation of sphingosine kinases 1 and 2 and secretion of sphingosine 1-phosphate requires Fyn kinase and contributes to mast cell responses. J. Biol. Chem. 281, 2515–2525.
Orida, N., Feldman, J. D., Katz, D. H., and Liu, F. T. (1983). IgE-mediated chemotaxis of rat basophilic leukemia cells towards specific antigen. J. Exp. Med. 157, 2166–2171.
Parekh, A. B., Fleig, A., and Penner, R. (1997). The store-operated calcium current Icrac: nonlinear activation by InsP3 and dissociation from calcium release. Cell 89, 973–980.
Park, C. Y., Hoover, P. J., Mullins, F. M., Bachhawat, P., Covington, E. D., Raunser, S., Walz, T., Garcia, K. C., Dolmetsch, R. E., and Lewis, R. S. (2009). STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 136, 876–890.
Petrie, R. J., Doyle, A. D., and Yamada, K. M. (2009). Random versus directionally persistent cell migration. Nat. Rev. Mol. Cell Biol. 10, 538–549.
Pierini, L., Holowka, D., and Baird, B. (1996). Fc epsilon RI-mediated association of 6-micron beads with RBL-2H3 mast cells results in exclusion of signaling proteins from the forming phagosome and abrogation of normal downstream signaling. J. Cell Biol. 134, 1427–1439.
Rivera, J., and Beaven, M. A. (1997). “Regulation of secretion from secretory cells by protein kinase C,” in Protein Kinase C, eds P. J. Parker, and D. L. Dekker (Austin, TX: RG Landes), 131–164.
Siraganian, R. P., Zhang, J., Suzuki, K., and Sada, K. (2002). Protein tyrosine kinase Syk in mast cell signaling. Mol. Immunol. 38, 1229–1233.
Smith, N. L., Hammond, S., Gadi, D., Wagenknecht-Wiesner, A., Baird, B., and Holowka, D. (2010). Sphingosine derivatives inhibit cell signaling by electrostatically neutralizing polyphosphoinositides at the plasma membrane. Self Nonself 1, 133–143.
Stumpo, D. J., Bock, C. B., Tuttle, J. S., and Blackshear, P. J. (1995). MARCKS deficiency in mice leads to abnormal brain development and perinatal death. Proc. Natl. Acad. Sci. U.S.A. 92, 944–948.
Takemura, H., Hughes, A. R., Thastrup, O., and Putney, J. W. Jr. (1989). Activation of calcium entry by the tumor promoter thapsigargin in parotid acinar cells. Evidence that an intracellular calcium pool and not an inositol phosphate regulates calcium fluxes at the plasma membrane. J. Biol. Chem. 264, 12266–12271.
Trifaro, J. M., Gasman, S., and Gutierrez, L. M. (2008). Cytoskeletal control of vesicle transport and exocytosis in chromaffin cells. Acta Physiol. (Oxf.) 192, 165–172.
Vasudevan, L., Jeromin, A., Volpicelli-Daley, L., De Camilli, P., Holowka, D., and Baird, B. (2009). The β- and γ-isoforms of type I PIP5K regulate distinct stages of Ca2+ signaling in mast cells. J. Cell Sci. 122, 2567–2574.
Vig, M., DeHaven, W. I., Bird, G. S., Billingsley, J. M., Wang, H., Rao, P. E., Hutchings, A. B., Jouvin, M. H., Putney, J. W., and Kinet, J. P. (2008). Defective mast cell effector functions in mice lacking the CRACM1 pore subunit of store-operated calcium release-activated calcium channels. Nat. Immunol. 9, 89–96.
Vig, M., Peinelt, C., Beck, A., Koomoa, D. L., Rabah, D., Koblan-Huberson, M., Kraft, S., Turner, H., Fleig, A., Penner, R., and Kinet, J. P. (2006). CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 312, 1220–1223.
Walsh, C. M., Chvanov, M., Haynes, L. P., Petersen, O. H., Tepikin, A. V., and Burgoyne, R. D. (2010). Role of phosphoinositides in STIM1 dynamics and store-operated calcium entry. Biochem. J. 425, 159–168.
Wang, Y., Deng, X., and Gill, D. L. (2010). Calcium signaling by STIM and Orai: intimate coupling details revealed. Sci. Signal. 3, pe42.
Wolfe, P. C., Chang, E. Y., Rivera, J., and Fewtrell, C. (1996). Differential effects of the protein kinase C activator phorbol 12-myristate 13-acetate on calcium responses and secretion in adherent and suspended RBL-2H3 mucosal mast cells. J. Biol. Chem. 271, 6658–6665.
Wu, M., Baumgart, T., Hammond, S., Holowka, D., and Baird, B. (2007). Differential targeting of secretory lysosomes and recycling endosomes in mast cells revealed by patterned antigen arrays. J. Cell. Sci. 120, 3147–3154.
Wu, M. M., Buchanan, J., Luik, R. M., and Lewis, R. S. (2006). Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J. Cell Biol. 174, 803–813.
Yuan, J. P., Zeng, W., Dorwart, M. R., Choi, Y. J., Worley, P. F., and Muallem, S. (2009). SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat. Cell Biol. 11, 337–343.
Zhang, S. L., Yu, Y., Roos, J., Kozak, J. A., Deerinck, T. J., Ellisman, M. H., Stauderman, K. A., and Cahalan, M. D. (2005). STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 437, 902–905.
Keywords: IgE receptors (FcεRI), store-operated Ca2+ entry, secretory lysosomes, phosphoinositides, chemotaxis
Citation: Holowka D, Calloway N, Cohen R, Gadi D, Lee J, Smith NL and Baird B (2012) Roles for Ca2+ mobilization and its regulation in mast cell functions. Front. Immun. 3:104. doi: 10.3389/fimmu.2012.00104
Received: 08 December 2011; Accepted: 16 April 2012;
Published online: 08 May 2012.
Edited by:
Toshiaki Kawakami, La Jolla Institute for Allergy and Immunology, USAReviewed by:
Alasdair Gilfillan, National Institutes of Health, USAJohn Ryan, Virginia Commonwealth University, USA
Anant Parekh, Oxford University, UK
Copyright: © 2012 Holowka, Calloway, Cohen, Gadi, Lee, Smith and Baird. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: David Holowka, Baker Laboratory, Department of Chemistry and Chemical Biology, Cornell University, Ithaca, NY 14853-1301, USA. e-mail:ZGFoMjRAY29ybmVsbC5lZHU=